
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePossible neuroprotective effects of amide alkaloids from Bassia indica and Agathophora alopecuroides: in vitro and in silico investigations†
Ahmed Othman ab,
Ahmed M. Sayedce,
Yhiya Amend and
Kuniyoshi Shimizu*a
ab,
Ahmed M. Sayedce,
Yhiya Amend and
Kuniyoshi Shimizu*a
aDepartment of Agro-environmental Sciences, Graduate School of Bioresources and Bioenvironmental Sciences, Kyushu University, Fukuoka 819-0395, Japan
bDepartment of Pharmacognosy, Faculty of Pharmacy, Al-Azhar University, Cairo 11371, Egypt
cDepartment of Pharmacognosy, Faculty of Pharmacy, Nahda University, Beni-Suef 62513, Egypt
dDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
eDepartment of Pharmacognosy, Faculty of Pharmacy, Almaaqal University, Basra 61014, Iraq
First published on 1st July 2022
Abstract
In Alzheimer's disease (AD), the accumulation of amyloid-β plaques, overactivity of MAO-B, and phosphorylated tau protein in the central nervous system result in neuroinflammation and cognitive impairments. Therefore, the multi-targeting of these therapeutic targets has emerged as a promising strategy for the development of AD treatments. The current study reports the isolation and identification of seven amide alkaloids, namely, N-trans-feruloyl-3-methoxytyramine (1), N-trans-feruloyltyramine (2), S-(−)-N-trans-feruloylnormetanephrine (3), S-(−)-N-trans-feruloyloctopamine (4), R-(+)-N-trans-feruloyloctopamine (5), N-trans-caffeoyltyramine (6), and S-(−)-3-(4-hydroxy-3-methoxyphenyl)-N-[2-(4-hydroxyphenyl)-methoxyethyl]acrylamide (7), from B. indica and A. alopecuroides, which are halophytic plants that have been reported to contain diverse phytochemicals. Additionally, the study explores the potential inhibition effects of the isolates on β-secretase, monoamine oxidase enzymes, and phosphorylated tau protein, and their anti-aggregation effects on amyloid-β fibrils. Compounds 1, 2, and 7 showed potent inhibitory activity against BACE1, MAO-B, and phosphorylated tau protein, as well as anti-aggregation activity against Aβ-peptides. Additionally, compound 6 displayed promising inhibition activity against MAO-B enzyme. Further in-depth in silico and modeling analyses (i.e., docking, absolute binding free energy calculations, and molecular dynamics simulations) were carried out to reveal the binding mode of each active compound inside the corresponding enzyme (i.e., MAO-B and BACE1). The results indicate that B. indica, A. alopecuroides, and the isolated amide alkaloids might be useful in the development of lead compounds for the prevention of neurodegenerative diseases, especially AD.
Introduction
Alzheimer's disease (AD) is a complex neurodegenerative disorder of the central nervous system that is considered to be a socioeconomic burden and a great challenge affecting millions of people, and for which there is no efficient therapy.1 Current available drugs are able to increase neurotransmitters in the brain, leading to improvement in cognitive functions; however, their efficiency is not constant for all patients, and they have a limited duration of action.2 Considering the complexity of AD and the limitations of a single treatment option, there is an urgent need for the discovery of new treatment options to controlling this disease.3 According to recent reports, approximately 55 million people worldwide suffer from dementia with the development of nearly 10 million new cases every year, and this number is predicted to reach 152 million by 2050.4 Hyperphosphorylated tau-protein, β-secretase, monoamine oxidase enzymes, and the accumulation of abnormal amyloid-β (Aβ) aggregates are key factors in the pathogenesis of AD and neurotoxicity.1,5 Therefore, the modulation and inhibition of these therapeutic targets are strongly suggested as an effective strategy to treat AD. Recently, bioactive compounds derived from plants have shown promising therapeutic activity in neurodegenerative diseases, making their evaluation as a source of alternative medicine for AD an effective approach.6Halophytes are plants with substantial economic potential and are potential sources of medication.7 Several species of halophytes have been utilized in folk medicine for their ability to produce various bioactive compounds, such as terpenes, saponins, and alkaloids.7 Nevertheless, the chemical and biological investigation of various species has still barely been reported.
Bassia indica and Agathophora alopecuroides (Chenopodiaceae) are halophytic herbs with therapeutic purposes that are widely distributed in the Egyptian ecosystem.8–11 In previous reports, chemical investigation of Chenopodiaceae species has revealed the presence of nitrogenous compounds, flavonoids, and saponins.12–17 Although phenolics and other secondary metabolites previously reported in B. indica displayed anti-acetylcholinesterase activity, their therapeutic potential in the treatment of AD has not yet been elucidated.8 Additionally, the herb A. alopecuroides is rich in phenolics, which can provide neuroprotective activity in fighting AD; however, its chemical and biological investigation remains unexplored.18
As a continuation of our ongoing projects for the utilization of the halophytes widely distributed in the Egyptian ecosystem and their therapeutic potential in the management of neurodegenerative diseases, the current study was conducted with the aim of isolating some nitrogenous compounds from B. indica and A. alopecuroides to determine their capacity in the inhibition of some therapeutic targets correlated with the pathogenesis of AD, as shown in Fig. 1. As a result, seven amide alkaloids, namely, lignanamides, were isolated from the halophytic herbs B. indica and A. alopecuroides. Based on the literature, calculation of their drug-likeness was carried out, and the isolated compounds were suggested to be good drug-like candidates.
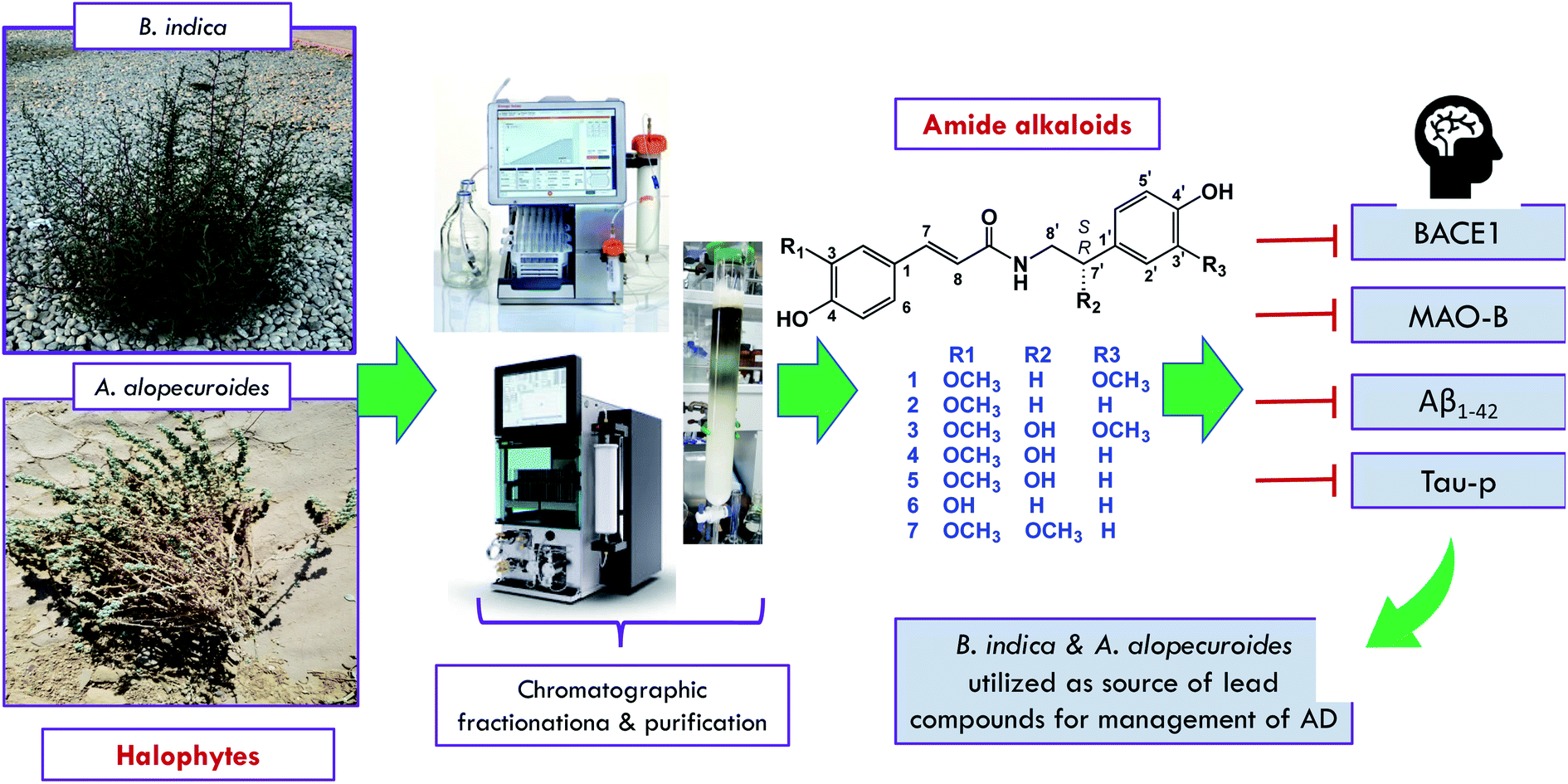 | ||
| Fig. 1 Outline of the present study. | ||
Materials and methods
Chemicals and standards
The human phospho-tau protein enzyme-linked immunosorbent assay kit used for screening phosphorylated tau (MBS013458) was obtained from MyBioSource, Inc. (San Diego, CA, USA). The capture and detection antibodies used in the MBS013458 kit were mouse monoclonal and rabbit polyclonal antibodies to tau proteins (phosphor S262). A BACE1 fluorescence human β-secretase (BACE1) inhibitor screening kit (K720-100), fluorescence β-amyloid1–42 ligand screening assay kit (K570-100), and inhibitor screening kit for monoamine oxidase B (K797-100) were purchased from BioVision, USA. Deionized water (18 MΩ cm) was obtained using a Milli-Q purification system from Millipore (Bedford, MA, USA). The n-hexane, dichloromethane, ethyl acetate, methanol, and acetonitrile used as solvents for extraction and purification were purchased from Wako Pure Chemical Industries (Osaka, Japan). NP-silica gel (75–150 μm), RP-C18 (38–63 μm), and open column chromatography equipment were purchased from Wako Pure Chemical Industries (Osaka, Japan). Diaion HP-20 purchased from Mitsubishi Chemical Co. (Tokyo, Japan) and a Biotage Selekt (Uppsala, Sweden) equipped with NP-silica gel or a RP-C18 flash chromatography column (Sfär C18 D Duo column, 30 μm, 30 and 60 g) were used for further fractionation. Purification of the compounds was achieved using a MPLC Pure C-850 Flash Prep® (Buchi, Flawil, Switzerland) system with UV and evaporative light scattering detector (ELSD) detection and an HPLC preparative column (Inertsil ODS-3, 5 μm, 20 × 250 mm) obtained from GL Sciences Inc., Japan. Reversed phase thin layer chromatography (RP-TLC, 60 RP-18 F254S, Merck, Darmstadt, Germany) was used for further purification. TLC silica gel 60 F254 plates were purchased from Merck (Darmstadt, Germany), and dimethyl sulfoxide (DMSO) and methanol-d4 (CD3OD) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA). Other analytical grade chemicals used in the present study were supplied by Merck Co. (Japan).Extraction and isolation
