
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic oxygen evolution from hydrogen peroxide by trans-[Co(en)2Cl2]@InBTB metal–organic framework catalytic system†
Sukbin Yoona,
In-Hwan Choia,
Youngmee Kim *b and
Seong Huh*a
*b and
Seong Huh*a
aDepartment of Chemistry and Protein Research Center for Bio-Industry, Hankuk University of Foreign Studies, Yongin 17035, Republic of Korea. E-mail: shuh@hufs.ac.kr; Fax: +82 31 330 4566; Tel: +82 31 330 4522
bDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 120-750, Republic of Korea. E-mail: ymeekim@ewha.ac.kr
First published on 12th May 2022
Abstract
The diethylammonium counter-cations of the [Et2NH2]3[In3(BTB)4] metal–organic framework (InBTB MOF, BTB = 1,3,5-benzenetribenzoate) with an anionic framework can be effectively exchanged with cationic trans-[Co(en)2Cl2]+ complex ions through a simple cation-exchange process. The heterogenized trans-[Co(en)2Cl2]+-encapsulated InBTB MOF (trans-[Co(en)2Cl2]@InBTB) catalytic system maintained the activity of the captured trans-[Co(en)2Cl2]+ complex ion for hydrogen peroxide decomposition in aqueous solution under mild reaction conditions. The captured trans-[Co(en)2Cl2]+ complex also exhibited trans–cis isomerization to produce either cis-[Co(en)2Cl2]@InBTB or cis-[Co(en)2(H2O)Cl]@InBTB based on IR spectroscopic investigation. The trans-[Co(en)2Cl2]@InBTB catalytic system showed high recyclability for oxygen evolution from hydrogen peroxide. The catalytic ability of trans-[Co(en)2Cl2]@InBTB was maintained up to seven times of recycling.
1. Introduction
The large channels present in metal–organic frameworks (MOFs) are ideal hosts for encapsulating various functional guests including catalytically active transition metal complexes,1,2 viologens,3 optical dyes,4–11 metallic nanoparticles,12–15 and drug molecules.16,17 The crystalline nature of the frameworks and channels provide highly ordered spatial arrangements of the encapsulated guests compared to other porous materials. The catalytic activities of captured guests can be efficiently preserved or enhanced and the optical activities of the dye guests can be systematically controlled through simple encapsulation into channels of MOFs. In particular, MOFs with negatively charged frameworks contain counter-cations in the channels. Negative framework charges often develop due to the charge unbalance between cationic metal ions and anionic bridging ligands. The counter-cations near the metal centres counterbalance the charge mismatch to satisfy charge neutrality. The presence of these counter-cations and anionic frameworks often renders MOFs more functional. For example, the counter-cations can be readily replaced with other functional cationic guests through a simple cation-exchange process. Several excellent examples of this process for the preparation of functional MOF systems have recently been reported.1–6A large cationic Reichardt's dye (RD) could be effectively encapsulated into the mesoscale channels of three-dimensional (3D) InBTB MOF ([Et2NH2]3[In3(BTB)4]·10DEF·14H2O, H3BTB = 1,3,5-benzenetribenzoic acid, DEF = N,N-diethylformamide) through cation-exchange to give RD@InBTB.4 The doubly interpenetrated framework structure of InBTB is shown in Fig. S1† (ESI). A partially encapsulated RD exhibited a quite distinct absorption behaviour compared to free RD crystals due to confinement effects. Similarly, organic dipolar chromophore, 4-(4-(diphenylamino)styryl)-1-dodecylpyridinium bromide (DPASD), has been orderly encapsulated into another phase of InBTB (= ZJU-28) to afford DPASD@ZJU-28 exhibiting highly active nonlinear optical (NLO) property.5 Several cationic dyes were also effectively incorporated into InBTB to give a series of dye@InBTB MOFs with good optical properties.6 These examples clearly suggest that the cation-exchange-driven generation of functional MOFs is a promising strategy to develop new MOF systems with diverse functions.
Additionally, the relatively strong electrostatic interaction between the cationic guests and anionic frameworks is very suited for simple immobilization of catalytically active cationic species inside MOF channels to generate recyclable catalytic systems.1,2 The encapsulated catalytic species can also be stabilized due to confinement effects. Thus, there are lots of opportunities to prepare recyclable heterogeneous catalytic systems using MOFs with anionic frameworks. In this sense, Rosseinsky et al. reported a system in which the catalytically active organometallic cation, [CpFe(CO)2(L)]+ (Cp = η5-C5H5−, L = weakly coordinating solvent), was successfully encapsulated into the channels of a 3D anionic InBTC MOF ([Et4N]3[In3(BTC)4], H3BTC = 1,3,5-benzenetricarboxylic acid) through cation-exchange process.1 This approach was employed to develop a series of new functional MOFs for diverse chemical applications.2,3
In search of a recyclable heterogeneous O2 evolution catalytic system, an attempt was made to encapsulate cationic O2 evolution catalyst into the channels of InBTB. The large mesoscale channels of InBTB allowed the incorporation of a functional cationic Co(III) complex ion, trans-[Co(en)2Cl2]+ where en is an ethylenediamine,18 through a simple cation-exchange method as shown in Fig. 1. Although there are two recent articles to review current progress of MOFs encapsulating various dyes and complex ions,19,20 the catalytic activity and trans–cis isomerization of the captured trans-[Co(en)2Cl2]+ ion in large mesoscale channels of MOF are worth of investigation because trans–cis isomerization of the captured cationic complex in nanoscopic space of MOF has not yet been explored.
