DOI:
10.1039/D2RA00719C
(Paper)
RSC Adv., 2022,
12, 13580-13592
Template synthesis, DNA binding, antimicrobial activity, Hirshfeld surface analysis, and 1D helical supramolecular structure of a novel binuclear copper(II) Schiff base complex†
Received
2nd February 2022
, Accepted 20th April 2022
First published on 5th May 2022
Abstract
A new binuclear copper(II) Schiff base complex [Cu2 L2− (NO3)2]·2CH3OH (1) [L = 2,6-bis((E)-(p-tolylimino)methyl)-4-methoxyphenol] was synthesized using a template method in which the tridentate N2O Schiff base ligand was derived from [1 + 2] condensation of 2,6-diformyl-4-methoxyphenol and p-methyl aniline in the presence of copper(II) ions as the template agent. The X-ray diffraction analyses revealed that this complex crystallizes in the monoclinic system with space group P21/n. The most remarkable structural feature of 1 is that it contains two types of 1D right-handed helical chains. The molecules are linked by intermolecular hydrogen bonds and π⋯π interactions, then a 3D supramolecular network was constructed. Moreover, the intermolecular interactions on the crystal packing of 1 have been further studied using Hirshfeld surface analysis and corresponding 2D fingerprint plots. Binding interaction of this complex with calf thymus DNA (CT-DNA) has been investigated using absorption and emission studies, viscosity experiments and circular dichroism studies. Complex 1 shows significant binding to the DNA. The results of fluorescence spectroscopy and UV absorption spectroscopy, CD spectroscopy and viscosity indicated that this complex interacted with CT-DNA in a groove binding mode where the binding constant was 1.3 ± 0.2 × 104 L mol−1. Our fluorimeteric study showed that the reaction between 1 and CT-DNA was exothermic (ΔH = 59.6 kJ mol−1; ΔS = 268.79 J mol−1 K−1). Antibacterial activities of the complex were screened by the disc diffusion method against three Gram-positive bacteria (Staphylococcus aureus ATCC 25923, Enterococcus faecalis ATCC 23212 and S. epidermidis ATCC 34384), and three Gram-negative bacteria (Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and Klebsiella pneumonia ATCC 70063). The results indicated that this complex demonstrated acceptable antibacterial activities.
1. Introduction
Helical structures retain a unique place in life since genetic information is encoded within helical DNA molecules. Much effort has been devoted in recent years to the design and fabrication of metal complexes exhibiting helical architectures, not only in modeling of biological systems, but also for applying them in the fields of supramolecular chemistry, asymmetric catalysis, and nonlinear optical material,1–8 and the aesthetically appealing structural topology. Reviews have shown the significance of such complexes9 and structures with a single helix,10 double helices,11 and triple helices,12 whether ligand directed,13 spacer controlled,14 or anion induced,15 employing many different kinds of N, O, and/or S containing coordinating ligands. Such helical molecules have been designed and assembled by the selection of basic components such as the coordination geometry of metal ions, the binding site of donating atoms, and the length of spacers.16–18 Single stranded helical structures are the most commonly observed form compared to double and triple stranded structures, either with enantiopure or achiral ligands. The secondary structure of helical complexes is determined by weak metal–ligand covalent interactions, halide ions such as iodide and supramolecular interactions such as hydrogen bonding and π⋯π stacking.19–24 The design and synthesis of small synthetic systems that can recognize specific sites of DNA through formation of non-covalently associated complexes are important areas of much current interest.25 Such physical complexation, more often, may produce important pharmacological effects by interfering with the biological processes in which DNA/RNA takes part. Such investigations have also provided insights as to the mechanism of action for antitumor antibiotics.26 The metal complexes can bind to DNA by different connection modes, such as π⋯π interactions which happen when the ligand contains planar heterocyclic ring systems, or when there is groove binding in large molecules through hydrogen bonding and van der Waals interactions.27 Beside, metal complexes of Schiff-bases have been emerged for the development of efficient variety of science.28–32
In the present study, we reported crystal structure of helical binuclear copper(II) Schiff base complex, [Cu2L2−(NO3)2]·2CH3OH (1) [L = 2,6-bis((E)-(p-tolylimino)methyl)-4-methoxyphenol] containing two types of 1D right-handed helical chains. We also examined the close intermolecular interactions between the molecules in the solid state of the crystal using Hirshfeld surface analysis. In addition, antibacterial activity of complex was investigated. Furthermore, DNA binding of 1 was studied using different physico-chemical methods. A list of abbreviations used in the text with explanations shows in Table S1.†
2. Experimental
2.1. Materials
High purity p-methyl aniline, copper(II) nitrate hydrate, 4-methoxy phenol, hexamethylenetetramine, trifluoroacetic acid were purchased from commercial sources and used as received. 2,6-Diformyl-4-methoxy phenol was synthesized by the reported procedure.51 Commercially pure chemicals such as Tris–HCl (Sigma Company, Madrid, Spain), were provided and used without purification. Highly polymerized calf thymus-DNA (CT-DNA) was procured from Sigma Company. All the experiments of DNA interaction were carried out in Tris–HCl buffer solutions and double distilled water (10 mM, pH 7.4). The stock solution of CT-DNA was procured by dissolving approximately 1–2 mg of CT-DNA fibers in 2 mL Tris–HCl buffer and stored at 4 °C for 24 hours. The concentration of DNA in stock solution was expressed in monomer units, as determined by spectrophotometry at 260 nm using the extinction coefficient (εp) of 6600 M−1 cm−1. DNA solutions were employed after no more than 4 days.
2.2. Instrumentation
In order to characterize and assess the structure of 1, the following instruments were used: the C, H, N data were obtained by using a Costech ECS 4010 elemental analyzer. FT-IR spectra were recorded on Bruker Vertex 70 spectrometer in KBr pellets from 400–4000 cm−1. The absorbance spectra were recorded for 1 using an HP spectrophotometer (Agilent 8453) equipped with a thermostated bath (Huber polysat cc1). The absorbance measurements were performed by keeping the complex concentration constant (2.5 × 10−5 M) while varying the DNA concentration from 0 to 8.0 × 10−5 M (ri = [DNA]/[complex] = 0.0–3.2). The spectra were recorded in the range of 200–500 nm. CD measurements were recorded on a JASCO (J-810) spectropolarimeter, keeping the concentration of DNA constant (5 × 10−5 M) while varying the complex concentration from 0 to 3.0 × 10−5 M (ri = [complex]/[DNA] = 0.0–0.6). Viscosity measurements were made using a viscosimeter (SCHOT AVS 450) preserved at 25 °C  0.5 °C in a constant temperature bath while the DNA concentration was fixed at 5 × 10−5 M and flow time was admeasured with a digital stopwatch. The data is reported as (η/η0)1/3 versus ri (ri = [complex]/[DNA] = 0.0–1.0), where η0 is the viscosity of the DNA solution alone. All fluorescence measurements were performed on a JASCO spectrofluorimeter (FP6200) using a quartz cell of 1 cm path length by keeping the concentration of the complex constant (1 × 10−5 mol L−1) while varying the DNA concentration from 0 to 3.4 × 10−5 M at three different temperatures (293, 310, 318 K). The samples were excited at 260 nm and the emission spectra were recorded in the range of 300 to 480 nm. The quenching experiments were conducted by adding stoichiometric small aliquots of potassium iodide stock solution (0.1 mol L−1) to 1 and CT–DNA–Cu complex solutions, respectively. The fluorescence intensity was recorded and the quenching constants (ksv) were calculated.
0.5 °C in a constant temperature bath while the DNA concentration was fixed at 5 × 10−5 M and flow time was admeasured with a digital stopwatch. The data is reported as (η/η0)1/3 versus ri (ri = [complex]/[DNA] = 0.0–1.0), where η0 is the viscosity of the DNA solution alone. All fluorescence measurements were performed on a JASCO spectrofluorimeter (FP6200) using a quartz cell of 1 cm path length by keeping the concentration of the complex constant (1 × 10−5 mol L−1) while varying the DNA concentration from 0 to 3.4 × 10−5 M at three different temperatures (293, 310, 318 K). The samples were excited at 260 nm and the emission spectra were recorded in the range of 300 to 480 nm. The quenching experiments were conducted by adding stoichiometric small aliquots of potassium iodide stock solution (0.1 mol L−1) to 1 and CT–DNA–Cu complex solutions, respectively. The fluorescence intensity was recorded and the quenching constants (ksv) were calculated.
2.3. Crystallographic measurements
Single crystal X-ray diffraction intensity data of 1 was performed at 120 K using Gemini four-circle diffractometer from Agilent Technologies, equipped with a mirror collimated Cu Kα radiation (1.54184 Å) and area detector Atlas. The structure was solved by direct methods using SUPERFLIP program33 and refined by full-matrix least-square technique on F2, using JANA 2006 (ref. 34) program with anisotropic displacement parameters for all non-hydrogen atoms. All hydrogen atoms were discernible in difference Fourier maps and could be refined to reasonable geometry. According to common practice, H atoms bonded to C were kept in ideal positions while positions of H atom bonded to O were refined with restrained geometry. In both cases Uiso(H) was set to 1.2Ueq(C,O). The molecular structure plots were prepared using the ORTEP III35 and Mercury.36 Crystallographic data and refinement parameters are summarized in Table 1, and the selected bond lengths and angles for 1 are displayed in Table 2. The selected hydrogen bonds are presented in Table 3.
