
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of an eco-friendly antibacterial agent for food packaging containing Houttuynia cordata Thunb. extract
Peifu Kong a,
Junichi Peter Abeb,
Akiko Nakagawa-izumib,
Mikio Kajiyamab and
Toshiharu Enomae*b
a,
Junichi Peter Abeb,
Akiko Nakagawa-izumib,
Mikio Kajiyamab and
Toshiharu Enomae*b
aDegree Programs in Life and Earth Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan. E-mail: enomae.toshiharu.fw@u.tsukuba.ac.jp
bFaculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan
First published on 31st May 2022
Abstract
This study aims to develop an antibacterial agent that can be used for food packaging. Essential oils of Houttuynia cordata Thunb., a well-known medical herb, were extracted by two methods: multi-solvent consecutive extraction method and single ethanol extraction with a pre-heating method. Consequently, the extract obtained by the single ethanol extraction with a pre-heating method was more satisfactory from the operational and economic aspects. Afterwards, one of the encapsulation techniques: co-precipitation method using β-cyclodextrins as wall materials, was applied to form capsules for the protection of the obtained extract. After the capsule synthesis, the results of scanning electron micrographs and X-ray diffraction showed β-cyclodextrin crystallites in the form of thinner plates became oriented upon co-precipitation. Combining the results of Fourier transform-infrared spectra and an antibacterial assay using Bacillus subtilis as an object microorganism, the extract was confirmed to be successfully encapsulated within hollow cavities of β-cyclodextrins. A significant inhibitory activity on the growth and breeding of Bacillus subtilis was observed after the addition of fabricated capsules, which suggests the capsules containing the Houttuynia cordata Thunb. extract can be used as eco-friendly antibacterial agents for food packaging.
1 Introduction
Packaging is forming a more and more inalienable part of our daily life. However, in the field of food packaging, microorganism contamination gives rise to the spread of disease and reduces the shelf life of food.1 Along with the sudden outbreak of the COVID-19 pandemic being life-threatening across the globe, increasing cases of food contact infection have accelerated the demand for antimicrobial packaging materials.To date, there has been considerable interest in developing biodegradable antimicrobial food packaging materials because of the pressure of environmental concerns stemming from the traditional plastic materials made from petroleum which are non-biodegradable and non-renewable. Nanoparticles of gold, silver and zinc oxide as nanofillers have been successfully exploited with biodegradable polymers (e.g., chitosan, cellulose, gelatin and alginate) to obtain antimicrobial composite films for food packaging.2–6 Wu et al.7 firstly reported that LAPONITE® immobilized silver nanoparticles can be synthesized with quaternized chitosan and the prepared silver nanoparticles-chitosan film was successfully applied for keeping litchis fresh. Tsai et al.8 revealed that the gold–silver nanoparticles could be successfully immobilized onto a cellulose film by heat treatment and an antibacterial activity of the gold–silver nanoparticles-cellulose film against Escherichia coli was presented. In another research reported by Kanmani et al.,9 active nanocomposite films were prepared by blending aqueous solutions of gelatin with different concentrations of silver nanoparticles using a solvent casting method and exhibited a strong antibacterial activity against some food-borne pathogens. However, there is a serious consumer concern about the risks posed by the introduction of metal nanoparticles into food packaging, which could place restrictions on their benefits as the metal nanoparticles can be neutralized on contact with food or diffuse rapidly from the surface of packaging into the food mass. Moreover, these metal nanoparticles may enter the human body through food, triggering certain physiological reactions.10 With the consideration of this security concern, the research group of Motelica et al.11–13 developed antimicrobial composite films containing a low concentration level of metal nanoparticles which were mixed with essential oils (EOs) previously. Due to a synergy of metal nanoparticles and EOs, the fabricated composite films exhibited an excellent antimicrobial coverage, and presented great UV light barrier property and low water vapor permeability. Besides, their fabricated films have been successfully used for the preservation of cheese and grapes. Nevertheless, the addition of a low concentration of metal nanoparticles is still remaining controversial due to their possible toxicity and potential hazards.13,14
A prospective alternative is produced from substances totally isolated from botanical sources such as EOs. EOs are volatile compounds from aromatic plants with a range of phytopharmacological properties, including antimicrobial, antiallergic, antitumor, antioxidative, and so forth. Extensive attention has been paid to EOs since they are natural products and have been considered as Generally Recognized as Safe food additives by the Food and Drug Administration.15,16 Since time immemorial, EOs have been utilized for aromatic and flavouring purposes and also in traditional medicines in Asia.17–19 Nevertheless, high volatility, high heat sensitivity, and easy oxidation impede their performances, and these defects may be sufficient to alter the quality of their applications.20,21
Encapsulation is required to prevent those performance reductions mentioned above and is crucial from a scientific, industrial and economical point of view,22,23 and has attracted a great deal of interest such as its current applications in medicine, catalysis, cosmetics, and foods. In the biomedical field, capsules prepared from biodegradable polymers as wall materials have been frequently utilized to drugs for controlled and sustained release.21,24–28 This technique could be generally divided into three categories, which are physical methods including co-precipitation and spray drying, physicochemical methods including coacervation and sol–gel encapsulation, and chemical methods such as in situ polymerization and the use of liposomes.29–31 Table 1 shows some applications of various encapsulation methods for EOs. Among them, the co-precipitation method using β-cyclodextrins (β-CD) has been generally utilized as β-CD have hydrophilic outer surfaces and hydrophobic hollow cavities which can embed EOs by use of their similar physical property.