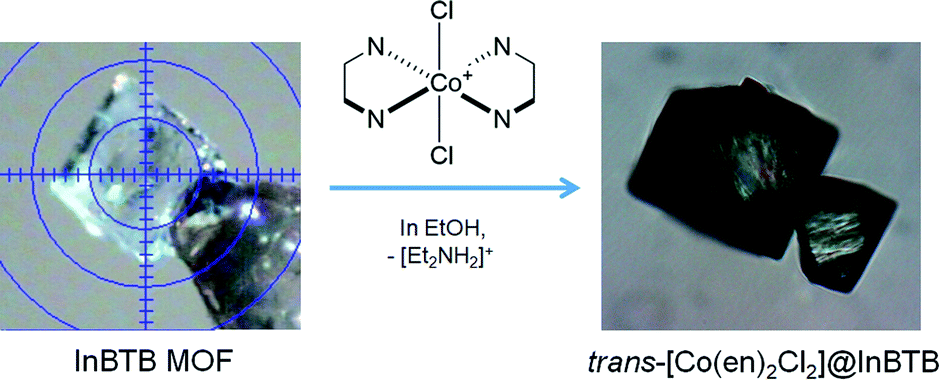 | ||
| Fig. 1 Schematic of the preparation of trans-[Co(en)2Cl2]@InBTB through the cation-exchange of [Et2NH2]+ with trans-[Co(en)2Cl2]+. | ||
2. Experimental methods
2.1. Synthesis of InBTB MOF and trans-[Co(en)2Cl2]Cl
The as-prepared InBTB was synthesized according to literature method.4 Trans-[Co(en)2Cl2]Cl was prepared according to literature method.182.2. Encapsulation of trans-[Co(en)2Cl2]Cl into InBTB MOF
The as-prepared InBTB MOF (10 mg) was added into 2 mM ethanol solution containing trans-[Co(en)2Cl2]Cl (10 mL). Encapsulation amount was measured by using UV/Vis spectrophotometer every 24 h for 120 h. Each measurement was performed by diluting 500 μL of the aliquot with 500 μL of ethanol in 1 mL quartz cuvette.2.3. Isomerization reaction of trans-[Co(en)2Cl2]Cl into cis-[Co(en)2Cl2]Cl in H2O
The trans–cis isomerization experiment was conducted by following the literature method.18 The trans-[Co(en)2Cl2]Cl (0.1 g) was dissolved in 3 mL of deionized water on a watch glass. The aqueous solution was kept at room temperature for 10 min. The watch glass was placed on the top of steam bath. After water was completely evaporated, violet solid product (cis-isomer) was formed.2.4. Isomerization of trans-[Co(en)2Cl2]@InBTB into cis-[Co(en)2Cl(H2O)]@InBTB or cis-[Co(en)2Cl2]@InBTB in H2O
The trans-[Co(en)2Cl2]@InBTB (5 mg) was transferred into a vial. Deionized water (5 mL) was added into the vial. The vial was sealed with a cap. Isomerization was carried out at 40 °C in water bath. The digital photo images of the sample in the vial were taken every 20 min for 120 h.2.5. Catalytic decomposition of H2O2 by trans-[Co(en)2Cl2]@InBTB
The trans-[Co(en)2Cl2]@InBTB (0.0293 g, 0.0140 mmol based on trans-[Co(en)2Cl2]+ ion) was transferred into a round bottom vial. Deionized water (20 mL) and 12.18 μL (0.140 mmol) of 34.5% H2O2 were added into the vial. The opening of the vial was covered loosely with aluminum foil. The reaction mixture was gently shaken at 40 °C in water bath. Titration was carried out periodically to monitor the decomposition of H2O2. Analyte was prepared by diluting 300 μL of aliquot with 22.5 mL of deionized water and acidifying with 79.5 μL of c-H2SO4. KMnO4 solution (0.08412 mM) was used as a titrant. After catalytic H2O2 decomposition, catalyst was filtered by using a P5-grade glass filter funnel and washed with deionized water. The retrieved catalyst was air-dried overnight and reused for recycling.2.6. Physical measurements
Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker New D8 Advance diffractometer (40 kV, 40 mA, step size = 0.02°). X-ray photoelectron spectroscopy (XPS) measurement was performed using an ESCA 2000 equipped with Al Kα as an X-ray source. The spectrum was calibrated by using C 1s signal at 284.6 eV. FT-IR spectra in attenuated total reflection (ATR) mode were recorded on a Jasco FT/IR-4100 spectrometer (Jasco, Japan). UV/Vis spectra were recorded on a Scinco S-3100 spectrophotometer (Scinco, Korea).2.7. Crystallography
The unit cell dimensions of trans-[Co(en)2Cl2]@InBTB was analyzed with a Bruker APEX-II diffractometer equipped with a monochromator using a Mo Kα (λ = 0.71073 Å) incident beam (CCDC 2142611).3. Results and discussion
3.1. Preparation of trans-[Co(en)2Cl2]@InBTB
