
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hemin-catalyzed oxidative oligomerization of p-aminodiphenylamine (PADPA) in the presence of aqueous sodium dodecylbenzenesulfonate (SDBS) micelles†
Nemanja Cvjetan a,
Reinhard Kissnerb,
Danica Bajuk-Bogdanovićc,
Gordana Ćirić-Marjanovićc and
Peter Walde*a
a,
Reinhard Kissnerb,
Danica Bajuk-Bogdanovićc,
Gordana Ćirić-Marjanovićc and
Peter Walde*a
aDepartment of Materials, Laboratory for Multifunctional Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: peter.walde@mat.ethz.ch
bDepartment of Chemistry and Applied Biosciences, Laboratory of Inorganic Chemistry, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland
cFaculty of Physical Chemistry, University of Belgrade, Studentski trg 12-16, 11158 Belgrade, Serbia
First published on 3rd May 2022
Abstract
In a previous report on the enzymatic synthesis of the conductive emeraldine salt form of polyaniline (PANI-ES) in aqueous solution using PADPA (p-aminodiphenylamine) as monomer, horseradish peroxidase isoenzyme C (HRPC) was applied as a catalyst at pH = 4.3 with H2O2 as a terminal oxidant. In that work, anionic vesicles were added to the reaction mixture for (i) guiding the reaction to obtain poly(PADPA) products that resemble PANI-ES, and for (ii) preventing product precipitation (known as the “template effect”). In the work now presented, instead of native HRPC, only its prosthetic group ferric heme b (= hemin) was utilized as a catalyst, and micelles formed from SDBS (sodium dodecylbenzenesulfonate) served as templates. For the elaborated optimal reaction conditions, complementary UV/vis/NIR, EPR, and Raman spectroscopy measurements clearly showed that the reaction mixture obtained after completion of the reaction contained PANI-ES-like products as dominating species, very similar to the products formed with HRPC as catalyst. HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonate) was found to have a positive effect on the reaction rate as compared to dihydrogenphosphate. This work is the first on the template-assisted formation of PANI-ES type products under mild, environmentally friendly conditions using hemin as a cost-effective catalyst.
1. Introduction
In a series of previous studies, it was shown that HRPC (horseradish peroxidase isoenzyme C) can be successfully used in the presence of hydrogen peroxide (H2O2) as a terminal oxidant in aqueous solution at pH = 4.3 and at room temperature for the synthesis of oligomeric and polymeric products rich in the conductive emeraldine salt form of linear polyaniline (PANI-ES) (Fig. 1). As monomers, either aniline,1–4 the aniline dimer p-aminodiphenylamine (PADPA)5 (Fig. 1), or mixtures of aniline and PADPA6 were used. Moreover, for obtaining reaction products that contain substantial amounts of the ideal PANI-ES repeating unit, i.e., half oxidized and protonated linear tetraaniline (Fig. 1), the presence of aggregates of anionic amphiphiles (mainly micelles or vesicles)1,7 or anionic polyelectrolytes8 as “templates” was shown to be essential. One of the roles these templates play in the reaction mixture is to accumulate the monomers by electrostatic and hydrophobic interactions, thereby increasing their local concentration. As a consequence, once the monomers are oxidized by HRPC/H2O2, the follow-up reactions are influenced by the templates in a positive way, leading to a desired coupling of the oxidized monomers so that finally PANI-ES repeating units are obtained.4,6,9–12 The use of anionic vesicles from AOT (the sodium salt of bis(2-ethylhexyl) sulfosuccinate) turned out to be particularly suitable for these HRPC-catalyzed reactions,1,5,13 but the reactions also proceeded well with micelles formed from SDBS (sodium dodecylbenzenesulfonate) (Fig. 1) using aniline as monomer.14 In the presence of SDBS micelles, however, HRPC is less stable than in the presence of AOT vesicles.14,15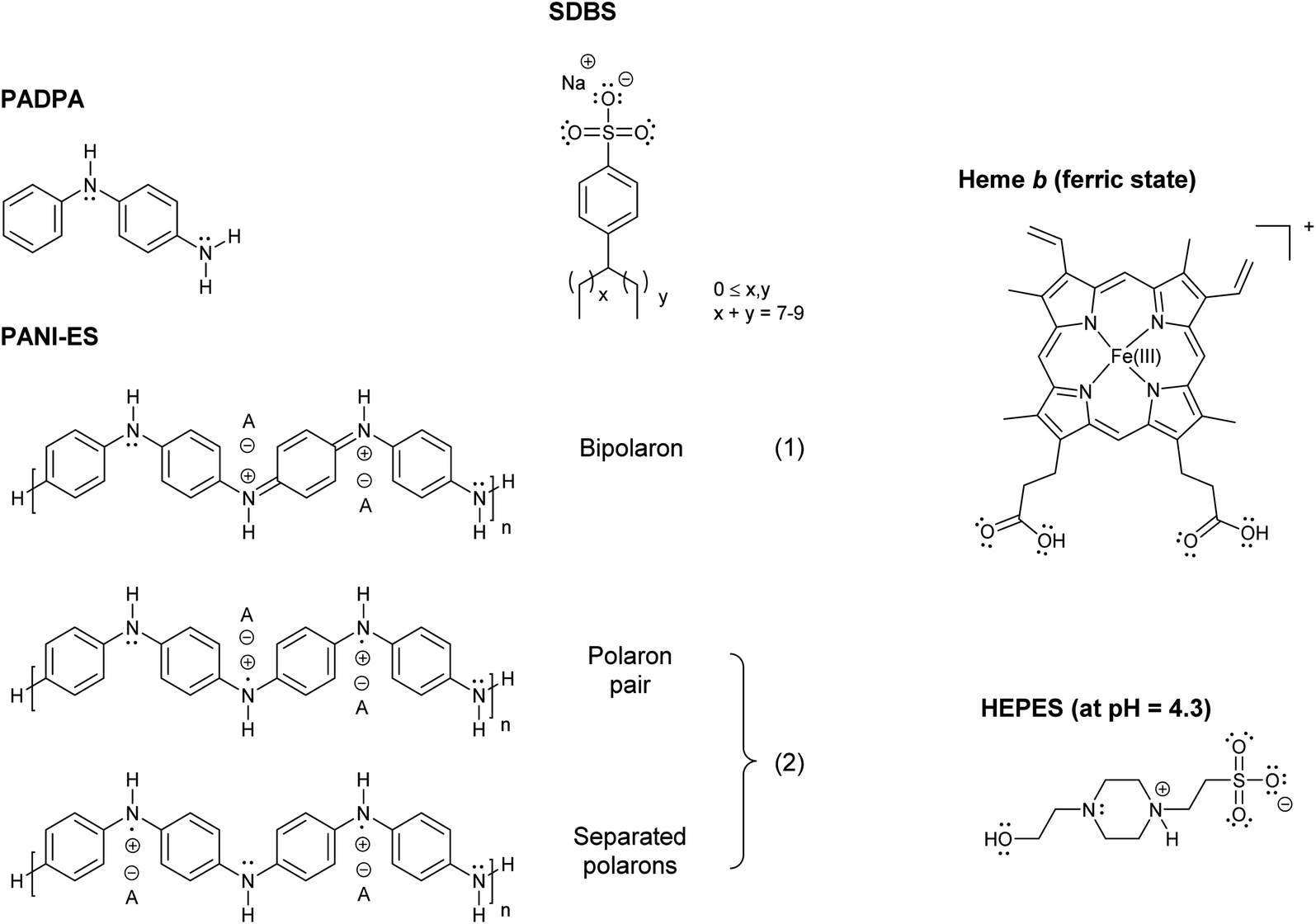 | ||
| Fig. 1 Chemical structures of PADPA (= p-aminodiphenylamine); PANI-ES, the emeraldine salt form of linear polyaniline in the bipolaron (1) and conductive polaron (2) states; SDBS (= sodium dodecylbenzenesulfonate), hard type;15 heme b in its ferric state (= Fe(III)protoporphyrin IX = por FeIII = hemin) with undissociated carboxylic acid groups at pH = 4.3;27 and HEPES (= 4-(2-hydroxyethyl)-1-piperazineethansulfonic acid) in the zwitterionic form, which dominates at pH = 4.3 (deprotonated sulfonic acid and protonated N-atom close to the sulfonate group).55 | ||
The idea behind the work presented now was to try to synthesize PANI-ES products from PADPA as monomer in the presence of SDBS micelles by using instead of HRPC its prosthetic group, ferric heme b‡ (Fig. 1).
In ferric heme b, Fe3+ is coordinated to the four nitrogen atoms of protoporphyrin IX (Fig. 1). Other designations and abbreviations for ferric heme b are iron(III) protoporphyrin IX, Fe(III)PPIX, or porFeIII (“por” standing for “protoporphyrin IX”). Ferric heme b is also known as hemin.16 The commercially available hemin form contains a chloride ion at the 5th coordination position (porFeIII-Cl).
The SDBS micelles used should play two essential roles in the reaction. They should not only serve as templates for the reaction, but also as a host for hemin by providing a local environment that ensures that hemin is in a catalytically active state. It is well known that heme b can form aggregates in an aqueous medium depending on the experimental conditions: π–π dimers, μ-oxo dimers, or large stacks of μ-oxo dimers.17–19 However, since only monomeric heme b is catalytically active, like at the active site of heme peroxidases20,21 and of other heme proteins,22–24 binding of hemin to the micelles must prevent hemin aggregation. Therefore, the challenges of the work were to investigate (i) whether suitable conditions can be found under which hemin in the presence of anionic SDBS micelles is in catalytically active (monomeric) state towards PADPA as substrate, and (ii) whether PANI-ES-like oligo- or poly(PADPA) products are formed under these conditions.
The use of micelles for hosting heme b was studied before, although investigations of the average localization of heme b in micelles are still rare.25–27 Based on previous reports, it is evident that only in the presence of certain types of micelle-forming surfactants monomeric hemin is obtained.17 So far, sodium dodecylsulfate (SDS) was most commonly used.25–30
Other possibilities for obtaining monomeric hemin in aqueous medium are to work at very low concentration,18–20 to use human serum albumin, to which heme b binds in monomeric state at one of the hydrophobic binding sites,31–33 or to form complexes with guanine (G)-rich DNAs (so-called G-quadruplex-DNAs).34–38 In the latter case, the activity of such complexes towards aniline was reported by Shen et al. (2014).39 Compared to G-quadruplex-DNAs or human serum albumin, however, SDBS micelles are not only simpler and cheaper hosts for hemin, but they can also serve as templates for the reaction (see above).