Table 1 Crystal data and structure refinement of 1
| Formula |
C46H42Cu2N6O10 · 2(CH4O) |
| CCDC no |
1456947 |
| Formula weight |
1030 |
| Temperature (K) |
120 |
| Wavelength (Å) |
1.5418 |
| Crystal system, space group |
Monoclinic, P21/n |
| Unit cell dimensions |
|
| a, b, c (Å) |
8.3185 (4), 17.6483 (7), 15.7257 (8) |
| β/(°) |
101.951 (4) |
| Volume (Å3) |
2258.61 (18) |
| Z, Dcalc/(Mg m−3) |
2, 1.515 |
| Absorption coefficient (mm−1) |
1.78 |
| F(000) |
1068 |
| Crystal size (mm3) |
0.30 × 0.25 × 0.14 |
| Crystal shape/colour |
Block, black |
| θ range for data collection/(°) |
5.0–66.7 |
| Limiting index |
−9,9/-14,20/-13,18 |
| Reflections collected/unique |
3911/3512 [R(int) = 0.023] |
| Absorption correction |
Analytical |
| Data/restraints/parameters |
3911/3/310 |
| Goodness-of-fit on F2 |
97 |
| Final R indices [I > 3σ (I)] |
R1 = 0.041, wR2 = 0.111 |
| R Indices (all data) |
R1 = 0.045, wR2 = 0.114 |
| Largest diff. peak, hole (e Å−3) |
0.58 and −0.78 |
Table 2 Selected bond lengths (Å) and angles (°) for 1a
| Bond length |
|
| Symmetry code: (i) −x, −y + 2, −z. |
| Cu1–O1 |
1.9529 (17) |
| Cu1–O1i |
1.9647 (16) |
| Cu1–N1i |
1.970 (2) |
| Cu1–N2 |
1.975 (2) |
| Cu1–O3i |
2.655 (18) |
| Cu1–O4 |
2.504 (16) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Bond angle |
|
| O1–Cu1–O1i |
78.82 (7) |
| O1–Cu1–N1i |
167.94 (8) |
| O1–Cu1–N2 |
90.55 (8) |
| O1i–Cu1–N1i |
90.30 (8) |
| O1i–Cu1–N2 |
169.36 (8) |
| N1i–Cu1–N2 |
100.32 (8) |
| O4–Cu1–O3i |
160.85 (57) |
| O3i–Cu1–N1i |
91.34 (67) |
| O4–Cu1–O1i |
84.13 (62) |
| O4–Cu1–O1 |
85.41 (62) |
| O4–Cu1–N1i |
98.75 (67) |
| O4–Cu1–N2 |
94.76 (68) |
| O3i–Cu1–O1 |
81.67 (6) |
| O3i–Cu1–O1i |
79.56 (61) |
| O3i–Cu1–N2 |
99.41 (7) |
Table 3 Intra and intermolecular hydrogen bonds in 1a
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
D–H⋯A |
| Symmetry code: (ii) −x − 1, −y + 2, −z. |
| C13–H1c13⋯O3 |
0.96 |
2.349 (17) |
3.240 (3) |
154.06 (15) |
| C17–H1c17⋯O4 |
0.96 |
2.31 (17) |
3.203 (3) |
153.82 (16) |
| O1m–H1o1m⋯O5 |
0.834 (19) |
1.963 (18) |
2.749 (3) |
156.8 (17) |
| C4–H1c4⋯O1m |
0.96 |
2.698 (29) |
3.549 (37) |
148.04 (16) |
| C15–H1c15⋯O1m |
0.96 |
2.519 (26) |
3.349 (35) |
144.59 (15) |
| C21–H1c21⋯O1m |
0.96 |
2.675 (30) |
3.592 (39) |
159.78 (15) |
| C7–H1c7⋯O5ii |
0.96 |
2.38 (2) |
3.284 (3) |
157.00 (15) |
| C6–H1c6··· O5ii |
0.96 |
2.53 (17) |
3.384 (3) |
148.17 (15) |
| C22–H2c22⋯O2 |
0.96 |
2.59 (17) |
3.499 (33) |
157.61 (18) |
| C20–H1c20⋯N3 |
0.96 |
2.596 (2) |
3.384 (32) |
139.52 (15) |
2.4. Hirshfeld surfaces analysis
Hirshfeld Surface (HS) analysis has become an attractive and growing technique in description of crystal structures.37–40 HS and the associated 2D fingerprint plots (FP) have been generated using Crystal Explorer 3.0 program.41 When the structure input file in the CIF format was uploaded into the CrystalExplorer software, all bond lengths to hydrogen were automatically modified to typical standard neutron values42 (C–H = 1.083 A, N–H = 1.009 A and O–H = 0.983 A). The 2D fingerprint plots were represented by using the standard 0.6–2.4 Å view with the de and di distance scales displayed on the graph axes, where de is the distance from the Hirshfeld surface to the nearest nucleus outside the surface and di is the corresponding distance to the nearest nucleus inside the surface. The function dnorm is a ratio that comprises the de, di and the van der Waals radii (rvdW) of the atom. The dnorm values were mapped onto the Hirshfeld surface using a red-blue-white colour scheme as follows: red regions with the negative values of dnorm represent the intermolecular contacts shorter than the sum of the van der Waals radii; blue regions with positive values of dnorm represent the intermolecular contacts longer than the sum of the van der Waals radii; white regions denote the distance of contacts exactly corresponding to the van der Waals separation with a dnorm values of zero.43 Furthermore, two colored properties namely, shape-index and curvedness surfaces can be specified based on the local curvature of the Hirshfeld surface, which indicates the interactions between neighboring molecules.44
2.5. Antimicrobial activity
The cultures of Staphylococcus epidermidis ATCC 34384, Staphylococcus aureus ATCC 25923 Enterococcus faecalis ATCC 29212, Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, and Klebsiella pneumonia ATCC 70063 were obtained from Hacettepe University, Department of Medical Microbiology. Bacterial strains were cultured overnight at 37 °C in nutrient broth. During the survey, these stock cultures were stored in the dark at 4 °C.
2.5.1 Disc diffusion method. For the investigation of the antibacterial activity, the synthesized complex, 1, was dissolved in dimethylsulfoxide (20% DMSO) to a final concentration of 10 mg mL−1 and sterilized by filtration through 0.45 μm millipore filters. Antimicrobial tests were then carried out by the disc diffusion method using 100 μL of suspension containing 108 CFU mL−1 bacteria, which was spread on a nutrient agar (NA) medium. The discs (6 mm in diameter) were impregnated with 25 μL of compound (200 μg per disc) at the concentration of 8 mg mL−1 and placed on the inoculated agar. DMSO impregnated discs were used as negative control. Sulfioxazole (300 μg per disk) were used as positive reference standards to determine the sensitivity of one strain/isolate in each microbial species tested. The inoculated plates were incubated at 37 °C for 24 h for bacterial strains isolates. The antimicrobial activity in the disc diffusion assay was examined by measuring the zone of inhibition against the test organisms. Each assay in this experiment was repeated twice.45,46
2.5.2 Micro dilution assays. The minimal inhibition concentration (MIC) values, except one, were also studied for the microorganisms sensitive to compound determined in the disc diffusion assay. The inocula of microorganisms were prepared from 12 h broth cultures and suspensions were adjusted to 0.5 McFarland standard turbidity. The test compound dissolved in dimethylsulfoxide (DMSO) was first diluted to the highest concentration (2000 μg mL−1) to be tested, and then serial, two-fold dilutions were made in a concentration range from 15.625 to 2000 μg mL−1 in 10 mL sterile test tubes containing nutrient broth. The MIC values of compound against bacterial strains were determined based on a micro-well dilution method.47 The 96-well plates were prepared by dispensing 95 μL of nutrient broth and 5 μL of the inoculums into each well. 100 μL from the test compound initially prepared at the concentration of 2000 μg mL−1 was added into the first wells. Then, 100 μL from each of their serial dilutions was transferred into eight consecutive wells. The last well containing 195 μL of nutrient broth without compound, and 5 μL of the inoculums on each strip, was used as negative control. The final volume in each well was 200 μL. The contents of the wells were mixed and the micro plates were incubated at 37 °C for 24 h. The compound tested in this study was screened twice against each microorganism. The MIC was defined as the lowest concentration of the compounds to inhibit the growth of microorganisms.48
2.6. Synthesis of [Cu2L2−(NO3)2]·2CH3OH (1)
A methanolic solution (10 mL) of p-methyl aniline (0.107 g, 1 mmol) was added dropwise to a solution of Cu(NO3)2·2H2O (0.162 g, 0.5 mmol) and 2,6-diformyl-4-methoxy phenol (0.09 g, 0.5 mmol) in the same solvent (15 mL). The mixture of reaction was heated for 24 h. Then, the deep green solution was kept in air for slow evaporation. After about a week, dark green colored crystals appeared. Yield: 0.722 g (70%), anal. calc. for C46H42Cu2N6O10, 2(CH4O): C, 55.97; H, 4.89; N, 8.16. Found: C, 56.00; H, 4.90; N, 8.16%. ΛM: (1 × 10−3 M in DMF) 73.86 Ω−1 cm2 mol−1). IR (KBr, cm−1): 3408 (O–H), 2831 (C–H), 1624, 1614 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1584 (CC); UV-vis [λmax, 454 nm, 270 nm (lit mol−1cm−1). MS (EI): (m/z) = 840 [Cu2L2]+, 814 [Cu2L2–OMe + 2H+]+, 747 [L2 + CH3OH]+
N), 1584 (CC); UV-vis [λmax, 454 nm, 270 nm (lit mol−1cm−1). MS (EI): (m/z) = 840 [Cu2L2]+, 814 [Cu2L2–OMe + 2H+]+, 747 [L2 + CH3OH]+
3. Results and discussion
3.1. Synthesis and characterization of [Cu2 L2− (NO3)2]. 2CH3OH (1)
1 was synthesized using reaction among Cu(II) salt, 2,6-diformyl-4-methoxyphenol and p-methyl aniline in methanol solvent at reflux conditions. The exact coordination sphere of 1 was determined by single-crystal X-ray crystallography. In FT-IR spectra, the peaks at 1614 and 1624 cm−1 is due to CN stretch.49 Conductivity measurements performed in DMF (Molar Conductance, ΛM (1 × 10−3 M in DMF) 73.86 Ω−1 cm2 mol−1) confirm the non-electrolyte nature of the complex.50 1 was stable in air at room temperature and soluble in common polar organic solvents such as ethanol, methanol, DMF, and DMSO. Moreover, the 1 was found to be stable in the solution phases used, as evidenced by the FT-IR spectra (Fig. S1†).