The trans-[Co(en)2Cl2]+ complex was encapsulated into the channels of the as-prepared InBTB in ethanol solution. The free trans-[Co(en)2Cl2]+ readily underwent trans–cis isomerization in deionized water at room temperature (RT) through aquation (Fig. 2a). The aquation facilitated the dissociation of chloride ligand and subsequent isomerization in water.21 Contrarily, the cis-isomer spontaneously converted back into trans-isomer in the ethanol solution. Therefore, trans-isomer seemed to be more stabilized in ethanol. Jacewicz et al. reported that the conversion of cis-[Co(en)2Cl2]+ into trans-[Co(en)2Cl2]+ is a first order reaction in methanol.22 This process was also strongly solvent dependent and the methanol solvent required high activation barrier. In this case, the dissociation of one of the –NH2 group of the en ligand was a key step for the trans–cis isomerization. The trans-form (green) and cis-form (violet) showed different colors, so the process could be easily monitored by naked eyes and UV/Vis spectroscopy. Thus, ethanol could be chosen as a solvent for the encapsulation of trans-[Co(en)2Cl2]+ to retard the trans–cis isomerization during encapsulation. The encapsulation amount after 5 days of immersion of as-prepared InBTB (10 mg) in 2 mM ethanol solution of trans-[Co(en)2Cl2]Cl (10 mL) was 5.55 μmol/10 mg-solid (Fig. 2b). The encapsulation amount of the monovalent trans-[Co(en)2Cl2]+ ion was slightly larger than the values, 3.3–4.2 μmol/10 mg-solid, observed for a series of RuL3@InBTB MOFs encapsulating divalent cationic [RuL3]2+ polypyridine complexes: [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine), [Ru(phen)3]2+ (phen = 1,10-phenanthroline), and [Ru(bpz)3]2+ (bpz = 2,2′-bipyrazine).2 This discrepancy was mainly rooted in the smaller ion size of trans-[Co(en)2Cl2]+ ion than [RuL3]2+ ions.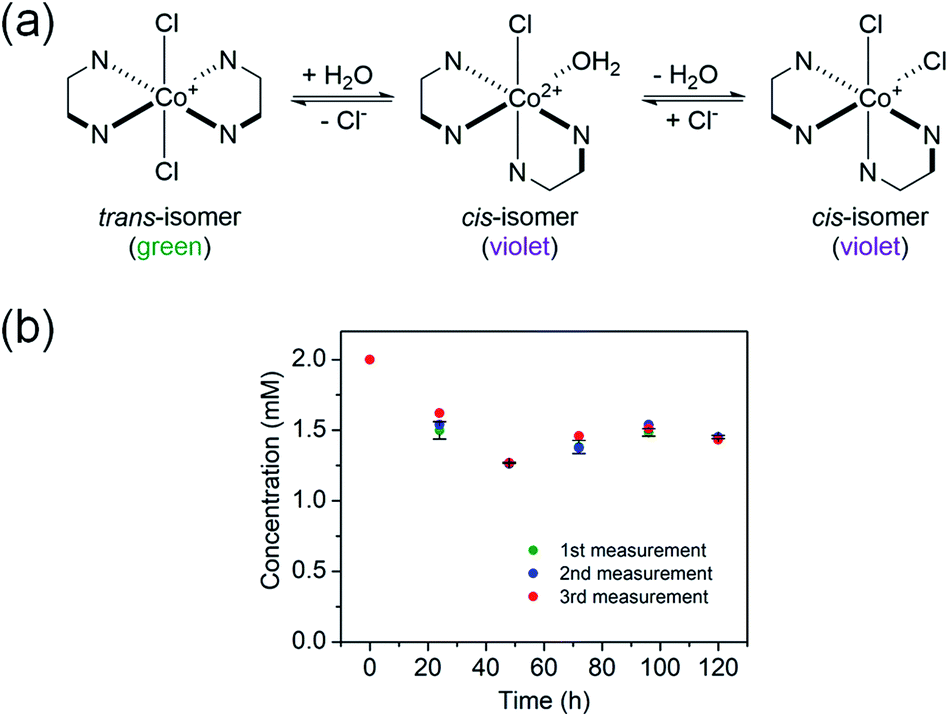 | ||
| Fig. 2 (a) The trans–cis isomerization process of trans-[Co(en)2Cl2]+ via aquation in water. (b) Time courses of the encapsulation of trans-[Co(en)2Cl2]+ ion by as-prepared InBTB in ethanol. The measurements were performed in triplicate. | ||
Although the crystallinity of InBTB was moderately preserved after encapsulation, single crystal analysis of trans-[Co(en)2Cl2]@InBTB was not successfully refined due to disordering of BTB ligand and relatively poor crystal quality compared to as-prepared InBTB. Nonetheless, the observed unit cell dimension change indicated the successful encapsulation of trans-[Co(en)2Cl2]+ ions. The unit cell dimension of a = 44.2269(19), b = 44.2269(19), c = 42.519(2) Å for InBTB changed into a = 44.879(8), b = 44.879(8), c = 42.029(7) Å for trans-[Co(en)2Cl2]@InBTB. While the trigonal unit cell dimensions of a- and b-axes for trans-[Co(en)2Cl2]@InBTB slightly increased but that of c-axis slightly decreased compared to as-prepared InBTB.4 The original unit cell dimensions of trigonal cell of InBTB tended to vary depending on the encapsulated guest ions as summarized in Table S1† (ESI). The approximate molecular dimension of trans-[Co(en)2Cl2]+ ion is 9.90 × 9.76 × 8.15 Å3. The dimension of the mesoscale channel of InBTB was estimated from the center-to-center distance of two alternating BTB ligands shown in Fig. S1† (ESI). The values are 27.034(1) and 15.486(1) Å.4 Therefore, the channel of InBTB is large enough to encapsulate trans-[Co(en)2Cl2]+ ion. It is worth noting that if the size of guest ions were suitable for the encapsulation into the channels, the spatial orientation of them would be more or less predetermined due to the fixed geometry of channels. This implied the possibility that guest ions were likely to be spatially arranged in a controlled manner on demand.