When hemin is placed in a non-protein environment, as in this work with SDBS micelles, a logical question arises: does the hemin/SDBS micelle system follow the same catalytic cycle as heme peroxidases do (but perhaps less efficiently)? Or, do alternative mechanisms play a role? Scheme S-1, ESI,† provides an overview of the peroxidase cycle of HRPC.1,20,40–48 In brief, the two-electron oxidation by H2O2 of porFeIII in the resting state of HRPC is followed by two one-electron oxidations of a reducing substrate, which is PADPA in the present case. Depending on the reducing substrate, non-enzymatic follow-up reactions lead to the final product(s), i.e., oligo- or/and poly(PADPA).1,5
Regardless of the mechanism by which PADPA is oxidized, the performance of the hemin/SDBS micelle system for the oxidation of PADPA to PANI-ES-like products was evaluated using the following criteria: (i) presence of spectroscopic features of the reaction products that are characteristic for the formation of PANI-ES; (ii) presence of high amounts of favorable PANI-ES products; (iii) high “quality” of the formed products in terms of amount of linear PANI-ES products vs. amount of unfavorable products containing branches or phenazine-type units; and (iv) no precipitation of the products formed. Therefore, the aqueous reaction mixtures were analyzed by three complementary methods, UV/vis/NIR, EPR, and in situ Raman spectroscopy measurements. Characteristic absorption bands for PANI-ES-like products are in the near-infrared (NIR) and visible (vis) regions of the spectrum, at λmax ≈ 800–1100 nm, assigned to the π → polaron transition,49 and at λmax ≈ 420 nm, assigned to the polaron → π* transition,49 with low absorption at λ ≈ 500–600 nm, indicative for the absence of extensive branching and absence of phenazine-type units.5,8 Due to the presence of unpaired electrons, conductive PANI-ES products in their polaron form (Fig. 1) have an EPR spectrum.50–52 Raman bands originating from C–N˙+ stretching vibrations of the polaron form of PANI-ES are at wavenumbers (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) in the range of 1320–1380 cm−1.11,53
) in the range of 1320–1380 cm−1.11,53
As a part of the work with the hemin/SDBS micelle system, we also explored the possible effect the type of salt used for the preparation of the aqueous pH = 4.3 solution might have on the rate of reaction and on the product distribution.§ Although all previous investigations with HRPC and PADPA were carried out with a sodium dihydrogenphosphate solution of pH = 4.3 (0.1 M), in the current work with heme b, a pH = 4.3 solution of 0.1 M HEPES was mainly used. The reason for this is the known positive effect HEPES molecules have on the activity of ferric heme b.54
2. Materials and methods
2.1. Commercial materials
HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 99%, for biochemistry, Mw = 238.30 g mol−1, lot A0233527, and hydrogen peroxide (H2O2) for analysis, 35 wt% solution in water, stabilized, lot: A0352305 were from Acros Organics. Sodium phosphate monobasic (sodium dihydrogenphosphate, NaH2PO4), ReagentPlus, ≥99.0%, lot BCBQ6142V, was purchased from Sigma-Aldrich. PADPA (p-aminodiphenylamine, N-phenyl-p-phenylenediamine), 98%, Mw = 184.24 g mol−1, lot MKBX9690V was from Aldrich Chemistry. PADPA was recrystallized from n-hexane as described before.52 Hemin from porcine, BioXtra, ≥97.0% (HPLC), lot BCCB6735, Mw = 651.94 g mol−1, was from Sigma. SDBS (dodecylbenzenesulfonic acid sodium salt, hard type), >95% (T), Mw = 348.48 g mol−1, was purchased from TCI Chemicals. Dimethyl sulfoxide (DMSO), Analytical reagent, lot 18K084026, was from VWR Chemicals, and n-hexane, for liquid chromatography, LiChrosolv, was from Merck. HRPC (horseradish peroxidase isoenzyme C), PEO-131, grade I, 271 U mg−1, RZ ≥ 3, lot 8153665000 was from Toyobo Enzymes. TMB (3,3′,5,5′-tetramethylbenzidine), lot BCBV1333 was purchased from Aldrich Chemistry. Polypropylene Eppendorf tubes (2 mL reaction tubes) were purchased from Greiner Bio-One GmbH.2.2. Preparation of stock solutions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 which gave a concentration of 77.6 μM.16 Such stock solution was kept at T = 4 °C. It remained very stable and could be used for at least for up to 2 years after preparation. For carrying out the reactions, stock solutions of lower HRPC concentration (0.8 μM) were freshly prepared before use. This was done by adding 10 μL of the 77.6 μM HRPC stock solution to 990 μL of the appropriate aqueous pH 4.3 solution.
000 M−1 cm−1 which gave a concentration of 77.6 μM.16 Such stock solution was kept at T = 4 °C. It remained very stable and could be used for at least for up to 2 years after preparation. For carrying out the reactions, stock solutions of lower HRPC concentration (0.8 μM) were freshly prepared before use. This was done by adding 10 μL of the 77.6 μM HRPC stock solution to 990 μL of the appropriate aqueous pH 4.3 solution.2.3. Preparation and analysis of reaction mixtures
3. Results and discussion
3.1. Determination of the “optimal reaction conditions” for poly(PADPA) formation with hemin as catalyst
With “optimal reaction conditions” we mean reaction conditions (i) at which the formed amount of favorable, dark-green, PANI-ES-like poly(PADPA) products is as high as possible, while (ii) the amount of unfavorable products containing branches and/or phenazine-type units is low. Moreover, (iii) the amount of products obtained from PADPA as monomer should allow UV/vis/NIR absorption measurements inside cuvettes with a path length of 0.1 cm without dilution of the reaction mixture; and (iv) precipitation of intermediates and products during the reaction or after completion of the reaction should not occur. These requirements were set by ourselves for practical reasons since they allow a clear and simple screening of different reaction conditions by direct UV/vis/NIR measurements without the involvement of possible effects originating from post-reaction treatments and from reaction product isolation. Our main interest was in finding reaction conditions that resulted in the appearance of absorption bands at λ ≈ 1000 and 420 nm (as expected for PANI-ES-like products) and absence of strong bands at λ ≈ 500–600 nm (i.e., absence of undesired branching and/or phenazine-type structure formation), see Introduction. Based on our previous work on the enzymatic oxidative oligomerization of PADPA,5 the initial concentrations for the reactions with hemin were kept at [PADPA]0 = 1.0 mM and [H2O2]0 = 1.0 mM. Furthermore, based on previous literature reports on the catalytic activity of hemin, the concentration of hemin used in all measurements was kept constant at [hemin] = 10 μM.57–59 This concentration is also useful for direct spectrophotometric measurements of hemin itself in the region of the Soret band (A400 (l = 1 cm) ≈ 0.5–1.2), so that information about possible changes in the aggregation state of hemin upon changing the reaction medium (in the absence of PADPA, i.e., without reaction) can be obtained.17For reference reactions with HRPC, the PADPA and H2O2 concentrations used were the same as in the case of the reactions with hemin, [PADPA]0 = [H2O2]0 = 1.0 mM, and the HRPC concentration applied was the one applied in our previous work with AOT vesicles as templates:5 [HRPC] = 30 nM.
As mentioned in the Introduction, SDBS micelles were used for hosting hemin and as templates for the reaction. The determination of the optimal SDBS concentration was carried out separately for the hemin- and the HRPC-catalyzed reactions. For both optimizations, an aqueous 0.1 M HEPES solution with pH = 4.3 was used. The spectra measured after a reaction time of t = 24 h in the presence of different SDBS concentrations are shown in Fig. S-1 and S-2, ESI.† The optimal SDBS concentration for the hemin system was found to be 5.0 mM, while for the HRPC system it is 3.0 mM. The same optimal SDBS concentrations were also obtained when an aqueous 0.1 M dihydrogenphosphate solution at pH = 4.3 was used instead of the HEPES solution (data not shown). For both systems, SDBS may play at least two roles, see Introduction. First, hemin (amphiphilic and positively charged at low pH if water molecules coordinate at the 5th and 6th coordination site) and HRPC (overall positively charged at pH = 4.3, pI(HRPC) = 8.8,21 are expected to bind to SDBS micelles. Therefore, the micelles localize the catalytic steps of the reaction. Second, cationic PADPA and (some of) the reaction intermediates and products are expected to bind to SDBS micelles, similarly to what was shown in the case of anionic AOT vesicles.11,52 Such binding results in increased substrate concentration in the area of the micelles if compared to the concentration in bulk solution. At optimal reaction conditions, no matter whether hemin or HRPC was used, the reaction products remained dispersed in the aqueous solution for several days without visible precipitation. This supports the existence of product-SDBS micelle interactions, with SDBS acting as counterion (dopant) of the formed PANI-ES-like products with their positively charged backbone. Moreover, in the presence of SDBS micelles, hemin at 10 μM is soluble in 0.1 M HEPES (or 0.1 M H2PO4−) at pH = 4.3, while without SDBS, at the same hemin concentration aggregates form (small particles that are visible by the naked eye), see Fig. S-3.†
3.2. Comparison of the reaction products obtained with hemin or HRPC as catalyst
 | ||
| Fig. 2 UV/vis/NIR absorption spectra of reaction mixtures prepared in HEPES solution containing hemin as catalyst, PADPA as monomer, and SDBS micelles, either with added H2O2 (1, solid line), or without added H2O2 (2, dashed line), recorded after a reaction time t = 24 h at RT. Reaction conditions: 0.1 M HEPES, pH = 4.3; [hemin] = 10 μM; [PADPA]0 = 1.0 mM; [SDBS] = 5.0 mM; [H2O2]0 = 1.0 mM (for 1). The contribution of hemin to A400 (l = 0.1 cm) is ≈ 0.066 or less; and to A500 and A610 (l = 0.1 cm) ≈ 0.012 and ≈ 0.005, respectively, see Fig. S-3.† | ||
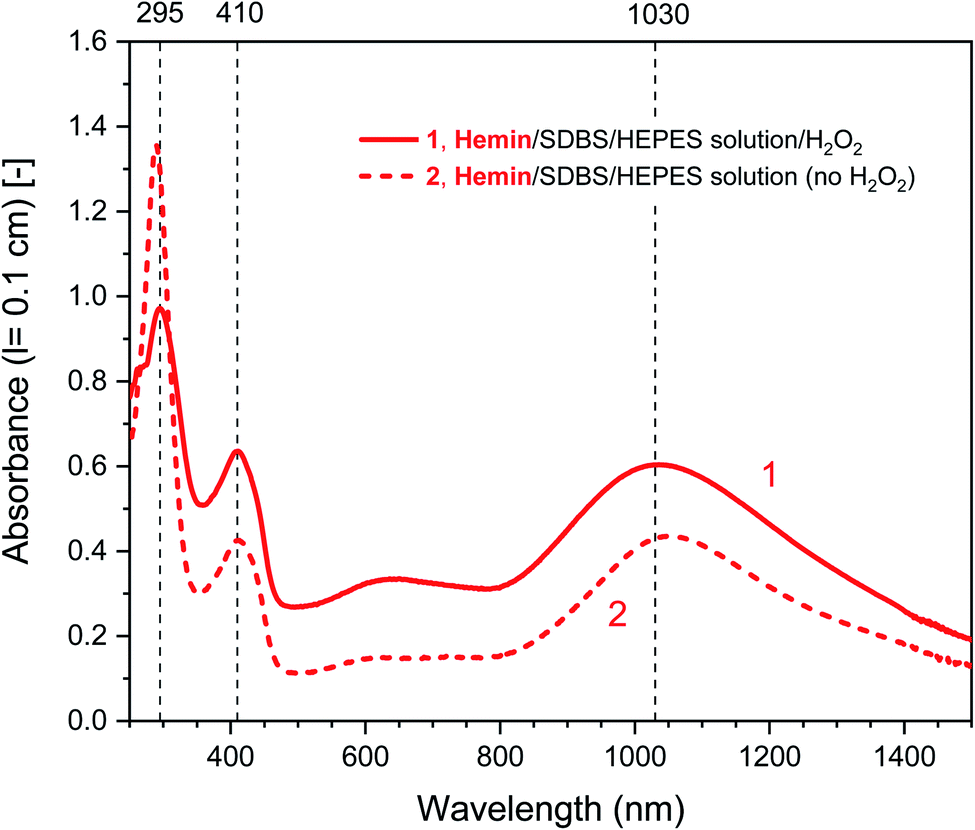
 | ||
| Fig. 3 UV/vis/NIR absorption spectra of reaction mixtures prepared in HEPES solution containing HRPC as catalyst, PADPA as monomer, and SDBS micelles, either with added H2O2 (1, solid line), or without added H2O2 (2, dashed line), recorded after a reaction time t = 24 h at RT. Reaction conditions: 0.1 M HEPES, pH = 4.3; [HRPC] = 30 nM; [PADPA]0 = 1.0 mM; [SDBS] = 3.0 mM; [H2O2]0 = 1.0 mM (for 1). | ||
Further comparative measurements were carried out for the hemin- and the HRPC-catalyzed reaction in the presence of 0.1 M dihydrogenphosphate instead of 0.1 M HEPES, again at pH = 4.3. The UV/vis/NIR absorption spectra of the reaction mixtures prepared with the H2PO4− solution and incubated for t = 24 h at RT are shown in Fig. S-5† together with the spectra obtained in the presence of the HEPES solution (data of Fig. 2 and 3). This comparison was made due to a previous report of Travascio and coworkers,54 in which it was demonstrated that the catalytic activity of a DNA aptamer–hemin complex in the presence of Triton X-100 may depend significantly on the salt type used, and that the use of HEPES at pH = 8.0 may result in a more efficient reaction as compared to phosphate (analyzed with ABTS2−, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate), as substrate). Our aim was to find out whether a similar salt-type dependence of the activity of hemin also exists in the case of PADPA as substrate in the presence of SDBS micelles. The measurements showed that this is indeed clearly the case (Fig. S-5†). Although there was no big difference in the absorption peak positions for the two systems (at λ ≈ 1030–1080, 400 nm, and 615 nm), two observations are obvious: (i) A≈1000 and A≈400 after t = 24 h were much higher for the reaction mixture prepared with the HEPES solution, while A≈500–600 was in both cases about the same; (ii) A≈300 after t = 24 h remained at a higher value for the reaction in the presence of dihydrogenphosphate as compared to the HEPES system (Fig. S-5†). These two observations clearly indicate higher conversion and a better “product quality” (higher A≈1000/A500 and A≈1000/A600 ratios) if the HEPES solution is used instead of the dihydrogenphosphate solution. For the reactions with HRPC, the band positions and intensities in the vis and NIR region of the absorption spectrum were very similar, although the spectrum recorded after t = 24 h at RT for the reaction run in the HEPES solution had slightly higher intensities at λ ≈ 400 nm and at the band maximum in the NIR region. However, the band maximum position and intensity were different (λmax = 980 nm, with A980 (0.1 cm) = 0.68, for the HEPES solution and λmax = 1074 nm, with A1074 (0.1 cm) = 0.63, for the dihydrogenphosphate solution) (Fig. S-5†). These latter intensities are similar to the value determined previously with AOT vesicles as templates (0.1 M NaH2PO4)5 instead of SDBS micelles with the same [PADPA]0, [H2O2]0, and [HRPC] (Fig. S-4,† A1100 (0.1 cm) ≈0.68 after t = 24 at RT).