The aerial parts of B. indica (Wight) A. J. Scott were collected from desert areas in Egypt (Ihnasia ancient city) in August 2019. The aerial parts of Agathophora alopecuroides (Delile) Fenzl ex Bunge were collected from desert areas southeast of Maadi, Egypt (29° 57′ 35′′ N, 31° 19′ 44′′ E). The plants were kindly identified by Prof. Ibrahim A. El-Garf, Professor of Botany, Faculty of Science, Cairo University, Egypt. The voucher specimens, BIC-2019-2 and AG-2019, were deposited at the herbarium of the Pharmacognosy Department, Faculty of Pharmacy, Al-Azhar University, Egypt. The extraction was carried out as described in our previous work.9Briefly, the shade-dried aerial parts of B. indica (1.3 kg) and A. alopecuroides (0.9 kg) were ground into a fine powder and then extracted by maceration with 80% aqueous methanol. Following evaporation under reduced pressure, 138.5 g and 100 g of crude extracts were obtained from B. indica and A. alopecuroides, respectively. The crude extract of B. indica was then suspended in H2O and sequentially partitioned with n-hexane, CH2Cl2, EtOAc, and n-BuOH, while the crude extract of A. alopecuroides was fractionated over Diaion HP-20 eluted with H2O–MeOH to obtain a 28.5 g methanol fraction.
For the purification of compounds from B. indica, following fractionation of the dichloromethane fraction (NP-silica gel; n-hex–EtOAc–MeOH; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:0–0:0:1), the fraction D3.2 (50.5 mg) was purified over RP-TLC eluted with H2O–MeOH (65:35) to obtain compound 1 (4.5 mg). Subfraction E3 of the ethyl acetate fraction (89 mg) was purified via MPLC with RP-C18 (4 g) eluted with H2O–MeOH (8:2 to 5:5, flow rate 8 mL min−1) to afford compound 2 (7.0 mg). Compounds 3 (0.9 mg), 4 (6.4 mg), and 5 (0.7 mg) were obtained from the combined E5C and E5D subfractions (178 mg) after purification over a preparative HPLC column (Inertsil ODS-3, 5 μm, 20 × 250 mm, 5 mL min−1) eluted with H2O–MeOH–FA 65:35:0.1.
:0:0–0:0:1), the fraction D3.2 (50.5 mg) was purified over RP-TLC eluted with H2O–MeOH (65:35) to obtain compound 1 (4.5 mg). Subfraction E3 of the ethyl acetate fraction (89 mg) was purified via MPLC with RP-C18 (4 g) eluted with H2O–MeOH (8:2 to 5:5, flow rate 8 mL min−1) to afford compound 2 (7.0 mg). Compounds 3 (0.9 mg), 4 (6.4 mg), and 5 (0.7 mg) were obtained from the combined E5C and E5D subfractions (178 mg) after purification over a preparative HPLC column (Inertsil ODS-3, 5 μm, 20 × 250 mm, 5 mL min−1) eluted with H2O–MeOH–FA 65:35:0.1.
For the purification of the compounds from A. alopecuroides, the methanol fraction was chromatographed using a Biotage Selekt packed with NP-silica gel eluted with n-hex–EtOAc–MeOH; 1:0:0–0:0:1 to obtain 11 fractions. Fraction 6 (160 mg) was purified using MPLC connected to an RP-C18 column (4 g) eluted with H2O–ACN–0.1% FA (flow rate 8 mL min−1) to afford compound 6 (20 mg). Moreover, compound 7 (9.1 mg) was obtained after purification of fraction 7 (212 mg) over RP-C18 (2 columns, 4 g each) eluted gradually with H2O–ACN in presence of 0.1% FA. In addition, compounds 2 and 4 (3.0 and 8.0 mg, respectively), which were purified from the aqueous methanol extract of B. indica in this study, were also purified from the methanol fraction of A. alopecuroides.
Compound 1: yellow amorphous powder; for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 342.1352 [M − H]− (calcd for C19H20NO5, 342.1341).
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | — | — | — | — | — | — | — |
| 2 | 7.09, d (1.9) | 7.12, d (1.9) | 7.00, d (1.9) | 7.15 d (2.0) | 7.09, d (1.9) | 7.01, d (1.9) | 7.11, d (2.0) |
| 3 | — | — | — | — | — | — | — |
| 4 | — | — | — | — | — | — | — |
| 5 | 6.77, d (8.1) | 6.79, d (8.2) | 7.09, d (8.2) | 6.80, d (8.2) | 6.77, d (8.2) | 6.78, d (8.4) | 6.79, d (8.1) |
| 6 | 7.00, dd (1.9, 8.1) | 7.03, dd (8.2, 1.9) | 6.71, dd (8.2, 1.9) | 7.04, dd (2.0, 8.2) | 6.99, dd (1.9, 8.2) | 6.91, dd (8.4, 1.9) | 6.98, dd (2.0, 8.1) |
| 7 | 7.43, d (15.7) | 7.44, d (15.7) | 7.25, d (15.5) | 7.46, d (15.6) | 7.41, d (15.7) | 7.40, d (15.7) | 7.30, d (15.6) |
| 8 | 6.39, d (15.7) | 6.40, d (15.7) | 6.38, d (15.5) | 6.47, d (15.6) | 6.37, d (15.7) | 6.35, d (15.6) | 6.52, d (15.6) |
| 9 | — | — | — | — | — | — | — |
| 1′ | — | — | — | — | — | — | — |
| 2′ | 6.81, d (2.0) | 7.07, d (8.6) | 6.88, d (1.8) | 7.24, d (8.3) | 7.20, d (8.6) | 7.07, d (8.3) | 7.11, d (8.3) |
| 3′ | — | 6.73, d (8.6) | — | 6.78, d (8.3) | 6.69, d (8.6) | 6.74, d (8.3) | 6.60, d (8.3) |
| 4′ | — | — | — | — | — | — | — |
| 5′ | 6.72, d (8.0) | 6.73, d (8.6) | 6.71, d (8.2) | 6.78, d (8.3) | 6.69, d (8.6) | 6.74, d (8.3) | 6.60, d (8.3) |
| 6′ | 6.66, dd (8.0, 2.0) | 7.07, d (8.6) | 6.90, dd (1.8, 8.2) | 7.24, d (8.3) | 7.20, d (8.6) | 7.07, d (8.3) | 7.11, d (8.3) |
| 7′ | 2.76, t (7.3) | 2.77, t (7.4) | 4.53, dd (7.3, 4.3) | 4.73, dd (4.88, 7.76) | 4.69, dd, (7.8, 5.1) | 2.76, t (7.4) | 4.16, m |
| 8′ | 3.48, t (7.3) | 3.48, t (7.4) | 3.46, d (13.0, 7.0) | 3.54, dd (13.6, 4.9) | 3.49, dd (11.3, 5.0) | 3.47, t (7.4) | 3.34, m |
| 3.21, dd (13.1, 7.0) | 3.45, dd (13.6, 4.9) | 3.38, dd (9.2, 6.3) | 3.33, m | ||||
| 3-OMe | 3.86, s | 3.89, s | 3.76, s | 3.89, s | 3.86, s | — | 3.80, s |
| 3′-OMe | 3.82, s | — | 3.72, s | — | — | — | — |
| 7′-OMe | — | — | — | — | — | — | 3.17, s |
| Solvent | CD3OD | CD3OD | DMSO-d6 | CD3OD | CD3OD | CD3OD | DMSO-d6 |
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 127.6 | 127.9 | 129.2 | 128.2 | 128.1 | 128.3 | 126.4 |
| 2 | 111.4 | 111.5 | 110.4 | 111.5 | 111.5 | 115.1 | 110.8 |
| 3 | 149.6 | 149.5 | 147.4 | 149.3 | 149.4 | 148.7 | 148.8 |
| 4 | 151.0 | 150.6 | 149.0 | 149.9 | 150.0 | 146.7 | 148.3 |
| 5 | 116.7 | 116.6 | 115.1 | 116.5 | 116.1 | 116.5 | 115.6 |
| 6 | 123.4 | 123.4 | 122.9 | 123.3 | 123.3 | 122.1 | 121.6 |
| 7 | 142.2 | 142.1 | 139.8 | 142.3 | 142.1 | 142.2 | 139.0 |
| 8 | 118.3 | 118.4 | 118.7 | 118.6 | 118.7 | 118.4 | 118.99 |
| 9 | 169.3 | 169.3 | 166.7 | 169.5 | 169.2 | 169.3 | 165.4 |
| 1′ | 132.0 | 131.3 | 134.9 | 134.7 | 134.4 | 131.3 | 129.8 |
| 2′ | 113.4 | 130.7 | 114.6 | 128.5 | 130.7 | 130.7 | 128.0 |
| 3′ | 144.0 | 116.3 | 145.8 | 116.1 | 116.3 | 116.3 | 115.2 |
| 4′ | 146.1 | 157.0 | 145.9 | 158.1 | 157.0 | 156.9 | 157.1 |
| 5′ | 116.2 | 116.3 | 115.2 | 116.1 | 116.3 | 116.3 | 115.2 |
| 6′ | 122.3 | 130.7 | 120.1 | 128.5 | 130.7 | 130.7 | 128.0 |
| 7′ | 36.2 | 35.8 | 71.7 | 73.4 | 73.5 | 35.8 | 81.5 |
| 8′ | 42.5 | 42.6 | 47.3 | 48.8 | 47.5 | 42.5 | 45.2 |
| 3-OMe | 56.3 | 56.4 | 55.7 | 56.4 | 56.4 | — | 55.8 |
| 3′-OMe | 56.3 | — | 55.5 | — | — | — | — |
| 7′-OMe | — | — | — | — | — | — | 55.5 |
| Solvent | CD3OD | CD3OD | DMSO-d6 | CD3OD | CD3OD | CD3OD | DMSO-d6 |
Compound 2: yellow amorphous powder; for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 312.1244 [M − H]− (calcd for C18H18NO4, 312.1236). IR (ZnSe): 3317, 1670, 1513, 1270.8 cm−1.