The successful incorporation of trans-[Co(en)2Cl2]+ ion was also confirmed by X-ray photoelectron spectroscopy (XPS) as shown in Fig. 3. The signals of Co 2p3/2 and Co 2p1/2 were observed at 780.73 and 796.11 eV, respectively.23 The estimated atom% ratio between In and Co elements was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.65. The powder X-ray diffraction (PXRD) pattern of trans-[Co(en)2Cl2]@InBTB indicated decreased crystallinity compared to that of as-prepared InBTB (Fig. S2, ESI†). We also observed similar phenomenon when we used EtOH for encapsulation of various cationic dyes.6 Nonetheless, we believe that the metal–ligand connectivity may not be seriously damaged because trans-[Co(en)2Cl2]+ ions did not leach out during catalytic reactions.
:0.65. The powder X-ray diffraction (PXRD) pattern of trans-[Co(en)2Cl2]@InBTB indicated decreased crystallinity compared to that of as-prepared InBTB (Fig. S2, ESI†). We also observed similar phenomenon when we used EtOH for encapsulation of various cationic dyes.6 Nonetheless, we believe that the metal–ligand connectivity may not be seriously damaged because trans-[Co(en)2Cl2]+ ions did not leach out during catalytic reactions.
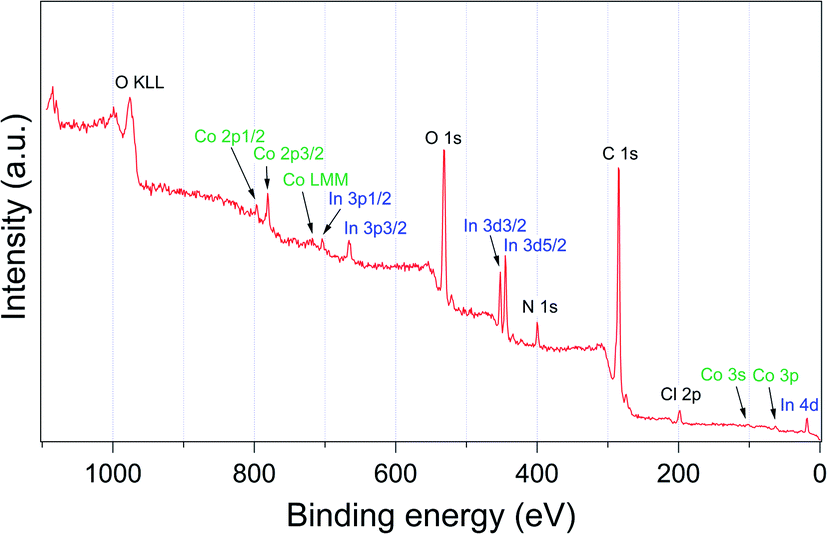 | ||
| Fig. 3 Survey XPS spectrum of trans-[Co(en)2Cl2]@InBTB. | ||
3.2. Trans–cis isomerization of the captured trans-[Co(en)2Cl2]+ ion of trans-[Co(en)2Cl2]@InBTB
Interestingly, the green color of trans-[Co(en)2Cl2]@InBTB changed very slowly into violet in the solid state over a couple of months. It was speculated that trans-isomer slowly isomerized into cis-form inside the channels. Although it was unable to identify water molecules in solid-state trans-[Co(en)2Cl2]@InBTB, it seemed that a small amount of water was present inside the channels and the aquation and isomerization slowly occurred in the confined spaces. This observation was quite remarkable because the trans–cis isomerization of metal complexes inside MOF channels has not been reported yet. It was believed that the mesoscale channels of InBTB behaved as a nanoscale flask.To further confirm the trans–cis isomerization of the trans-[Co(en)2Cl2]+ ion, trans-[Co(en)2Cl2]+ was treated in deionized water at 40 °C and its spectral changes were monitored for 1 h by UV/Vis spectroscopy (Fig. 4a). As expected, the gradual decrease of absorption band at 619 nm for trans-[Co(en)2Cl2]+ ion and a gradual increase of a new signal at 505 nm for cis-[Co(en)2Cl(H2O)]2+ ion clearly indicated the instantaneous occurring of the aquation-induced isomerization.21 Interestingly, once hydrogen peroxide (H2O2) was added in the above solution very similar spectral changes were observed (Fig. 4b). Thus, the trans–cis isomerization also occurred in the presence of hydrogen peroxide.