3.2. Electronic absorption spectroscopy
The electronic spectrum of 1 was recorded in EtOH. The maximal absorption peaks at 270 nm and 454 nm arising from intraligand π–π* transitions and charge transfer transition, respectively.51–53 Also, the weak band around 658 nm for 1 is assignable to the 2Eg–2T2g transition that is characteristic of octahedral geometry around Cu(II) ion.54
3.3. Description of crystal structure [Cu2 L2− (NO3)2]·2CH3OH (1)
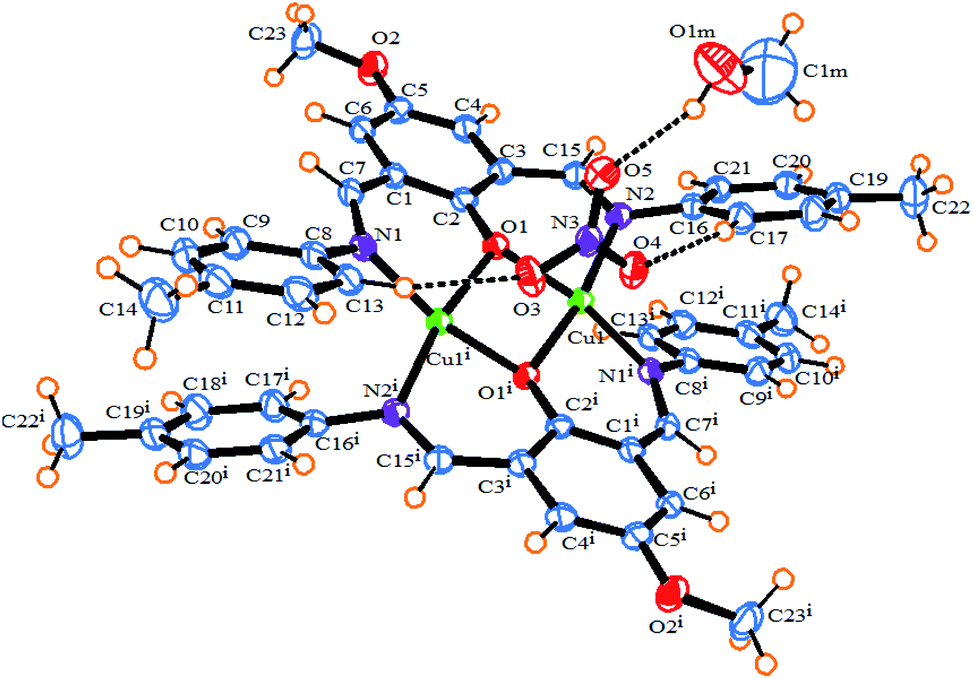
The ORTEP diagram of 1 is shown in Fig. 1. The crystallographic parameters, selected bond distances and angles around the metal centers, and selected hydrogen bonds are listed in Tables 1–3, respectively. The X-ray determination result of 1 reveals that it crystallizes in a monoclinic crystal system with space group P21/n and each asymmetric unit contains half of the crystallographically unique molecule (Fig. 1). Each Cu(II) center displays a six-coordinate distorted octahedral geometry with CuN2O4 configuration (Fig. 2), where the basal plane is composed of two phenoxo-bridged μ2-O1 atoms and two imino nitrogen atoms (N1 and N2) from two deprotonated Schiff base ligands (L−). Moreover, the axial positions are occupied by the two bridging oxygen atoms from two nitrate anions (O3 and O4). In the CuN2O4 coordination sphere, the axial Cu–O contacts of μ2-NO3 bridged anions (Cu–O3, 2.655 (18) Å) and Cu–O4, 2.504 (16) Å) are much longer than (Cu–O1, 1.965 (2) Å and Cu–O1, 1.953 (2) Å), demonstrating the Jahn–Teller effect55 (Table 2). The Cu–N bond lengths in this complex are 1.970 and 1.975 and are similar to a previously reported six-coordinated copper(II) complex with nitrogen donor atoms.56–59
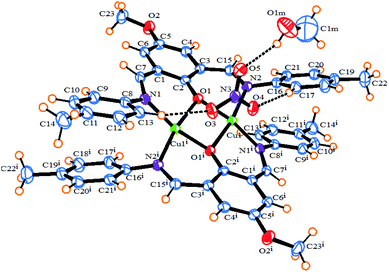 |
| | Fig. 1 An ORTEP diagram of 1 with thermal ellipsoids at 55% probability. Inter and intra-molecular hydrogen bonds shown as dotted lines. | |
 |
| | Fig. 2 Coordination environments of the Cu(II) in 1. | |
Also, the bond lengths and angles of 1 are in close agreement with those reported for other six-coordinated copper(II) complexes.60,61 The distance of dinuclear center is 3.027(5) Å. This long distance between two Cu(II) centers suggests that there is no interaction between these two metal centers.61 We found two intermolecular C–H⋯O hydrogen bonds between the hydrogen atoms of aromatic rings of ligands and oxygen atoms of bridging nitrate anions as well as one intramolecular π⋯π stacking interaction between the aromatic rings of two ligands with a distance of 3.423 Å in the complex which, play an important role in stabilizing the structure. The O⋯H distances and C–H⋯O angles are 2.349 (17) Å, 154.06° for C13–H1⋯O3 and 2.31 (17) Å, 153.82° for C17–H1⋯O4, respectively. As shown in Fig. S2b,† the non-coordinated oxygen atom O(5) of the bridging NO3− forms bifurcated hydrogen bonds with the hydrogen atoms attached to the imino and phenyl carbon atoms of Schiff base ligand, C7–H1⋯O5 and C6–H1⋯O5, respectively (Table 3). Furthermore, there are π–π stacking interactions between the aromatic rings of Schiff base ligands belonging to neighboring units in complex 1, leading to the neighboring complex molecules connected together along the a axis (Fig. S2a†).