As an important result from this HEPES vs. dihydrogenphosphate comparison is that under the conditions used the presence of HEPES has a positive effect on the outcome of the hemin-catalyzed formation of poly(PADPA). Whether this positive “HEPES effect” is a direct consequence of the influence the HEPES molecules have on the catalytic activity of hemin, or whether it is an effect which originates from HEPES-substrate interactions, was investigated by using TMB (3,3′,5,5′-tetramethylbenzidine) as substrate ([TMB]0 = [H2O2]0 = 0.3 mM, pH = 4.3, see Fig. S-6†). The initial rate of TMB oxidation was about 3.6 times faster in the presence of HEPES as compared to dihydrogenphosphate. This supports a direct effect of HEPES on the catalytic activity of hemin, possibly by coordinating to the iron atom at the 5th coordination site. For the results presented in the following, the reaction mixtures usually contained 0.1 M HEPES at pH = 4.3 (optimal conditions). Only for comparative measurements, a 0.1 M H2PO4− solution at pH = 4.3 was used as well.
Different control measurements were carried out for both the hemin- and the HRPC-catalyzed reactions in the presence of 0.1 M HEPES and PADPA (1.0 mM) at pH 4.3, whereby one of the other components present in the optimal reaction mixtures was omitted, either SDBS, catalyst (hemin or HRPC), or hydrogen peroxide. In the case of the HRPC system, the reaction mixture did not turn green after t = 24 h at RT if one of the mentioned components was absent (no absorption in the near-infrared region of the spectrum, i.e., no formation of PANI-ES-like products), see Fig. S-7.† Without H2O2 (but with HRPC and SDBS), no reaction occurred (the spectrum measured after t = 24 h was the spectrum of PADPA (Fig. 3, dashed line); without HRPC (but with H2O2 and SDBS), the absorption at λ ≈ 300 nm due to PADPA remained high after t = 24 h (no significant PADPA conversion); without SDBS (but with HRPC and H2O2), a reaction occurred but most of the brown products that formed precipitated, as already observed before.5 Therefore, the oxidation and oligomerization or polymerization of PADPA into PANI-ES-like oligo- or poly(PADPA) products with HRPC requires the presence of H2O2 as well as SDBS (Fig. S-7†). In the case of the hemin system, the situation is different. For two control measurements, the outcome was as expected: (i) in the absence of hemin (with H2O2 and SDBS), or (ii) in the absence of SDBS (with hemin and H2O2), no significant amounts of the green PANI-ES-like products were obtained (no significant absorption at λ ≈ 1000 nm, presence of a weak absorption centered around λ = 600 nm) (Fig. S-8,† compare also with the spectrum shown in Fig. S-1,† for “no SDBS”). More interesting and somewhat surprising, however, was the outcome of the control measurements without H2O2 (but with hemin and SDBS). The reaction proceeded quite well without added H2O2, resulting after t = 24 h at RT in an absorption spectrum with λmax = 1051 nm with A1051 (0.1 cm) = 0.44 and λmax = 412 nm with A412 (0.1 cm) = 0.43, as compared to λmax = 1030 nm with A1030 (0.1 cm) = 0.60 and λmax = 410 nm with A410 (0.1 cm) = 0.64, for the reaction run under the “optimal conditions”, i.e., in the presence of H2O2, hemin and SDBS (Fig. 2, dashed line and Fig. S-8†). This observed reactivity of hemin without H2O2 indicates not only (i) how insightful control measurements can be, but also (ii) that – under the conditions used (0.1 M HEPES, pH = 4.3) – the oxidation of PADPA with hemin in the absence of added H2O2 cannot proceed via a peroxidase-like mechanism (see Scheme S-1†), where H2O2 first oxidizes the heme group in a two-electron oxidation reaction to yield compound I, which then initiates the oxidation of the substrate (PADPA) in two consecutive one-electron oxidation reactions. Compound I formation – if compound I forms at all – must occur in a different way in the absence of H2O2. For the hemin-catalyzed reaction without added H2O2, the absorption at λ ≈ 600 nm after t = 24 h at RT was lower than in the case of the reaction run under the “optimal conditions” (with H2O2) (Fig. 2). The ratio of A1051/A600 for the reaction mixture where no H2O2 was added was 3.0, as compared to A1030/A600 = 1.9 for the reaction run under “optimal conditions” (with H2O2). Understanding how the hemin-catalyzed oxidation of PADPA in the absence of H2O2 occurs is an interesting challenge for detailed future investigations (see below).
For a direct comparison, Fig. S-9† shows the UV/vis/NIR absorption spectra of the reaction mixtures containing either hemin or HRPC and incubated for t = 24 h at RT (in the HEPES solution with or without H2O2).
The same set of control measurements for the hemin- and HRPC-catalyzed reactions were also carried out in the presence of aqueous 0.1 M dihydrogenphosphate solution instead of 0.1 M HEPES at pH 4.3 (see Fig. S-10 and S-11†). Qualitatively, similar results were obtained when using the dihydrogenphosphate solution as compared to using the HEPES solution: For HRPC, there was no significant reaction without H2O2 (Fig. S-11†); for hemin, a clear reaction also occurred without H2O2. Moreover, the absorption spectrum of the oligo- or poly(PADPA) products obtained after t = 24 h at RT was about the same for the reaction run in the H2PO4− solution with H2O2 as compared to without H2O2 (Fig. S-10†). Obviously, the addition of H2O2 had no beneficial effect at all if the reaction was run in the dihydrogenphosphate solution at pH = 4.3. This is different to what we observed with the HEPES solution, where the added H2O2 still was advantageous (see Fig. S-5†): compared to the reactions run in the dihydrogenphosphate solution with hemin and PADPA, the reactions in HEPES solution were more efficient (clearly higher intensity in the NIR region of the absorption spectrum of the products obtained).
Using TMB as substrate and measuring the initial rate of TMB oxidation in the presence of hemin, qualitatively the same results were obtained as with PADPA, see Fig. S-12 and S-13:† (i) with the HEPES solution, TMB was also oxidized without added H2O2, but the TMB oxidation was much faster with added H2O2 (Fig. S-12†); (ii) with the dihydrogenphosphate solution, the same initial rate of TMB oxidation was measured, independent on whether H2O2 was added or not (Fig. S-13, ESI†); and (iii) with the HEPES solution, the initial rate of TMB oxidation in the presence of H2O2 was ≈3.6 times faster than with the H2PO4− solution (please compare Fig. S-12 and S-13†).
Overall, the “best” conditions for oxidizing PADPA to PANI-ES-like oligo- or poly(PADPA) products with hemin is to use the HEPES solution with added H2O2 at the optimal reaction conditions already mentioned above: 0.1 M HEPES, pH = 4.3; [SDBS] = 5.0 mM; [hemin] = 10 μM, [PADPA]0 = 1.0 mM; [H2O2]0 = 1.0 mM.