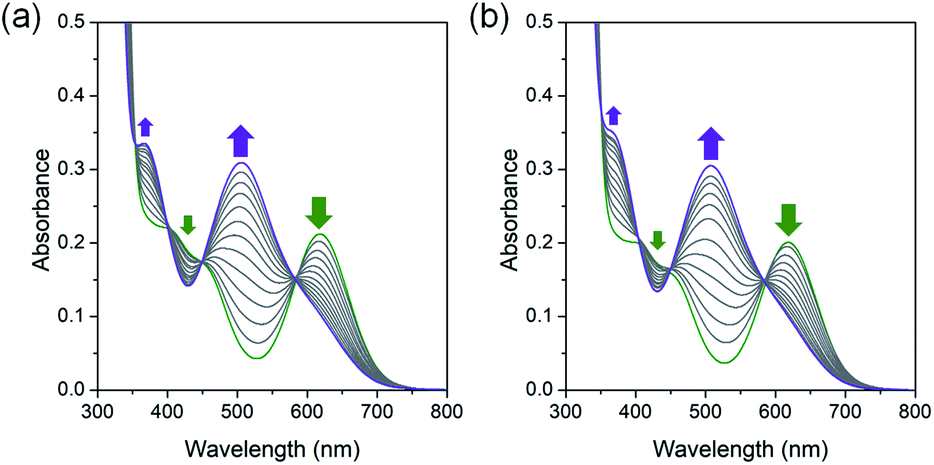 | ||
| Fig. 4 UV/Vis spectral change during the trans–cis isomerization process of trans-[Co(en)2Cl2]+ ion. (a) trans-[Co(en)2Cl2]Cl in deionized water at 40 °C for 1 h. (b) Trans-[Co(en)2Cl2]Cl in deionized water with hydrogen peroxide at 40 °C for 1 h. Green and violet arrows indicate the absorption signals from trans-[Co(en)2Cl2]+ and cis-[Co(en)2Cl(H2O)]2+ ions, respectively. | ||
3.3. Hydrogen peroxide decomposition by trans-[Co(en)2Cl2]@InBTB
The generation of O2 from hydrogen peroxide is an important process in biochemistry and water treatment.24 The catalytic process generating oxygen source from hydrogen peroxide also became increasingly important for partial oxidation of propene to produce propylene oxide by titanium silicate (TS-1) catalyst.25 Thus, the catalytic activity and recyclability of the new heterogenized O2 evolution system were investigated together with the trans–cis isomerization of the captured trans-[Co(en)2Cl2]+ species inside the MOF channels. Cowie and Wadi reported that polymer-bound trans-[Co(en)2Cl(NH2-polymer)]2+ derivatives displayed good catalytic activities for O2 evolution from hydrogen peroxide.26 The NH2-polymer derivatives (PMI-TETRAEN and PHpI-TETRAEN) were poly(monomethyl-co-dimethyl itaconate) and poly(mono-n-heptyl-co-di-n-heptyl itaconate) functionalized with tetraethylenepentamine. Nevertheless, the recyclability of this polymer-bound system was not fully demonstrated. In this regard, the catalytic activity and recyclability of the heterogenized trans-[Co(en)2Cl2]@InBTB catalytic system were tested for hydrogen peroxide decomposition.The trans-[Co(en)2Cl2]@InBTB displayed good catalytic activity for the decomposition of hydrogen peroxide to produce oxygen. The catalytic system was stable enough to recycle seven times without significant loss of catalytic activities (Fig. 5a and S3, ESI†). To our surprise, the conversion of hydrogen peroxide was gradually increased upon recycling up to sixth run. In general, the recycling of heterogeneous catalyst tended to show gradual decrease of activity because small amount of catalyst can be lost during recovery. Furthermore, the color of recycled catalyst changed into violet color even after first round of reaction (Fig. 5b). This color change may be attributable to the trans–cis isomerization of the captured trans-[Co(en)2Cl2]+ species. It was speculated that a small portion of the captured trans-[Co(en)2Cl2]+ kept transforming to cis-isomers, either cis-[Co(en)2Cl(H2O)]2+ or cis-[Co(en)2Cl2]+, during the catalytic reaction. When all captured trans-[Co(en)2Cl2]+ converted into cis-isomers during the recycling experiments, then the reaction rate enhancement was no longer possible. Therefore, the reaction conversion of seventh run was marginally smaller than that of the sixth run. The most widely accepted reaction mechanism for the decomposition of hydrogen peroxide by transition metal active sites suggested potential generation of Co(III)-hydroperoxy species through H–OOH bond activation.24,27 Based on this proposed reaction mechanism, the above mentioned cis-isomers were likely to be more reactive than trans-isomer. Also, oxygen bubbles formed right on the surfaces of MOF crystals were able to be observed during the reaction (Fig. 5b). This inferred that the captured trans-[Co(en)2Cl2]+ species maintained its catalytic activity very well. The concentration of the catalyst was 10 mol% relative to the substrate in the catalytic reaction. Therefore, the turnover number (TON) can be easily calculated from the conversion values. The highest TON for trans-[Co(en)2Cl2]@InBTB during recycling experiment was 6.8 at the 6th recycling. The aforementioned polymer-bound trans-[Co(en)2Cl(NH2-polymer)]2+ derivatives exhibited conversions of ∼8% after 9 h with the catalyst concentration of 11.6 mol%.26 If we compare the conversions after 9 h, trans-[Co(en)2Cl2]@InBTB showed higher activities than trans-[Co(en)2Cl(NH2-polymer)]2+ derivatives as shown in Fig. 5a.