This 1D chain structures which are formed along the a axis, are linked together through C–H⋯N (C20–H1⋯N3 2.596(2) Å, C–H⋯O (C4–H1⋯O1m 2.698(29) Å, C15–H1⋯O1m 2.519(26) Å, C21–H1⋯O1m 2.675(30) Å) and O–H⋯O hydrogen bonds (O1m–H1⋯O5 2.614(4) Å) to form 2D supramolecular structure (Fig. S2b†). The resulting 2D structures are joined into the 3D packing framework connected by C–H⋯O (C22–H2⋯O2 2.59(17) Å) (Fig. S2c†). The most remarkable structural feature of 1 is that it contains two types of 1D right-handed helical chains. One is along the b axis, whose the [Cu2 L2− (NO3)2] monomers are connected through C–H⋯O hydrogen bonds between the C–H group of a phenyl ring and O-methoxy group of Schiff base ligands (C22–H2⋯O2 2.59(17) Å) belonging to neighboring units in complex 1 (Fig. 3). The other is along the perpendicular to ac plane (b* axis) by the hydrogen bonding between the nitrogen atom of the coordinated nitrate group and C–H group of a phenyl ring Schiff base ligand (C20–H1⋯N3 2.596(2) Å) (Fig. S3†).
 |
| | Fig. 3 (a) Hydrogen bonds network. (b) Space-filling view of the 1D right-handed (R) helical chains along b axis. | |
3.4. Hirshfeld surface analysis of [Cu2 L2− (NO3)2]·2CH3OH (1)
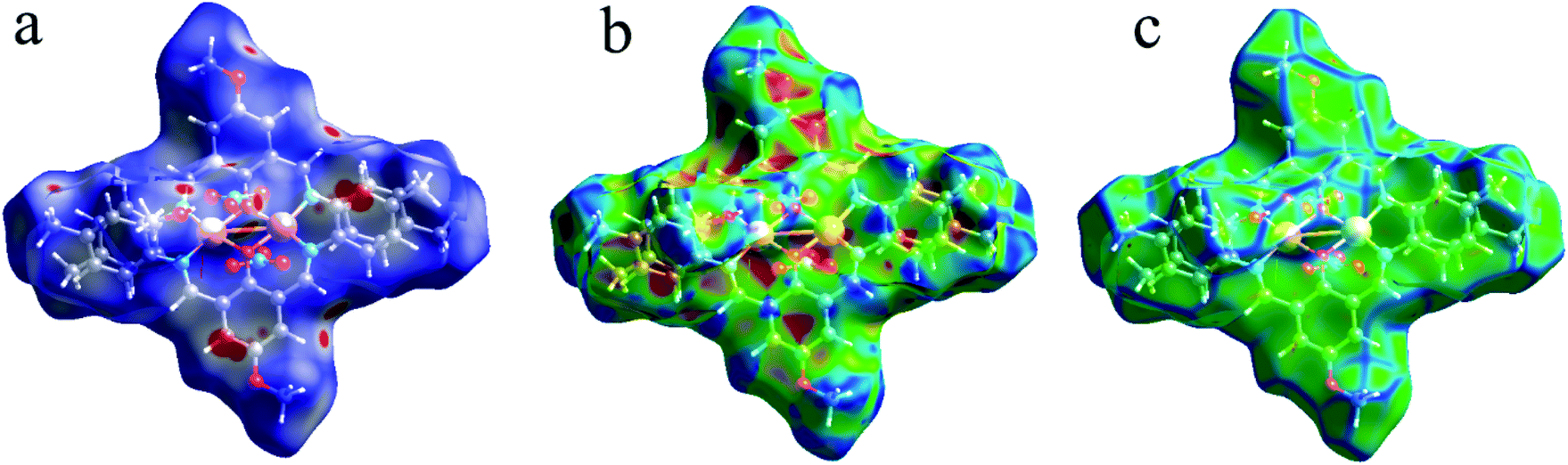
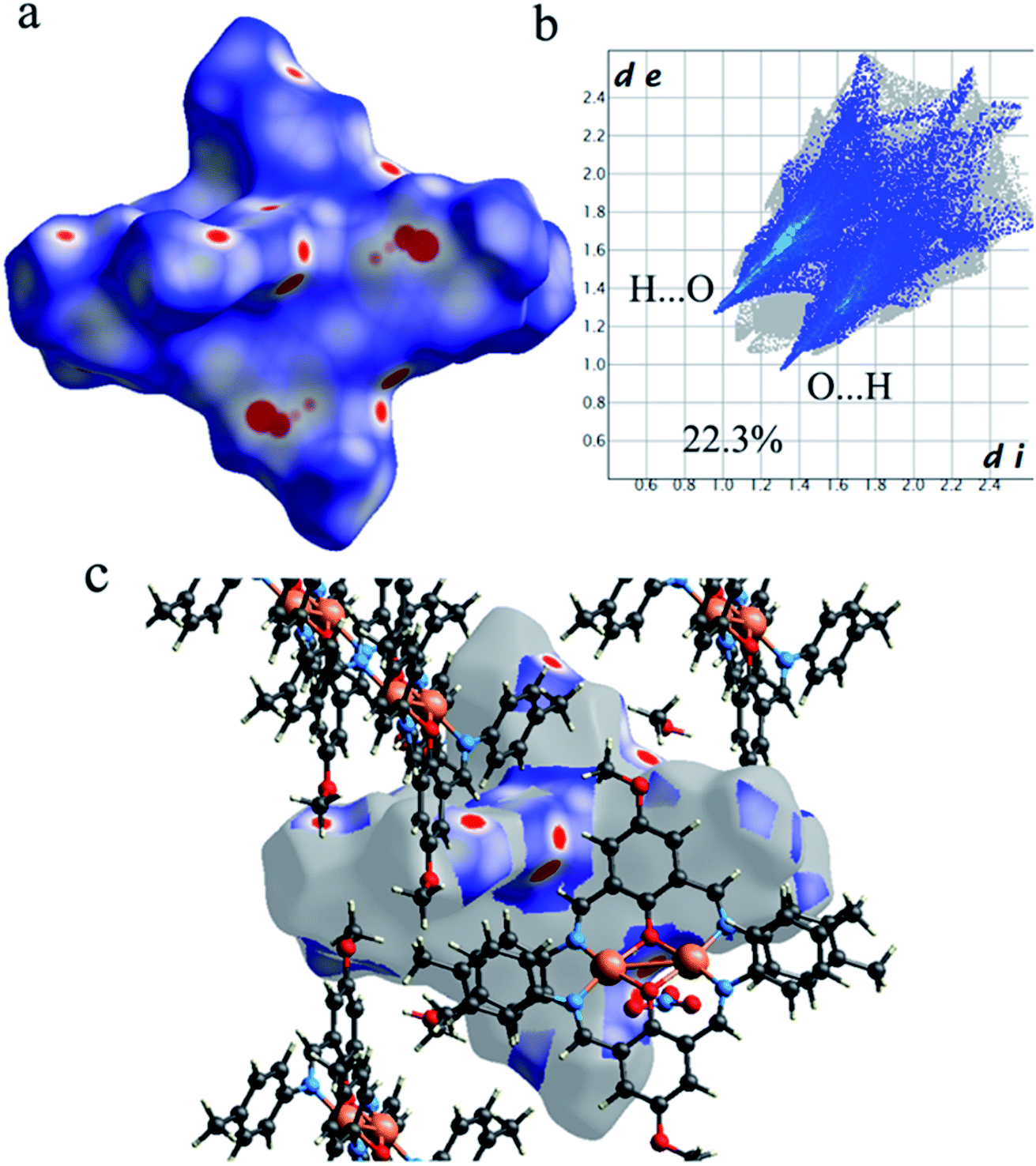
As shown in Fig. 4, the Hirshfeld surfaces (dnorm, shape index, and curvedness) of 1 are mapped over dnorm (ranging from −0.303 (red) to 1.370 (blue) Å), shape index (ranging from −1 to 1.0 Å), and curvedness (ranging from −4.0 to 0.4) Å. The molecular surfaces of 1 are shown as transparent to allow the visualization of the molecular moiety, around which they were calculated. The red spots on the dnorm surface of 1 indicate the intermolecular close contacts involved in the hydrogen bonds such as, C–H⋯O (Fig. 5), N–H⋯O, while the white regions represent contacts around the van der Waals separation looking like H⋯H interactions and the blue areas are devoid of such close contacts. The intermolecular interactions involved in the molecular structure are visible on the 2D fingerprint plots, which can be decomposed to quantify particular contributions of intermolecular interactions in the molecular packing of a solid network.
 |
| | Fig. 4 Hirshfeld surfaces mapped with (a) dnorm, (b) shape index and (c) curvedness for 1. | |
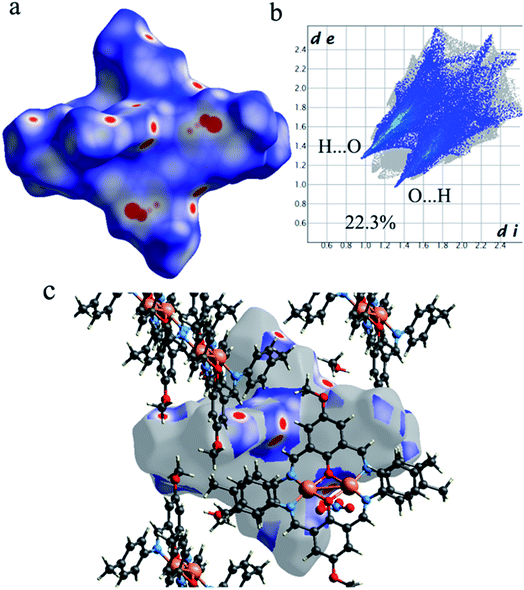 |
| | Fig. 5 (a) The Hirshfeld surfaces mapped with dnorm function, (b) the Hirshfeld surfaces mapped with dnorm function and (c) corresponding 2D fingerprint for the C–H⋯O interactions in 1. | |
Complementary regions are evident in the fingerprint plots, indicating that a molecule acts as a donor (de > di) and another one as an acceptor (de < di). The 2D fingerprint plots of 1 show that the main intermolecular interactions between its molecules are H⋯H, H⋯N, H⋯O, H⋯C, and C⋯C intermolecular interactions (Fig. 6).