Compound 3: white amorphous powder; [α]25D − 35.00 (c 0.01, MeOH); for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 358.1301 [M − H]− (calcd for C19H20NO6, 358.1291).
Compound 4: light-yellow amorphous powder; [α]25D − 35.10 (c 0.02, MeOH); for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 328.1196 [M − H]− (calcd for C18H18NO5, 328.1185). IR (ZnSe): 3360, 2965, 1660, 1593, 1278 cm−1.
Compound 5: yellow amorphous powder; [α]25D + 24.29 (c 0.01, MeOH); for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 328.1195 [M − H]− (calcd for C18H18NO5, 328.1185).
Compound 6: white amorphous powder; for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 298.1089 [M − H]− (calcd for C17H16NO4, 298.1079). IR (ZnSe): 3398, 2970, 1650, 1590 cm−1.
Compound 7: brownish-yellow amorphous powder; [α]25D − 12.23 (c 0.01, MeOH); for 1H and 13C NMR data, see Tables 1 and 2; ESIMS m/z 342.1356 [M − H]− (calcd for C19H20NO5, 342.1341). IR (ZnSe): 3400, 2914, 1650, 1503, 1268 cm−1.
Identification of the isolated compounds
Optical rotations were determined using a Jasco P-2200 polarimeter (Jasco, Tokyo, Japan). The NMR spectra of the isolated compounds were recorded using a Bruker DRX-600 spectrometer (Bruker Daltonics, Billerica MA, USA) with tetramethylsilane (TMS) as an internal standard. The chemical shifts were expressed as δ values. The HR-MS of the compounds was determined using a quadrupole time-of-flight (qTOF) mass spectrometer (Agilent Technologies, USA). FT-IR measurements of samples were obtained using an FTIR-620 spectrophotometer (JASCO International, Co. Ltd., Tokyo, Japan).In vitro enzyme assays
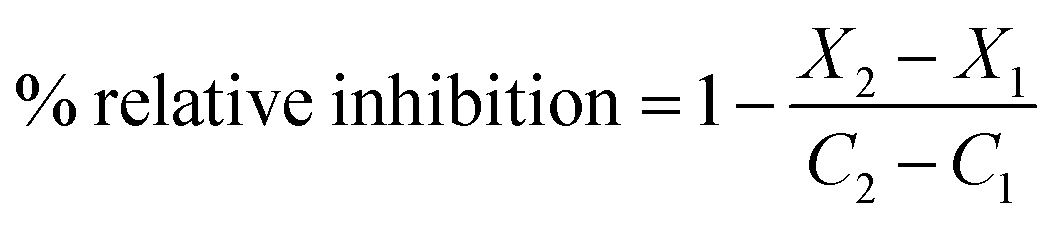
The assay was expressed in terms of relative fluorescence units (RFU), where X2: fluorescence of test sample measured at time T2; X1: fluorescence of test sample measured at time T1; C2: fluorescence of a control measured at time T2; and C1: fluorescence of a control measured at time T1.
| % relative inhibition = (RFU of PC × RFU of S)/(RFU of S) × 100 |
where, slope of EC is the slope of the enzyme control, and the slope of S is the slope of the sample screen.
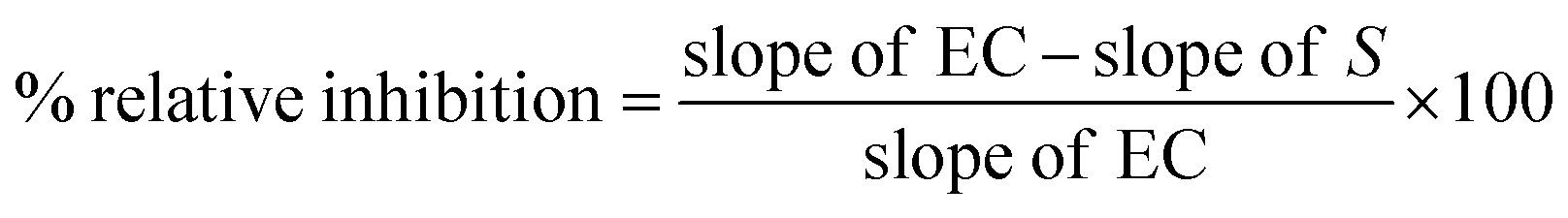
Statistical analysis
All experimental results in the current study were obtained from three repeated biological experiments. The results are expressed as the means ± SD (n = 3). The statistical significance of differences between means was established using ANOVA with Duncan's post hoc tests. p values < 0.05 indicate statistical significance.In silico investigations
Molecular docking
AutoDock Vina software was used in all molecular docking experiments.19 All isolated compound structures were docked into the MAO-B and BACE1 crystal structures (PDB codes 6FWC and 4WY1, respectively). The binding site for each protein was determined according to the enzyme's co-crystallized ligand. The co-ordinates of the grid box were: x = −1.12; y = −18.86; z = 33.74 for BACE1, and x = 51.37; y = 155.39; z = 27.85 for MAOB. The size of the grid box was set to 10 Å. Exhaustiveness was set to 24. Ten poses were generated for each docking experiment. The docking poses were analysed and visualized using PyMOL software.19Molecular dynamics simulations
Desmond v. 2.2 software was used for performing molecular dynamics simulation (MDS) experiments.20–22 This software applies the OPLS force field. Protein systems were built using the System Builder option, where the protein structure was embedded in an orthorhombic box of TIP3P water together with 0.15 M Na+ and Cl− ions in 20 Å solvent buffer. Afterward, the prepared systems were energy minimized and equilibrated for 10 ns. Desmond software automatically parameterizes inputted ligands during the system building step according to the OPLS (optimized potentials for liquid simulations) force field. For simulations performed using NAMD,23 the parameters and topologies of the compounds were calculated either using the Charmm27 force field with the online software Ligand Reader and Modeler (https://www.charmm-gui.org/?doc=input/ligandrm)24 or using the VMD plugin Force Field Toolkit (ffTK). Afterward, the generated parameters and topology files were loaded into VMD to readily read the protein–ligand complexes without errors and then conduct the simulation step.Binding free energy calculations