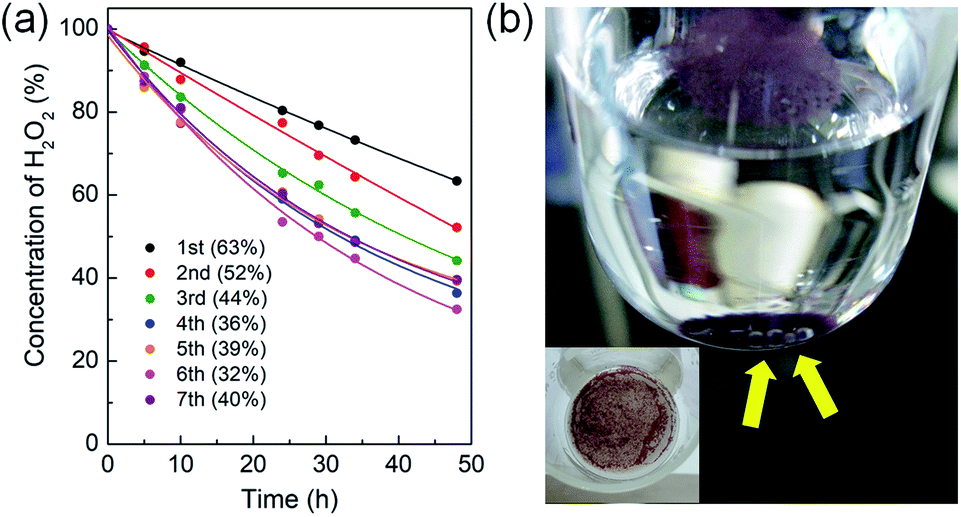 | ||
| Fig. 5 (a) The conversions of the hydrogen peroxide decomposition catalyzed by trans-[Co(en)2Cl2]@InBTB at 40 °C. The catalyst was recycled up to seven times. (b) The color-changed trans-[Co(en)2Cl2]@InBTB catalyst during the decomposition of hydrogen peroxide. Yellow arrows indicate the oxygen bubbles formed on the surfaces of MOF crystals during the decomposition of hydrogen peroxide. Inset shows violet solids recovered using a glass frit after reaction. | ||
As a control experiment of the color change of trans-[Co(en)2Cl2]@InBTB during the reaction, the reaction condition was emulated in the absence of hydrogen peroxide in water at two different temperatures, 25 and 40 °C. As depicted in Fig. 6a and b, the higher the temperature, the faster the color change. At 25 °C, the green color did not change after 2 h. However, the color of the crystals changed into violet after about 1 h at 40 °C. This implied that the color change was attributable to the trans–cis isomerization of the captured trans-[Co(en)2Cl2]+, because the isomerization process was temperature dependent.22 The color change of H2O2-treated trans-[Co(en)2Cl2]@InBTB is also clearly discernible as shown in Fig. S4† (ESI).
 | ||
| Fig. 6 Trans-[Co(en)2Cl2]@InBTB in deionized water at 25 °C (a) and 40 °C (b). | ||
The Fourier-transform infrared (FT-IR) spectra of the as-prepared InBTB and trans-[Co(en)2Cl2]@InBTB are shown in Fig. 7. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of the DEF solvate in the as-prepared InBTB at 1657 cm−1 almost disappeared for trans-[Co(en)2Cl2]@InBTB due to the exchange with trans-[Co(en)2Cl2]+ ions or ethanol solvent.28 Contrarily, the deformation signal of –NH2 group in en ligand was newly observed at 1581 cm−1 for trans-[Co(en)2Cl2]@InBTB.21 We also measured the FT-IR spectra of cis-[Co(en)2Cl2]Cl and trans-[Co(en)2Cl2]Cl complexes as shown in Fig. S5† (ESI). The stretching frequencies of –NH2 group are slightly different from each other: 1577 cm−1 for cis-[Co(en)2Cl2]Cl and 1594 cm−1 for trans-[Co(en)2Cl2]Cl due to different molecular symmetry. As we proposed earlier, H2O2-treated trans-[Co(en)2Cl2]@InBTB showed a signal at 1578 cm−1 indicating the possible trans–cis isomerization. Thus, the signal observed at 1581 cm−1 for trans-[Co(en)2Cl2]@InBTB indicates the formation of cis-isomers even in solid state without H2O2 treatment. The retrieved trans-[Co(en)2Cl2]@InBTB after catalytic reaction did not show significant IR spectral change compared to trans-[Co(en)2Cl2]@InBTB. Despite the possible isomerization of the captured trans-[Co(en)2Cl2]+ ion, the FT-IR study indicated that the trans-[Co(en)2Cl2]@InBTB was a highly robust catalytic system for hydrogen peroxide decomposition.