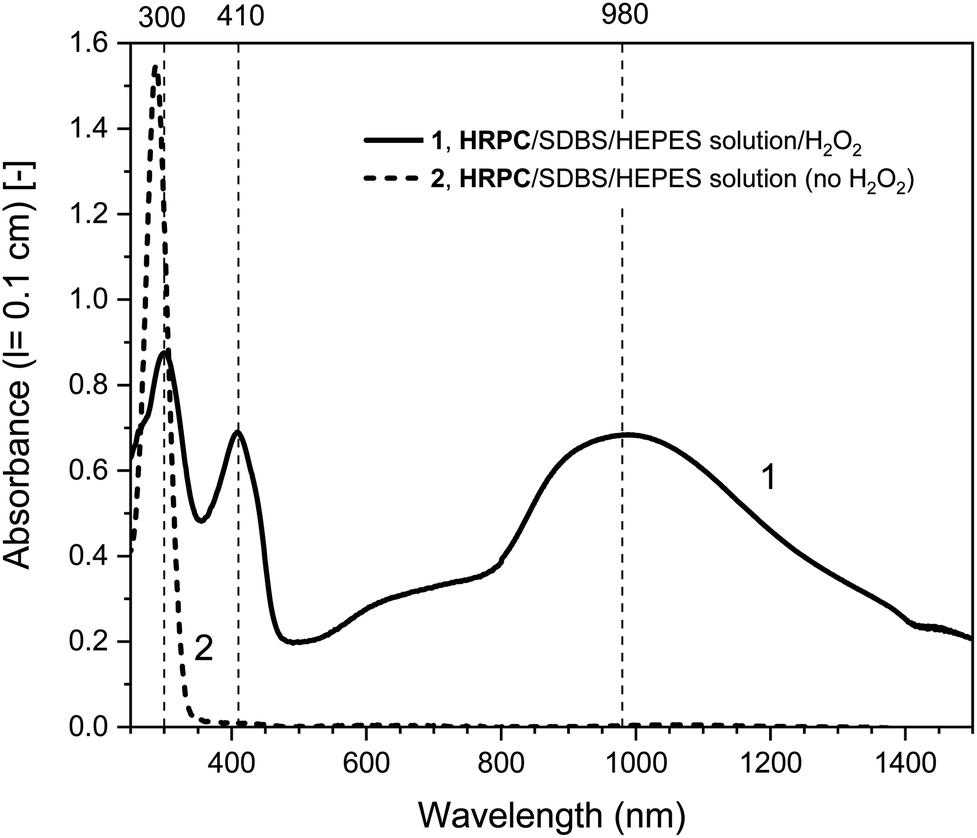
In Fig. 4, the time-dependent changes of the UV/vis/NIR absorption spectrum for the hemin- and the HRP-catalyzed oxidative oligomerization of PADPA in the presence of SDBS micelles and with added H2O2 in the HEPES solution are shown.
 | ||
| Fig. 4 UV/vis/NIR absorption measurements during the reaction of PADPA with either hemin (A) or HRPC (B) as catalyst and added H2O2. Reaction conditions: (A) 0.1 M HEPES pH 4.3 solution, [SDBS] = 5.0 mM, [hemin] = 10.0 μM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM; (B) 0.1 M HEPES pH 4.3 solution, [HRP] = 30.0 nM, [SDBS] = 3.0 mM, [PADPA]0 = 1.0 mM [H2O2]0 = 1.0 mM. For the sake of clarity, only 4 measurements (the most prominent ones) per system are shown; a complete data set can be found in Fig. S-14 and S-15.† Total reaction time t ≈ 24 h, RT. | ||
With hemin, the development of the UV/vis/NIR absorption spectrum over time is relatively simple. There is an increase of absorption above λ ≈ 380 nm over time with a clear appearance of bands centered around λ ≈ 1030 and ≈ 410 nm (indication of the formation of favorable PANI-ES-type products). A broad band in the region of λ ≈ 615 nm is also clearly developing, which indicates formation of unfavorable products containing phenazine-type units. There is also a decrease of absorbance at λ ≈ 300 nm, which is an indication of PADPA being consumed during the reaction. The rate of reaction but not the pattern of the spectral evolution was affected by the type of salt present (HEPES vs. dihydrogenphosphate) and the presence or absence of H2O2 (see Fig. S-16–S-18†). The fastest initial rate of spectral development in the hemin-catalyzed system was observed when the HEPES salt and H2O2 were used, i.e., the “optimal reaction conditions”.
With HRPC, the development of the UV/vis/NIR absorption spectrum with time was rather different from the spectral development of the reaction mixtures containing hemin. During the first 30 min, there was a sharp increase in absorbance of the bands positioned at λ ≈ 410 and ≈ 915 nm, which indicates the presence of PANI-ES-type products. In addition, there was a decrease in absorbance at A296, which shows that PADPA was consumed during the reaction. After 30 min, there was a slow decrease of A915 and at the same time an increase of absorption intensity in the NIR region, at λ = 1200–1400 nm, with an isosbestic point at λ ≈ 1100 nm. In addition, during the reaction, a shoulder at λ ≈ 700 nm was always present. Please note that the spectrum obtained after t = 24 h was slightly different from the one obtained for the same reaction conditions shown in Fig. 2. This difference originates from the different reaction containers used in the two cases (see Materials and methods).
Fig. 5 shows a direct comparison of the time-dependent changes of A1030 for the reaction mixtures prepared with the HEPES solution with hemin as catalyst, with (1) or without (2) H2O2. Data for the HRPC system with H2O2 (3) are also shown (note that without H2O2 no reaction occurred, see Fig. 3). In the case of the HRPC-catalyzed reaction, A1030 leveled off after t ≈ 15 min already, with a continuous slow decrease beyond this time due to a broadening of the spectrum into the NIR region (see Fig. 4B). For the hemin-catalyzed system, a plateau value was reached after t ≈ 8 hours in the presence of H2O2, while without added H2O2, the reaction clearly was much slower but A1030 kept gradually increasing (at least up to t = 24 h). This latter comparison is again a clear proof that for the hemin-catalyzed reaction the addition of H2O2 is not necessary for the reaction to proceed (see Fig. 2), but that the rate of reaction with addition of H2O2 is much faster than without added H2O2.
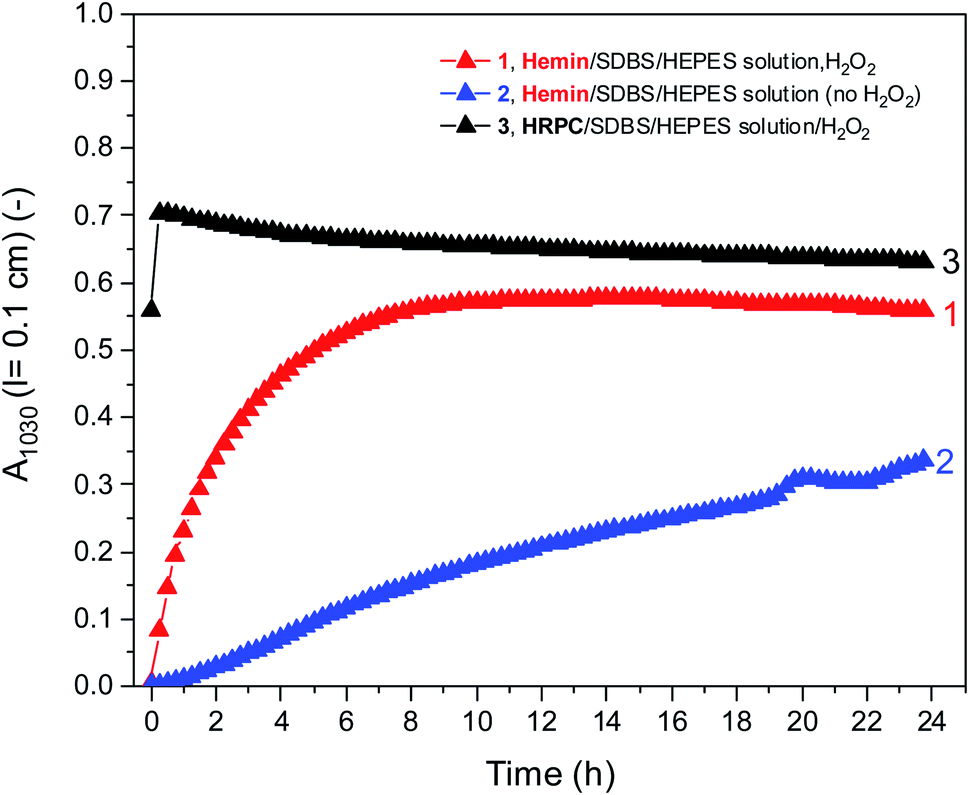 | ||
| Fig. 5 Comparison of the changes of A1030 during the reaction of PADPA in 0.1 M HEPES solution (pH = 4.3) with either hemin or HRPC as catalyst at RT. Reaction conditions: (1) [hemin] = 10.0 μM, [SDBS] = 5 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM; (2) [hemin] = 10.0 μM, [SDBS] = 5 mM, [PADPA]0 = 1.0 mM, no H2O2]; (3) [HRPC] = 30 nM, [SDBS] = 3 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM. Data points were obtained by running the reaction inside quartz cuvettes with a pathlength of l = 0.1 cm and recording the spectrum every 15 min for up to t ≈ 24 h at RT. | ||
For the sake of completeness, kinetic measurements were also carried out by using the 0.1 M H2PO4− solution instead of the 0.1 M HEPES solution, see Fig. S-19.† These measurements confirmed the lower activity of hemin in the H2PO4− solution, in which the addition of H2O2 had no beneficial effect at all (see also Fig. S-10†). For all further measurements by using EPR and Raman spectroscopy as analytical methods, the focus was on reaction mixtures prepared in 0.1 HEPES at pH = 4.3. Nevertheless, the results obtained by using 0.1 M H2PO4− at pH = 4.3 are also mentioned, with the corresponding data shown in the ESI.
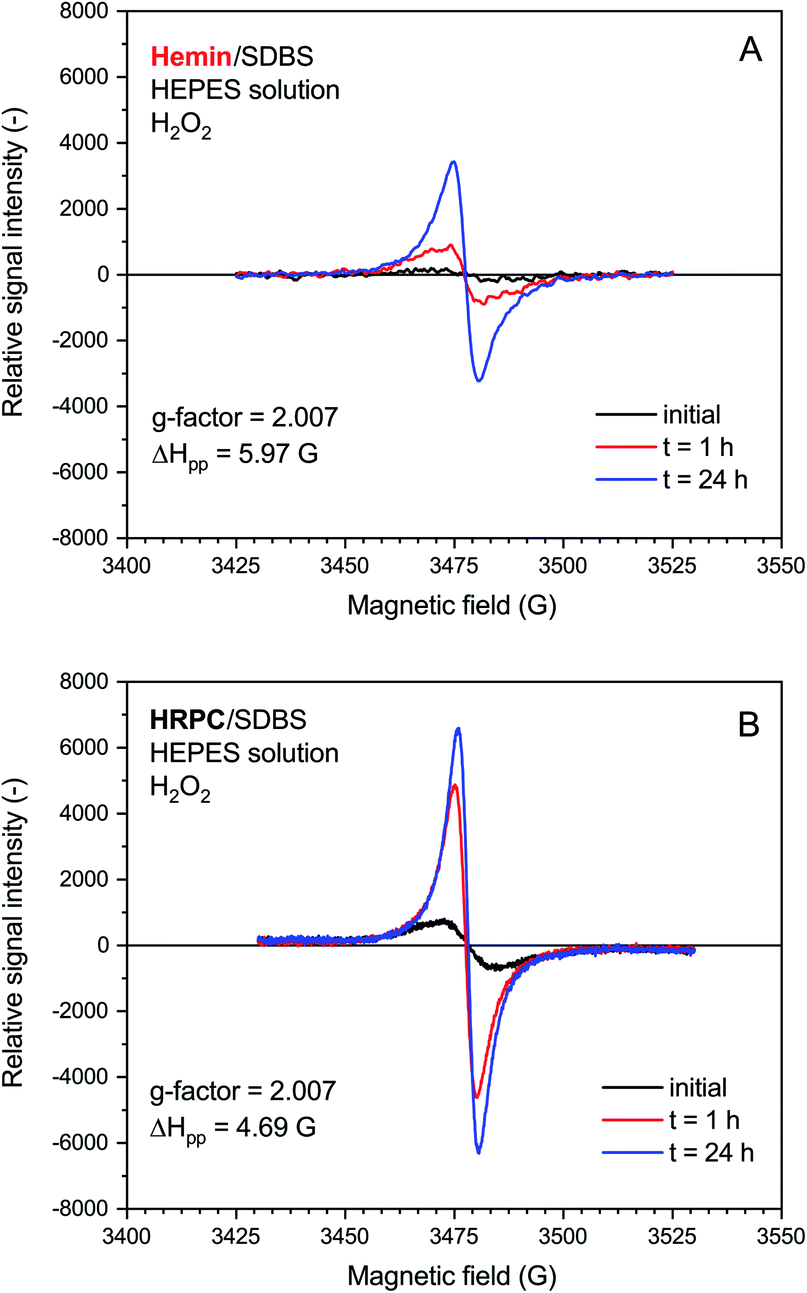 | ||
| Fig. 6 EPR spectroscopy measurements for reaction mixtures prepared with 0.1 M HEPES, pH = 4.3, with either hemin (A) or HRPC (B) as catalyst. Reaction conditions: (A) [hemin] = 10.0 μM, [SDBS] = 5.0 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM; (B) [HRPC] = 30.0 nM, [SDBS] = 3 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM. The reactions were run at RT inside Eppendorf reaction tubes and analyzed after t ≈ 2 min, 1 h or 24 h. G-factor and ΔHpp values are calculated for the spectrum recorded at t = 24 h. | ||
The rate of increase of the intensity of the EPR signal with time was slower if hemin was used as catalyst, as compared to HRPC. This is in agreement with the kinetic data obtained from the UV/vis/NIR absorption measurements, given that the presence of an absorption band in the NIR region of the spectrum is due to the presence of unpaired electrons that give rise to an EPR spectrum (see Introduction). The relative quantity of radicals produced till t = 24 h was significantly higher for the reaction run with HRPC as compared to hemin. The calculated g-factor values in both systems are identical (2.007, see Fig. 6) and are in good agreement with previously reported values for the emeraldine salt form of PANI.61,62 The EPR signal width (ΔHpp) for the spectrum recorded after t = 24 h indicates that the radical species are more uniform in the HRPC-catalyzed reaction (ΔHpp = 4.69 G) than for the reaction run with hemin (ΔHpp = 5.97 G), see Fig. 6.
