
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of enol phosphates directly from ketones via a modified one-pot Perkow reaction†
Huichuang Guo,
Yulong Zhang,
Zhenya Li,
Peichao Zhao,
Ning Li and
Enxue Shi *
*
State Key Laboratory of NBC Protection for Civilian, Beijing 102205, P. R. China. E-mail: exshi@sina.com
First published on 17th May 2022
Abstract
A modified Perkow reaction, named Perkow-Shi reaction, was developed based on the one-pot α-tosyloxylation of ketones following by addition of P(III)-reagents and 4 Å molecular sieves. Diversity of enol phosphates, as well as enol phosphonates, enol phosphinates, and enol phosphoramidates were synthesized in high yields directly from the ubiquitously available ketones instead of the unfavourable α-chloroketones under a mild and environmental friendly condition.
Phosphoenols (P(O)–OC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) are the key structural motif of many agrochemicals and bioactive natural products (Fig. 1). For example, more than ten kinds of organophosphorus pesticides such as dichlorvos (DDVP), mevinphos and monocrotophos are typical enol phosphates (EPs).1 The endogenetic phosphoenolpyruvate (PEP) plays a vital role in transformation of adenosine diphosphate (ADP) into adenosine triphosphate (ATP) in living organisms;2 the naturally occurring cyclophostin,3 cyclipostins4 and salinipostins,5 characterized by a unique bicyclic core with a unusual seven-membered cyclic phosphoenol triester, are merging as a novel class of multi-target promising therapeutic candidates with diversity of biological properties; phosphaisocoumarins, a phosphorus analogue of isocoumarins, have displayed versatile and interesting biological activities.6
C) are the key structural motif of many agrochemicals and bioactive natural products (Fig. 1). For example, more than ten kinds of organophosphorus pesticides such as dichlorvos (DDVP), mevinphos and monocrotophos are typical enol phosphates (EPs).1 The endogenetic phosphoenolpyruvate (PEP) plays a vital role in transformation of adenosine diphosphate (ADP) into adenosine triphosphate (ATP) in living organisms;2 the naturally occurring cyclophostin,3 cyclipostins4 and salinipostins,5 characterized by a unique bicyclic core with a unusual seven-membered cyclic phosphoenol triester, are merging as a novel class of multi-target promising therapeutic candidates with diversity of biological properties; phosphaisocoumarins, a phosphorus analogue of isocoumarins, have displayed versatile and interesting biological activities.6
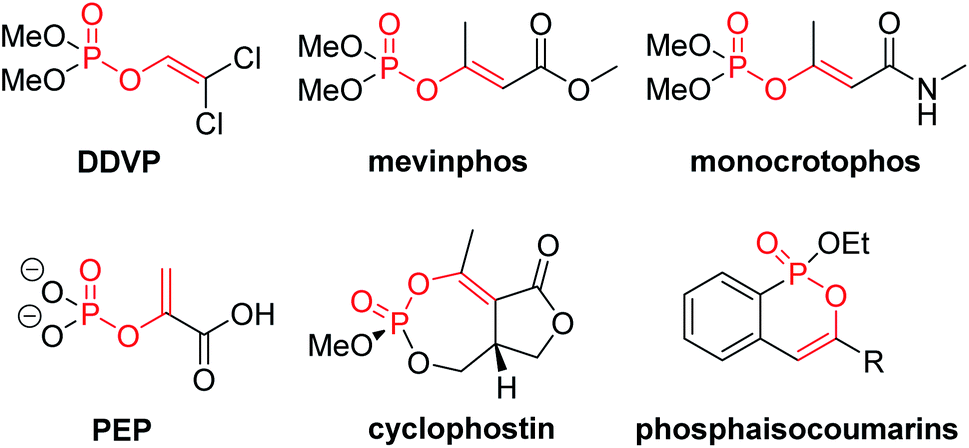 | ||
| Fig. 1 Selected examples of enol phosphates. | ||
Furthermore, as a kind of outstanding vinyl electrophiles and nucleophiles possessing high stability and accessibility, functionalized EPs are of great importance due to their versatile synthetic applications as EPs mainly have three active centres: the electrophilic phosphoryl group and enol oxygen-attached carbon atom, and the electrophilic carbon–carbon double bond, which makes them capable of participating in diverse type of phosphorylation and transition metal catalyzed cross-coupling reaction as well as cycloadditions leading to new P–O, C–O, C–H, C–C, C–X bonds formation (Fig. 2).