O stretching frequency of the DEF solvate in the as-prepared InBTB at 1657 cm−1 almost disappeared for trans-[Co(en)2Cl2]@InBTB due to the exchange with trans-[Co(en)2Cl2]+ ions or ethanol solvent.28 Contrarily, the deformation signal of –NH2 group in en ligand was newly observed at 1581 cm−1 for trans-[Co(en)2Cl2]@InBTB.21 We also measured the FT-IR spectra of cis-[Co(en)2Cl2]Cl and trans-[Co(en)2Cl2]Cl complexes as shown in Fig. S5† (ESI). The stretching frequencies of –NH2 group are slightly different from each other: 1577 cm−1 for cis-[Co(en)2Cl2]Cl and 1594 cm−1 for trans-[Co(en)2Cl2]Cl due to different molecular symmetry. As we proposed earlier, H2O2-treated trans-[Co(en)2Cl2]@InBTB showed a signal at 1578 cm−1 indicating the possible trans–cis isomerization. Thus, the signal observed at 1581 cm−1 for trans-[Co(en)2Cl2]@InBTB indicates the formation of cis-isomers even in solid state without H2O2 treatment. The retrieved trans-[Co(en)2Cl2]@InBTB after catalytic reaction did not show significant IR spectral change compared to trans-[Co(en)2Cl2]@InBTB. Despite the possible isomerization of the captured trans-[Co(en)2Cl2]+ ion, the FT-IR study indicated that the trans-[Co(en)2Cl2]@InBTB was a highly robust catalytic system for hydrogen peroxide decomposition.
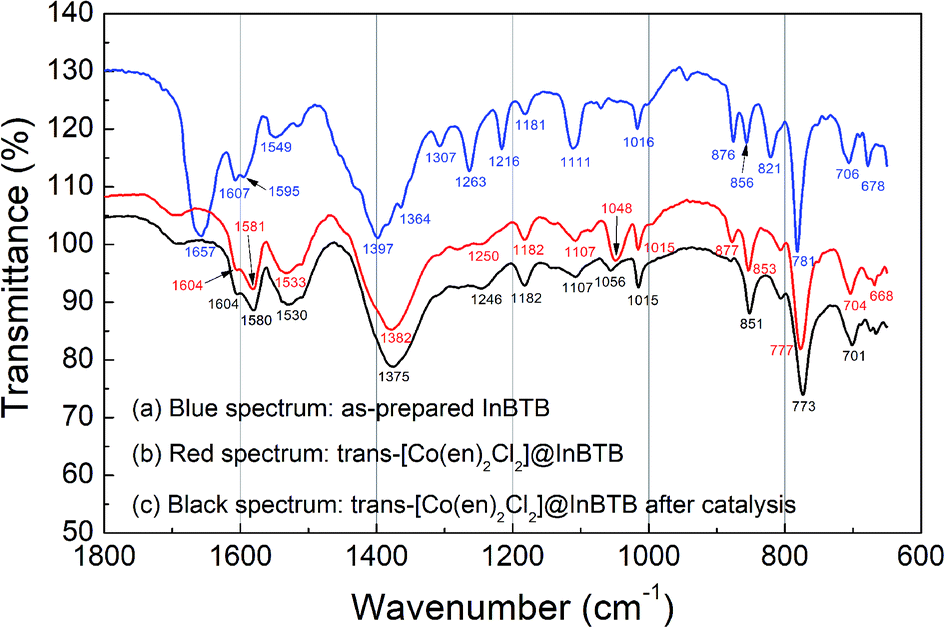 | ||
| Fig. 7 FT-IR spectra of the as-prepared InBTB (a), trans-[Co(en)2Cl2]@InBTB (b), and the retrieved trans-[Co(en)2Cl2]@InBTB after catalytic reaction (c). | ||
4. Conclusions
The cationic trans-[Co(en)2Cl2]+ complex was encapsulated into the channels of InBTB through a simple cation-exchange method. The trans-[Co(en)2Cl2]@InBTB was found to be highly recyclable O2 evolution catalytic system using hydrogen peroxide. The decomposition of hydrogen peroxide could be repeated seven times without significant loss of activity. The activity was gradually enhanced upon recycling possibly because of trans–cis isomerization via aquation to produce captured cis-[Co(en)2Cl(H2O)]2+ or cis-[Co(en)2Cl2]+ due to stronger trans effect of Cl− ligand compared to N-donor atom of the en ligand. The latter species were likely to be more active than trans-[Co(en)2Cl2]+ for the decomposition of hydrogen peroxide. These results were interesting because the mesoscale channels of InBTB behaved as a nanoscale flask for the unprecedented trans–cis isomerization of catalytically active transition metal complex in confined spaces. It is believed that employing this basic yet efficient strategy, there will be numerous opportunities to create novel functional MOF systems.Conflicts of interest
The authors have no conflicts of interest to disclose.Acknowledgements
This work was supported by grants (2018R1D1A1B07043017 and 2018R1D1A1B07045327) of the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology, Republic of Korea.Notes and references
- A. Grigoropoulos, G. F. S. Whitehead, N. Perret, A. P. Katsoulidis, F. M. Chadwick, R. P. Davies, A. Haynes, L. Brammer, A. S. Weller, J. Xiao and M. J. Rosseinsky, Chem. Sci., 2016, 7, 2037–2050 RSC.