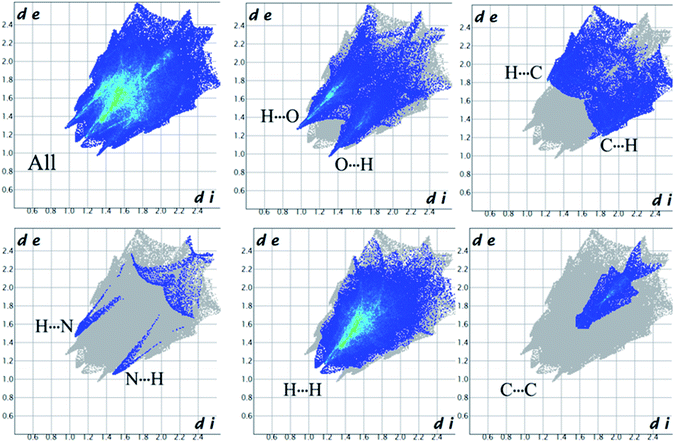 |
| | Fig. 6 The 2D fingerprint plots for all interactions present in 1. | |
Moreover, the O⋯H/H⋯O, and N⋯H/H⋯N interactions are represented by a spike in the bottom right (acceptor) area (O⋯H 8.5%, N⋯H 0.8%) and a spike at the top left (donor) area (H⋯O 13.8%, H⋯N 0.9%) of the 2D fingerprint plot. The H⋯C/C⋯H intermolecular interactions comprise 5.9% of the total Hirshfeld surface area which emerge as very short spike at the top left (H⋯C 6.2%) and bottom right (C⋯H 7.6%) of the 2D fingerprint plot. The H⋯H contacts appear in the largest region accompanied by the short and tenuous spike at the middle of fingerprint plot, which has the most significant contribution to the total Hirshfeld surfaces (53.7%). The 2D fingerprint plots of 1 show that the π⋯π (C⋯C) stacking interactions comprise 5.8% of the total Hirshfeld surface area. Furthermore, the presence of π⋯π (C⋯C) stacking interactions in the crystal packing of 1 are clearly confirmed by characteristic ‘bow-tie’ patterns in the shape index function (Fig. 7). Indeed, in the shape index, the highlighted red and blue triangles (highlighted circles in Fig. 7) prove the presence of the π⋯π stacking interactions in the crystal structure of 1, where the red triangle denotes the aromatic ring atoms of the molecule outside the surface, and the blue one represents the aromatic ring atoms inside the surface.
 |
| | Fig. 7 The Hirshfeld surfaces mapped with shape index function for the C⋯C (π⋯π) and chelate ring⋯chelate ring stacking interactions in 1. | |
The quantities of interactions with percentages are shown as a histogram in Fig. 8. Fig. 8 shows the H⋯H and N⋯H contacts provide the highest and lowest share of the total intermolecular interactions in the title complex.
 |
| | Fig. 8 Relative contributions of various intermolecular interactions contributing to the Hirshfeld surfaces of 1. | |
3.5. Antibacterial activity results
The test compound was screened in vitro for their antibacterial activity against three Gram-negative species (Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumonia) and three Gram-positive species (Staphylococcus epidermidis, Staphylococcus aureus, Enterococcus faecalis) of bacterial strains by the disc diffusion and micro dilution methods. The antibacterial activities obtained by the two methods are given in Tables 4 and 5.
Table 4 Measured inhibition zone diameter (mm) of 1 and antibiotic by disc diffusion methoda
| Diameter inhibition zone (mm, 200 μg per disk) |
| Bacteria strains |
E. coli ATCC 25922 |
P.aeruginosa ATCC 27853 |
K. pneumoniae ATCC 70063 |
S. aureus ATCC 25923 |
E. faecalis ATCC 23212 |
S. epidermidis ATCC 34384 |
| Sulfisoxazole (300 μg per disk) < 10: weak; >10 moderate; >16: significant. |
| Cu complex |
10 |
8 |
12 |
10 |
12 |
11 |
| Sulfisoxazole |
20 |
9 |
20 |
24 |
17 |
15 |
Table 5 The MICs of antibacterial activity of 1
| Bacteria strains |
MIC μg mL−1 (mM) |
| E. coli ATCC 25922 |
P. aeruginosa ATCC 27853 |
K. pneumoniae ATCC 70063 |
S. aureus ATCC 25923 |
E. faecalis ATCC 23212 |
S. epidermidis ATCC 34384 |
| Cu complex |
250 (0.446) |
250 (0.446) |
62.5 (0.111) |
250 (0.446) |
125 (0.223) |
125 (0.223) |
| Sulfisoxazole |
23.4 (0.088) |
375 (1.403) |
23.4 (0.088) |
23.4 (0.088) |
93.75 (0.35) |
93.75 (0.35) |
The disc diffusion assay results demonstrate (Table 4) that 1 has exhibited the moderate inhibition effect against most of the test bacteria (Fig. S4†). The ligands derived from the different amines and derivatives of salicylaldehyde have higher antibacterial activity than their respective complexes.62 The remarkable activity of such ligands may arise from existence of two hydroxyl groups in these ligands which may play an important role in antibacterial activity. However, Cu(II) complex (1) in this paper has no hydroxyl group in its structure and has a lower antibacterial activity than similar Cu(II) Schiff base complex in previous works.63,64
3.6. DNA binding studies
3.6.1. Electronic absorption spectra. Electronic absorption spectroscopy is one of the most useful techniques for studying binding mode of metal complexes to DNA.65,66 Absorption spectra of 1 in the absence and presence of DNA (pH 7.4) showed a shoulder in the 200–350 nm regions and a peak at range of 400–500 nm (Fig. 9).
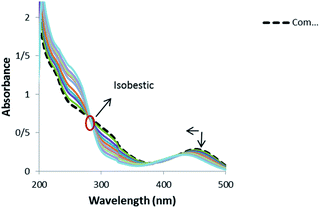 |
| | Fig. 9 Absorption spectra of 1 (1.25 × 10−5 M) in the absence and presence of increasing amounts of CT-DNA: ri = [CT-DNA]/[complex]: 0.0, 0.05, 0.1, 0.13, 0.17, 0.21, 0.25, 0.29, 0.33, 0.37, 0.65. | |
Band 454 gently decreased with the increasing concentration of DNA and a blue shift was observed from 454 to 433 nm. Also, an isobestic point at 280 nm provided evidence for the new complex–DNA formation.67 These observations offer that there exists a strong interaction between the complex and DNA. No red shift was observed in the UV spectra, which indicates that the binding mode is not intercalation and the hypochromism indicates that the binding mode of 1 to DNA might be groove binding. The similar hypochromism without red shift was observed for the complexes of copper(II) and nickel(II) containing the drug mesalamine that bound to DNA through groove mode.68,69
In order to further illustrate the binding strength of 1 with CT-DNA, the intrinsic binding constant Kb was determined from the spectral titration data using the following equation:
| |
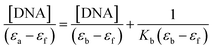 | (1) |
where, [DNA] is the concentration of DNA.
εa,
εf and
εb correspond to
Aobs/[compound], the extinction coefficient for the free complex and its entirely DNA-bound combination, respectively. In the plots of [DNA]/(
εa _
εf)
versus [DNA],
Kb was obtained by the ratio of the slope to intercept (Fig. S5
†). The binding constant,
Kb, for
1 was 1.3 ± 0.2 × 10
4 L mol
−1. This observation indicated the binding capability of the complex toward DNA with an affinity lower than the classical intercalators, due to the two chlorides with the DNA base pairs and relatively bulky structure of ligand molecules. Such a bulky structure cannot allow them to stack between the base pairs and inhibits any other bond formation (hydrogen bonds,
etc.) with DNA.
70 This
Kb value is similar to those reported for well-established groove binding agent ([Ni(phen)
3]
+2, 1.4 × 10
4 M
−1),
71 ([Ho(phen)
2Cl
3]·H
2O (1.36 × 10
4 M
−1),
72 (Cu–Sn
2 complex, 1.67 × 10
4 M
−1).