Binding free energy calculations (ΔG) were performed using the free energy perturbation (FEP) method. This method was described in detail in a recent article by Kim and co-workers.23 Briefly, this method calculates the binding free energy ΔGbinding according to the following equation: ΔGbinding = ΔGcomplex − ΔGligand. Each ΔG value is estimated from a separate simulation using NAMD software. Interestingly, all input files required for simulation by NAMD can be prepared using the online website CharmmGUI (https://charmmgui.org/?doc=input/afes.abinding). Subsequently, we can use these files in NAMD to produce the required simulations using the FEP calculation function in NAMD. The equilibration was achieved in the NPT ensemble at 300 K and 1 atm (1.01325 bar) with Langevin piston pressure (for “complex” and “ligand”) in the presence of the TIP3P water model. Then, 10 ns FEP simulations were performed for each compound, and the last 5 ns of the free energy values was measured for the final free energy values.20 Finally, the generated trajectories were visualized and analyzed using VMD software. It worth noting that Ngo and co-workers in their recent benchmarking study found that the FEP method for the determination of ΔG was the most accurate method in terms of predicting enzyme inhibitors.24Results
Identification of the isolated compounds
The dried aerial parts of B. indica and A. alopecuroides were extracted with 80% methanol followed by suspending the obtained crude extracts in H2O. Solvents of successively greater polarities, including n-hexane, CH2Cl2, EtOAc, and n-BuOH, were used for partitioning the crude methanol extract of B. indica, while a Diaion HP-20 chromatography column was used for the fractionation of the crude methanol extract of A. alopecuroides. MPLC connected to either RP-C18 flash columns or a preparative HPLC column was used for the final purification of compounds. A total of seven amide alkaloids (phenylpropanoid amides) were isolated and identified (Fig. 2). The biosynthetic pathway of this class of compounds is shown in Fig. 3.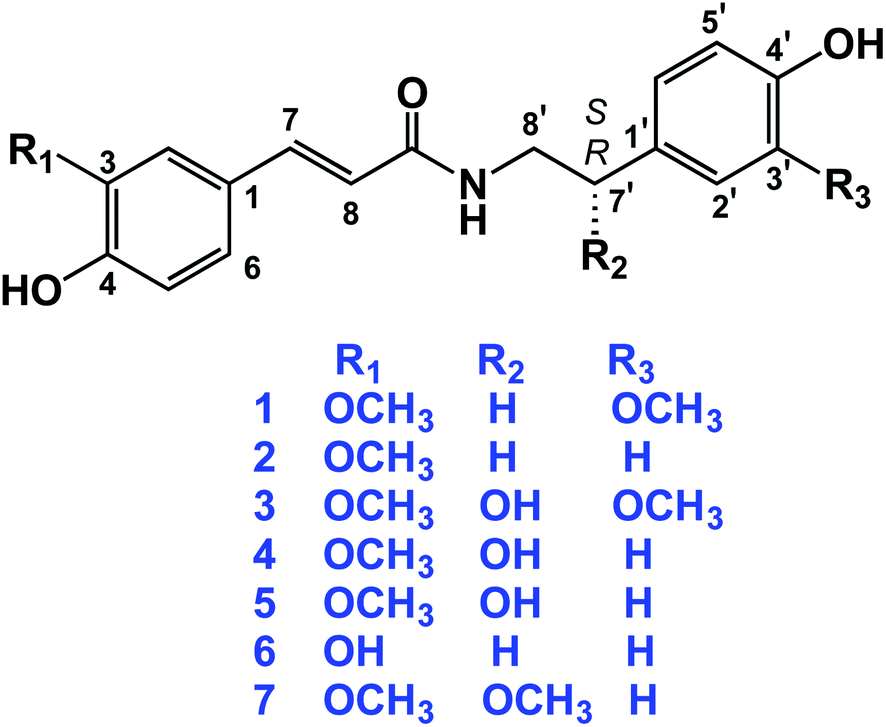 | ||
| Fig. 2 Structures of the isolated compounds 1–7. | ||
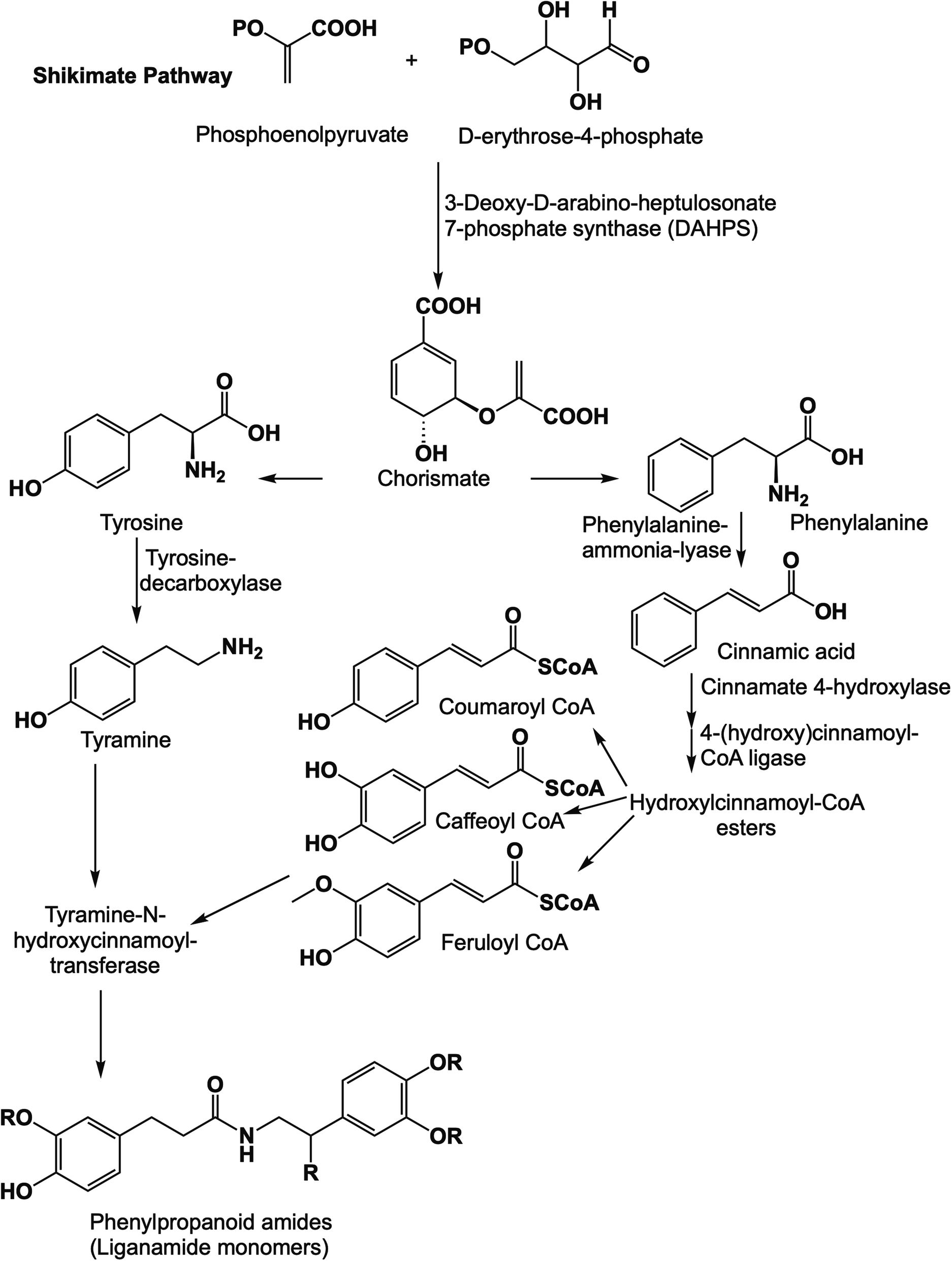 | ||
| Fig. 3 The biosynthetic pathway to phenylpropanoid amides. | ||
N-trans-feruloyl-3-methoxytyramine (1) was obtained as a yellow amorphous powder. The molecular formula of 1 was determined to be C19H21NO5 based on the HR-ESI-MS [M − H]− ion peak at m/z 342.1352 and was confirmed from the 1H and 13C-NMR spectral data (Tables 1 and 2). The 1H-NMR data of 1 revealed a pair of downfield doublets at δH 7.43 (d, J = 15.7 Hz) and 6.39 (d, J = 15.7 Hz), indicating an α,β-unsaturated ketone with the (E)-configuration. Additionally, signals in the aromatic region indicated the presence of two sets of ABX patterns, each of which was assigned as a trisubstituted benzene ring. The chemical shifts at δH 7.09 (d, J = 1.9 Hz), 6.77 (d, J = 8.1 Hz), and 7.00 (dd, J = 1.9, 8.1 Hz) were assigned as H-2, H-5, and H-6, respectively. On the other hand, the set of aromatic signals resonating at δH 6.81 (d, J = 2.0 Hz), 6.72 (d, J = 8.0 Hz), and 6.66 (dd, J = 8.0, 2.0 Hz) were assigned as H-2′, H-5′, and H-6′ of the methoxy-tyramine moiety. Moreover, the presence of the two signals in the aliphatic region resonating at δH 2.76 (t, J = 7.3 Hz) and 3.48 (t, J = 7.3 Hz) indicated the two methylene groups of the methoxy-tyramine moiety. The 13C-NMR spectral data of 1 showed resonances for all 19 C-atoms, comprising two CH2, eight CH, two CH3 and seven quaternary C-atoms. The downfield signal at δC 169.3 (C-9) indicated the presence of the amide C-atom, while the signals at δC 142.2 (C-7) and 118.3 (C-8) indicated that an olefinic bond was present in the molecule. The downfield signals at δC 127.6 (C-1), 111.4 (C-2), 149.6 (C-3), 151.0 (C-4), 116.7 (C-5), 123.4 (C-6), 132.0 (C-1′), 113.4 (C-2′), 144.0 (C-3′), 146.1 (C-4′), 116.2 (C-5′), and 122.3 (C-6′) suggested the presence of two phenyl rings that comprise six CH and six quaternary C-atoms, of which six were oxygenated carbon atoms. Additionally, each pair of directly connected hydrogen and carbon atoms was determined using HSQC as shown in Fig. S4.† In the HMBC spectrum, cross peaks between H-7 (δ 7.43, d, J = 15.7 Hz) and C-9 (δ 169.3), H-2 (δ 7.09, d, J = 1.9 Hz) and C-7 (δ 142.2), and H-5 (δ 6.77, d, J = 8.1 Hz) and C-1 (δ 127.6) confirmed the trisubstituted benzene ring and α,β-unsaturated carbons. Moreover, the ethyl linkage connected to the trisubstituted benzene ring was proved by the correlations of H-8′/C-1′ and H-7′/H-6′, as shown in Fig. S5.† Additionally, the interaction of H-8′ with C-9 confirmed the attachment of the dopamine-derived part of the compound to substituted cinnamic acid part, which finally confirmed the structure of 1. In the COSY experiment, the interactions of H-5/H-6, H-7/H-8, and H-7′/H-8′ further confirmed the structure of 1.