- I.-H. Choi, S. Yoon, S. Huh, S.-J. Kim and Y. Kim, Chem.–Eur. J., 2020, 26, 14580–14584 CrossRef CAS PubMed.
- S. Yoon, H.-C. Kim, Y. Kim and S. Huh, Bull. Korean Chem. Soc., 2021, 42, 326–332 CrossRef CAS.
- E.-Y. Cho, J.-M. Gu, I.-H. Choi, W.-S. Kim, Y.-K. Hwang, S. Huh, S.-J. Kim and Y. Kim, Cryst. Growth Des., 2014, 14, 5026–5033 CrossRef CAS.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542–10545 CrossRef CAS PubMed.
- I.-H. Choi, S. B. Yoon, S. Huh, S.-J. Kim and Y. Kim, Sci. Rep., 2018, 8, 9838 CrossRef PubMed.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- M. Handke, T. Adachi, C. Hu and M. D. Ward, Angew. Chem., Int. Ed., 2017, 56, 14003–14006 CrossRef CAS PubMed.
- D.-M. Chen, N.-N. Zhang, C.-S. Liu and M. Du, ACS Appl. Mater. Interfaces, 2017, 9, 24671–24677 CrossRef CAS PubMed.
- W. Zhang, R.-Z. Zhang, Y.-Q. Huang and J.-M. Yang, Cryst. Growth Des., 2018, 18, 7533–7540 CrossRef CAS.
- R. Grünker, V. Bon, A. Heerwig, N. Klein, P. Müller, U. Stoeck, I. A. Baburin, U. Mueller, I. Senkovska and S. Kaskel, Chem. - Eur. J., 2012, 18, 13299–13303 CrossRef PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- H. Liu, L. Chang, C. Bai, L. Chen, R. Luque and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5019–5023 CrossRef CAS PubMed.
- B. Gole, U. Sanyal, R. Banerjee and P. S. Mukherjee, Inorg. Chem., 2016, 55, 2345–2354 CrossRef CAS PubMed.
- M. Mon, M. A. Rivero-Crespo, J. Ferrando-Soria, A. Vidal-Moya, M. Boronat, A. Leyva-Pérez, A. Corma, J. C. Hernández-Garrido, M. López-Haro, J. J. Calvino, G. Ragazzon, A. Credi, D. Armentano and E. Pardo, Angew. Chem., Int. Ed., 2018, 57, 6186–6191 CrossRef CAS PubMed.
- L. Wang, M. Zheng and Z. Xie, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- D.-C. Zhong, L.-Q. Liao, J.-H. Deng, Q. Chen, P. Liana and X.-Z. Luo, Chem. Commun., 2014, 50, 15807–15810 RSC.
- J. C. Bailar, Inorg. Synth., 1946, 2, 222–225 Search PubMed.
- P. Mialane, C. Mellot-Draznieks, P. Gairola, M. Duguet, Y. Benseghir, O. Oms and A. Dolbecq, Chem. Soc. Rev., 2021, 50, 6152–6220 RSC.
- Y. Zhou, J. Lu, Y. Zhou and Y. Liu, Environ. Pollut., 2019, 252, 352–365 CrossRef CAS PubMed.
- J. P. Lanorio and J. G. Lanorio, J. Lab. Chem. Educ., 2018, 6, 159–163 CAS.
- D. Jacewicz, J. Pranczk, D. Wyrzykowski, K. Żamojć and L. Chmurzyński, React. Kinet., Mech. Catal., 2014, 113, 321–331 CrossRef CAS.
- T. E. Khalil, S. M. Soliman, N. A. Khalil, A. El-Faham, S. Foro and A. El-Dissouky, Appl. Organomet. Chem., 2022, 36, e6565 CrossRef CAS.
- J. de Laat and H. Gallard, Environ. Sci. Technol., 1999, 33, 2726–2732 CrossRef CAS.
- A. Thetford, G. J. Hutchings, S. H. Taylor and D. J. Willock, Proc. R. Soc. A, 2011, 467, 1885–1899 CrossRef CAS.
- J. M. G. Cowie and N. M. A. Wadi, Polymer, 1985, 26, 1571–1574 CrossRef CAS.
- J. To, A. A. Sokol, S. A. French and C. R. A. Catlow, J. Phys. Chem. C, 2008, 112, 7173–7185 CrossRef CAS.
- I.-H. Choi, Y. Kim, D. N. Lee and S. Huh, Polyhedron, 2016, 105, 96–103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Framework structure, PXRD patterns, photo images of crystals, IR spectra, and table of unit cell parameters. See https://doi.org/10.1039/d2ra02208g |
| This journal is © The Royal Society of Chemistry 2022 |