
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into rare-earth-catalysed C–H alkylation of sulfides: sulfide facilitating alkene insertion and beyond†
Yu Zhou,
Ping Wu,
Fanshu Cao,
Lei Shi,
Ni Zhang,
Zuqian Xue and
Gen Luo *
*
Institutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China. E-mail: luogen@ahu.edu.cn
First published on 5th May 2022
Abstract
The catalytic C–H alkylation with alkenes is of much interest and importance, as it offers a 100% atom efficient route for C–C bond construction. In the past decade, great progress in rare-earth catalysed C–H alkylation of various heteroatom-containing substrates with alkenes has been made. However, whether or how a heteroatom-containing substrate would influence the coordination or insertion of an alkene at the catalyst metal center remained elusive. In this work, the mechanism of Sc-catalysed C–H alkylation of sulfides with alkenes and dienes has been carefully examined by DFT calculations, which revealed that the alkene insertion could proceed via a sulfide-facilitated mechanism. It has been found that a similar mechanism may also work for the C–H alkylation of other heteroatom-containing substrates such as pyridine and anisole. Moreover, the substrate-facilitated alkene insertion mechanism and a substrate-free one could be switched by fine-tuning the sterics of catalysts and substrates. This work provides new insights into the role of heteroatom-containing substrates in alkene-insertion-involved reactions, and may help guide designing new catalysis systems.
Introduction
Developing efficient methodologies for synthesis and functionalization of sulfides is of great interest and importance, as sulfide motifs exist widely and play important roles in bioactive molecules, natural products, functional materials, and organocatalysts.1 The most straightforward and atom-economical route for the synthesis of alkylated sulfide derivatives is the catalytic C–H alkylation of sulfides with alkenes. However, such C–H alkylation approaches met with limited success,2 and in most cases only alkenes with an electron-withdrawing group showed reactivity. Recently, Hou and coworkers reported that half-sandwich scandium catalysts such as (C5Me5)Sc(CH2C6H4NMe2-o)2 could serve as a unique platform for the C–H alkylation of a wide range of methyl sulfides with various alkenes and dienes, which represents the first example of regioselective catalytic α-C(sp3)–H addition of sulfides to unactivated alkenes (Scheme 1a).3 Based on experimental observations, a possible reaction mechanism was proposed as shown in Scheme 1b. The C–H activation of the methyl group of sulfide 1 by the cationic scandium monoalkyl species A, which was generated by the reaction of the scandium dialkyl complex with [Ph3C][B(C6F5)4],4 could give an intermediate I as the catalytically active species. In the reaction with α-alkenes (Scheme 1b, left), the insertion of an alkene 2 into the Sc–C bond in I would give II, which after the protonation (C–H activation) of another sulfide 1 would release the hydrothiomethylation product 3 and regenerate the active species I. In the case of 1,5-dienes (Scheme 1b, right), starting from I, the reaction would proceed through sequential insertion of the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and the subsequent protonation, giving the corresponding thiomethyl-functionalized cyclopentane derivative 3′. It is also worth noting that a significant kinetic isotope effect (KIE ≈ 5) was observed experimentally, which suggests that C–H activation of the methyl group in the sulfides should be involved in the rate-determining step.3
C bonds and the subsequent protonation, giving the corresponding thiomethyl-functionalized cyclopentane derivative 3′. It is also worth noting that a significant kinetic isotope effect (KIE ≈ 5) was observed experimentally, which suggests that C–H activation of the methyl group in the sulfides should be involved in the rate-determining step.3
 | ||
| Scheme 1 (a) Sc-catalysed α-C–H functionalization of sulfides with alkenes and dienes and (b) proposed mechanism. | ||
Although the above proposed mechanism could schematically explain the products obtained experimentally, many details remained unclear, such as structural information on the active catalyst species, key intermediates and transition states, the energy profiles of the reaction processes, and rationale for the observation that C–H activation is the rate-determining step. Moreover, although the C–H functionalization of various heteroatom-containing substrates with alkenes catalysed by rare-earth complexes have been widely reported,5–11 whether or how a heteroatom-containing substrate would influence the coordination or insertion of an alkene at the catalyst metal center remained elusive. In order to clarify these important issues, we performed density functional theory (DFT) studies on the Sc-catalysed C–H alkylation of sulfides with alkenes and dienes. We found that alkene insertion took place through a sulfide-facilitated mechanism, preferably proceeding at a catalyst species bearing a sulfide ligand. Moreover, the C–H alkylation of other heteroatom-containing substrates such as anisoles and pyridines with alkenes may also take place through the similar mechanism. In addition, it is also noting that the substrate-facilitated alkene insertion mechanism and the substrate-free one could be switched by combining different factors, such as substrate, catalyst ligand, metal center and so on. These new findings reported here clearly revealed the role of the heteroatom-containing substrate and effectively enrich the mechanism of rare-earth catalyzed C–H functionalization.