| Author | Core material | Wall material | Preparation method | Capsule diameter (μm) | Application |
|---|---|---|---|---|---|
| Arana-Sánchez et al.41 | Oregano oil | β-CD | Co-precipitation | <30 | Antimicrobial and antioxidant agents |
| Kfoury et al.42 | Nine selected EOs | Six kinds of cyclodextrin | Co-precipitation | No data | Radical scavenging efficacy |
| Karaaslan et al.43 | Pepper seed oil | Gum arabic and maltodextrin | Spray drying | 0.5–80 | Antimicrobial and peroxidative stability |
| Hsieh et al.44 | Citronella oil | Chitosan | Simple coacervation | 10–250 | Controlled release |
| K. Rutz et al.45 | Palm oil | Chitosan and xanthan; chitosan and pectin | Complex coacervation | <5 | Controlled release |
| Songkro et al.46 | Citronella oil | Gelatin and gum Arabic | Complex coacervation | <8 | Mosquito repellents |
| Vega et al.47 | Nine selected EOs | Hybrid organic-inorganic silica | Sol–gel | Not specified | Antibacterial agents |
| Lee et al.48 | Floral oil | Melamine and formalin | In situ polymerization | 12–15 | Not specified |
| Sebaaly et al.49 | Clove oil | Soybean | Liposome | 200–320 | Stability improvement of eugenol |
Houttuynia cordata Thunb. referred-to as “HC” hereafter, a perennial herb, is a plant of the Saururaceae family predominantly distributed in Asia. This plant, universally known across Japan as “Dokudami”, has been used as a traditional remedy for the treatment of various diseases or symptoms in local areas of Japan and China.32–34 Japanese Pharmacopoeia guidebook in the 18th edition (2021) lists it and describes that the aerial parts of HC can ameliorate the symptoms of constipation and chronic skin diseases, and its decoction can facilitate diuretic and anti-inflammatory effects.35 Das et al.36 reported the bioactive phytocompounds from HC can be applied as potential inhibitors for three main replication proteins of SARS-CoV-2, which control the replication process of COVID-19 infection. Furthermore, the traditional Chinese medicine “Lianhua Qingwen” including HC has been proved to be an effective therapy for the treatment of mild COVID-19 patients and employed since year 2020.37 Some reports have revealed that HC contains sundry active constituents such as EOs, flavonoids, alkaloids, organic and fatty acids, sterols and microelements, which possess diverse pharmacological activities including diuretic, antibacterial, antiviral, antitumor, anti-inflammatory, antioxidative, anti-diabetic, anti-allergic, and anti-mutagenic effects.38,39 A large number of reports have confirmed that the antimicrobial effect of HC primarily comes from EOs which show a significant inhibitory effect on SARS coronavirus, HSV, Bacillus cereus, Bacillus subtilis (B. subtilis), Staphylococcus aureus, Vibrio parahaemolyticus, Penicillium, Aspergillus niger, and so on.33,40 The conventional process to obtain EOs from aromatic plants is steam distillation, which is time-consuming and requires plenty of samples.33 Herein, organic solvent extraction as a feasible alternative was applied, and B. subtilis was chosen as the object microorganism to evaluate the antibacterial activity of its extract.
The aim of this research is to develop a capsule of EOs extracted from HC as an antibacterial agent for food packaging. The study is composed of the following three phases: (1) EOs extraction from HC using the two methods: multi-solvent consecutive extraction (MCE) method and single ethanol extraction with a pre-heating (SEEP) method, (2) synthesis of capsules by co-precipitation using β-CD and (3) characterization and antibacterial assessment of the fabricated capsules.
2 Experimental
2.1. Plant materials and EOs extraction
HC was picked in the campus of Univ. of Tsukuba around September and stored in a freezer (approx. −20 °C). The storage time of all HC used in this study was within one month.2.2. Identification of EOs extracted from HC
As discussed in Section 3.1 of the “Result and Discussion” part later, based on an antibacterial evaluation for all obtained extracts by the two extraction methods, the eeHCP(SEEP) was most satisfactory from the operational and economic aspects. Therefore, only eeHCP(SEEP) was used for the identification of EOs and subsequent encapsulation procedure and characterization.The identification of EOs in eeHCP(SEEP) was conducted using a gas chromatography-mass spectrometry (GC-MS). Here, the eeHC(SEEP) was set as a control. The condition details were as follows: injecting 2 μL of an eeHCP(SEEP) or eeHC(SEEP) solution into a GC-MS-QP2010Plus (Shimadzu, Japan) with a Rtx-5MS column (30 m × 0.25 μm, 0.25 μm in film thickness). The oven temperature of GC was programmed described by Pan et al.32 with a few modifications: kept at 60 °C for 2 min, then increased to 125 °C at a rate of 10 °C min−1 and maintained for 5 min, to 165 °C at 10 °C min−1 and held for 10 min, to 185 °C at 2 °C min−1, and finally to 240 °C at 10 °C min−1 and maintained for 10 min. The injector temperature was 260 °C and the flow rate of carrier helium gas was 1.5 mL min−1 with a split ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mass spectrometer was operated in the electron impact (EI) mode at 70 eV and mass spectra were acquired using a scan mode with a mass scan range of 50–650 m/z. The temperature of the ion source and interface was 250 °C and 280 °C, respectively. The solvent delay was set to 3.5 min. Identification of the phytocompounds of EOs was based on the comparison of their mass spectra with those of National Institute Standard and Technology version-05 and 05s (NIST 05 and NIST 05s) library data.