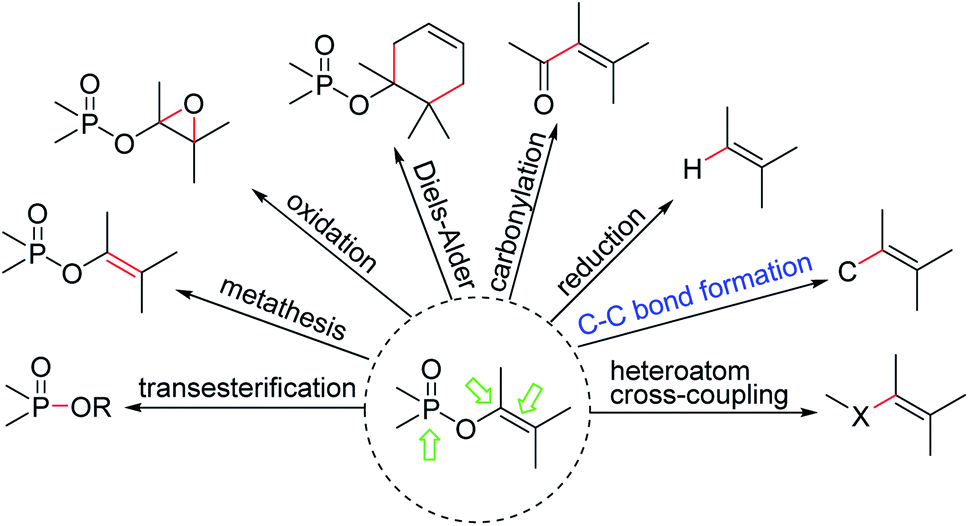 | ||
| Fig. 2 Representative reactions involving enol phosphates. | ||
In particular, the transesterification and acidolysis of EPs with cleavage of the enol linkage affording tris-substituted phosphates, mixed anhydrides namely acyl phosphates and pyrophosphates;7 and intramolecular olefin metathesis of terminal EPs affording new type of cyclic phosphoenols;8 and catalytic epoxidation of EPs with NaOCl affording phosphonyl epoxides;9 and Diels–Alder reaction of EPs with cyclodiene affording the corresponding adducts;10 and palladium catalyzed carbonylation of EPs with CO affording a range of enoates;11 and reduction and alkylation of EPs with a variety of hydride transfer reagents affording alkenes widely exploited in total synthesis;12 and Suzuki–Miyaura, Stille, Negishi, Heck, and related cross-coupling reactions of EPs affording more highly substituted alkenes;13 and also substitution by simple heteroatom nucleophiles affording the formation of alkenes with C-heteroatom (C–O, C–N, C–S, C–B, C–Sn, C–S) linkages,14 have attracted a lot of attentions as these functional groups are fundamental components of a large variety of natural products, versatile precursors, and important building blocks.
To date, numerous methods to synthesize EPs have been reported which can be broadly categorized as follows: (1) the earliest recorded and still extensively used Perkow reaction between α-haloketones and phosphites;15 (2) the phosphorylation of in situ generated enolate of ketones by various kinds of metalation process;16 (3) other approaches such as hydrophosphoryloxylation of alkynes,17 tandem reaction of α-aryloxyacetophenones,18 and so on.19 However, whilst the classical Perkow reaction is much limited by the availability of the toxic and lachrymatory α-haloketones and the lack of effective regio- and stereocontrol, the non-Perkow methods always suffer from harsh reaction conditions including use of strong bases (mostly metallic) and unstable phosphorochloridates some of which are generally inconvenient to prepare such as P–C and/or P–N containing materials. Therefore, a more environmentally benign synthetic route to EPs is still much desirable.
On the other hand, with respect to the known Perkow reaction mechanism,20 we envisioned that substrates of ketones with a proper α-positioned leaving group (LG) instead of the unfavourable halogen should also probably be feasible of proceeding Perkow reaction. Nevertheless, to our best knowledge, only two attempts have been reported: one from Paleta that when treating the 3-trifluoroacyloxy quinolinedione with P(OEt)3 in refluxing toluene for 7 h, a Perkow product was obtained in 36% yield;21a the other from Denney that when stirring α-keto p-toluenesulfonates with phosphites in refluxing ether for several days, a mixture of unidentified vinylphosphates and methylphosphonates was obtained.21b Herein we wish to report a modified one-pot Perkow reaction based on the α-tosyloxylation of ketones following by addition of P(III)-reagents and 4 Å molecular sieves (Scheme 1).