Computational details
Geometry optimizations and frequency calculations of all structures were performed with Gaussian 16 software package12 at the B3PW91 (ref. 13)/BSI level. In BSI, the 6-31G(d) basis set was considered for C, H, O, N, and S atoms, and SDD14 basis sets were used for Sc, Y, and Lu (MWB60) atoms. The frequency calculations confirmed a stationary point as a minimum (no imaginary frequency) or a transition state (one imaginary frequency) and obtained the thermodynamic corrections to Gibbs free energy (298.15 K, 1 atm). Intrinsic reaction coordinate (IRC) calculations were performed to verify a direct connection between two minima for a transition state. All structures were optimized without any symmetry restrictions. To obtain more reliable relative energies, single-point energy calculations were performed at the level of M06 (ref. 15)/BSII with SMD implicit solvent model16 for considering solvation effect of toluene. In BSII, the 6-311+G(d,p) basis set was used for all nonmetal atoms, and SDD basis sets were used for Sc, Y, and Lu (MWB60) atoms. The theoretical strategy adopted here has been successfully applied in similar systems.10 In addition, several other widely used functional/basis set/solvation model combinations have also been tested for key processes and no significant effect on the obtained results was observed (see Fig. S6†), suggesting the reliability of the current calculation strategy. In this work, relative Gibbs free energies in toluene solution (including gas-phase corrections) were used to discuss reaction pathways. Distortion–interaction analysis17 of transition states and natural bond orbital (NBO) calculations were performed at the M06/BSII level. Three-dimensional molecular graphics were visualized with CYLview software.18Results and discussion
Some possible reaction processes and energy profile of the Sc-catalysed α-C–H alkylation of nC5H11SMe (1a) with 1-octene (2a)3 are shown in Fig. 1. The reaction proceeds through the coordination of the sulfur atom of 1a to the Sc atom in the cationic scandium species A, giving an adduct B. The following C–H activation of the α-methyl group in the coordinated 1a via the transition state TS1 (ΔG‡ = 26.4 kcal mol−1) could form a three-membered metallacycle intermediate C with an N,N-dimethyl-o-toluidine (RH) moiety. In addition, the N-dissociated pathway of the C–H activation by coordinating another 1a was calculated to overcome a high energy barrier (via TS1′, Fig. S1†). This is mainly due to the stronger Sc⋯N interaction than Sc⋯S one based on HSAB theory, which makes the N-ligated pathway more favourable (via TS1, Fig. 1). For comparison, the C–H activation of the α-methylene group in 1a is also considered and calculated to be unfavourable due to steric hindrance (Fig. S2†). The release RH from C generates the proposed naked cationic scandium species C′ with a high relative free energy (ΔG = 12.5 kcal mol−1), so it should be excluded. Alternatively, the ligand exchange between 1a and RH in C could easily occur to form the species D with a coordinated 1a.11c–e Starting from D, the ligand exchange between 2a and 1a could take place to form a π-complex E. Subsequently, the alkene ligand in E may insert into the Sc–C bond via TS2 (ΔG = 24.6 kcal mol−1) to give the five-membered ring intermediate F, as proposed in literature.3 The re-coordination of a sulfide 1a to the Sc atom easily takes place to give the more stable species G. The protonation of the Sc–alkyl bond via C–H activation of the coordinated-1a in G would finally release the alkylation product 3a. | ||
| Fig. 1 Energy profile for Sc-catalysed C–H alkylation of nC5H11SMe (1a) with 1-octene (2a) calculated at the level of M06/6-311+G(d,p)/SDD (toluene, SMD)//B3PW91/6-31G(d)/SDD. The natural charge of the metal center (QSc) is given in blue with parentheses. | ||
It is noted that alkene insertion transition state TS2 has the highest free energy in potential energy surface. In addition, we noted that the coordination of one more molecule of 1a to D could give a more stable species D′ (ΔG = −3.1 kcal mol−1). Further coordinated sulfides will lead to an increase in energy because of steric factors (see Fig. S3† for more details). According to the energetic span model,19 therefore, the species D′ with two coordinated sulfides should be regarded as the catalytically active species and the energy barrier of the alkene insertion via TS2 should be 27.7 kcal mol−1 (D′ → TS2). However, the energy barrier is significantly higher than that of the subsequent C–H activation process in G (via TS3, ΔG‡ = 22.8 kcal mol−1). This is not in agreement with the experimental observation of a significant kinetic isotope effect (KIE ≈ 5), which suggested that the C–H activation of the methyl group in the sulfide should be involved in the rate-determining step.3