:1. The mass spectrometer was operated in the electron impact (EI) mode at 70 eV and mass spectra were acquired using a scan mode with a mass scan range of 50–650 m/z. The temperature of the ion source and interface was 250 °C and 280 °C, respectively. The solvent delay was set to 3.5 min. Identification of the phytocompounds of EOs was based on the comparison of their mass spectra with those of National Institute Standard and Technology version-05 and 05s (NIST 05 and NIST 05s) library data.
2.3. Encapsulation procedure
The co-precipitation method using β-CD was applied according to the methodology described by Ayala-Zavala et al.51 with a few modifications. A total of 2.5 g (±0.01 g) of β-CD (Wako 1st Grade, FUJIFILM Wako Chemicals, Japan) were dissolved in 50 mL of a co-solvent of ethanol and distilled water (1:2 v/v) mixture at 55 °C (±2 °C) and 700 rpm on a hot stirrer plate. In addition, another co-solvent was prepared with 0.1 g (±0.01 g) oil-in-water emulsifier gum Arabic (referred-to as “GA” hereafter) (AMC. CO., Japan) added with the aim of increasing the dispersibility of EOs in the co-solvent. After cooling the co-solvent to 45 °C, 2 mL of the eeHCP(SEEP) was added dropwise using a syringe. Subsequently, the mixture was stirred at 45 °C (±2 °C) for 2 h and then maintained overnight at 4 °C (±2 °C). The cold precipitates (supposed to be β-CD-eeHCP(SEEP) and β-CD-GA-eeHCP(SEEP) capsules) were recovered via the suction filtration and rinsed with 60% ethanol to remove the residual eeHCP(SEEP), and then the capsules were dried in a desiccator at 23 °C for 72 h and stored in an airtight glass at 23 °C before subsequent characterization. As a control, blank β-CD capsules without the addition of eeHCP(SEEP) were prepared in the same condition and steps as mentioned above.
2.4. Physicochemical characterization of capsules
2.5. Antibacterial assay
2.6. Statistical analysis
This study was carried out based on a completely randomized design with equal replication. All values were conducted in triplicate and reported as the mean ± standard deviation. For antibacterial evaluation of various extracts obtained by the MCE method and SEEP method, the area of inhibitory zone was measured with ImageJ 1.53e software in consideration of the asymmetrical appearance. The results were analysed for statistical significance via one-way analysis of variance (ANOVA) with Statistical Package of the Social Science (SPSS) version 28.0 (IBM Corporation, USA) software. Significant differences at a level of p <0.05 were identified by the least significant difference (LSD) test.3 Results and discussion
3.1. Antibacterial activity of various extracts
Table 2 shows the antibacterial activity statistical analysis of various extracts through the growth inhibition against B. subtilis. No inhibitory zone was observed with the blank paper disk which was impregnated with 99.5% ethanol and then air-dried for 5 min, indicating that the step of air-drying for 5 min was sufficient to eliminate the impact from solvents on the growth of B. subtilis. Among the extracts obtained by the MCE method, it is obvious that the best inhibition against B. subtilis was achieved by the eeeHC(MCE), followed by the eeHC(MCE), and then the heHC(MCE). It is assumed that greases and some proteins can be removed primarily with hexane; some active phytocompounds of EOs containing a certain amount of hydroxyl or carboxyl groups resemble ethyl ether in polarity can be removed with ethyl ether and lastly the remaining constituents such as alkaloids and organic acids are supposed to be separated out in ethanol. The experimental results demonstrated the above notion that the eeeHC(MCE) containing a large amount of EOs exhibited the superb inhibitory effect on the growth of B. subtilis. However, the polarity of some active phytocompounds of EOs such as 2-undecanone and β-myrcene are positioned between ethyl ether and hexane and might be partially extracted in hexane, accounting for why heHC(MCE) showed a certain inhibitory effect as well. A prominent inhibitory effect was also noted in eeHC(MCE), probably due to the amount of ethyl ether used was too small to extract all EOs, resulting in a partial flow of active phytocompounds into the eeHC(MCE), and consequently an inhibitory effect appeared in the eeHC(MCE). Therefore, if the extraction time of hexane was reduced and the amount of ethyl ether was increased, the antibacterial activity of eeeHC(MCE) might have greater enhancement, then further experiment is required to confirm whether it is true or false.| Sample | Area of inhibition zone (mm2) |
|---|---|
| Blank paper disk | 0 |
| heHC(MCE) | 130 ± 15 |
| eeeHC(MCE) | 750 ± 29 |
| eeHC(MCE) | 316 ± 14 |
| eeHCP(SEEP) | 712 ± 24 |
| eeHC(SEEP) | 234 ± 25 |
As to the SEEP method, the inhibitory effect of eeHCP(SEEP) was much superior to that of eeHC(SEEP) which was set as a control, presumably because the process of heating for a short time was considered to be a simple operation like steam distillation, which might reduce the EOs leaching resistance during the vortex.50 This clarifies why the inhibitory effect of eeHCP(SEEP) (approx. 710 mm2 of inhibition zone) was close to that of eeeHC(MCE) (approx. 750 mm2 of inhibition zone). Nevertheless, from the perspective of operational, economic and environmental aspects, eeHCP(SEEP) seems to be more advantageous, and therefore only eeHCP(SEEP) was chosen for the subsequent encapsulation procedure.