A very weak EPR signal was also observed in the reaction mixture containing hemin without added H2O2, see Fig. 7. The relative intensity after t = 24 h was, however, much lower than for the reaction run with H2O2 (compare Fig. 7 with Fig. 6). This is again in qualitative agreement with the UV/vis/NIR absorption measurements, see Fig. 2. The g-factor determined for the spectrum recorded after t = 24 h is identical with the ones described in Fig. 6. Interestingly, ΔHpp is more similar to the one determined for the spectrum of the HRPC system than for the spectrum of the hemin system with added H2O2.
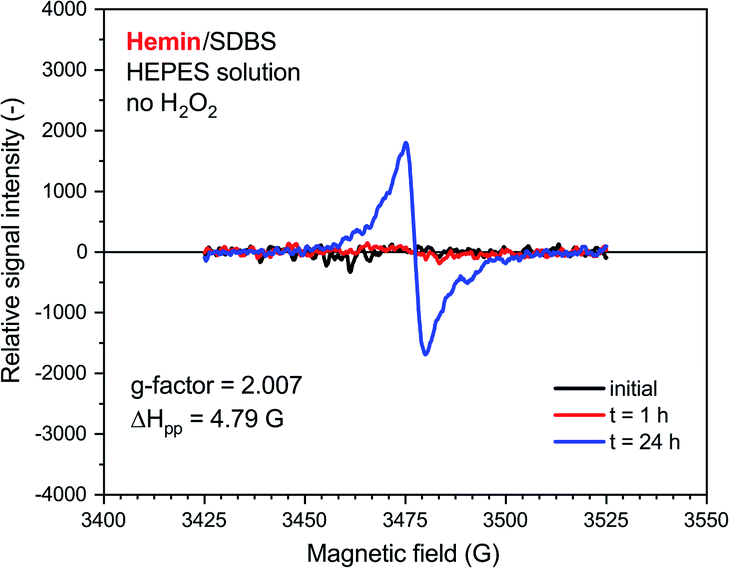 | ||
| Fig. 7 EPR spectroscopy measurements for a reaction mixture prepared with 0.1 M HEPES, pH = 4.3, with hemin without added H2O2. Reaction conditions: [hemin] = 10.0 μM, [SDBS] = 5.0 mM, [PADPA]0 = 1.0 mM, no H2O2. The reaction was run at RT inside an Eppendorf reaction tube and analyzed after t ≈ 2 min (initial), 1 h or 24 h. G-factor and ΔHpp values are calculated for the spectrum recorded at t = 24 h. | ||
EPR measurements were also carried out for reaction mixtures containing hemin in 0.1 M H2PO4− solution (pH = 4.3) instead of 0.1 M HEPES (pH = 4.3), both in the presence and absence of H2O2, see Fig. S-20.† The data are again in good qualitative agreement with the UV/vis/NIR measurements, i.e. there was no beneficial effect of H2O2 on the EPR signal intensity for the reaction with hemin in the reaction mixture containing dihydrogenphosphate. Furthermore, when the reaction mixture was carried out in the 0.1 M dihydrogenphosphate solution (pH = 4.3) the relative intensity of the EPR signal obtained when HRPC was used as catalyst was much higher than when hemin was used.
In Fig. 8, Raman spectra obtained for the reaction mixture containing hemin are shown for reaction times t = 5 min, 1 h, and 24 h. For the reaction mixture analyzed after t = 5 min, the Raman spectrum contained numerous bands that originate from starting materials and early reaction intermediates. As time progressed, the spectrum changed, and several bands that are typical for PANI-ES-like oligomeric products of PADPA appeared, as discussed in the following.
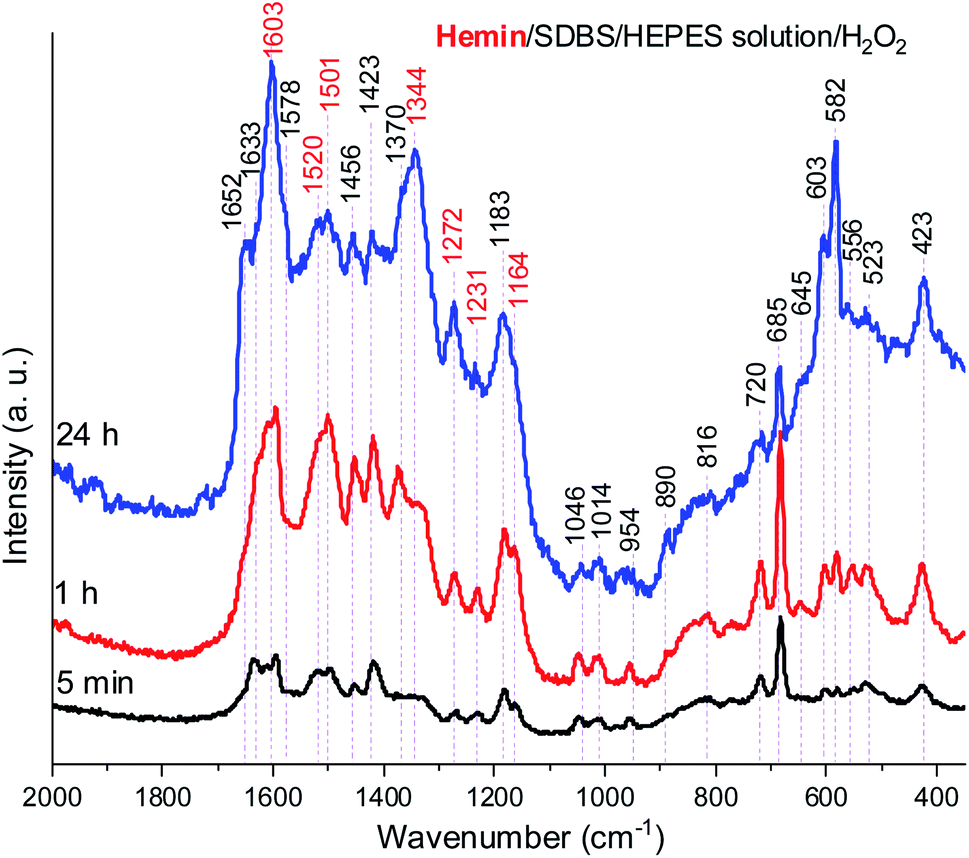 | ||
| Fig. 8 Raman spectra of a reaction mixture prepared with an aqueous 0.1 M HEPES solution at pH 4.3, with [hemin] = 10.0 μM, [SDBS] = 5 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM, recorded at t = 5 min, 1 h, and 24 h at RT. The bands characteristic for PANI-ES products are labeled in red, see text; a.u., arbitrary units. | ||
A number of bands that are characteristic for structural units of ordinary (“standard”) PANI can be observed at = 1620 cm−1 (m, C–C stretching vibrations of benzenoid (B) rings, ν(C–C)B), 1603 cm−1 (m, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C stretching vibrations of quinonoid (Q) and semiquinonoid (SQ) rings, ν(CC)Q and ν(C–C)SQ), 1520 cm−1 (m, N–H bending vibration, δ(N–H), commonly related to SQ (polaron) structures in PANI), 1501 cm−1 (m, CN stretching in quinonediimine units, ν(CN)Q), 1340 cm−1 (w, C–N˙+ stretching vibration in SQ (polaron) units, ν(C–N˙+)SQ, characteristic for the conducting form of PANI, the emeraldine salt PANI-ES), 1272 cm−1 (w, C–N stretching vibrations in B rings, ν(C–N)B˙), 1231 cm−1 (w, ν(C–N)B), 1181 cm−1 (m, C–H bending in-plane vibration of B ring, δ(C–H)B), and 1164 cm−1 (m, C–H bending in-plane vibration of Q ring, δ(C–H)Q).53 The Raman spectrum also indicates formation of structural units different from those of “standard” PANI, by the bands observed at = 1633 cm−1 (m, attributable to C–C ring-stretching vibrations in phenazine-, N-phenylphenazine- and/or phenoxazine-type units, mixed with the C–C stretching vibration of the B ring, ν(C–C)B), 1578 cm−1 (sh, phenazine-type of units), 1456 cm−1 (w, assignable to C–C ring stretching/CN stretching in quinonoid type of units that are probably present in short chains/branches and in substituted phenazine- and N-phenylphenazine-type segments), 1423 cm−1 (m, assignable to the ring-stretching vibration in phenazine-, N-phenylphenazine- and phenoxazine-type units, combined with a contribution from DMSO), 1370 cm−1 (vw, ν(C–N+) ring stretching vibration in substituted N-phenylphenazine-type units and to phenoxazine-type units/(C–N˙+)SQ vibration in polaron-SQ structures of low delocalization), and 582 cm−1 w, a band at similar position in spectra of PANI has been attributed to substituted phenoxazine-like units.53,63,64 Such phenoxazine-like units could have been formed by the oxidative intramolecular cyclization of substituted ortho-aminophenol units, formed as products of hydrolysis of fully oxidized pernigraniline-like oligomers,65 and substituted phenazine- and N-phenylphenazine-type units.53 Contributions of C–S stretching vibration ν(C–S) and SO2 deformation vibration δ(SO2) probably originating from the anion of SDBS (DBS) to the band at 582 cm−1 are also possible.66 The weak band in the spectrum at t = 5 min observed at 603 cm−1 could be assigned to δ(SO2) vibrations, originating from SDBS,66 and benzenoid (B) ring deformation in PANI-like segments of oligo- or poly(PADPA).¶53,67 In the Raman spectrum recorded at t = 1 h, one can observe that the band at = 1370 cm−1, attributed to ν(C–N˙+)SQ of polarons with lower delocalization/N-phenylphenazine and phenoxazine units, is well developed, being even stronger than the band at = 1340 cm−1 assigned to ν(C–N˙+)SQ of more delocalized polarons. However, in the Raman spectrum for t = 24 h, the intensity ratio of these two bands is reversed, and the band at = 1370 cm−1 becomes a shoulder of the band at = 1344 cm−1.