We then try to explore other potential routes for alkene insertion. Notably, considering that the unique heteroatom affinity of rare-earth metal center and the opening coordination space in the structure of TS2, it encourages us to examine whether the alkene insertion can be accomplished with the aid of a molecule of sulfide. To our delight, the alkene insertion significantly reduces in energy by the assistance of a sulfide via 1,2-insertion transition state TS2′,20 because the coordination of a sulfide ligand would lower the positive charge of the Sc center (QSc = 1.25 in TS2 vs. QSc = 1.00 in TS2′) and thereby stabilize the transition state. Consequently, the energy barrier of alkene insertion can be reduced from 27.7 to 19.0 kcal mol−1 (D′ → TS2′, Fig. 1, pink line) instead of being a rate-determining step. In addition, the alkene insertion assisted by other potential coordinating molecules in current system (i.e., N,N-dimethyl-o-toluidine and 1-octene) as well as assisted by two sulfides were also considered and were ruled out due to higher relative energies (Fig. 2). Therefore, the newly established sulfide-facilitated mechanism seems more reasonable and could well explain the experimental result of KIE. We also calculated the KIE for C–H activation,21 in which the CH3 group to be activated in C–H activation was replaced by CD3. The computed KIE of 4.57 via TS3 matches the experimental datum (KIE ≈ 5),3 which provides further support for the mechanism scenario here. The computational results suggest that the reaction mainly involve the formation of catalytically active species, alkene insertion, and protonation steps. To be noted, the active species should be a cationic scandium species with two coordinated sulfide molecules (D′) rather than the proposed cationic species without sulfide (C′)3,11a,b or with one coordinated sulfide reported previously (D).11c–e The sulfide substrate not only involves in C–H activation processes (the formation of active species and the final protonation) but also serves as a Lewis acidic ligand to stabilize related intermediates and transition states in alkene insertion process.
 | ||
| Fig. 2 Calculated transition states of alkene insertion assisted by different coordination molecules (R1 = nC5H11; R2 = nC6H13; Ar = o-Me-phenyl). Relative free energies are given in kcal mol−1. All H atoms in 3D structures are omitted for clarity. | ||
With such a sulfide-facilitated mechanism in hand, we turn to the mechanism of C–H alkylation of sulfides with unconjugated dienes. The reaction of 1,5-heptadiene (2b) with nC5H11SMe (1a) giving thiomethyl-functionalized cyclopentane product 3b was computationally investigated (Fig. 3). Similar in the case of 1-octene, the terminal CC double bond insertion occurs through the sulfide-facilitated mechanism (via TS4′) is significantly favourable than the sulfide-free pathway (via TS4) by 8.1 kcal mol−1. By contrast, the following CC insertion (cyclization) in J′ takes place through a sulfide-free pathway (TS5) instead of the sulfide-facilitated mechanism (TS5′) due to steric effects (vide infra). It is worth noting that the direct protonation of Sc–alkyl bond in J′ by hydrogen abstraction of the coordinated 1a via TS5′′ needs to overcome an energy barrier of 22.3 kcal mol−1 (Fig. 2, gray line), which is much higher than that of the cyclization process (via TS5, ΔG‡ = 13.1 kcal mol−1). This is well in line with the experimental observation of the exclusive formation of the thiomethylated cyclopentane product rather than a simple alkylation product with a free olefinic bond. The results clearly show that, in the case of diene, the reaction operates through the sulfide-facilitated alkene insertion, followed by the sulfide-free alkene insertion (cyclization) and protonation steps to complete catalytic cycle. The C–H activation is the rate-determining step, as in the case of 1-octene.