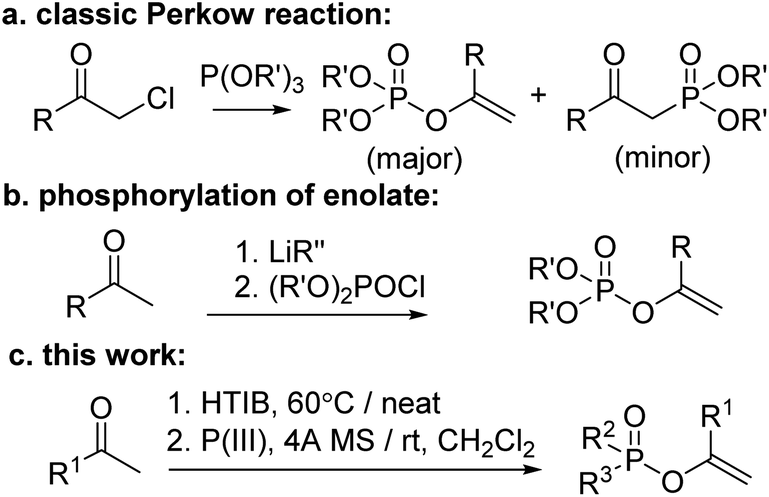 | ||
| Scheme 1 Synthesis of enol phosphates. | ||
We initially selected (EtO)3P and α-functionalized acetones as the test reaction. Solvent-free condition was first focused in view of its high regioselectivity.15b As shown in Table 1, among the eight kinds of LGs examined, α-tosyloxylated acetone afforded the best result that when carried out at room temperature for 3 h, it did proceed to give the desired Perkow product 3ba in 95% yield (Table 1, entry 5). Contrary to those completely unreactive α-carboxylated (Table 1, entries 1 and 2) and α-phosphoroxylated acetones (Table 1, entry 8) even with higher temperature and prolonged reaction time, all the α-sulfoxylated acetones (Table 1, entries 3–7) gave acceptable conversion after suitable reaction time respectively. To be mentioned, compared with the reported typical α-tosyloxylation at 60 °C conducted in solvent such as MeCN, THF, and DCM, solvent-free reaction between CH3COCH2OTs and HTIB was found to be much more effective by remarkably shortening the reaction time from about 12 h to 3 h at 60 °C or even less than 1 h for those aromatic ketones.

In light of the significant progress in α-tosyloxylation of ketones especially with those iodine(III)-mediated reagents such as the most popular hydroxy(tosyloxy)-iodobenzene (HTIB),22 the readily accessible and economic tosylate was thus chosen as the prior α-LG for the next investigation.
Inspired by the above achievements, we then turned into the possibility of the one-pot manipulation. Considering the convenience of monitoring, we then carried out the next experiments using aromatic acetophenone with HTIB following by the addition of (EtO)3P. As shown in Table 2, after α-tosyloxylation with subsequent Perkow reaction at 40 °C, only 24% yield of product 3aa was achieved whereas two side-products (EtO)2PHO (63%) and (EtO)3PO (13%), were observed by LC-MS and NMR analysis in the crude reaction mixtures (Table 2, entry 1). Encouragingly, when adding a small amount of solvent to acquire sufficient stirring, a much better result in DCM was afforded giving 49% yield of product 3aa (Table 2, entry 4).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | T (°C) | 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HTIB:P(OEt)3 :HTIB:P(OEt)3 |
Additive | Conversionb (%) | ||
| 3aa | (EtO)2PHO | (EtO)3PO | |||||
| a Reaction conditions: HTIB (1 mmol).b Determined by LC-MS and 31P NMR after 2 h reaction.c React for 5 h. | |||||||
| 1 | — | 40 | 1:1:1 |
— | 24 | 63 | 13 |
| 2 | MeCN | 40 | 1:1:1 |
— | 25 | 70 | 5 |
| 3 | THF | 40 | 1:1:1 |
— | 0 | 87 | 13 |
| 4 | DCM | 40 | 1:1:1 |
— | 49 | 43 | 8 |
| 5 | EA | 40 | 1:1:1 |
— | 42 | 45 | 13 |
| 6 | DCM | 40 | 1:1:1 |
4 Å MS | 84 | 7 | 9 |
| 7 | DCM | 40 | 1.1:1:1 |
4 Å MS | 95 | 3 | 2 |
| 8 | DCM | 25 | 1.1:1:1 |
4 Å MS | 99 | 0 | 1 |
| 9c | DCM | 0 | 1.1:1:1 |
4 Å MS | 97 | 2 | 1 |

Next, we take DCM as the solvent for optimization. According to the reaction mechanism, (EtO)2PHO was supposed to be the hydrolysis product of (EtO)3P by H2O formed in the reaction system, whereas (EtO)3PO should be the oxidation product of (EtO)3P by the reactive hypervalent iodine reagents HTIB, which was verified by our following modifications experiments. As presumed, when adding equimolecular water-capturing 4 Å molecular sieves (MS) powder, the majority of (EtO)2PHO were avoided of generation (Table 2, entry 6). And when slightly reducing the HTIB loading at the same time, both (EtO)2PHO and (EtO)3PO were then mostly suppressed as expected (Table 2, entry 7). Very pleasingly, the reaction conducted at room temperature was found efficient enough to give nearly quantitative conversion of phosphite into product (Table 2, entry 8). Lower temperature at 0 °C would lead to longer reaction time for full conversion and then result more by-product formation (Table 2, entry 9). Therefore, a satisfactory yield of target EP product could be achieved by an one-pot Perkow procedure as following: ketone (1.1 eq.) and HTIB (1 eq.) react 1 h at 60 °C neatly, then added at room temperature 4 Å MS (1 eq.) and phosphite (1 eq.) in DCM, and further react about 2 h.