Although eeHCP(SEEP) exhibited satisfactory results, the use of ethanol as solvent led to a large number of constituents extracted from HC. Thus, vortex (extraction) time ought to be an exceptionally critical factor. To further investigate the impact of vortex time, an antibacterial assay was performed using a certain amount of B. subtilis. Fig. 1 shows the antibacterial activity of eeHCP(SEEP) with an increased vortex time and eeHC(SEEP) was set as a comparison. By vortex at high frequencies, EOs were extracted rapidly due to the pre-treatment of heating and their property of high volatility. Therefore, the vortex time should be adjusted to warrant a maximum possible, while not excessive or insufficient EOs extraction, otherwise other compounds would emerge or inadequate EOs contents might reduce the concentration of EOs in the extract, thus reducing the overall antibacterial activity. Evidently, for both eeHCP(SEEP) and eeHC(SEEP), a maximum area of the inhibitory zone was attained at a vortex time of 3 min, and hence the eeHCP(SEEP) obtained at this vortex time was chosen for the subsequent encapsulation procedure. In addition, Fig. 2 shows a comparison in the area of inhibitory zone of eeHCP(SEEP) after incubation for 24 h and 72 h. The area was slightly smaller after incubation for 72 h than that for 24 h, ensuring a strong antibacterial activity of eeHCP(SEEP) against B. subtilis.
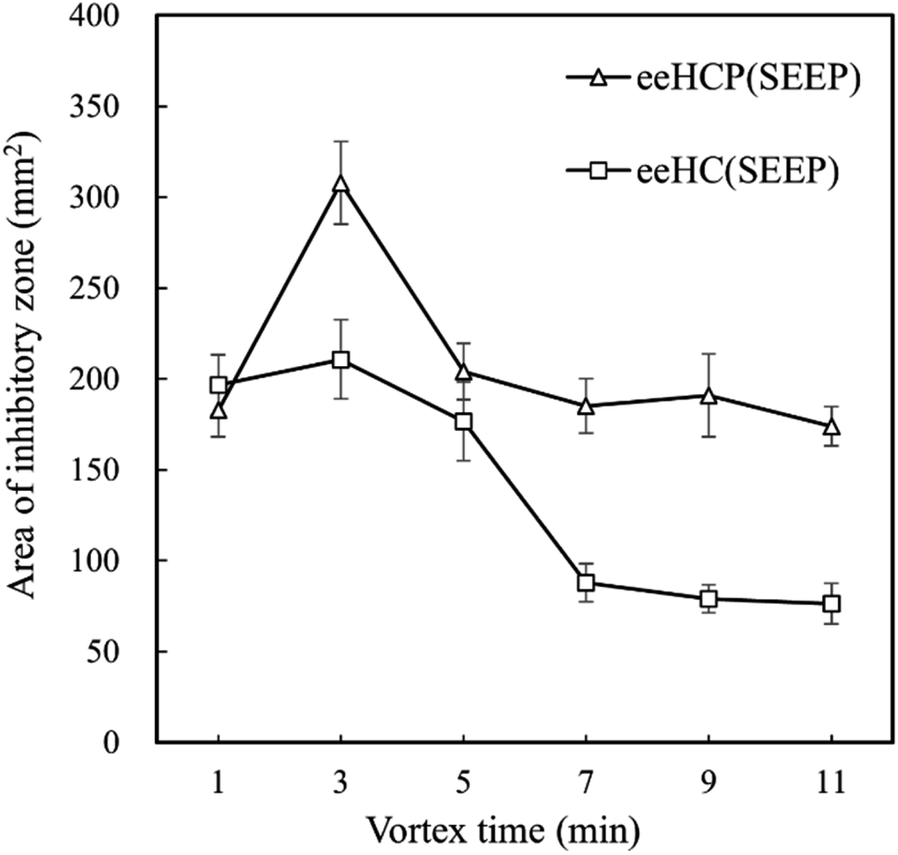 | ||
| Fig. 1 Area of inhibitory zone of eeHCP(SEEP) and eeHC(SEEP) versus vortex time after incubation at 37 °C for 24 h. | ||
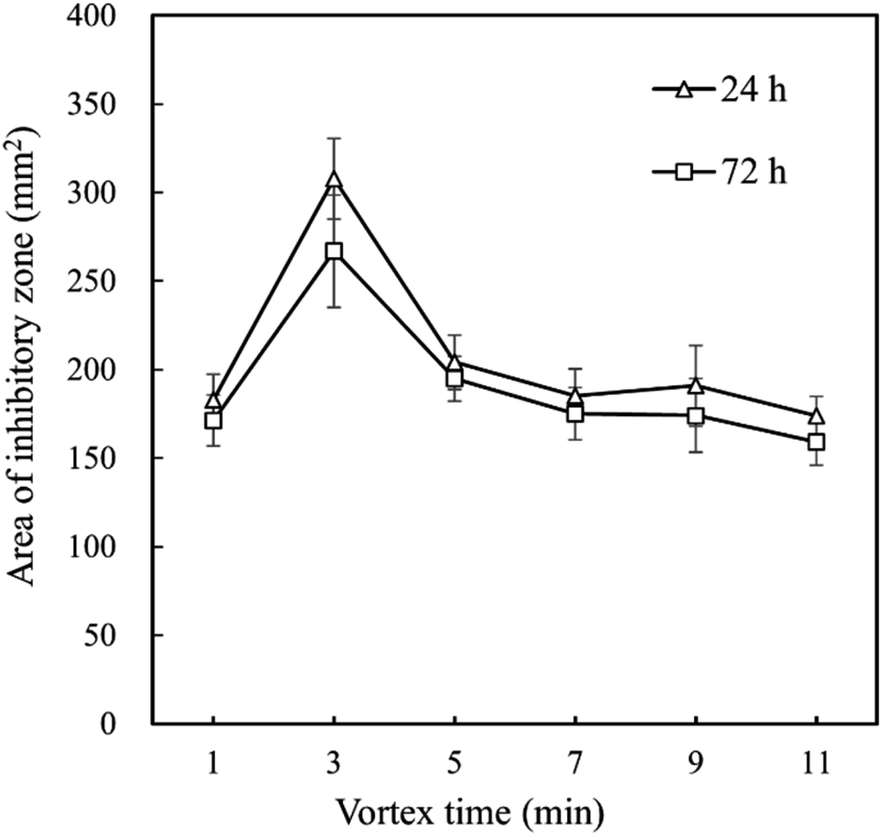 | ||
| Fig. 2 Difference in the area of inhibitory zone of eeHCP(SEEP) versus vortex time after incubation at 37 °C for 24 h and 72 h. | ||
3.2. GC-MS analysis
eeHCP(SEEP) and eeHC(SEEP) which was set as a control were subjected to GC-MS analysis. A total of 168 phytocompounds from eeHCP(SEEP) and 185 phytocompounds from eeHC(SEEP) were identified by GC-MS, separately. Fig. 3 shows total ion chromatograms (TIC) of eeHCP(SEEP) and eeHC(SEEP). The integrated peak area of top 50 phytocompounds which occupied more than 98% of the overall area in each of TIC, are selected for display in Table 3. Altogether, 26 identical phytocompounds were identified. Visually, the TIC of eeHCP(SEEP) was similar to that of eeHC(SEEP), but there was a slight difference. The intensity of phytocompounds detected from eeHCP(SEEP) was distinctly