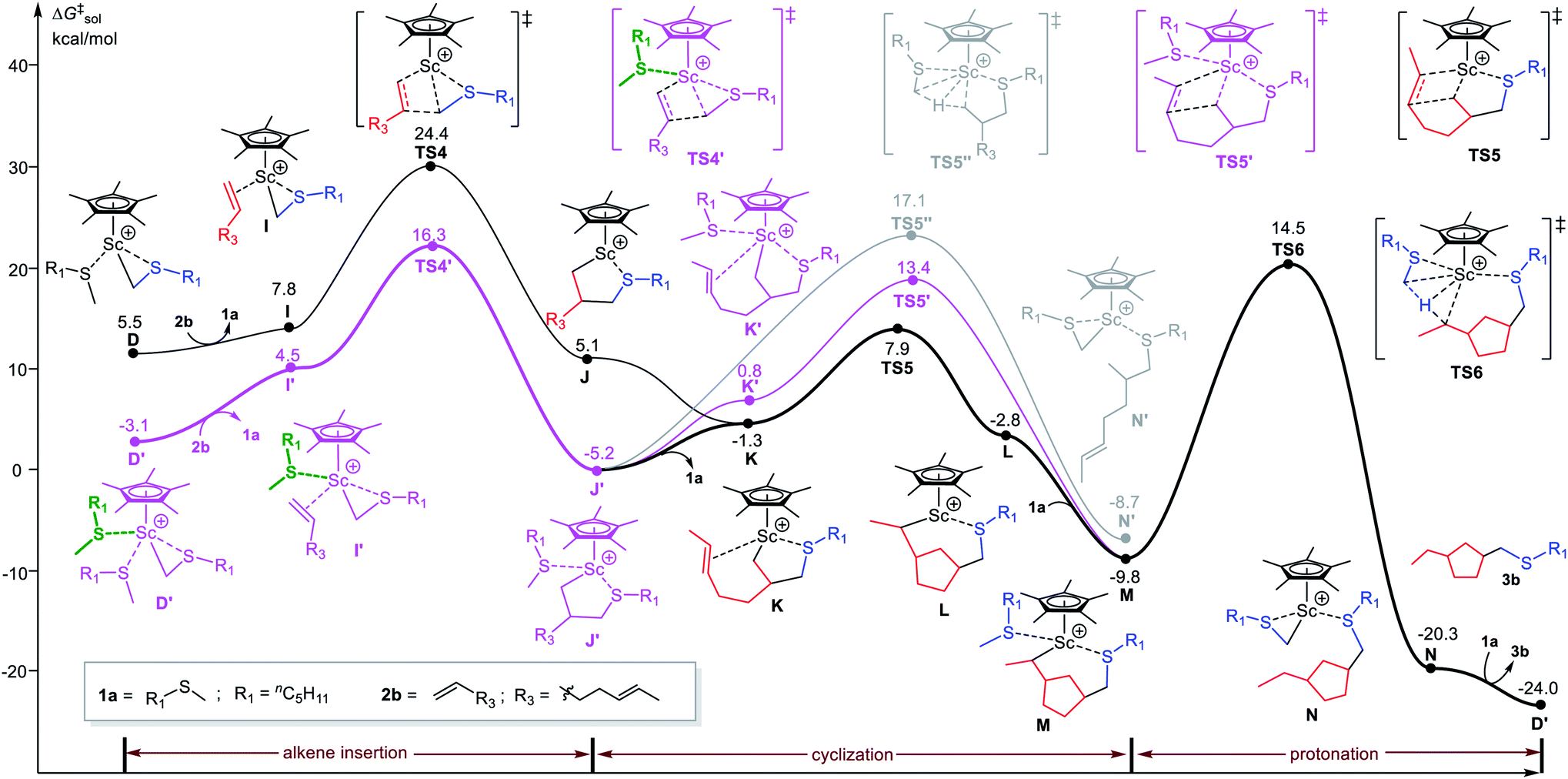 | ||
| Fig. 3 Energy profile for Sc-catalyzed C–H alkylation of nC5H11SMe (1a) with 1,5-heptadiene (2b). The free energies (kcal mol−1) are relative to the species A as shown in Fig. 1. | ||
To have a better understand of the discrepancy of mechanism for the two sequential alkene insertion processes mentioned above, we performed more calculations on alkene insertion (cyclization) processes by modifying diene or catalyst (ligand and metal center). The results are shown in Fig. 4b–d, and the case of 1,5-heptadiene catalysed by (C5Me5)ScR2 is illustrated again in Fig. 4a for comparison. As shown in Fig. 4b, by reducing steric hindrance of diene, the sulfide-facilitated alkene insertion mechanism and the sulfide-free one become competitive with each other when 1,5-hexadiene is used instead of 1,5-heptadiene in the (C5Me5)Sc-catalysed cyclization process. When the steric hindrance is further reduced by using the scandium catalyst with smaller ligand C5H5 (Fig. 4c) or the C5Me5-ligated catalyst with the larger yttrium or lutetium center (than scandium) (Fig. 4d and S5†), it obviously shows that the sulfide-facilitated mechanism becomes more favourable than the sulfide-free pathway. Therefore, these results clearly indicate that the mechanisms of alkene insertion could be switched by tuning the sterics, including substrate, catalyst ligand, metal center and so on. There is a subtle balance between steric and electronic factors, leading to different mechanisms. When the