73 It was detected that the binding mode between the complex and DNA was groove binding.
3.6.2. Viscosity measurements. Hydrodynamic methods that are sensitive to length change (i.e., viscosity and sedimentation) are considered to be one of the least ambiguous and most exacting tests of a binding mode in solution in the absence of crystallographic structural data. The sensitivity of viscosity measurement largely depends on the changes in the length of DNA that occur as a consequence of its different binding modes with guest molecules. Intercalating agents of ligand such as the classical intercalations are expected to elongate the double helix to incorporate the small molecules in between the bases leading to an increment in the viscosity of DNA.65 Reciprocally, a partial and/or non-classical intercalation of ligand could bend (or kink) the DNA helix, reducing its effective length and, simultaneously, its viscosity.74,75 The values of (η/η0)1/3 were depicted against [complex]/[DNA] that η and η0 are the relative viscosities of DNA in the presence and absence of 1, respectively (Fig. S6†). The plot of Fig. S6† reveals that 1 shows relatively small changes in DNA viscosity, indicating that they bind weakly to DNA, which is consistent with DNA groove binding suggested above.76
3.6.3. CD spectral studies. Due to the sensitivity of CD signal to subtle variation of DNA chiral conformation, CD bands are a useful technique to follow the complex–DNA interaction via investigating the changes in the DNA morphology. In particular, B-DNA shows two conservative CD bands in the UV region: a positive band at 278 nm due to base stacking and a negative band at 246 nm because of the right-handed helicity. Both the bands are quite sensitive to the interaction mode with small molecules. Since different DNA structures have different CD spectra, this technique is a powerful procedure to understand the conformational changes of DNA. The CD spectra of DNA in the presence of 1 were illustrated in Fig. 10.
 |
| | Fig. 10 CD spectra of DNA (5 × 10−5 M) in 10 mM Tris–HCl buffer, in the presence of increasing amounts of 1 (ri = [complex]/[DNA] = 0.0, 0.6, 1.6). | |
The CD spectra of DNA with different values of the compound exhibit decreases in the positive peak and also decreases in the negative band, which clearly indicates a non-classical intercalation between the complex and DNA.70 Decrease in molar ellipticity at these bands (275 nm and 248 nm) suggest the distortion in native conformation of B-DNA due to its interaction with the complex 1. These spectral variations show the presence of some C form DNA features in native conformation of DNA upon interaction. Moreover, reduction in 248 nm CD band is considered a key marker of C form of DNA.77,78 However, it seems that the perturbation in DNA conformation is limited to few base pairs because when complete B to C transition occurs, then the CD band at 248 nm shows a 66% decrease in its intensity. However, not much decrease was observed in the concerned band (40.1%). Therefore, there are possibilities for the formation of an intermediate form of DNA having features of both B and C conformation. Similar results have been observed in the case of cationic lipid and neutral lipid binding with DNA, which could be ascribed to a non-cooperative augment in DNA.79,80 Moreover, increase in winding angle (or decrease in propeller twist) causes widening in DNA groove that enables proper positioning of small molecules in the groove pocket.81
3.6.4. Fluorescence spectra. Fixed amounts (2.5 × 10−5 mol L−1) of 1 were titrated with increasing amounts of CT-DNA. The complex emits luminescence in Tris–HCl buffer with two maximums that appear at 357 and 408 nm. The fluorescence titrations spectra of 1 in the absence and presence of CT-DNA at 298 K are given in Fig. S7.† The fluorescence intensity of 1 decreased with an increase in CT-DNA concentration. As shown in Fig. S7,† the fluorescence intensity of the compound is quenched steadily with the increasing concentration of the CT-DNA. Similar results were observed for [Cu(adefovir)2Cl2] complex that interacted with DNA by groove binding.71 This phenomenon of the quenching of luminescence of 1 by DNA may be ascribed to the photoelectron transfer from the guanine base of DNA to the excited levels of the complex.82,83
3.6.4.1. Fluorescence studies. Quenching is carried out by different mechanisms, which are usually classified as dynamic and static. Dynamic quenching refers to a process in which the fluorophore (here the complex) and the quencher (DNA) come into contact during the transient existence of the exited state, but static quenching refers to fluorophore–quencher complex formation. In general, dynamic and static quenching can be known by their differing dependence on temperature and excited-state lifetime. The dynamic quenching process increases with an increase in the temperature because upon this condition the compounds move faster causing more collision probability. As the formation of composite is responsible for the static quenching, an increase in the temperature diminishes the stability of the composite and causes the fluorescence quenching to descend.84 Since in both cases the fluorescence intensity is related to the concentration of the quencher (DNA), the quenched fluorophore can be used as an indicator for the quenching agent.85 Fluorescence quenching is described by the Stern–Volmer equation (eqn (2)).| | |
F0/F = 1 + kq τ0 [Q] = 1 + ksv [Q]
| (2) |
where F0 and F represent the fluorescence intensities in the absence and in the presence of quencher, kq is the quenching rate constant of the biomolecule, kSV is the dynamic quenching constant, τ0 is the average lifetime of the molecule without quencher and [Q] is the concentration of quencher (here CT-DNA). Of course, these assumptions should be modified by considering the point that quenching can occur through different mechanisms: the inner filter effect, collisional quenching, and binding-related changes in fluorescence. Thus emission needs to be corrected for inner filter effect by the following equation:| | |
Fcor = Fobs × e(Aex+Aem)/2
| (3) |
That Fcor and Fobs are the fluorescence intensities corrected and observed, respectively. Moreover, Aex and Aem are the absorption of the system at the excitation and the emission wavelength, respectively. Then the Fcor is used in eqn (2). The results are shown in Table 6. In this experiment, with an increase in the temperature from 283 K to 310 K, the Stern–Volmer constant (ksv) decreased from (3.59 ± 0.02) ×103 M−1 to (2.70 ± 0.02)×103 M−1 for 1 (Fig. S8† and Table 6). It means that the quenching mechanism in the case of 1 is static. Also, kq is much larger than 2.0 × 1010 L mol−1 s−1, suggesting a static quenching process. The binding constant (Kb) for the complex formation between 1 and DNA was assayed using eqn (4):86,87
| | |
1/ΔF = 1/ (Kb. ΔF0)[Q] + 1/ΔF0
| (4) |
Where Δ
F is fluorescence intensity change,
Kb is the binding constant, and Δ
F0 is maximum fluorescence intensity change. The binding constant can be obtained by plotting 1/Δ
F vs. 1/[Q] and then dividing intercept by slope.
Table 6 Stern–Volmer dynamic quenching constant of 1 to CT-DNA at different temperatures
| T (K) |
kSV(±0.02)×10−3 |
kq (±0.02) × 10−11 |
R2 |
| 283 |
3.59 |
3.59 |
0.99 |
| 298 |
3.15 |
3.15 |
0.99 |
| 310 |
2.70 |
2.70 |
0.98 |
In the present study, the binding constants of 1 were obtained at various temperatures (Table 7). The value of Kb for 1 at room temperature is comparable to N,N-bis (3β-acetoxy-5α-cholest-6-yl-idene)hydrazine (4.7 × 103 M−1)88 and resistomycin (3.23 × 103 M−1)89 which binds to DNA in an groove binding mode. The plot of logKb versus 1/T (Fig. S9†) allows to acquire the enthalpy (ΔH), entropy (ΔS) (Eq. (5)) and free energy (ΔG) change by the van't Hoff equation, (eqn (6)), assuming that the enthalpy change (ΔH) is independent of temperature over the range of applied temperatures.
| | |
lnKb = −ΔH/RT + ΔS/R
| (5) |
Table 7 The binding constants and thermodynamic parameters for the copper complex-DNA at different temperatures
| T (K) |
Kb(±0.02) (M−1) |
ΔG (±0.3) (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (J mol−1K−1) |
| 283 |
1.09 × 103 |
−16.4 |
— |
— |
| 298 |
3.74 × 103 |
−20.5 |
59.6 ± 0.3 |
268.8 ± 0.3 |
| 310 |
1.00 × 104 |
−23.7 |
— |
— |
Table 7 exhibits the thermodynamic values of 1. According to the data of enthalpy changes (ΔH) and entropy changes (ΔS), the model of interaction between the complex and biomolecule like DNA can be concluded: (1) ΔH > 0 and ΔS > 0, hydrophobic forces; (2) ΔH < 0 and ΔS < 0, van der Waals interaction and hydrogen bonds; (3) ΔH < 0 and ΔS > 0, electrostatic interactions.89 It can be seen that the negative value of ΔG revealed the DNA interaction process is spontaneous; the positive ΔH and ΔS values indicated that hydrophobic forces play main roles in binding the 1 to DNA.