higher than that of eeHC(SEEP) during the retention time from 4 to 20 min, while the opposite result was noticed from 39 to 46 min. Six typical phytocompounds a, b, c, d, e and f labelled in Fig. 3 correspond to 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl- (No 5), decanal (No 3), 2-furancarboxaldehyde, 5-(hydroxymethyl)- (No 2), hydroquinone (No 7), 4-tridecanone (No 1) and 9-octadecenamide, (Z)- (No 22) are shown in Table 3. Of these, peak area of phytocompounds a (5.63%), b (7.48%), c (8.96%) and e (19.00%) were higher in eeHCP(SEEP) than a (3.42%), b (3.53%), c (3.89%) and e (8.91%) in eeHC(SEEP) respectively, whereas d (2.71%) and f (0.55%) were lower in eeHCP(SEEP) than d (7.41%) and f (6.39%) in eeHC(SEEP). According to the reports of Yang et al.33 and Wu et al.,34 decanal and its derivatives play an extremely crucial role in acting antibacterial activity, which therefore clarifies why the antibacterial activity of eeHCP(SEEP) was much greater than that of eeHC(SEEP). One additional finding was that some phytocompounds detected in eeHCP(SEEP) but not in eeHC(SEEP) had structural similarities to the phytocompounds detected in eeHC(SEEP) but not in eeHCP(SEEP), such as No 32 and No 65, with the main difference being the variation of the individual functional groups, which could be elucidated by that some of the substances underwent structural changes as a result of the pre-heating in the eeHCP(SEEP) preparation.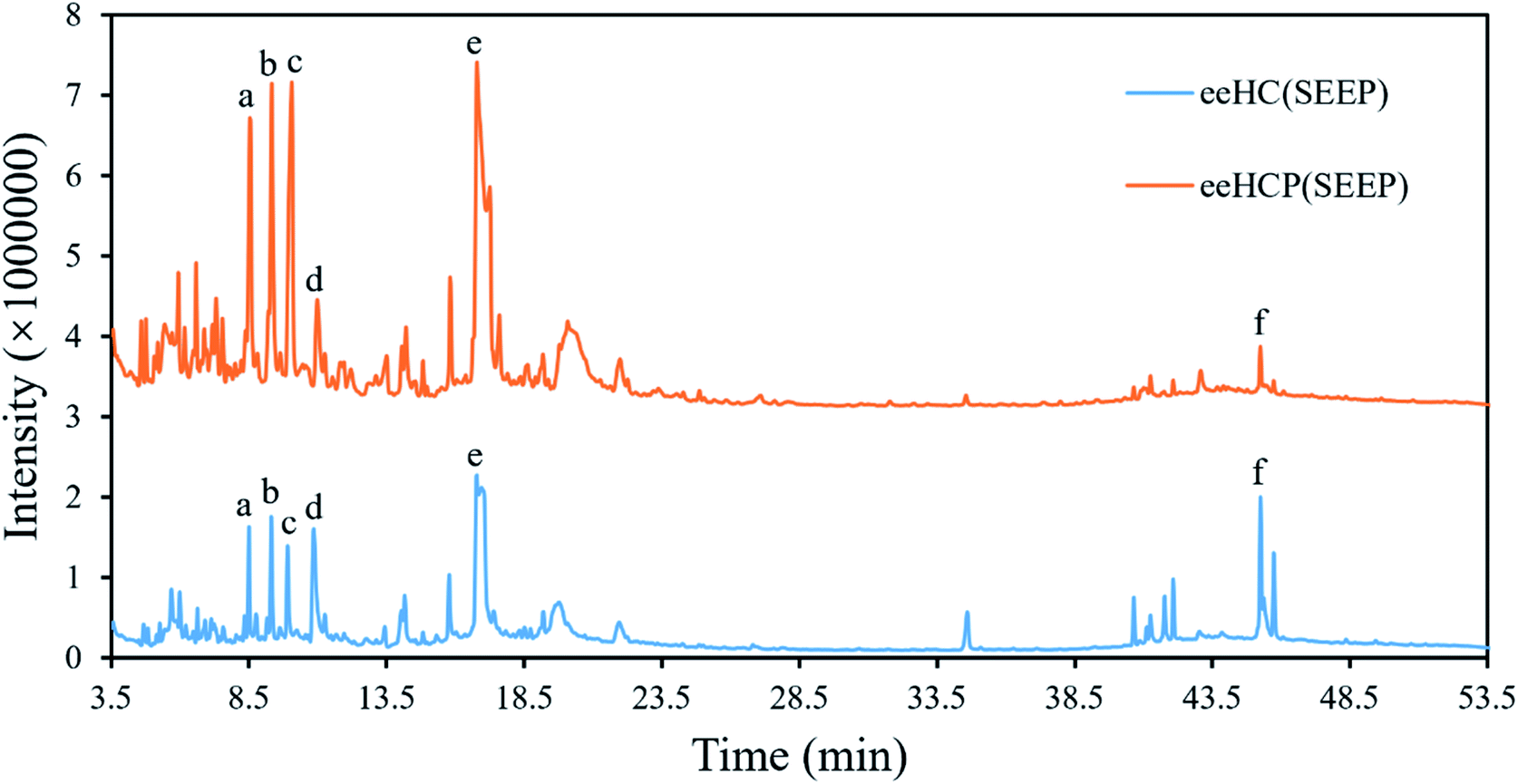 | ||
| Fig. 3 Total ion chromatogram of eeHCP(SEEP) and eeHC(SEEP). | ||
| No | Phytocompound name | Peak area (%) | |
|---|---|---|---|
| eeHCP(SEEP) | eeHC(SEEP) | ||
| a Note: 1 A Peak area of 0% does not mean non-existence but not included in the top 50 phytocompounds of eeHCP(SEEP) or eeHC(SEEP).b 2 The peak area ratio of each phytocompound was calculated based on the sum of the peak area of the top 50 phytocompounds assumed to be 100%. | |||
| 1 | 4-Tridecanone | 19.00 ± 0.85 | 8.91 ± 0.48 |
| 2 | 2-Furancarboxaldehyde, 5-(hydroxymethyl)- | 8.96 ± 0.52 | 3.89 ± 0.41 |
| 3 | Decanal | 7.48 ± 0.47 | 3.53 ± 0.40 |
| 4 | Benzoic acid, 3-hydroxy- | 6.36 ± 0.46 | 14.69 ± 1.88 |
| 5 | 4H-Pyran-4-one, 2,3-dihydro-3,5-dihydroxy-6-methyl- | 5.63 ± 1.05 | 3.42 ± 0.14 |
| 6 | Megastigmatrienone | 3.59 ± 0.02 | 0.58 ± 0.04 |
| 7 | Hydroquinone | 2.71 ± 0.12 | 7.41 ± 0.37 |
| 8 | 2,4-Dihydroxy-2,5-dimethyl-3(2H)-furan-3-one | 2.22 ± 0.08 | 1.56 ± 0.07 |