The molecular formula of N-trans-feruloyl tyramine (2) was established to be C18H19NO4 due to a deprotonated molecular ion peak at m/z 312.1244 [M − H]− (calcd for C18H18NO4, 312.1236). The IR spectrum showed characteristic absorption bands for the hydroxyl group at 3317 cm−1, amide carbonyl group at 1670 cm−1, and conjugated double bond at 1513 cm−1. The structural data of 2 were compared with those of 1, and the HR-MS data revealed the loss of a methoxy group in compound 2. The 1H-NMR data of compound 2 revealed the presence of two sets of aromatic protons. One set was an ABX system resonating at δH 7.12 (d, J = 1.9 Hz), 6.79 (d, J = 8.2 Hz), and 7.03 (dd, J = 1.9, 8.2 Hz), while the other set was an AA′-BB′ system resonating at δH 7.07 (d, J = 8.6 Hz) and 6.73 (d, J = 8.6 Hz), which suggested one trisubstituted and one disubstituted benzene ring. Additionally, the 13C-NMR data showed that compound 2 is similar to 1 in possessing one phenylpropanoid moiety and one tyramine moiety, except for the absence of one methoxy group in compound 2. In HMBC, the correlation of H-7′ (δ 2.77, t, J = 7.4 Hz) with the 13C atom signals at δ 131.3, 130.7 and 42.6 assigned to C-1′, C-2′, and C-8′, respectively, indicated the presence of a disubstituted ethyl benzene moiety. The complete assignment of compound 2 was aided by HSQC, HMBC, and COSY experiments, as well as comparison with published literature.25 The complete NMR data for 2 is shown in Tables 1 and 2.
S-(−)-N-trans-feruloyl octopamine (4), [α]25D − 35.10 (c 0.02, MeOH), was obtained as a light-yellow amorphous powder. The optical rotation measurement of this compound indicated that 4 is the S-isomer.26 The molecular formula was determined to be C18H19NO5 on the basis of the [M − H]− ion peak at m/z 328.1196 in the HR-ESI-MS experiment and from the 13C-NMR data. As shown in Tables 1 and 2, the 1H- and 13C-NMR spectral data indicated similarity between compounds 4 and 2 except for the presence of one chiral centre in the aliphatic side chain of the tyramine moiety. The signals resonating at δH 4.73, dd (J = 4.9, 7.8 Hz) and δC 73.4 were assigned to the chiral carbon in the molecule (C-7′). The structure of 4 was confirmed using HSQC, HMBC, and 1H–1H COSY experiments. In the HMBC experiment, the cross peaks between H-7′ (δ 4.73) and the carbon signals at δ 48.8 (C-8′) and δ 134.7 (C-1′) further confirmed the location of hydroxyl group at C-7′. Additionally, the correlations of H-7′ (δ 4.73)/H-8′ (δ 3.45 and 3.54) in the COSY experiment further confirmed the position of the hydroxyl group at C-7′.27
N-trans-caffeoyl tyramine (6) was obtained as a white amorphous powder. The 1H- and 13C-NMR spectral data of 6 (Tables 1 and 2) were similar to those of compound 2, except that 6 does not possess a methoxy group. Finally, compound 6 was identified as N-trans-caffeoyl tyramine.28,38
The molecular formula of S-(−)-3-(4-hydroxy-3-methoxyphenyl)-N-[2-(4-hydroxyphenyl)methoxyethyl]acrylamide (7), [α]25D − 12.23 (c 0.01, MeOH), was determined to be C19H21NO5 based on the negative ion peak in HR-MS at m/z 342.1356 (calcd for C19H20NO5, 342.1341). The 1H- and 13C-NMR spectra data of compound 7 were similar to that of compound 1, except in the position of one methoxy group, which was assigned as being attached at C-7′ in compound 7 instead of C-3′ as in compound 1. The methoxy group at C-7′ was assigned to the peaks at δH 3.17, s and δC 81.5 ppm.29
S-(−)-N-trans-feruloylnormetanephrine (3), [α]25D − 35.00 (c 0.01, MeOH), and the rarely occurring R-(+)-N-trans-feruloyloctopamine (5), [α]25D + 24.29 (c 0.01, MeOH), were reported in our previous work,9 and their NMR spectral data are shown in Tables 1 and 2. In addition, the isolated compounds 1, 2, 4, 6 and 7 were subjected to in silico investigation to provide more scientific data regarding their binding mechanisms to some molecular targets implicated in AD.
To the best of our knowledge, compound 7 is first reported in the Chenopodiaceae family, while compounds 2, 4, and 6 are first reported in the genus Agathophora. Moreover, compounds 2, 4, 6, and 7 were first isolated from the plant A. alopecuroides. In continuation of our ongoing projects for the discovery of bioactive compounds targeting AD, as part of our efforts to utilize these potentially economically valuable halophytic plants, the isolated lignanamides in the present study were evaluated in terms of their inhibition capacity on BACE1, MAO-B, Aβ1–42 aggregation, and hyperphosphorylated tau protein.
Inhibitory effects on BACE1
BACE-1 (β-site amyloid precursor protein cleaving enzyme 1) is a key protein that plays a vital role in the abnormal production of amyloid beta (Aβ), which is one of the major factors in AD.30 Hence, the isolated compounds were tested for their inhibitory activity against BACE1. As shown in Table 3, the results showed that compounds 1 and 7 displayed promising BACE1 inhibition activity with IC50 < 6 μg mL−1. Moreover, compounds 2 and 6 exhibited inhibition activity with IC50 values of 11.6 ± 0.39 and 10.2 ± 0.34 μg mL−1, respectively.| Compound | BACE1 | MAO-B | Aβ1–42 aggregation | Tau-p (pg mL−1) |
|---|---|---|---|---|
| B. indica | 28.9 ± 0.97 | 8.7 ± 0.29 | 40.6 ± 1.36 | 6.62 ± 0.14 |
| A. alopecuroides | 15.4 ± 0.52 | 1.7 ± 0.06 | 3.01 ± 0.10 | 2.27 ± 0.12 |
| 1 | 5.39 ± 0.18 | 0.71 ± 0.02 | 0.3 ± 0.01 | 1.62 ± 0.13 |
| 2 | 11.6 ± 0.39 | 2.24 ± 0.08 | 1.2 ± 0.04 | 2.12 ± 0.11 |
| 4 | 36.1 ± 1.21 | 5.92 ± 0.2 | 13.1 ± 0.44 | 5.53 ± 0.14 |
| 6 | 10.2 ± 0.34 | 2.62 ± 0.09 | 6.05 ± 0.2 | 6.14 ± 0.19 |
| 7 | 5.64 ± 0.19 | 1.52 ± 0.05 | 2.62 ± 0.09 | 2.79 ± 0.23 |
| LY2811376 | 0.8 ± 0.03 | — | — | 2.09 ± 0.08 |
| Selegiline | — | 0.55 ± 0.02 | — | — |
| Tacrine | — | — | 1.93 ± 0.06 | — |
| Control | — | — | — | 10.8 ± 0.68 |
Inhibition activity on MAO-B
Monoamine oxidase-B (MAO-B) has been deemed a potential therapeutic target for AD. Higher MAO-B activity is associated with aberrant levels of gamma amino butyric acid (GABA), neurotoxicity, neuroinflammation, and Aβ production.5 Therefore, the discovery of bioactive compounds with the capability to inhibit monoamine oxidase-B could play a crucial role in AD treatment. In this context, the crude methanol extracts of the halophytic herbs B. indica and A. alopecuroides exhibited promising therapeutic activity against MAO-B enzyme with IC50 values of 8.7 ± 0.29 and 1.7 ± 0.06 μg mL−1, respectively (Table 3). It is worth noting that the isolated lignanamide compounds in this study displayed potential inhibition activity against MAO-B enzyme, with IC50 values ranging from 0.71 to 5.92 μg mL−1. Notably, compounds 1, 2, 6 and 7 showed the greatest potential MAO-B inhibition effect with IC50 values of 0.71 ± 0.02, 2.24 ± 0.08, 2.62 ± 0.09, and 1.52 ± 0.05 μg mL−1, respectively, which are comparable to that of selegiline (IC50 = 0.55 ± 0.02).Inhibition activity on Aβ1–42 aggregation
One of the key factors in neurodegeneration is amyloid-β accumulation, which leads to the aggregation of neurotic senile plaques.1 Amyloid-β is neurotoxic and is correlated with the severity of AD symptoms, cognitive impairments, and neuronal cell death.31 Considering the fact that various therapeutic strategies have been concerned with the inhibition of Aβ-aggregate formation, we evaluated the isolated compounds in terms of their inhibition effect on Aβ-aggregation. Our results indicated that the methanol extract of A. alopecuroides has potential inhibition activity against neurotoxic Aβ-formation (IC50 = 3.01 ± 0.10 μg mL−1). Among the isolates, compounds 1, 2, and 7 exhibited extraordinary activity against Aβ-aggregation, with IC50 values of 0.3 ± 0.01, 1.2 ± 0.04, and 2.62 ± 0.09 μg mL−1, respectively, which are comparable to that of the positive control (IC50 = 1.93 ± 0.06 μg mL−1) as shown in Table 3.Inhibition activity on tau protein
Tau is a microtubule-associated protein that plays an important role in cell integrity.32 Tau protein hyperphosphorylation leads to its dissociation from microtubules and subsequently to abnormal tau aggregation and the formation of neurotoxic neurofibrillary tangles (NFTs).33 Therefore, approaches for blocking tau-mediated neurotoxicity include the inhibition of tau aggregation. In the present study, the results of the in vitro evaluation of the crude extracts of B. indica and A. alopecuroides against tau-protein revealed that the halophytic herb A. alopecuroides reduces the concentration of phosphorylated tau-protein to 2.27 ± 0.12 pg mL−1, which is comparable to the value observed for the control, which was nearly 5 times greater (10.8 ± 0.68 pg mL−1). In addition, the assay results indicated that the isolated compounds showed variable activity against phosphorylated tau-protein. Notably, compounds 1, 2, and 7 were the most potent candidates in terms of their capacity to reduce the concentration of phosphorylated tau-protein, with values of 1.62 ± 0.13, 2.12 ± 0.11, 2.79 ± 0.23 pg mL−1, respectively, which were comparable to that of control group, which was nearly five- to seven-times greater, as shown in Table 3.Based on the above evidence, the methoxy group in the tyramine moiety of the isolated molecules such as that in compounds 1 and 7 seems to play a crucial rule, not only in terms of MAO-B enzyme inhibition activity, but also for the inhibition activities toward BACE1 enzyme and Aβ1–42 aggregation.