With the optimized conditions in hand, we then evaluated the scope and limitations of this modified Perkow reaction for structure-diversity EPs. As for the ketone substrates without α-branched groups such as aromatic acetophenone and aliphatic acetone, all the common acyclic P(III) reagents possessing P–O, P–C, and/or P–N bonds as well as those cyclic ones gave the corresponding EPs in high yields. Various types of aromatic ketones with electron-donating or electron-withdrawing substituted groups especially those easily available hetero-aromatic ketones whose α-chloro species are generally commercially inaccessible, were all applied well to the present enolphosphorylation. In other words, our one-pot Perkow reaction methodology exhibited extreme tolerance in preparation of diversely structural enol phosphates, enol phosphonates, enol phosphinates, and enol phosphoramidates (Table 3).
|
|---|
| a Reaction conditions: HTIB (1 mmol), 4 (1.1 mmol), P(III) (1 mmol). Isolated yield. |
 |

However, the reaction showed no preference of E/Z geometry in EP products. None of the conditions altering temperature, solvent and reaction time were found effective in controlling the stereochemistry and in most cases only mixtures of the E- and Z-isomers in nearly 1:1 ratio were obtained with a slightly decreased yields mainly due to the relatively low reactive α-tosyloxylated secondary carbons (Table 4).
|
|---|
| a Reaction conditions: HTIB (1 mmol), 5 (1.1 mmol), P(OEt)3 (1 mmol). Isolated yield. |
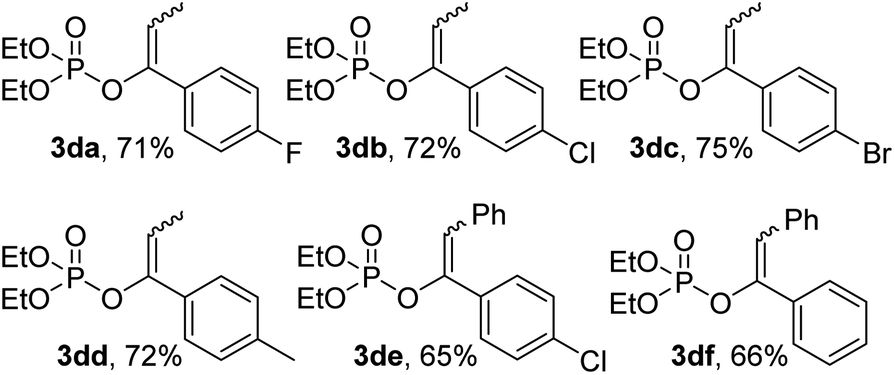 |

To be further mentioned, for those asymmetric aliphatic ketones with two possible reactive sites, for example, butan-2-one, pentan-2-one and heptan-2-one, only mixed products were obtained with very low yields (about 10–20%). Moreover, the α-tosyloxylation was found to mainly occurred at the more substituted carbons of the asymmetric ketones, which is consistent with literature results.23
A plausible mechanism is shown in Scheme 2. Similar to the classical Perkow reaction, it should proceed through a pathway that the nucleophilic P(III) atom of phosphite attacks the carbonyl C atom of in situ generated α-tosyloxylated ketone forming a triatomic heterocyclic intermediate which followed by the ring cleavage transforms into a phosphonium salt and then by the O-dealkylation gives the EP product. According to the nucleofugality model by Jaramillo,24 the ω index of TsO (1.63 eV) is about two times of Cl (0.77 eV), suggesting TsO could be even more reactive than Br (0.90 eV) and I (1.34 eV) α-substituted ketones, both of which however have been proven to be less regioselective for Perkow reaction vs. Arbuzov reaction. It demonstrates that Perkow reaction may not just follow in agreement with the order of departure ability of the α-LGs from the substrate, which deserves further investigation.