| 9 | Benzaldehyde, 2-hydroxy-4-methyl- | 1.81 ± 0.06 | 2.80 ± 0.43 |
| 10 | 2H-Pyran-2,6(3H)-dione | 1.40 ± 0.46 | 0.78 ± 0.05 |
| 11 | Benzoic acid, 4-hydroxy- | 1.67 ± 0.10 | 1.26 ± 0.03 |
| 12 | n-Decanoic acid | 1.59 ± 0.16 | 0.88 ± 0.05 |
| 13 | 14-Bromo-2-methyltetradec-1-en-3-ol | 1.41 ± 0.07 | 2.10 ± 0.00 |
| 14 | 2,5-Dimethyl-4-hydroxy-3(2H)-furanone | 1.36 ± 0.08 | 0.85 ± 0.04 |
| 15 | β-Myrcene | 1.07 ± 0.07 | 2.81 ± 0.73 |
| 16 | Methyl 2-furoate | 1.05 ± 0.04 | 0.58 ± 0.02 |
| 17 | Decanoic acid, ethyl ester | 0.93 ± 0.04 | 1.13 ± 0.07 |
| 18 | Ethanamine, 2-methoxy-N-(2-methoxyethyl)-N-methyl- | 0.87 ± 0.05 | 0.70 ± 0.02 |
| 19 | Benzenecarboxylic acid | 0.70 ± 0.12 | 1.20 ± 0.05 |
| 20 | Oxime-, methoxy-phenyl- | 0.74 ± 0.06 | 0.39 ± 0.04 |
| 21 | 3-Hydroxy-4-methoxybenzoic acid | 0.64 ± 0.14 | 0.33 ± 0.02 |
| 22 | 9-Octadecenamide, (Z)- | 0.55 ± 0.09 | 6.39 ± 0.33 |
| 23 | 2-Undecanone | 0.56 ± 0.03 | 1.51 ± 0.51 |
| 24 | Dodecanal | 0.51 ± 0.03 | 0.34 ± 0.02 |
| 25 | 1,2-Cyclopentanedione | 0.52 ± 0.01 | 0.43 ± 0.09 |
| 26 | Octadecanamide | 0.34 ± 0.02 | 2.99 ± 0.30 |
| 27 | 1,2,3,5-Cyclohexanetetrol, (1α, 2β, 3α, 5β)- | 4.69 ± 0.40 | 0 |
| 28 | 2-Furancarboxaldehyde, 5-methyl- | 3.29 ± 1.01 | 0 |
| 29 | 4-Ethylcatechol | 1.77 ± 0.10 | 0 |
| 30 | 3-Furancarboxylic acid | 1.67 ± 0.01 | 0 |
| 31 | Thiophene, 2-formyl-2,3-dihydro- | 1.58 ± 0.03 | 0 |
| 32 | 2,5-Furandione, dihydro-3-methylene- | 1.49 ± 0.06 | 0 |
| 33 | 3-Hydroxy-β-damascone | 1.41 ± 0.11 | 0 |
| 34 | Ethanone, 1-(2-hydroxy-5-methylphenyl)- | 1.21 ± 0.04 | 0 |
| 35 | Ethanamine, N-ethyl-N-nitroso- | 1.13 ± 0.05 | 0 |
| 36 | Bicyclo[3.1.1]heptan-3-ol, 2,6,6-trimethyl-, (1α,2β,3α,5β)- | 1.00 ± 0.07 | 0 |
| 37 | Ribitol | 0.93 ± 0.04 | 0 |
| 38 | 2,13-Dibora-1,3,6,9,12,14,17,20-octaoxacyclodocosane, 2,13-diethyl- | 0.74 ± 0.03 | 0 |
| 39 | Benzamide | 0.60 ± 0.04 | 0 |
| 40 | 2(5H)-Furanone | 0.62 ± 0.01 | 0 |
| 41 | Benzofuran, 2,3-dihydro- | 0.55 ± 0.07 | 0 |
| 42 | Furan, 2-[(methylthio)methyl]- | 0.55 ± 0.03 | 0 |
| 43 | Benzyl β-d-glucoside | 0.53 ± 0.03 | 0 |
| 44 | Octyl-β-D-glucopyranoside | 0.46 ± 0.02 | 0 |
| 45 | 1H-Indene, 2,3-dihydro-1,1,5,6-tetramethyl- | 0.42 ± 0.02 | 0 |
| 46 | Naphthalene, 1,2-dihydro-2,5,8-trimethyl- | 0.41 ± 0.01 | 0 |
| 47 | Pyrimidine-4,6-diol, 5-methyl- | 0.36 ± 0.02 | 0 |
| 48 | Nonane, 1,1-diethoxy- | 0.36 ± 0.01 | 0 |
| 49 | 9,12,15-Octadecatrienoic acid, methyl ester, (Z,Z,Z)- | 0.28 ± 0.07 | 0 |