Molecular modeling and in silico-based analysis
To gain deeper insight into the possible modes of interaction between the active compounds and both MAO-B and BACE1, we carried out several modeling and in silico-based investigations. First, the structures of active compounds (1, 2, 4, 6, and 7) were docked into the active sites of both enzymes. Ten binding poses were generated for each compound and each enzyme. Usually, poses with lowest affinity scores are the true binding poses. To confirm this assumption, we re-docked the structure of the co-crystalized inhibitors into the active site of each enzyme. The generated poses with the lowest binding affinity in kcal mol−1 were the ones most similar to the original crystalized binding mode (RMSDs = 0.953 Å and 1.142 Å for MAOB and BACE1, respectively; Fig. 4). Moreover, we calculated the absolute binding free energies (ΔGbinding) of each generated binding pose for each compound, and the top-scoring binding poses generated from the docking step were also the best in terms of ΔGbinding (Table 4). This step was achieved with the aid of a molecular dynamics simulation (MDS)-based free energy perturbation method.24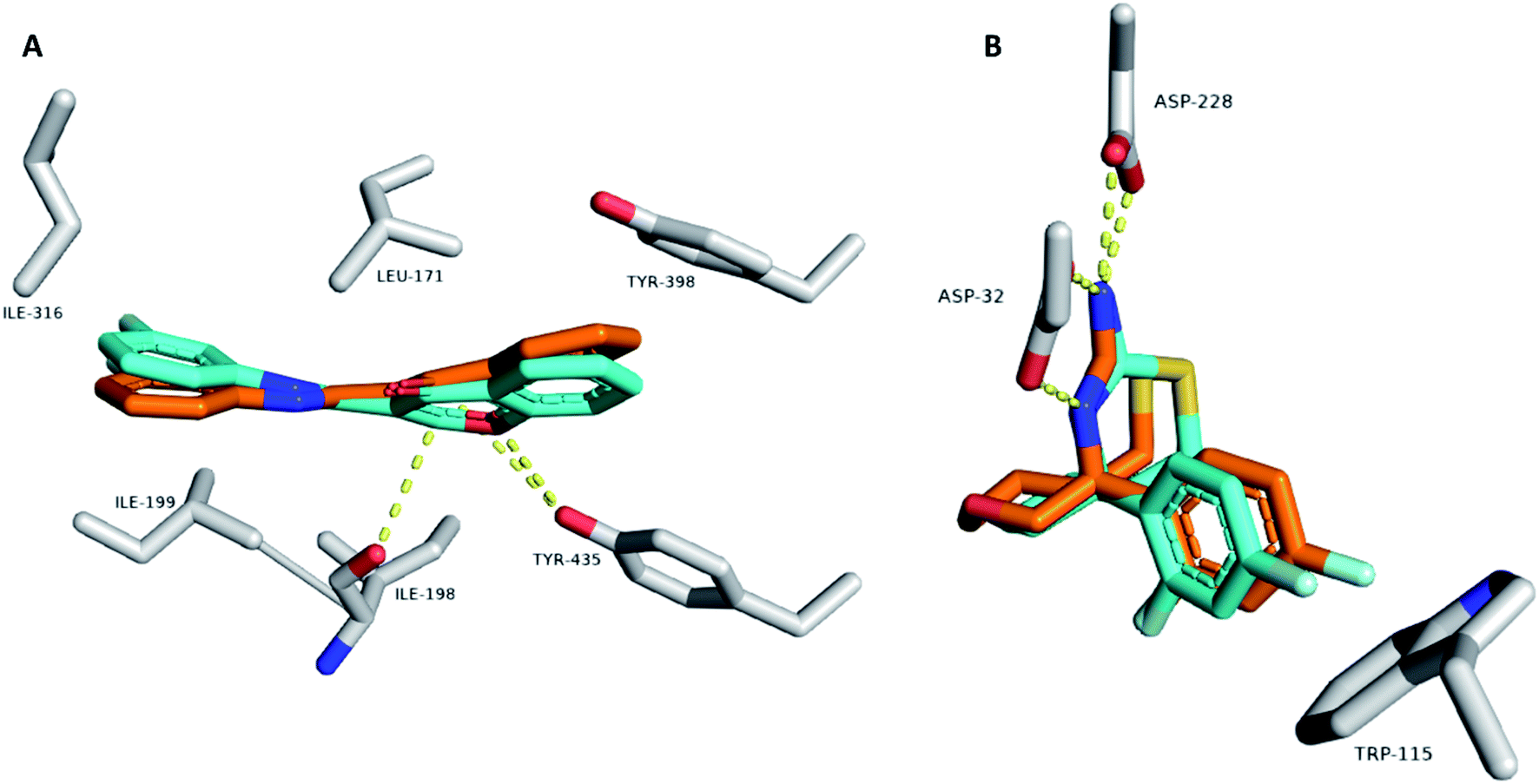 | ||
| Fig. 4 Binding modes of the co-crystalized inhibitor structures inside the active sites of both MAOB and BACE1 ((A) and (B), respectively). Cyan-colored structures represent the original binding modes inside the crystalized proteins (PDB codes 6FWC and 4WY1, respectively). Orange-colored structures represent the re-docked binding modes with RMSD values of 0.953 Å and 1.142 Å, respectively, from the original crystalized ones. | ||
| Structure | BACE1 (Vina score) | MAO-B (Vina score) | BACE1 (ΔGbinding) | MAO-B (ΔGbinding) |
|---|---|---|---|---|
| 1 | −9.6 | −10.3 | −8.9 | −8.9 |
| 2 | −9.2 | 10.2 | −9.5 | −9.2 |
| 4 | −5.1 | −9.8 | −5.5 | −9.4 |
| 6 | −8.9 | −10.2 | −7.9 | −9.9 |
| 7 | −9.0 | −9.9 | −8.1 | −9.8 |
| MAOB inhibitor | — | −9.8 | — | −9.5 |
| BACE1 inhibitor | −9.1 | — | −9.4 | — |
Second, each generated binding pose selected from the previous step was subjected to a 50 ns MDS run to obtain more insight into the dynamic binding stability and behavior of each structure inside the corresponding binding sites.
MAOB's active site is a tunnel-shaped binding site, and hence, each docked structure fitted perfectly inside it. To study the binding, the key binding interactions of each structure during the MDS runs and the highly-populated poses for each structure were extracted from the MDS trajectories of each MDS experiment.
As shown in Fig. 5A–E, each structure showed very good alignment with the structure of the co-crystalized inhibitor, and hence, they showed identical biding interactions. Each structure was able to achieve stable H-bonding with ILE-198 and TYR-435. Furthermore, they achieved multiple stable hydrophobic interactions with LEU-171, ILE-198, ILE-199, ILE-316, and TYR-398.