catalyst–substrate combined structure has less steric hindrance, it prefers to undergo heteroatom-containing substrate facilitated alkene insertion mechanism due to the heteroatom affinity of rare-earth metal center. On the contrary, the heteroatom-containing substrate-free mechanism is more likely to occur. These findings are helpful for us to better understand how the heteroatom-containing substrate influence the coordination and insertion of an alkene at the catalyst center and are also expected to guide the design of new rare-earth catalysis reactions.
 | ||
| Fig. 4 Energy profiles for cyclization processes for the case of 1,5-heptadiene catalyzed by (C5Me5)ScR2 (a) and the case of 1,5-heptadiene catalysed by (C5Me5)ScR2 (b), (C5H5)ScR2 (c), and (C5Me5)YR2 (d), respectively. | ||
To examine the potential applicability of the heteroatom-containing substrate facilitated alkene insertion mechanism, we extended the investigation to C–H alkylation of O-(anisole)8a and N-containing (pyridine)6a substrates with alkenes catalysed by the same scandium catalyst (C5Me5)Sc(CH2C6H4NMe2-o)2 (Fig. 5) as used in the sulfide system.3 It is found that, in the case of anisole (Fig. 5, left), the alkene insertion via the anisole-free pathway (TS7) has a relative free energy of 21.5 kcal mol−1, while the anisole-facilitated pathway via TS7′ has a relative free energy of 17.1 kcal mol−1. Therefore, similar to the sulfide system, the anisole-facilitated alkene insertion mechanism is much more favourable than the