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
In summary, we have developed a concise method for the synthesis of enol phosphates, as well as enol phosphonates, enol phosphinates, and enol phosphoramidates based on an modified Perkow reaction, herein named Perkow–Shi reaction, which employed the ubiquitously commercial ketones and the readily available α-tosyloxylation reagent HTIB by a kind of one-pot methodology. The E/Z-selectivity of the modified Perkow reaction may be achieved by the introduction of chirality on the α-tosyloxylated carbon, which are under investigation in our lab.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Fest and K. J. Schmidt, The chemistry of organophosphorus pesticides, Springer Science & Business Media, 2012 Search PubMed.
- (a) C. T. Walsh, T. E. Benson, D. H. Kim and W. J. Lees, Cell Chem. Biol., 1996, 3, 83–91 CAS; (b) P. M. Krizkova, S. Prechelmacher, A. Roller and F. Hammerschmidt, J. Org. Chem., 2017, 82, 10310–10318 CrossRef PubMed.
- (a) T. Kurokawa, K. Suzuki, T. Hayaoka, T. Nakagawa, T. Izawa, M. Kobayashi and N. Harada, J. Antibiot., 1993, 46, 1315–1318 CrossRef CAS; (b) R. K. Malla, S. Bandyopadhyay, C. D. Spilling, S. Dutta and C. M. Dupureur, Org. Lett., 2011, 13, 3094–3097 CrossRef CAS PubMed.
- L. Vertesy, B. Beck, M. Brönstrup, K. Ehrlich, M. Kurz, G. Müller, D. Schummer and G. J. Seibert, J. Antibiot., 2002, 55, 480–494 CrossRef CAS PubMed.
- C. J. Schulze, G. Navarro, D. Ebert, J. DeRisi and R. G. Linnington, J. Org. Chem., 2015, 80, 1312–1320 CrossRef CAS.
- (a) G. F. Ruda, P. E. Wong, V. P. Alibu, S. Norval, K. D. Read, M. P. Barrett and I. H. Gilbert, J. Med. Chem., 2010, 53, 6071–6078 CrossRef CAS PubMed; (b) B. Li, B. Zhou, H. Lu and L. Ma, Eur. J. Med. Chem., 2010, 45, 1955–1963 CrossRef CAS PubMed; (c) X. Li, D. Zhang, H. Pang, F. Shen, H. Fu, Y. Jiang and Y. Zhao, Org. Lett., 2005, 7, 4919–4922 CrossRef CAS PubMed.
- R. W. Lichtenthaler, Chem. Rev., 1961, 61, 607–649 CrossRef.
- A. Whitehead, J. D. Moore and P. R. Hanson, Tetrahedron Lett., 2003, 44, 4275–4277 CrossRef CAS.
- M. Koprowski, J. Łuczak and E. Krawczyk, Tetrahedron, 2006, 62, 12363–12374 CrossRef CAS.
- F. Kienzle and P. Rosen, Helv. Chim. Acta, 1979, 62, 442–447 CrossRef CAS.
- (a) M. Sasaki, S. Honda, T. Noguchi, H. Takakura and K. Tachibana, Synlett, 2000, 2000, 838–840 CrossRef; (b) L. Bartali, D. Scarpi, A. Guarna, C. Prandi and E. G. Occhiato, Synlett, 2009, 2009, 913–916 CrossRef; (c) E. G. Occhiato, D. Scarpi and C. Prandi, Heterocycles, 2010, 80, 697–724 CrossRef CAS; (d) O. Lagerlund, M. L. Mantel and M. Larhed, Tetrahedron, 2009, 65, 7646–7652 CrossRef CAS.
- (a) T. Calogeropoulou, G. B. Hammond and D. F. Wiemer, J. Org. Chem., 1987, 52, 4185–4190 CrossRef CAS; (b) K. C. Nicolaou, R. J. Aversa, J. Jin and F. Rivas, J. Am. Chem. Soc., 2010, 132, 6855–6861 CrossRef CAS.