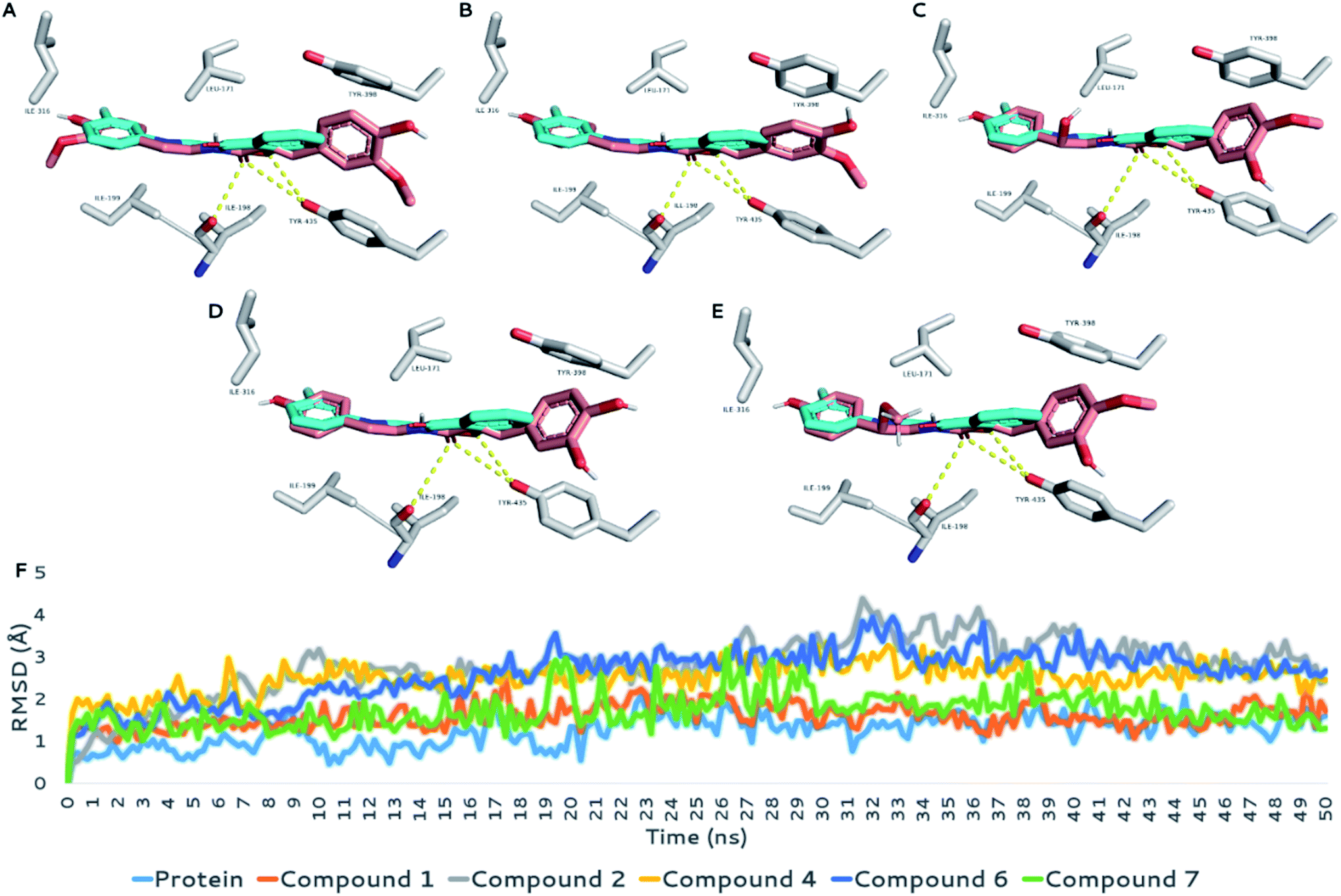 | ||
| Fig. 5 (A–E) Binding modes of the structures of compounds 1, 2, 4, 6, and 7, respectively, inside the binding site of MAOB (brick-red coloured structures) in alignment with that of the co-crystalized inhibitor (cyan-coloured structure). (F) RMSD values of the structures of compounds 1, 2, 4, 6, and 7 inside the binding site of MAOB, along with that of the co-crystalized inhibitor, over 50 ns MDS runs. | ||
Their average RMSDs inside the binding site over the course of MDS were convergent and ranged from 1.7 Å to 2.4 Å, indicating stable binding interactions with the enzyme binding sites. Accordingly, the calculated ΔGbinding values were convergent (ranging from −8.9 to −9.8 kcal mol−1; Table 4), and in turn, their in vitro inhibitory activities were also convergent (Table 3).
These structure- and biophysics-based data revealed the potential of the isolated amide alkaloids as very promising scaffolds for the future development of new MAOB inhibitors.
With regard to BACE1, the structures under investigation (compounds 1, 2, 4, 6, and 7) showed three different binding modes inside the active site of the enzyme. As shown in Fig. 6, the first binding mode for compounds 1, 2, and 6 showed an extended structure inside the binding site, in which they interacted with the enzyme differently than the co-crystalized inhibitor. However, this binding mode was stable over the course of the simulation (average RMSD = 1.6 Å to 1.9 Å; ΔGbinding ranging −7.9 to −9.5 kcal mol−1; Table 4), showing stable H-bonds with GLY-11, TRP-76, and THR-23 and two hydrophobic interactions with TYR-71 and TRP-115, similarly to the co-crystalized inhibitor. Accordingly, their calculated IC50 values (Table 3) indicated good inhibitory activity against the enzyme activity in vitro (Fig. 6A, B and D).
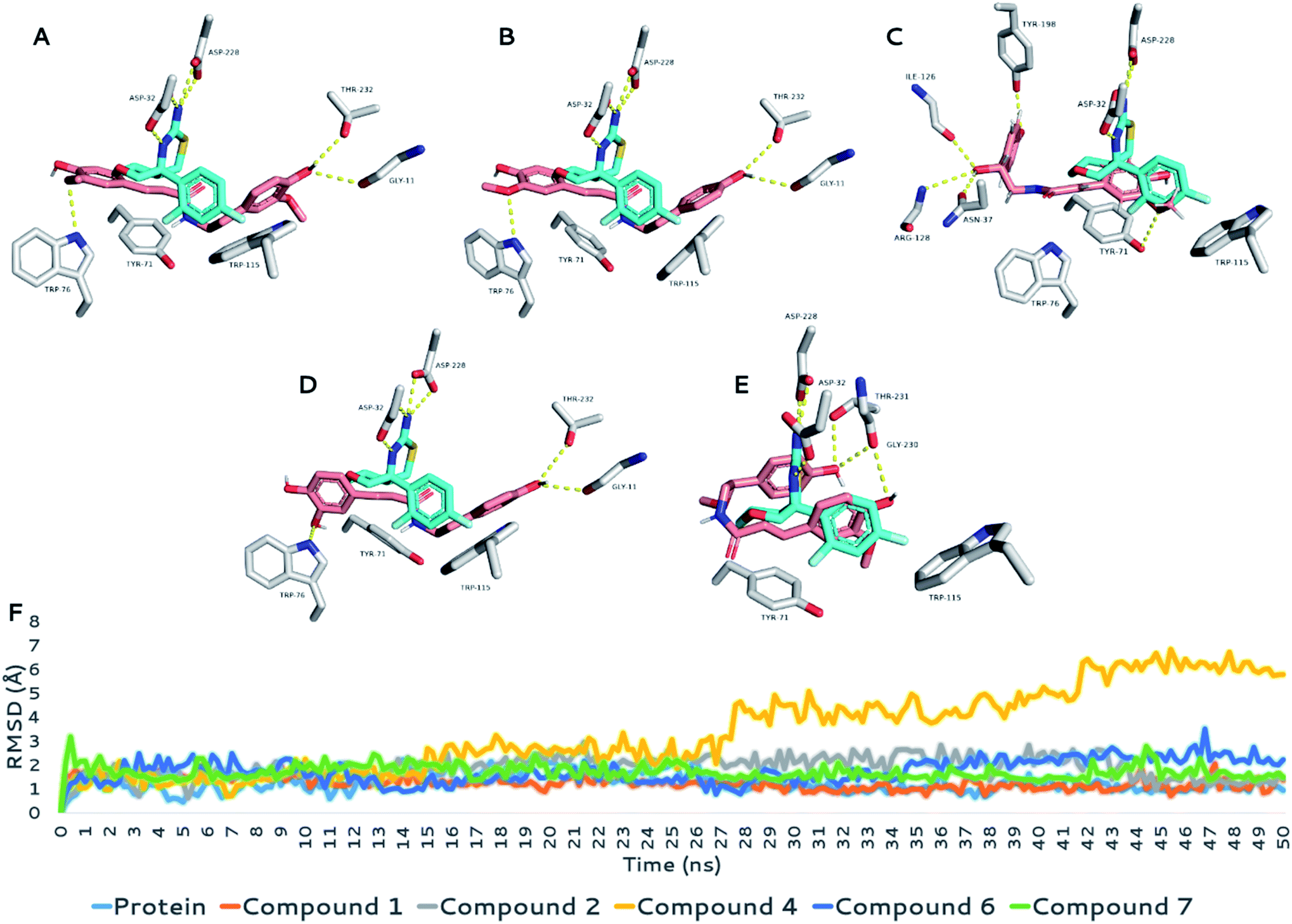 | ||
| Fig. 6 (A–E) Binding modes of the structures of compounds 1, 2, 4, 6, and 7, respectively, inside the binding site of BACE1 (brick-red coloured structures) in alignment with that of the co-crystalized inhibitor (cyan-coloured structure). (F) RMSD values of the structures of compounds 1, 2, 4, 6, and 7 inside the binding site of BACE1 along with that of the co-crystalized inhibitor over 50 ns MDS runs. | ||
The second binding mode was that of compound 4 (Fig. 6C), in which the hydroxyl group at C-7′ forced the whole structure to take on another orientation due to the steric clash with TRP-115. However, this orientation, which was the best among the generated docking poses in terms of docking score and ΔGbinding, was relatively the least stable one in comparison with those of the other compounds. Its average RMSD over the course of MDS was 5.4 Å, and its calculated ΔGbinding was −5.5 kcal mol−1, indicating relatively lower binding affinity than that of the remaining compounds. The third binding mode was that of compound 7, in which the structure of the compound took on an orientation that made it perfectly aligned with the structure of the co-crystalized inhibitor (Fig. 6E). Interestingly, the binding mode of compound 7 was almost identical to that of compound 4 at the beginning of the MDS; however, it began to change to the current one at 2 ns, in which it was not able to be stabilized by H-bonding (i.e., with ASN-37, ILE-126, and ARG-128) similarly to compound 4 because of the additional methyl group of the C-7′ hydroxyl group.