4. Conclusions
The work described in this paper involved the synthesis and characterization a binuclear copper(II) Schiff base complex, [Cu2 L2− (NO3)2]·2CH3OH (1) from reaction of 2,6-diformyl-4-methoxy phenol, p-methyl aniline and Cu(NO3)2·2H2O in methanol solvent. The complex was characterized by FT-IR, elemental analysis (CHN), UV-vis spectroscopy and molar conductivity measurement. Furthermore, structure of 1 was determined by single crystal X-ray analysis. This complex is neutral and found to have a distorted octahedral geometry with the six donor atoms (N2O4) of the ligand. The crystal structure and Hirshfeld surface analysis represent the presence of various intermolecular interactions such as H⋯H, H⋯N, H⋯O, H⋯C and C⋯C in assembling the molecules of 1 into a 3D supramolecular network. The 2D fingerprint plots of 1 show that the H⋯H interactions have the largest share of the total intermolecular interactions with contributions of 53.7%. Also, the shape index function accompanied by its 2D fingerprint plot indicate the presence of the π⋯π interactions (5.8%) in the crystal lattice of compound 1. Moreover, the existence of two 1D helical chains in 1 was observed, one along the b axis and other one along the perpendicular to ac plane (b* axis) via C–H⋯O and C–H⋯N hydrogen bonds. This complex exhibited moderate antibacterial activity against the three Gram-positive bacteria: Staphylococcus aureus ATCC 25923, Enterococcus faecalis ATCC 23212, S. epidermidis ATCC 34384, and against the three Gram-negative bacteria: Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853 and Klebsiella pneumonia ATCC 70063. Finally, DNA binding of 1 was investigated using UV-vis spectroscopy, fluorescence quenching, CD spectrum and viscosity methods. Fluorescence quenching and UV-Vis spectroscopy results represent that 1 binds to CT-DNA with binding constant of 1.3 ± 0.2 × 104 L mol−1. The results of spectroscopic techniques suggested that 1 binds to CT-DNA through groove binding.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support by Ilam University, Iran. Crystallography part was supported by the project 14-03276S of the Czech Science Foundation.
References
- Y. Cui, O. R. Evans, H. L. Ngo, P. S. White and W. Lin, Angew. Chem., Int. Ed., 2002, 41, 1159 CrossRef CAS PubMed.
- P. J. Deuss, R. den Heeten, W. Laan and P. C. J. Kamer, Chem.–Eur. J., 2011, 17, 4680 CrossRef CAS PubMed.
- H. Hou, Y. Wei, Y. Song, Y. Zhu, L. Li and Y. Fan, J. Mater. Chem., 2002, 12, 838 RSC.
- A. C. Kathalikkattil, K. K. Bisht, N. Aliaga-Alcalde and E. Suresh, Cryst. Growth Des., 2011, 11, 1631 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- A. F. Trindade, P. M. P. Gois and C. A. M. Afonso, Chem. Rev., 2009, 109, 418 CrossRef CAS PubMed.
- J. Zhang, S. Chen, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2008, 130, 12882 CrossRef CAS PubMed.
- Q. Zhang, X. Bu, Z. Lin, M. Biasini, W. Beyermann and P. Feng, Inorg. Chem., 2007, 46, 7262 CrossRef CAS PubMed.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 CrossRef CAS PubMed.
-
(a) A. J. Blake, N. R. Champness, P. A. Cooke and J. E. B. Nicolson, Chem. Commun., 2000, 665 RSC;
(b) O. S. Jung, Y. J. Kim, Y. A. Lee, H. K. Chae, H. G. Jang and J. Hong, Inorg. Chem., 2001, 40, 2105 CrossRef CAS PubMed.
-
(a) J. M. Lehn and A. Rigault, Angew. Chem., Int. Ed. Engl., 1988, 27, 1095 CrossRef;
(b) J. Sanches-Quesada, C. Seel, P. Prados and J. de Mendoza, J. Am. Chem. Soc., 1996, 118, 277 CrossRef.
-
(a) E. J. Enemark and T. D. P. Stack, Angew. Chem., Int. Ed. Engl., 1995, 34, 996 CrossRef CAS;
(b) S. Sailaja and M. V. Rajasekharan, Inorg. Chem., 2000, 39, 4586 CrossRef CAS;
(c) J. J. Lessmann and W. D. Horrocks, Jr, Inorg. Chem., 2000, 39, 3114 CrossRef CAS PubMed.
- E. C. Constable, S. M. Elder, J. Healy and M. D. Ward, J. Am. Chem. Soc., 1990, 112, 4590 CrossRef CAS.
- M. J. Hannon, S. Bunce, A. J. Clarke and N. W. Alcock, Angew. Chem., Int. Ed. Engl., 1999, 38, 1277 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani, D. M. Proserpio and A. Sironi, Inorg. Chem., 1998, 37, 5941 CrossRef CAS.
- E. C. Constable, Tetrahedron, 1992, 48, 10013 CrossRef CAS.
- N. C. Jana, K. Pramanik, A. Bauzá, P. Brandão, M. Patra, A. Frontera and A. Panja, Polyhedron, 2017, 124, 262 CrossRef CAS.
- J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CrossRef CAS PubMed.
- N. Delsuc, F. Godde, B. Kauffmann, J.-M. Leger and I. Huc, J. Am. Chem. Soc., 2007, 129, 11348 CrossRef CAS PubMed.
- P. García-Holley, N. Ortiz-Pastrana, R. A. Toscano, M. Flores-Álamo and N. Barba-Behrens, Polyhedron, 2016, 104, 127 CrossRef.
- H. Y. Hu, J. F. Xiang, Y. Yang and C. F. Chen, Org. Lett., 2008, 10, 1275 CrossRef CAS PubMed.
- D. B. Hassan, W. Rekik, A. Otero-de-la Roza, F. B. Mefteh, T. Roisnel, T. Bataille and H. Naïli, Polyhedron, 2016, 119, 238 CrossRef.
- D. R. Xiao, E. B. Wang, H. Y. An, Y. G. Li and L. Xu, Cryst. Growth Des., 2007, 7, 506 CrossRef CAS.
- L. D. Chen, L. X. Zhou, Y. Q. Zheng and H. L. Zhu, Polyhedron, 2017, 126, 150 CrossRef CAS.
-
(a) F. R. Keene, J. A. Smith and J. G. Collins, Coord. Chem. Rev., 2009, 253, 2021 CrossRef CAS;
(b) K. Dhara, J. Ratha, M. Manassero, X.-Y. Wang, S. Gao and P. Banerjee, J. Inorg. Biochem., 2007, 101, 95 CrossRef CAS PubMed;
(c) V. Uma, A. Castineiras and B. U. Nair, Polyhedron, 2007, 26, 3008 CrossRef CAS.
- U. Basu, I. Khan, A. Hussain, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2012, 51, 2658 CrossRef CAS PubMed.
-
(a) S. Anbu and M. Kandaswamy, Polyhedron, 2011, 30, 123 CrossRef CAS;
(b) S. Roy, A. K. Patra, S. Dhar and A. R. Chakravarty, Inorg. Chem., 2008, 47, 5625 CrossRef CAS PubMed.
- P. Moradi and M. Hajjami, New J. Chem., 2021, 45, 2981 RSC.
- A. Jabbari, B. Tahmasbi, M. Nikoorazm and A. Ghorbani-Choghamarani, Appl. Organomet. Chem., 2018, 32, e4295 CrossRef.
- M. Nikoorazm, Z. Rezaei and B. Tahmasbi, J. Porous Mater., 2020, 27, 671 CrossRef CAS.
- P. Moradi and M. Hajjami, RSC Adv., 2021, 11, 25867 RSC.
- A. Ghorbani-Choghamarani, B. Tahmasbi, F. Arghand and S. Faryadi, RSC Adv., 2015, 5, 92174 RSC.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, JANA 2006, Structure Determination Software Programs, Institute of Physics, Praha, Czech Republic, 2006 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- Y. Abbasi Tyula, A. Zabardasti, H. Goudarziafshar, M. Sadeghi Roudsari, M. Dusek and V. Eigner, J. Mol. Struct., 2017, 1150, 383 CrossRef.
- J. K. Zareba, M. J. Białek, J. Janczak, J. Zon and A. Dobosz, Cryst. Growth Des., 2014, 14, 6143 CrossRef CAS.
- Y. Abbasi Tyula, A. Zabardasti, H. Goudarziafshar, M. Kucerakova and M. Dusek, Appl. Organomet. Chem., 2018, 32, e4141 CrossRef.
- M. Wilk, K. N. Jarzembska, J. Janczak, J. Hoffmann and V. Videnova-Adrabinska, J. Mol. Struct., 2014, 1074, 240 CrossRef CAS.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CRYSTALEXPLORER 3.0, University of Western Australia, Perth, Australia, 2012 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- Q. L. Liu, L. J. Yang, Y. H. Luo and B. W. Sun, New J. Chem., 2016, 40, 3892 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
- A. W. Bauer, W. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493 CrossRef CAS PubMed.
- A. BalabanGündüzalp and H. Özbay, Russ. J. Inorg. Chem., 2012, 57, 257 CrossRef.
- N. Özbek, S. Alyar, H. Alyar and N. Karacan, Spectrochim. Acta, Part A, 2013, 108, 123 CrossRef PubMed.
- F. Zhang, L. L. Gan and C. Zhou, Bioorg. Med. Chem. Lett., 2010, 20, 1881 CrossRef CAS PubMed.
- K. Nakamoto, Infrared And Raman Spectra Of Inorganic And Coordination Compounds, Part B: Applications In Coordination, Organometallic And Bioinorganic Chemistry, John Wiley & Sons Inc, New York, New Jersey, 6th edn, 2009 Search PubMed.