- (a) T. M. Gøgsig, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2009, 11, 4886–4888 CrossRef; (b) K. C. Nicolaou, G. Shi, J. Gunzner, P. Gartner and Z. Yang, J. Am. Chem. Soc., 1997, 119, 5467–5468 CrossRef CAS; (c) J. Jiang, R. DeVita, G. Doss, M. Goulet and M. Wyvratt, J. Am. Chem. Soc., 1999, 121, 593–594 CrossRef CAS; (d) A. L. Hansen, J. P. Ebran, T. M. Gøgsig and T. Skrydstrup, Chem. Commun., 2006, 39, 4137–4139 RSC; (e) J. W. Coe, Org. Lett., 2000, 2, 4205–4208 CrossRef CAS.
- (a) K. C. Nicolaou, Z. Yang, G. Shi, J. L. Gunzner, K. A. Agrios and P. Gartner, Nature, 1998, 392, 264–269 CrossRef CAS PubMed; (b) F. Kang, Z. Sui and W. V. Murray, Eur. J. Org. Chem., 2009, 2009, 461–479 CrossRef; (c) J. E. Milne and P. J. Kocienski, Synthesis, 2003, 2003, 584–592 CrossRef; (d) E. G. Occhiato, F. Lo Galbo and A. Guarna, J. Org. Chem., 2005, 70, 7324–7330 CrossRef CAS; (e) Y. Okuda, M. Sato, K. Oshima and H. Nozaki, Tetrahedron Lett., 1983, 24, 2015–2018 CrossRef CAS.
- (a) W. Perkow, K. Ullericr and F. Meyer, Naturwissenschaften, 1952, 39, 353 CrossRef; (b) Y. Cao, Z. Gao, J. Li, X. Bi, L. Yuan, C. Pei, Y. Guo and E. Shi, RSC Adv., 2020, 10, 29493–29497 RSC.
- (a) H. Nakatsuji, Y. Ashida, H. Hori, Y. Sato, A. Honda, M. Taira and Y. Tanabe, Org. Biomol. Chem., 2015, 13, 8205–8210 RSC; (b) W. J. Kerr, D. M. Lindsay, V. K. Patel and M. Rajamanickam, Org. Biomol. Chem., 2015, 13, 10131–10135 RSC; (c) H. Li, Y. Zhu, D. Lu and Y. Gong, Org. Biomol. Chem., 2018, 16, 5907–5912 RSC.
- A. Dalie, W. Zhang, B. Pan and Y. Zhao, Eur. J. Org. Chem., 2021, 2021, 314–317 Search PubMed.
- R. Song, Y. Liu, J. Wu, Y. Xie, G. Deng, X. Yang, Y. Liu and J. Li, Synthesis, 2012, 44, 1119–1125 CrossRef CAS.
- P. H. Lee, S. Kim, A. Park and B. C. Chary, Angew. Chem., 2010, 122, 6958–6961 CrossRef.
- A. E. Ferao, J. Phys. Chem. A, 2017, 121, 6517–6522 CrossRef PubMed.
- (a) O. Paleta, K. Pomeisl and S. Kafka, Beilstein J. Org. Chem., 2005, 1, 17 Search PubMed; (b) D. B. Denney, N. Gershman and J. Giaci, J. Org. Chem., 1966, 31, 2833–2837 CrossRef CAS.
- (a) R. M. Moriarty, R. K. Vaid and G. F. Koser, Synlett, 1990, 1990, 365–383 CrossRef; (b) G. F. Koser, Aldrichimica Acta, 2001, 34, 89–102 CAS; (c) G. F. Koser, R. H. Wettach and C. S. Smith, J. Org. Chem., 1980, 45, 1543–1544 CrossRef CAS; (d) O. Prakash and S. Goyal, Synthesis, 1992, 1992, 629–630 CrossRef; (e) J. C. Lee and J. H. Choi, Synlett, 2001, 2001, 234–235 CrossRef; (f) G. F. Koser, A. G. Relenyi, A. N. Kalos, L. Rebrovic and R. H. Wettach, J. Org. Chem., 1982, 47, 2487–2489 CrossRef CAS.
- J. Hu, M. Zhu, Y. Xu and J. Yan, Synthesis, 2012, 44, 1226–1232 CrossRef CAS.
- P. Jaramillo, L. R. Domingo and P. Pérez, Chem. Phys. Lett., 2006, 420, 95–99 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02340g |
| This journal is © The Royal Society of Chemistry 2022 |