C and C–C stretching vibrations of quinonoid (Q) and semiquinonoid (SQ) rings, ν(CC)Q and ν(C–C)SQ), 1520 cm−1 (m, N–H bending vibration, δ(N–H), commonly related to SQ (polaron) structures in PANI), 1501 cm−1 (m, CN stretching in quinonediimine units, ν(CN)Q), 1340 cm−1 (w, C–N˙+ stretching vibration in SQ (polaron) units, ν(C–N˙+)SQ, characteristic for the conducting form of PANI, the emeraldine salt PANI-ES), 1272 cm−1 (w, C–N stretching vibrations in B rings, ν(C–N)B˙), 1231 cm−1 (w, ν(C–N)B), 1181 cm−1 (m, C–H bending in-plane vibration of B ring, δ(C–H)B), and 1164 cm−1 (m, C–H bending in-plane vibration of Q ring, δ(C–H)Q).53 The Raman spectrum also indicates formation of structural units different from those of “standard” PANI, by the bands observed at = 1633 cm−1 (m, attributable to C–C ring-stretching vibrations in phenazine-, N-phenylphenazine- and/or phenoxazine-type units, mixed with the C–C stretching vibration of the B ring, ν(C–C)B), 1578 cm−1 (sh, phenazine-type of units), 1456 cm−1 (w, assignable to C–C ring stretching/CN stretching in quinonoid type of units that are probably present in short chains/branches and in substituted phenazine- and N-phenylphenazine-type segments), 1423 cm−1 (m, assignable to the ring-stretching vibration in phenazine-, N-phenylphenazine- and phenoxazine-type units, combined with a contribution from DMSO), 1370 cm−1 (vw, ν(C–N+) ring stretching vibration in substituted N-phenylphenazine-type units and to phenoxazine-type units/(C–N˙+)SQ vibration in polaron-SQ structures of low delocalization), and 582 cm−1 w, a band at similar position in spectra of PANI has been attributed to substituted phenoxazine-like units.53,63,64 Such phenoxazine-like units could have been formed by the oxidative intramolecular cyclization of substituted ortho-aminophenol units, formed as products of hydrolysis of fully oxidized pernigraniline-like oligomers,65 and substituted phenazine- and N-phenylphenazine-type units.53 Contributions of C–S stretching vibration ν(C–S) and SO2 deformation vibration δ(SO2) probably originating from the anion of SDBS (DBS) to the band at 582 cm−1 are also possible.66 The weak band in the spectrum at t = 5 min observed at 603 cm−1 could be assigned to δ(SO2) vibrations, originating from SDBS,66 and benzenoid (B) ring deformation in PANI-like segments of oligo- or poly(PADPA).¶53,67 In the Raman spectrum recorded at t = 1 h, one can observe that the band at = 1370 cm−1, attributed to ν(C–N˙+)SQ of polarons with lower delocalization/N-phenylphenazine and phenoxazine units, is well developed, being even stronger than the band at = 1340 cm−1 assigned to ν(C–N˙+)SQ of more delocalized polarons. However, in the Raman spectrum for t = 24 h, the intensity ratio of these two bands is reversed, and the band at = 1370 cm−1 becomes a shoulder of the band at = 1344 cm−1.
The relative intensities of the two bands characteristic of conducting PANI-ES at = 1603 cm−1 and 1344 cm−1 increased as the reaction time increased, so that they are present as the most intense bands in the Raman spectrum recorded at t = 24 h, along with the band at 582 cm−1. The strong band at 1344 cm−1 indicates a conductive nature of the poly(PADPA) product. Strengthening of the band at 1344 cm−1 indicates an increase in the relative amount of delocalized polaron structures and consequently an increase in electrical conductivity of the oligo- or poly(PADPA) products over time. On the other hand, the relative intensity of the bands atypical for standard PANI, at 1633 cm−1 and 1420 cm−1, decreased with reaction time in relation to the intensity of the mentioned PANI-ES bands. Additional interesting changes occur in the spectrum at t = 24 h related to the spectrum at t = 1 h: the bands attributed to “atypical segments” at ≈ 1456 cm−1 and 1420 cm−1 become at t = 24 h noticeably weaker compared to the bands of standard PANI-ES (e.g., at = 1603 cm−1 and 1344 cm−1), the band at = 1633 cm−1 disappears, and we observe a new band at = 1652 cm−1 (which is also noticeably weaker compared to the bands of ordinary PANI at = 1603 cm−1 and 1344 cm−1) and a shoulder at = 1578 cm−1. The band at = 1652 cm−1 could be attributed to the stretching of quinonoid CO groups/C–C ring-stretching vibrations in phenoxazine-type units64 with possible contributions of phenazine- and N-phenylphenazine-type units; CO groups can be formed by the partial hydrolysis of iminoquinonoid CN bonds.65 The shoulder at = 1578 cm−1 can also be attributed to substituted phenoxazine- and phenazine-type units.53 The bands at = 1652, 1578, and 585 cm−1 indicate certain overoxidation of oligo- or poly(PADPA) produced at long reaction times (t = 24 h). The relative intensities of the bands attributed to DMSO (at = 1046, 720, and 685 cm−1) gradually decrease with reaction time, e.g., the band at = 685 cm−1 is very strong and the most intense band in the spectrum at t = 5 min, while it is weak at t = 24 h. This occurs due to the increase of the amount of oligo- or poly(PADPA) chains (as the reaction proceeds) on the surface of the reaction mixture “drop” which consequently leads to a reduction of surface concentration of DMSO. Thereby, as the reaction proceeds, oligo- or poly(PADPA) chains are becoming dominating species for Raman scattering instead of DMSO. We have observed an increase in the relative intensity of the band at = 603 cm−1 (assigned to δ(SO2)/B ring deformation in PANI-like segments) with an increase in reaction time (it is weak at t = 5 min, medium at t = 1 h, and strong at t = 24 h). This feature could be explained by an increase in oligo- or poly(PADPA) yield, and therefore, an increase in the surface concentration of DBS anions, which could serve as counterions (dopant ions), compensating positive charges of the poly(PADPA) backbone. The prominent increase in the relative intensity of the band at = 582 cm−1 with increasing reaction time is observed. At t = 24 h, it becomes very strong, bearing an intensity comparable to that of the band at = 1344 cm−1. Such band development might come from an increase in the amount of phenoxazine-type units at longer reaction time due to the side processes connected with ortho-coupling of PADPA molecules. An additional factor could be an increase in DBS anions surface concentration. It should be noted that bands at similar positions, = 576 cm−1 (strong) and ≈ 607 cm−1 (weak), are present in the solid-state Raman spectrum of PANI doped with dodecyl benzenesulfonic acid (PANI-DBSA).68
The Raman spectra of the reaction mixture containing HRPC as a catalyst for the reaction times t = 7 min, 1 h and 24 h (Fig. 9) qualitatively resemble the Raman spectra obtained with hemin as a catalyst. Nevertheless, there are certain differences which will be specified and discussed in the following.
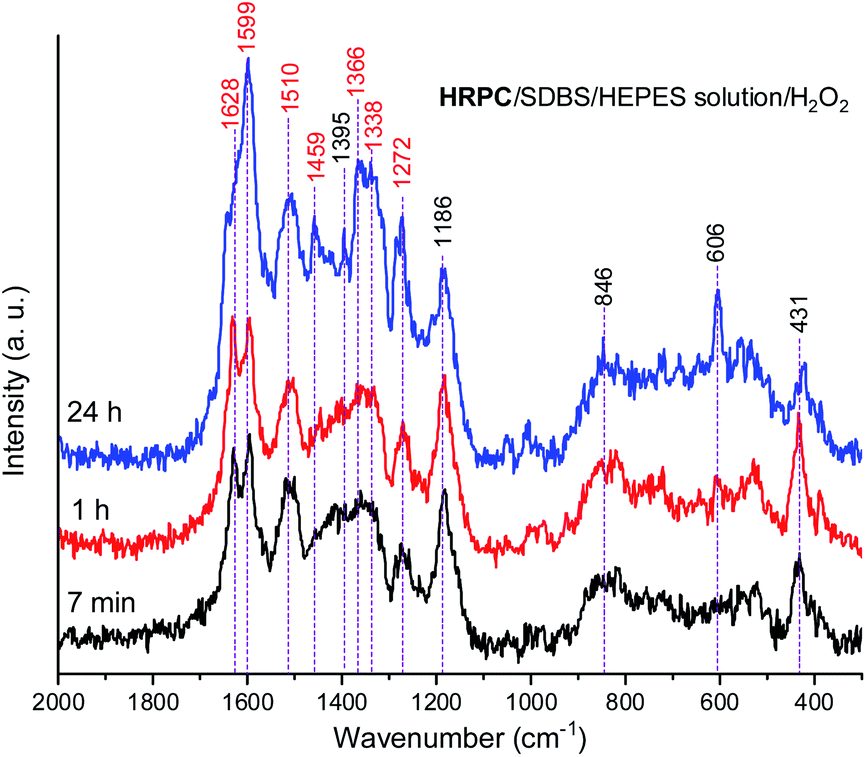 | ||
| Fig. 9 Raman spectra of a reaction mixture prepared with an aqueous 0.1 M HEPES solution at pH 4.3, with [HRPC] = 30.0 nM, [SDBS] = 3.0 mM, [PADPA]0 = 1.0 mM, [H2O2]0 = 1.0 mM, recorded at t = 7 min, 1 h, and 24 h at RT. The bands characteristic for PANI-ES products are labeled in red, see text; a.u., arbitrary units. | ||
In both systems, the one containing hemin and the other containing HRPC after a reaction time t = 24 h, the bands attributed to standard PANI-ES ( ≈ 1600 cm−1 and ≈ 1340 cm−1) are stronger compared to the bands of atypical units ( ≈ 1420–1440 cm−1) and both spectra contain a band at = 606 cm−1. It is interesting to note that the Raman spectra obtained in the reaction mixture containing HRPC as catalyst do not contain a band at ≈ 580 cm−1 (attributable to phenoxazine-type units), and they do not show a strong “phenazine band” at ≈ 1420 cm−1 in the initial phase of the reaction (t = 7 min), as it is the case for the reaction mixture with hemin as a catalyst. In addition, the spectra of the system with HRPC do not show a band at ≈ 680 cm−1, as was the case when hemin was used (Fig. 8). This indicates that the consumption of PADPA and the formation of poly(PADPA) oligo- or polymeric chains at the surface of the reaction mixture “drop” analyzed was faster in the system with HRPC than in the one containing hemin as a catalyst, which was also expected. This is in good agreement with the results from the UV/vis/NIR and EPR measurements (see above). When comparing the Raman spectra recorded at t = 24 h of the reaction mixtures with either HRPC or hemin as a catalyst, one more difference is evident. The spectrum for the system with HRPC contains a broad band in the region of the ν(C–N+) vibrations, with two peaks of similar intensity at around = 1340 cm−1 (ν(C–N˙+)SQ vibration of more delocalized polaron structures) and = 1366 cm−1 (ν(C–N+) ring stretching vibration in substituted N-phenylphenazine- and phenoxazine-type units and ν(C–N˙+)SQ vibration in polaron structures of low delocalization) (Fig. 9), while the spectrum of the system containing hemin (Fig. 8) has a strong ν(C–N˙+)SQ band, at = 1340 cm−1 and a shoulder at ≈ 1370 cm−1.