- F. Arjmand, A. Jamsheera and D. Mohapatra, J. Photochem. Photobiol., B, 2013, 121, 75 CrossRef CAS PubMed.
- N. Sultana, A. Naz, M. S. Arayne and M. A. Mesaik, J. Mol. Struct., 2010, 969, 17 CrossRef CAS.
- S. Naskar, S. Biswas, D. Mishra, B. Adhikary, L. R. Falvello, T. Soler, C. H. Schwalbe and S. K. Chattopadhyay, Inorg. Chim. Acta, 2004, 357, 4257 CrossRef CAS.
- R. Gup and B. Kırkan, Spectrochim. Acta, Part A, 2005, 62, 1188 CrossRef PubMed.
- H. Liu, H. Wang, F. Gao, D. Niu and Z. Lu, J. Coord. Chem., 2007, 60, 2671 CrossRef CAS.
- C. D. Stevenson and Y. S. Kim, J. Am. Chem. Soc., 2000, 122, 3211 CrossRef CAS.
- P. Chakraborty, I. Majumder, H. Kara, S. K. Chattopadhyay, E. Zangrando and D. Das, Inorg. Chim. Acta, 2015, 436, 139 CrossRef CAS.
- G. A. Van Albada, A. Mohamadou, I. Mutikainen, U. Turpeinen and J. Reedijk, Eur. J. Inorg. Chem., 2004, 18, 3733 CrossRef.
- M. A. Ali, A. H. Mirza, A. M. S. Hossain and M. Nazimuddin, Polyhedron, 2001, 20, 1045 CrossRef CAS.
- Z. Chu, W. You and W. Huang, J. Mol. Struct., 2009, 920, 277 CrossRef CAS.
- M. Rezaeivala, R. Golbedaghi and M. Khalili, Russ. J. Coord. Chem., 2016, 42, 66 CrossRef CAS.
- H. Keypour, M. Shayesteh, M. Rezaeivala and K. Sayin, J. Mol. Struct., 2016, 1112, 110 CrossRef CAS.
- H. Keypour, M. Shayesteh, R. Golbedaghi, A. Chehregani and A. G. Blackman, J. Coord. Chem., 2012, 65, 1004 CrossRef CAS.
- H. Keypour, M. Shayesteh, M. Rezaeivala, F. Chalabian and L. Valencia, Spectrochim. Acta, Part A, 2013, 101, 59 CrossRef CAS PubMed.
- Z. C. Liu, B. D. Wang, Z. Y. Yang, Y. Li, D. D. Qin and T. R. Li, Eur. J. Med. Chem., 2009, 44, 4477 CrossRef CAS PubMed.
- S. Ramakrishnan and M. Palaniandavar, J. Chem. Sci., 2005, 117, 179 CrossRef CAS.
- N. Shahabadi and N. H. pour Moghadam, J. Lumin., 2013, 134, 629 CrossRef CAS.
- Z. Q. Liu, Y. T. Li, Z. Y. Wu and S. F. Zhang, Inorg. Chim. Acta, 2009, 362, 71 CrossRef CAS.
- N. Shahabadiab, S. Moradi Filia and M. Shahlaeic, J. Coord. Chem., 2015, 68, 3667 CrossRef.
- S. Kathiresan, R. Dhivya, M. Vigneshwar, M. Rajasekaran, J. Ranjanic, J. Rajendhran, S. Srinivasan, S. Mugesh, M. Murugan, P. Athappan and J. Annaraj, J. Coord. Chem., 2016, 69, 238 CrossRef CAS.
- S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2011, 40, 3524 RSC.
- S. Niroomand, M. Khorasani-Motlagh, M. Noroozifar and A. Moodi, J. Photochem. Photobiol., B, 2012, 117, 132 CrossRef CAS PubMed.
- M. N. Dehkordi, A.-K. Bordbar, P. Lincoln and V. Mirkhani, Spectrochim. Acta, Part A, 2012, 90, 50 CrossRef CAS PubMed.
- F. Arjmand and M. Muddassir, J. Photochem. Photobiol., B, 2010, 101, 37 CrossRef CAS PubMed.
- B. D. Wang, Z. Y. Yang, D. W. Zhang and Y. Wang, Spectrochim. Acta, Part A, 2006, 63, 213 CrossRef PubMed.
- C.-Y. Zhou, J. Zhao, Y. B. Wu, C. X. Yin and Y. Pin, J. Inorg. Biochem., 2007, 101, 10 CrossRef CAS PubMed.
- S. Kashanian, A. T. Ghobadi, H. Roshanfekr and Z. Shariati, Mol. Biol. Rep., 2013, 40, 1173 CrossRef CAS PubMed.
- Z. Zhang, W. Huang, J. Tang, E. Wang and S. Dong, Biophys. Chem., 2002, 97, 7 CrossRef CAS PubMed.
- Y. Zhou and Y. Li, Biophys. Chem., 2004, 107, 273 CrossRef CAS PubMed.
- C. S. Braun, G. S. Jas, S. Choosakoonkriang, G. S. Koe, J. G. Smith and C. R. Middaugh, Biophys. J., 2003, 84, 1114 CrossRef CAS.
- N. J. Zuidam, Y. Barenholz and A. Minsky, FEBS Lett., 1999, 457, 419 CrossRef CAS PubMed.
- C. Zimmer and U. Wähnert, Prog. Biophys. Mol. Biol., 1986, 47, 31 CrossRef CAS PubMed.
- N. Shahabadi, M. Falsafi and S. Moradi Fili, J. Coord. Chem., 2015, 68, 1387 CrossRef CAS.
- L. F. Tan, H. Chao, H. Li, Y. J. Liu, B. Sun, W. Wei and L.-N. Ji, J. Inorg. Biochem., 2005, 99, 513 CrossRef CAS PubMed.
- P. X. Xi, Z. H. Xu, X. H. Liu, F.-j. Cheng and Z.-z. Zeng, Spectrochim. Acta, Part A, 2008, 71, 523 CrossRef PubMed.
- A. S. El-Tabl, F. A. El-Saied, W. Plass and A. N. Al-Hakimi, Spectrochim. Acta, Part A, 2008, 71, 90 CrossRef PubMed.
- I. Berlman, Handbook of fluorescence spectra of aromatic molecules, Elsevier, 2012 Search PubMed.
- M. M. Islam and G. S. Kumar, J. Mol. Struct., 2008, 875, 382 CrossRef CAS.
- M. M. Islam and G. S. Kumar, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 829 CrossRef CAS PubMed.
- Z. Tabassum, M. Muddassir, O. Sulaiman and F. Arjmand, J. Lumin., 2012, 132, 2178 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for the structure reported in this article have been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 1456947. Copies of this information may be obtained free of charge from the CCDC (E-mail: E-mail: deposit@ccdc.cam.ac.uk; or www: https://www.ccdc.cam.ac.uk). CCDC 1456947. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra00719c |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Michal Dušekc and
Václav Eignerc
*b,
Michal Dušekc and
Václav Eignerc
 0.5 °C in a constant temperature bath while the DNA concentration was fixed at 5 × 10−5 M and flow time was admeasured with a digital stopwatch. The data is reported as (η/η0)1/3 versus ri (ri = [complex]/[DNA] = 0.0–1.0), where η0 is the viscosity of the DNA solution alone. All fluorescence measurements were performed on a JASCO spectrofluorimeter (FP6200) using a quartz cell of 1 cm path length by keeping the concentration of the complex constant (1 × 10−5 mol L−1) while varying the DNA concentration from 0 to 3.4 × 10−5 M at three different temperatures (293, 310, 318 K). The samples were excited at 260 nm and the emission spectra were recorded in the range of 300 to 480 nm. The quenching experiments were conducted by adding stoichiometric small aliquots of potassium iodide stock solution (0.1 mol L−1) to 1 and CT–DNA–Cu complex solutions, respectively. The fluorescence intensity was recorded and the quenching constants (ksv) were calculated.
0.5 °C in a constant temperature bath while the DNA concentration was fixed at 5 × 10−5 M and flow time was admeasured with a digital stopwatch. The data is reported as (η/η0)1/3 versus ri (ri = [complex]/[DNA] = 0.0–1.0), where η0 is the viscosity of the DNA solution alone. All fluorescence measurements were performed on a JASCO spectrofluorimeter (FP6200) using a quartz cell of 1 cm path length by keeping the concentration of the complex constant (1 × 10−5 mol L−1) while varying the DNA concentration from 0 to 3.4 × 10−5 M at three different temperatures (293, 310, 318 K). The samples were excited at 260 nm and the emission spectra were recorded in the range of 300 to 480 nm. The quenching experiments were conducted by adding stoichiometric small aliquots of potassium iodide stock solution (0.1 mol L−1) to 1 and CT–DNA–Cu complex solutions, respectively. The fluorescence intensity was recorded and the quenching constants (ksv) were calculated.