anisole-free mechanism in the Sc-catalysed C–H alkylation of anisole system. As expected, in the case of pyridine, similar results are obtained that alkene insertion undergoes the pyridine-facilitated mechanism (Fig. 5, right), which remind us to modify the previously proposed mechanism of C–H alkylation of pyridines with alkenes.11a,b
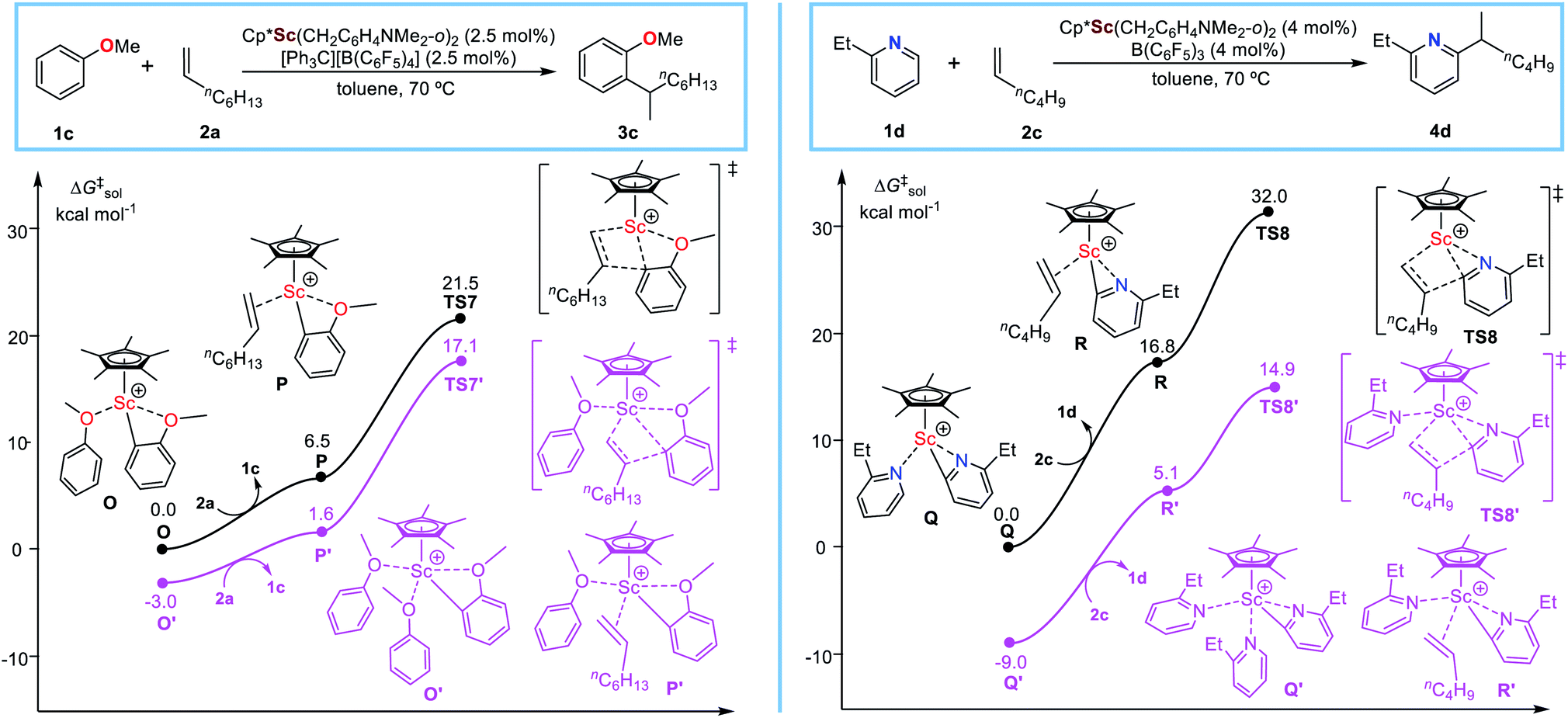 | ||
| Fig. 5 Energy profile for alkene insertion processes involved in Sc-catalysed C–H alkylation of other heteroatom (N and O)-containing substrates with alkenes. | ||
The result suggests that the heteroatom-containing substate facilitated alkene insertion mechanism potentially has wide applicability in the field of rare-earth catalysed C–H alkylation, which is complementary or modified to those mechanisms proposed previously5a,11 and thus effectively enrich the mechanism framework of rare-earth catalysed C–H functionalization chemistry. In addition, it is worth mentioning that Cui, Hou, and Marks groups reported the interaction between the heteroatom (e.g., N, O, P, S, Se) in functionalized olefin monomer and the rare-earth catalyst could significantly improve the polymerization activity.22 The promotion effect of the heteroatom in olefin polymerization (continuous alkene-insertion-involved reactions) is similar to that in the C–H alkylation systems discussed here. Therefore, the heteroatom-containing substrate facilitated alkene insertion mechanism is also expected to apply in polymerization of heteroatom-functionalized olefins.