Due to the results obtained for the hemin-catalyzed reaction analyzed by UV/vis/NIR and EPR measurements (see above), Raman spectra of the reaction mixture containing hemin without added H2O2 were also recorded, at t = 5 min, 1 h, and 24 h, see Fig. 10.
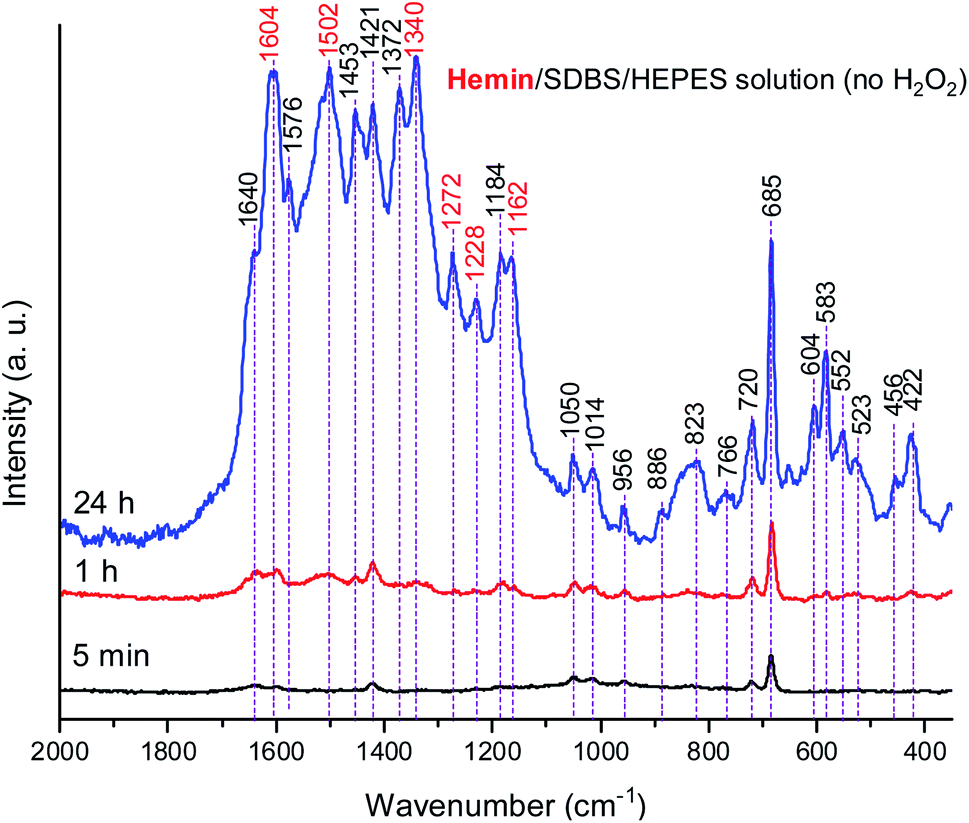 | ||
| Fig. 10 Raman spectra of a reaction mixture prepared with an aqueous 0.1 M HEPES solution at pH 4.3, with [hemin] = 10.0 μM, [SDBS] = 5 mM, [PADPA]0 = 1.0 mM, no H2O2, recorded at t = 5 min, 1 h, and 24 h at RT. The bands characteristic for PANI-ES products are labeled in red, see text; a.u., arbitrary units. | ||
A comparison of the Raman spectra of the reaction mixtures without H2O2 (Fig. 10) and with H2O2 (Fig. 8) – both recorded after t = 24 h – shows that the absorption band pattern in both cases is very similar. However, differences in the band intensities are obvious when spectra recorded after the same reaction time are compared. This is directly related to the lower PADPA monomer consumption for the reaction without H2O2 (lower band intensities) as compared to the one with H2O2 (higher band intensities).
Comparing the Raman spectra recorded at t = 24 h for the two hemin systems (with and without H2O2) in terms of ratios of intensities of the polaron ν(C–N˙+)SQ band at ≈ 1340 cm−1, which is characteristic of standard conductive PANI-ES, and the bands at ≈ 1453 and 1420 cm−1, attributed to segments atypical for standard PANI-ES, I1340/I1453, and I1340/I1420, respectively, it is clear that these ratios are higher for the hemin system containing H2O2 than the one without H2O2. Also, at t = 24 h, the bands at = 1372 cm−1 and 1576 cm−1 are present as distinct bands in the spectrum of the hemin reaction mixture without H2O2, while in the spectrum of the hemin reaction mixture with H2O2 they appear as shoulders, only. The bands at = 604 and 583 cm−1 have lower relative intensities in the spectrum of the hemin system without H2O2 compared to these bands in the spectrum of the hemin system with H2O2.
Although it was not the main focus of the work, Raman spectroscopy measurements were also carried out for reaction mixtures prepared with 0.1 M dihydrogenphosphate solution instead of 0.1 M HEPES solution (both at pH = 4.3), see Fig. S-21–S-24.† As a general observation, the data obtained is in good agreement with the UV/vis/NIR and EPR data for the same reaction conditions and the explanations given above.
3.3. Discussion of the main points of interest
So far, important control measurements of reactions run without H2O2 clearly demonstrated that the hemin-catalyzed formation of PANI-ES-like products from PADPA in 0.1 M HEPES solution at pH = 4.3 (in the presence of SDBS micelles) also occurs to some extent without H2O2 (Fig. 2), although the reaction is much slower than with H2O2 (Fig. 5 and S-12†). It seems that the oxidation of PADPA (or TMB) with hemin as catalyst occurs via two mechanisms, one involving H2O2 as terminal oxidant, the other with O2 (which was present in all reaction mixtures due to their exposure to air). Preliminary measurements with PADPA as substrate were carried out without H2O2 under “oxygen-free” conditions (see Fig. S-25, S-26 and accompanying text for details†). The observation made is that in the absence of H2O2 oxygen is necessary for the reaction to proceed. Although the mechanism involving oxygen is not clear at the moment and deserves further investigations, one possibility how the reaction might proceed is that Fe(III)PPIX is reduced to Fe(II)PPIX by PADPA, while at the same time PADPA is oxidized to the PADPA radical. Fe(II)PPIX is then reoxidized to Fe(III)PPIX by oxygen.|| Alternatively, the mechanism might be completely different, and it may include the formation of hydrogen peroxide in situ.71
4. Conclusion and outlook
The work carried out showed that hemin can be used successfully as catalyst for the oxidation of PADPA in a slightly acidic aqueous micellar solution of SDBS to yield products with PANI-ES structural units that are typical for the conductive form of polyaniline. Apart from applications of the products obtained, future studies may involve the investigation of the binding of hemin to the micelles, the use of other surfactant aggregates, or an investigation of the reaction mechanism and a possible substrate selectivity. Beyond this, experimental proof of catalytically active hemin in aggregates of amphiphiles is also of interest for origin-of-life research. Porphyrins like PPIX are molecules that are not only ubiquitously present as prosthetic groups in contemporary proteins,72 but may have been present already in prebiotic times.73,74 Due to the likely prebiotic existence of simple membrane-forming amphiphiles,75 these amphiphiles might have associated with metal porphyrins to form catalytically active assemblies for accelerating localized chemical transformations. In this way, amphiphile-heme aggregates could have served as a kind of “primitive enzymes”, even before the suggested heme RNA quadruplex systems possibly formed.76 Although this is pure speculation, the idea is worth considering in scenarios on the prebiological emergence of functional compartment systems, in steps that preceded the first living cells at the origin of life on earth.77Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by the Horizon 2020 Marie Skłodowska-Curie Actions Innovative Training Network (ITN) GA no. 813873, ProtoMet “Protometabolic pathways: exploring the chemical roots of systems biology”. G. Ć.-M. and D. B.-B. thank the Ministry of Education, Science and Technological Development of the Republic of Serbia for the financial support (Contract number 451-03-68/2022-14/200146).References
- K. Junker, G. Zandomeneghi, Z. Guo, R. Kissner, T. Ishikawa, J. Kohlbrecher and P. Walde, RSC Adv., 2012, 2, 6478–6495 RSC.
- R. Wang and X. Huang, ACS Omega, 2021, 6, 20699–20709 CrossRef CAS PubMed.
- V. Rumbau, J. A. Pomposo, J. A. Alduncin, H. Grande, D. Mecerreyes and E. Ochoteco, Enzyme Microb. Technol., 2007, 40, 1412–1421 CrossRef CAS.
- W. Liu, A. L. Cholli, R. Nagarajan, J. Kumar, S. Tripathy, F. F. Bruno and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 11345–11355 CrossRef CAS.
- S. Luginbühl, M. Milojević-Rakić, K. Junker, D. Bajuk-Bogdanović, I. Pašti, R. Kissner, G. Ćirić-Marjanović and P. Walde, Synth. Met., 2017, 226, 89–103 CrossRef.
- Y. Zhang, S. Serrano-Luginbühl, R. Kissner, M. Milojević-Rakić, D. Bajuk-Bogdanović, G. Ćirić-Marjanović, Q. Wang and P. Walde, Langmuir, 2018, 34, 9153–9166 CrossRef CAS PubMed.
- W. Liu, J. Kumar, S. Tripathy and L. A. Samuelson, Langmuir, 2002, 18, 9696–9704 CrossRef CAS.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78 CrossRef CAS.
- P. Walde and Z. Guo, Soft Matter, 2011, 7, 316–331 RSC.
- G. V. Otrokhov, O. V. Morozova, I. S. Vasil’eva, G. P. Shumakovich, E. A. Zaitseva, M. E. Khlupova and A. I. Yaropolov, Biochemistry (Moscow), 2013, 78, 1539–1553 CrossRef CAS PubMed.
- A. Janoševic Ležaić, S. Luginbühl, D. Bajuk-Bogdanović, I. Pašti, R. Kissner, B. Rakvin, P. Walde and G. Ćirić-Marjanović, Sci. Rep., 2016, 6, 30724 CrossRef PubMed.
- P. Xu, A. Singh and D. L. Kaplan, Enzymatic catalysis in the synthesis of polyanilines and derivatives of polyanilines, Springer, Berlin, Heidelberg, 2006 Search PubMed.
- M. Kurisu, H. Aoki, T. Jimbo, Y. Sakuma, M. Imai, S. Serrano-Luginbühl and P. Walde, Commun. Chem., 2019, 2, 117 CrossRef.
- T. Fujisaki, K. Kashima, S. Serrano-Luginbühl, R. Kissner, D. Bajuk-Bogdanović, M. Milojević-Rakić, G. Ćirić-Marjanović, S. Busato, E. Lizundia and P. Walde, RSC Adv., 2019, 9, 33080–33095 RSC.
- Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405 CrossRef CAS PubMed.
- H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187–251 CrossRef CAS.
- C. Asher, K. A. de Villiers and T. J. Egan, Inorg. Chem., 2009, 48, 7994–8003 CrossRef CAS PubMed.
- D. Kuter, G. A. Venter, K. J. Naidoo and T. J. Egan, Inorg. Chem., 2012, 51, 10233–10250 CrossRef CAS PubMed.
- D. Kuter, V. Streltsov, N. Davydova, G. A. Venter, K. J. Naidoo and T. J. Egan, Inorg. Chem., 2014, 53, 10811–10824 CrossRef CAS PubMed.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
- H. B. Dunford, Peroxidases and catalases: biochemistry, biophysics, biotechnology and physiology, John Wiley & Sons, 2010 Search PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- G. Bertini, H. B. Gray, H. Gray, J. S. Valentine, E. I. Stiefel and E. Stiefel, Biological inorganic chemistry: structure and reactivity, University Science Books, 2007 Search PubMed.
- S. Mazumdar, J. Phys. Chem., 1990, 94, 5947–5953 CrossRef CAS.
- S. Mazumdar and S. Mitra, in Structures and Biological Effects (Structure and Bonding, vol. 81), Springer-Verlag, Berlin, Heidelberg, 1993, pp. 115–145 Search PubMed.
- D. K. Das and O. K. Medhi, Indian J. Chem., 2005, 44A, 2228–2232 CAS.
- J. Simplicio, Biochemistry, 1972, 11, 2525–2528 CrossRef CAS PubMed.
- S. Mazumdar, O. K. Medhi and S. Mitra, Inorg. Chem., 1988, 27, 2541–2543 CrossRef CAS.
- A. Boffi, T. K. Das, S. della Longa, C. Spagnuolo and D. L. Rousseau, Biophys. J., 1999, 77, 1143–1149 CrossRef CAS PubMed.
- G. H. Beaven, S.-H. Chen, A. D'Albis and W. B. Gratzer, Eur. J. Biochem., 1974, 41, 539–546 CrossRef CAS PubMed.
- E. Monzani, B. Bonafè, A. Fallarini, C. Redaelli, L. Casella, L. Minchiotti and M. Galliano, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1547, 302–312 CrossRef CAS.
- P. Ascenzi, A. di Masi, G. Fanali and M. Fasano, Cell Death Discovery, 2015, 1, 15025 CrossRef CAS PubMed.
- P. Travascio, A. J. Bennet, D. Y. Wang and D. Sen, Chem. Biol., 1999, 6, 779–787 CrossRef CAS PubMed.
- P. Travascio, P. K. Witting, A. G. Mauk and D. Sen, J. Am. Chem. Soc., 2001, 123, 1337–1348 CrossRef CAS PubMed.
- D. Sen and L. C. H. Poon, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 478–492 CrossRef CAS PubMed.
- L. Stefan, F. Denat and D. Monchaud, Nucleic Acids Res., 2012, 40, 8759–8772 CrossRef CAS PubMed.
- S. Hagiwara, A. Momotake, T. Ogura, S. Yanagisawa, A. Suzuki, S. Neya and Y. Yamamoto, Inorg. Chem., 2021, 60, 11206–11213 CrossRef CAS PubMed.
- W. Shen, H. Deng and Z. Gao, RSC Adv., 2014, 4, 53257–53264 RSC.
- E. Derat and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8185–8198 CrossRef CAS PubMed.
- E. Derat and S. Shaik, J. Am. Chem. Soc., 2006, 128, 13940–13949 CrossRef CAS PubMed.
- E. Derat, S. Shaik, C. Rovira, P. Vidossich and M. Alfonso-Prieto, J. Am. Chem. Soc., 2007, 129, 6346–6347 CrossRef CAS PubMed.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS PubMed.
- N. C. Veitch and A. T. Smith, Adv. Inorg. Chem., 2000, 51, 107–162 CrossRef.
- J. E. Penner-Hahn, K. Smith Eble, T. J. McMurry, M. Renner, A. L. Balch, J. T. Groves, J. H. Dawson and K. O. Hodgson, J. Am. Chem. Soc., 1986, 108, 7819–7825 CrossRef CAS PubMed.
- M. A. Ator, S. K. David and P. R. Ortiz de Montellano, J. Biol. Chem., 1987, 262, 14954–14960 CrossRef CAS.
- A. Gumiero, E. J. Murphy, C. L. Metcalfe, P. C. Moody and E. L. Raven, Arch. Biochem. Biophys., 2010, 500, 13–20 CrossRef CAS PubMed.
- M. Ortmayer, K. Fisher, J. Basran, E. M. Wolde-Michael, D. J. Heyes, C. Levy, S. L. Lovelock, J. R. Anderson, E. L. Raven and S. Hay, ACS Catal., 2020, 10, 2735–2746 CrossRef CAS PubMed.
- G. M. do Nascimento and M. A. d. Souza, Spectroscopy of nanostructured conductive polymers, in Nanostructured Conductive Polymers, John Wiley and Sons, Chichester, 2015 Search PubMed.
- A. V. Kulikov, V. R. Bogatyrenko, O. V. Belonogova, L. S. Fokeeva, A. V. Lebedev, T. A. Echmaeva and I. G. Shunina, Russ. Chem. Bull., 2002, 51, 2216–2223 CrossRef CAS.
- V. I. Krinichnyi, H. K. Roth, M. Schrödner and B. Wessling, Polymer, 2006, 47, 7460–7468 CrossRef CAS.
- K. Junker, S. Luginbühl, M. Schüttel, L. Bertschi, R. Kissner, L. D. Schuler, B. Rakvin and P. Walde, ACS Catal., 2014, 4, 3421–3434 CrossRef CAS.
- G. Ćirić-Marjanović, M. Trchová and J. Stejskal, J. Raman Spectrosc., 2008, 39, 1375–1387 CrossRef.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS PubMed.
- R. K. P. Śledź, M. Chruszcz, M. D. Zimmerman, W. Minor and K. Woźniak, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 482–492 CrossRef PubMed.
- K. A. de Villiers, C. H. Kaschula, T. J. Egan and H. M. Marques, JBIC, J. Biol. Inorg. Chem., 2007, 12, 101–117 CrossRef PubMed.
- A. A. Moosavi-Movahedi, F. Semsarha, H. Heli, K. Nazari, H. Ghourchian, J. Hong, G. H. Hakimelahi, A. A. Saboury and Y. Sefidbakht, Colloids Surf., A, 2008, 320, 213–221 CrossRef CAS.
- Z. Moosavi-Movahedi, H. Gharibi, H. Hadi-Alijanvand, M. Akbarzadeh, M. Esmaili, M. S. Atri, Y. Sefidbakht, M. Bohlooli, K. Nazari, S. Javadian, J. Hong, A. A. Saboury, N. Sheibani and A. A. Moosavi-Movahedi, J. Biomol. Struct. Dyn., 2015, 33, 2619–2632 CrossRef CAS PubMed.
- Z. Moosavi-Movahedi, E. S. Kalejahi, M. Nourisefat, P. Maghami, N. Poursasan and A. A. Moosavi-Movahedi, Colloids Surf., A, 2017, 522, 233–241 CrossRef CAS.
- P. Walde, K. Kashima and G. Ćirić-Marjanović, Front. Bioeng. Biotechnol., 2019, 7, 165 CrossRef PubMed.
- E. Dmitrieva and L. Dunsch, J. Phys. Chem. B, 2011, 115, 6401–6411 CrossRef CAS PubMed.
- L. Dennany, P. C. Innis, S. T. McGovern, G. G. Wallace and R. J. Forster, Phys. Chem. Chem. Phys., 2011, 13, 3303–3310 RSC.
- G. M. do Nascimento, J. E. Pereira da Silva, S. I. Córdoba de Torresi and M. L. A. Temperini, Macromolecules, 2002, 35, 121–125 CrossRef CAS.
- A. G. Brolo and A. C. Sanderson, Can. J. Chem., 2004, 82, 1474–1480 CrossRef CAS.
- G. Ćirić-Marjanović, M. Trchová, E. N. Konyushenko, P. Holler and J. Stejskal, J. Phys. Chem. B, 2008, 112, 6976–6987 CrossRef PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, 2004 Search PubMed.
- M. Cochet, G. Louarn, S. Quillard, M. I. Boyer, J. P. Buisson and S. Lefrant, J. Raman Spectrosc., 2000, 31, 1029–1039 CrossRef CAS.
- A. Shakoor and T. Z. Rizvi, J. Raman Spectrosc., 2010, 41, 237–240 CAS.
- S. Ravichandran, S. Nagarajan, A. Kokil, T. Ponrathnam, R. M. Bouldin, F. F. Bruno, L. Samuelson, J. Kumar and R. Nagarajan, Langmuir, 2012, 28, 13380–13386 CrossRef CAS PubMed.
- I. Márquez, J. L. Olloqui-Sariego, M. Molero, R. Andreu, E. Roldán and J. J. Calvente, Inorg. Chem., 2021, 60, 42–54 CrossRef PubMed.
- A. C. Brezny, H. S. Nedzbala and J. M. Mayer, Chem. Commun., 2021, 57, 1202–1205 RSC.
- E. Sitte and M. O. Senge, Eur. J. Org Chem., 2020, 2020, 3171–3191 CrossRef CAS PubMed.
- H. L. Pleyer, H. Strasdeit and S. Fox, Origins Life Evol. Biospheres, 2018, 48, 347–371 CrossRef CAS PubMed.
- S. Fox and H. Strasdeit, Astrobiology, 2013, 13, 578–595 CrossRef CAS PubMed.
- D. Deamer, J. P. Dworkin, S. A. Sandford, M. P. Bernstein and L. J. Allamandola, Astrobiology, 2002, 2, 371–381 CrossRef CAS PubMed.
- C. Anastasi, F. F. Buchet, M. A. Crowe, A. L. Parkes, M. W. Powner, J. M. Smith and J. D. Sutherland, Chem. Biodiversity, 2007, 4, 721–739 CrossRef CAS PubMed.
- P.-A. Monnard and P. Walde, Life, 2015, 5, 1239–1263 CrossRef CAS PubMed.
- A. Puustinen and M. Wikström, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 6122–6126 CrossRef CAS PubMed.
- T. L. Mollan and A. I. Alayash, Antioxid. Redox Signaling, 2013, 18, 2251–2253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02198f |
| ‡ Although it has been suggested to use capital letters for specifying isolated, free hemes, e.g. heme B, and lowercase letters for hemes bound or associated to proteins (i.e., heme b), in this work we do not make this distinction and always use “heme b” for both cases.78 The extent of amphiphilicity of heme b depends on the degree of dissociation of the two carboxylic acids. At the pH value of the aqueous solutions used in this work, it is likely that the two carboxylic acids are not ionized at all. |
| § For the pH = 4.3 conditions used for the reaction, the “buffer” salts used, HEPES or dihydrogenphosphate, do not act as buffers since pH = 4.3 is too far away from the corresponding pKa values of the two salts. |
| ¶ Sulfonates often have two bands in the range = 610–500 cm−1 due to SO2 deformation vibrations.66 |
| || Heme Fe2+ undergoes autooxidation in the presence of oxygen to Fe3+.79 |
| This journal is © The Royal Society of Chemistry 2022 |