| 50 | 1,3-Cyclohexanedione, 2-methyl- | 0.28 ± 0.05 | 0 |
| 51 | (1R,3R,4R,5R)-(−)-Quinic acid | 0 | 5.74 ± 0.10 |
| 52 | Pentadecanoic acid | 0 | 1.84 ± 0.16 |
| 53 | 1,3-Benzenediol, 4-ethyl- | 0 | 1.70 ± 0.26 |
| 54 | Hexadecanamide | 0 | 1.62 ± 0.16 |
| 55 | 1,2,3-Benzenetriol | 0 | 1.73 ± 0.03 |
| 56 | Octadecanoic acid | 0 | 1.55 ± 0.19 |
| 57 | Phytol | 0 | 1.34 ± 0.06 |
| 58 | Cyclopentene, 1,2-dimethyl-4-methylene- | 0 | 1.19 ± 0.03 |
| 59 | Thiophene, 2-methoxy-5-methyl- | 0 | 1.09 ± 0.11 |
| 60 | Phenol | 0 | 1.07 ± 0.10 |
| 61 | Dichloroacetic acid, tridec-2-ynyl ester | 0 | 0.98 ± 0.06 |
| 62 | 2-Furancarboxylic acid | 0 | 0.97 ± 0.05 |
| 63 | 2-Benzyl-2-methyl-1,3-oxathiolane | 0 | 0.84 ± 0.10 |
| 64 | Ethanamine, N-ethyl-N-nitroso- | 0 | 0.90 ± 0.03 |
| 65 | 2,5-Furandione, 3-methyl- | 0 | 0.85 ± 0.02 |
| 66 | 4H-Pyran-4-one, 3,5-dihydroxy-2-methyl- | 0 | 0.78 ± 0.07 |
| 67 | 1,4:3,6-Dianhydro-α-d-glucopyranose | 0 | 0.83 ± 0.02 |
| 68 | 4-Benzoyloxy-1-morpholinocyclohexene | 0 | 0.71 ± 0.03 |
| 69 | 9,12-Octadecadienoic acid, methyl ester | 0 | 0.68 ± 0.02 |
| 70 | 1,3,6-Octatriene, 3,7-dimethyl-, (E)- | 0 | 0.55 ± 0.03 |
| 71 | 3-Chloropropionic acid, 2,6-dimethylnon-1-en-3-yn-5-yl ester | 0 | 0.53 ± 0.01 |
| 72 | Isolongifolan-8-ol | 0 | 0.38 ± 0.05 |
| 73 | Bendiocarb | 0 | 0.32 ± 0.07 |
| 74 | 3-Ethyl-3-heptanol | 0 | 0.33 ± 0.04 |
| Total (%) | 100 | 100 | |
3.3. Morphological and dimensional analyses of capsules
The β-CD-eeHCP(SEEP) and β-CD-GA-eeHCP(SEEP) capsules obtained by the co-precipitation method were in powder form after drying and the SEM images of them are shown in Fig. 4. Plain β-CD (Fig. 4a) as purchased was characterized as homogeneous massive or lamellar plates with well-grown crystals in varying sizes. However, the shape of the blank β-CD capsules (Fig. 4b) without the addition of eeHCP(SEEP) was not consistent with that of plain β-CD and appeared to be more granular in shape. Actually, the blank β-CD capsules were obtained through a process in which plain β-CD was dissolved in the co-solvent and then precipitated, suggesting a condition like precipitation rate caused some alteration in the particle shape of recovered β-CD, in consistency with the findings of Nakai et al.52 who also confirmed that water molecules exert an influence on the crystal structure of cyclodextrins. This is why the surface of the β-CD-eeHCP(SEEP) capsules (Fig. 4c) and β-CD-GA-eeHCP(SEEP) (Fig. 4d) resembled that of blank β-CD capsules rather than the plain β-CD. It is observed that β-CD-eeHCP(SEEP) capsules and β-CD-GA-eeHCP(SEEP) capsules had extremely similar cubic and irregular structures with the dimensions of approx. 