Conclusions
In summary, the mechanism of Sc-catalysed C–H alkylation of sulfides with alkenes and dienes has been carefully examined by DFT calculations. The results suggest that the reaction mainly involves several steps: the formation of catalytically active species, alkene insertion (sequential insertion for diene), and protonation. The catalytically active species is the cationic species with two coordinated sulfide molecules rather than the sulfide-free or one sulfide coordinated cationic species proposed previously. Interestingly, it is found that the alkene insertion takes place through a sulfide-facilitated mechanism, during which the sulfide substrate serves as a ligand to stabilize key intermediates and transition states in alkene insertion process. More importantly, such a substrate-facilitated alkene insertion mechanism also works for Sc-catalysed C–H alkylation of other heteroatom (O and N)-containing substrates. The promotion effect of the heteroatom-containing substrate on alkene insertion is obviously complementary or modified to those mechanisms proposed previously. In addition, the results also show that the substrate-facilitated alkene insertion mechanism and the substrate-free one could be switched by fine-tuning the sterics of catalysts and substrates. The new insights into the role of heteroatom-containing substrate on alkene insertion and the adjustable mechanisms not only greatly enrich the mechanism framework of rare-earth catalysed C–H functionalization chemistry but also may help guide designing new catalysis systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22003001), Anhui Provincial Natural Science Foundation (2108085Y04), and the Innovation and Entrepreneurship Project of Overseas Returnees in Anhui Province (2020LCX006). We gratefully appreciate access to the High-Performance Computing Platform of Anhui University and Hefei Advanced Computing Center for computational resources. The authors thank Prof. Zhaomin Hou at RIKEN for helpful discussions.Notes and references
- (a) D. Poirier, S. Auger, Y. Mérand, J. Simard and F. Labrie, J. Med. Chem., 1994, 37, 1115–1125 CrossRef CAS PubMed; (b) D. Meng, W. Chen and W. Zhao, J. Nat. Prod., 2007, 70, 824–829 CrossRef CAS PubMed; (c) Z. Ke, C. K. Tan, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2014, 136, 5627–5630 CrossRef CAS PubMed; (d) M. Nakano and K. Takimiya, Chem. Mater., 2017, 29, 256–264 CrossRef CAS.
- (a) J. Chen, R. L. Kirchmeier and J. M. Shreeve, Inorg. Chem., 1996, 35, 6676–6681 CrossRef CAS PubMed; (b) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638–3641 CrossRef CAS PubMed; (c) H. Yang, H. Yan, P. Sun, Y. Zhu, L. Lu, D. Liu, G. Rong and J. Mao, Green Chem., 2013, 15, 976–981 RSC; (d) H. Cao, D. Liu, C. Liu, X. Hu and A. Lei, Org. Biomol. Chem., 2015, 13, 2264–2266 RSC; (e) S. Kamijo, G. Takao, K. Kamijo, T. Tsuno, K. Ishiguro and T. Murafuji, Org. Lett., 2016, 18, 4912–4915 CrossRef CAS PubMed; (f) D. Xia, Y. Li, T. Miao, P. Li and L. Wang, Green Chem., 2017, 19, 1732–1739 RSC; (g) Y.-F. Liu, L. Zheng, D.-D. Zhai, X.-Y. Zhang and B.-T. Guan, Org. Lett., 2019, 21, 5351–5356 CrossRef CAS PubMed.
- Y. Luo, Y. Ma and Z. Hou, J. Am. Chem. Soc., 2018, 140, 114–117 CrossRef CAS PubMed.
- X. Li, M. Nishiura, K. Mori, T. Mashiko and Z. Hou, Chem. Commun., 2007, 4137–4139 RSC.
- For reviews, see: (a) M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS PubMed; (b) R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 CrossRef CAS.
- For examples of rare-earth catalysed C–H bond addition of pyridines to alkenes, see: (a) B.-T. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086–18089 CrossRef CAS PubMed; (b) B.-T. Guan, B. Wang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2013, 52, 4418–4421 CrossRef CAS PubMed; (c) G. Song, W. W. N. O and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209–12212 CrossRef CAS PubMed; (d) G. Song, B. Wang, M. Nishiura and Z. Hou, Chem.–Eur. J., 2015, 21, 8394–8398 CrossRef CAS PubMed; (e) Y. Luo, H.-L. Teng, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2017, 56, 9207–9210 CrossRef CAS PubMed; (f) Y. Chen, D. Song, J. Li, X. Hu, X. Bi, T. Jiang and Z. Hou, ChemCatChem, 2018, 10, 159–164 CrossRef CAS.
- For examples of rare-earth catalysed C–H bond addition of anilines to alkenes, see: (a) X. Xu, X. Zheng and X. Xu, ACS Catal., 2021, 11, 14995–15003 CrossRef CAS; (b) J. Su, Y. Luo and X. Xu, Chem. Commun., 2021, 57, 3688–3691 RSC; (c) J. Su, Y. Cai and X. Xu, Org. Lett., 2019, 21, 9055–9059 CrossRef CAS PubMed; (d) H. Gao, J. Su, P. Xu and X. Xu, Org. Chem. Front., 2018, 5, 59–63 RSC; (e) G. Song, G. Luo, J. Oyamada, Y. Luo and Z. Hou, Chem. Sci., 2016, 7, 5265–5270 RSC.