The current binding mode of compound 7 was stabilized with a network of H-bonds with GLY-230 and THR-231, in addition to two hydrophobic interactions with TYR-71 and TRP-115. This binding mode achieved the highest binding affinity toward the binding site of BACE1 (ΔGbinding = −8.1 kcal mol−1), and hence, its inhibitory activity was the second-most-potent (IC50 = 5.64 ± 0.19 μg mL−1) after that of compound 1 (IC50 = 5.39 ± 0.18 μg mL−1).
Conclusions
AD is well known for its multifactorial nature; consequently, it demands bioactive molecules with multimodal action that can act on more than one target to counteract possible drug–drug interactions during combined therapy. The present study describes the isolation and spectroscopic characterization of seven phenylpropanoid amides from the aerial parts of B. indica and A. alopecuroides in addition to the potential utilization of the crude extracts obtained from both plants and the isolated compounds in the management of AD through the multi-targeting of BACE1, MAO-B, Aβ1–42 aggregation, and hyperphosphorylated tau protein. The majority of the isolates exhibited promising inhibition activity against all tested targets. Moreover, the current study indicated that compounds 1, 2, and 7 exhibited potent inhibition effects against BACE1, MAO-B, Aβ1–42 aggregation, and hyperphosphorylated tau protein, followed by compounds 4 and 6.Taking into account previous reports that have discussed the possible neuroprotective activity of N-trans-feruloyltyramine through the stimulation of neurogenesis and neurotrophins,34 in addition to the role of N-trans-caffeoyltyramine and N-trans-coumaroyltyramine in the reduction of BACE1 gene expression,6 our study results suggested the contribution of amide alkaloids as effective lead compounds for the development of AD therapies. Furthermore, the study reveals the beneficial effects of B. indica and A. alopecuroides in the prevention as well as the potential treatment of neurodegenerative diseases owing to their phenyl amide contents.
Modeling and biophysics-based simulation studies showed interesting structural information regarding the putative interaction modes of these compounds inside the active sites of both MAOB and BACE1. This new information could be very helpful during the development of new inhibitor molecules using the scaffolds of the amide alkaloids reported here. In the future, further in vivo experiments will be needed to support the primary results obtained in our study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to Associate Prof. Shingo Yokota, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, Japan, for carrying out the IR measurements. The first author (A. O.) would like to express his deepest gratitude to the Egyptian Cultural Affairs and Missions' Sector, Ministry of Higher Education, Egypt for the scholarship and financial support.References
- Y. C. Kuo and R. Rajesh, Expert Rev. Neurother., 2019, 19, 623–652 CrossRef CAS PubMed.
- M. Tolar, S. Abushakra and M. Sabbagh, Alzheimers. Dement., 2020, 16, 1553–1560 CrossRef PubMed.
- L. Blaikie, G. Kay and P. K. T. Lin, MedChemComm, 2019, 10, 2052–2072 RSC.
- World Health Organization, Dementia, 2021 Search PubMed.
- J. H. Park, Y. H. Ju, J. W. Choi, H. J. Song, B. K. Jang, J. Woo, H. Chun, H. J. Kim, S. J. Shin, O. Yarishkin, S. Jo, M. Park, S. K. Yeon, S. Kim, J. Kim, M. H. Nam, A. M. Londhe, J. Kim, S. J. Cho, S. Cho, C. Lee, S. Y. Hwang, S. W. Kim, S. J. Oh, J. Cho, A. N. Pae, C. J. Lee and K. D. Park, Sci. Adv., 2019, 5, eaav0316 CrossRef CAS PubMed.
- J. R. Martinez, G. Selo, M. Á. Fernandez-Arche, B. Bermudez and M. D. García-Gimenez, J. Nat. Prod., 2021, 84, 2447–2453 CrossRef PubMed.
- S. S. Arya, S. Devi, K. Ram, S. Kumar, N. Kumar, A. Mann, A. Kumar and G. Chand, Ecophysiology, Abiotic Stress Responses and Utilization of Halophytes, 2019, pp. 271–287 Search PubMed.
- A. Othman, Y. Amen and K. Shimizu, Fitoterapia, 2021, 152, 104907 CrossRef CAS PubMed.
- A. Othman, Y. Amen, M. Matsumoto, M. Nagata and K. Shimizu, Nat. Prod. Res., 2021, 1–9 CrossRef PubMed.
- V. Täckholm, Students' Flora of Egypt, 1974 Search PubMed.
- L. Boulos, Kew Bull., 1992, 47, 283 CrossRef.
- T. Kuljanabhagavad, P. Thongphasuk, W. Chamulitrat and M. Wink, Phytochemistry, 2008, 69, 1919–1926 CrossRef CAS PubMed.
- R. S. El Dine, H. M. Abdallah, Z. A. Kandil, A. A. Zaki, S. I. Khan and I. A. Khan, Planta Med., 2019, 85, 274–281 CrossRef PubMed.
- R. Tundis, F. Menichini, F. Conforti, M. R. Loizzo, M. Bonesi, G. Statti and F. Menichini, J. Enzyme Inhib. Med. Chem., 2009, 24, 818–824 CrossRef CAS PubMed.
- P. Ninfali, E. Antonini, A. Frati and E.-S. Scarpa, Phytother. Res., 2017, 31, 871–884 CrossRef CAS PubMed.
- K. M. Khan, G. M. Maharvi, A. Abbaskhan, S. Hayat, M. T. H. Khan, T. Makhmoor, M. I. Choudhary, F. Shaheen and Atta-ur-Rahman, Helv. Chim. Acta, 2003, 86, 457–464 CrossRef CAS.
- R. R. Abd El-Latif, R. M. A. Mansour, M. Sharaf and A. Farag, J. Asian Nat. Prod. Res., 2014, 16, 434–439 CrossRef CAS PubMed.
- N. Hamza, B. Berke, A. Umar, C. Cheze, H. Gin and N. Moore, J. Ethnopharmacol., 2019, 238, 111841 CrossRef PubMed.
- D. Seeliger and B. L. de Groot, J. Comput.-Aided Mol. Des., 2010, 24, 417–422 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- S. Release, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY Search PubMed.
- K. J. Bowers, D. E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Scalable algorithms for molecular dynamics simulations on commodity clusters, In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, IEEE, New York, NY, USA, 2006, p. 43, DOI:10.1109/SC.2006.54.
- S. Kim, H. Oshima, H. Zhang, N. R. Kern, S. Re, J. Lee, B. Roux, Y. Sugita, W. Jiang and W. Im, J. Chem. Theory Comput., 2020, 16, 7207–7218 CrossRef CAS PubMed.
- S. T. Ngo, N. M. Tam, M. Q. Pham and T. H. Nguyen, J. Chem. Inf. Model., 2021, 61, 2302–2312 CrossRef CAS PubMed.
- C. A. B. Amaro, M. González-Cortazar, M. Herrera-Ruiz, R. Román-Ramos, L. Aguilar-Santamaría, J. Tortoriello and E. Jiménez-Ferrer, Molecules, 2014, 19, 11366–11384 CrossRef PubMed.
- H. Huang, Q. Chao, R. X. Tarll, H. Sun, D. Wang and S. Zhao, Planta Med., 1999, 65, 92–93 CrossRef CAS PubMed.
- J. Han, L. Li, L. Han, X. Huang and T. Yuan, Biochem. Syst. Ecol., 2015, 61, 399–401 CrossRef CAS.
- Y. H. Song, D. W. Kim, M. J. Curtis-Long, C. Park, M. Son, J. Y. Kim, H. J. Yuk, K. W. Lee and K. H. Park, Eur. J. Med. Chem., 2016, 114, 201–208 CrossRef CAS PubMed.
- J. Sun, H. X. Huo, J. Zhang, Z. Huang, J. Zheng, Q. Zhang, Y. F. Zhao, J. Li and P. F. Tu, Biochem. Syst. Ecol., 2015, 58, 265–269 CrossRef CAS.
- C. Qi, Q. Zhou, W. Gao, M. Liu, C. Chen, X. N. Li, Y. Lai, Y. Zhou, D. Li, Z. Hu, H. Zhu and Y. Zhang, Phytochemistry, 2019, 165, 112041 CrossRef CAS PubMed.
- M. H. Melchor, F. G. Susana, G. S. Francisco, H. I. Beltran, R. F. Norma, J. A. Lerma-Romero, P. Y. Lopez-Camacho and B. I. Gustavo, RSC Adv., 2018, 8, 39667–39677 RSC.
- S. Chinnathambi, Aggregation species of Amyloid-β and Tau oligomers in Alzheimer's Disease: Role in therapeutics and diagnostics, 2022, vol. 12 Search PubMed.
- K. Wojtunik-Kulesza, T. Oniszczuk, J. Mołdoch, I. Kowalska, J. Szponar and A. Oniszczuk, Int. J. Mol. Sci., 2022, 23, 1212 CrossRef CAS PubMed.
- Z. Khan, S. M. Hong, J. W. Lee, E. Y. Moon, J. Huh, K. A. Chang and S. Y. Kim, J. Funct. Foods, 2021, 80, 104432 CrossRef CAS.
- A. Diana, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, W. W. Keith and K. D. Kopple, J. Med. Chem., 2002, 45(12), 2615–2623 CrossRef CAS PubMed.
- Y. C. Wu, G. Y. Chang, F. N. Ko and C. M. Teng, Planta Med., 1995, 61(02), 146–149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02275c |
| This journal is © The Royal Society of Chemistry 2022 |