- For examples of rare-earth catalysed C–H bond addition of anisoles to alkenes, see: (a) J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828–12832 CrossRef CAS PubMed; (b) B. Tang, X. Hu, C. Liu, T. Jiang, F. Alam and Y. Chen, ACS Catal., 2019, 9, 599–604 CrossRef CAS; (c) X. Shi, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2016, 138, 6147–6150 CrossRef CAS PubMed.
- For examples of rare-earth catalysed C–H bond addition of aldimines/amines to alkenes, see: (a) X. Cong, G. Zhan, Z. Mo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2020, 140, 5531–5537 CrossRef PubMed; (b) X. Cong, Q. Zhuo, N. Hao, Z. Mo, G. Zhan, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2022, 61, e202115996 CrossRef CAS PubMed; (c) A. Nako, J. Oyamada, M. Nishiura and Z. Hou, Chem. Sci., 2016, 7, 6429–6434 RSC.
- For examples of rare-earth catalysed C–H bond addition of quinolines to alkenes/alkynes, see: (a) S.-J. Lou, G. Luo, S. Yamaguchi, K. An, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2021, 143, 20462–20471 CrossRef CAS PubMed; (b) S.-J. Lou, Q. Zhuo, M. Nishiura, G. Luo and Z. Hou, J. Am. Chem. Soc., 2021, 143, 2470–2476 CrossRef CAS PubMed; (c) S.-J. Lou, L. Zhang, Y. Luo, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 140, 18128–18137 CrossRef PubMed.
- Theoretical studies on rare-earth catalysed C–H alkylation with alkenes, see: (a) G. Luo, Y. Luo, J. Qu and Z. Hou, Organometallics, 2012, 31, 3930–3937 CrossRef CAS; (b) G. Zhou, G. Luo, X. Kang, Z. Hou and Y. Luo, Organometallics, 2018, 37, 2741–2748 CrossRef CAS; (c) F. Liu, G. Luo, Z. Hou and Y. Luo, Organometallics, 2017, 36, 1557–1565 CrossRef CAS; (d) G. Luo, F. Liu, Y. Luo, G. Zhou, X. Kang, Z. Hou and L. Luo, Organometallics, 2019, 38, 1887–1896 CrossRef CAS; (e) P. Wang, G. Luo, J. Yang, X. Cong, Z. Hou and Y. Luo, J. Org. Chem., 2021, 86, 4236–4244 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) A. D. Beck, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (b) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- For reviews, see: (a) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC; (b) F. Liu, Y. Liang and K. N. Houk, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed; (c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, https://www.cylview.org Search PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- The insertion of 1-octene into Sc-alkyl bond via 1,2-fashion is more favourable than 2,1-mode due to electronic factors (Fig. S4†), also see ref. 11a and c.
- The semiclassical KIE was calculated using the Eyring model, KIE = kH/kD = exp[(ΔGD‡ − ΔGH‡)/RT], in which the free energy barriers (ΔG‡) were calculated at the level of M06/6-311+G(d,p)/SDD (toluene, SMD)//B3PW91/6-31G(d)/SDD (298.15 K), see: Y. Sun, H. Tang, K. Chen, L. Hu, J. Yao, S. Shaik and H. Chen, J. Am. Chem. Soc., 2016, 138, 3715–3730 CrossRef CAS PubMed.
- (a) D. Liu, C. Yao, R. Wang, M. Wang, Z. Wang, C. Wu, F. Lin, S. Li, X. Wan and D. Cui, Angew. Chem., Int. Ed., 2015, 54, 5205–5209 CrossRef CAS PubMed; (b) C. Wang, G. Luo, M. Nishiura, G. Song, A. Yamamoto, Y. Luo and Z. Hou, Sci. Adv., 2017, 3, e1701011 CrossRef PubMed; (c) J. Chen, Y. Gao, B. Wang, T. L. Lohr and T. J. Marks, Angew. Chem., Int. Ed., 2017, 56, 15964–15968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Distortion–interaction analysis, other energy profiles, coordinates of all optimized structures. See https://doi.org/10.1039/d2ra02180c |
| This journal is © The Royal Society of Chemistry 2022 |