80 μm in diameter and exhibited clustering, as was likewise reported by Sauceau et al.53 The original intention of the addition of GA as an emulsifier to the co-solvent was to improve the dispersion of the EOs in the co-solvent, but in terms of experimental results, the same objective was achieved without the addition of GA. This was attributed to the ethanol in the co-solvent. Not only did the ethanol improve the solubility of β-CD (only about 1.5 g β-CD can be dissolved in 50 mL of water-solvent in the same condition described in Section 2.3) and thus increased the efficiency of encapsulation, but also it contributed to the dispersion of the EOs in the co-solvent. However, it is also important that a high content of ethanol may assist the dissolution of some EOs phytocompounds in the co-solvent rather than being embedded within hollow cavities of β-CD, so an optimum ratio of water to ethanol needs further experimentation. | ||
| Fig. 4 SEM images of plain β-CD (a), blank β-CD capsules (b), β-CD-eeHCP(SEEP) capsules (c) and β-CD-GA-eeHCP(SEEP) capsules (d). | ||
3.4. XRD analysis
XRD is a useful method for the detection of crystalline states. Fig. 5 shows the XRD patterns of capsules produced by the co-precipitation method. The XRD pattern of blank β-CD capsules (Fig. 5b) was almost identical to those of β-CD-eeHCP(SEEP) capsules (Fig. 5c) and β-CD-GA-eeHCP(SEEP) (Fig. 5d), while it was similar to that of plain β-CD (Fig. 5a) in the peak position (2θ) but with different peak intensity mutually. The diffraction angle 2θ of the plain β-CD had intense and sharp peaks at 11.32, 14.47 17.42 and 18.53°. However, in the patterns of blank β-CD capsules, β-CD-eeHCP(SEEP) capsules and β-CD-GA-eeHCP(SEEP) capsules, these peaks were substantially compressed, despite heightened peaks at 10.43, 12.24 and 19.45°. This is because crystallites of the β-CD become oriented upon precipitation, just as discussed in the SEM section, and the presence or absence of eeHCP(SEEP) did not alter the crystal orientation of the capsules, in line with the results reported by Munhuweyi et al.54 who observed the same XRD patterns of capsules produced via employing two different kinds of EOs.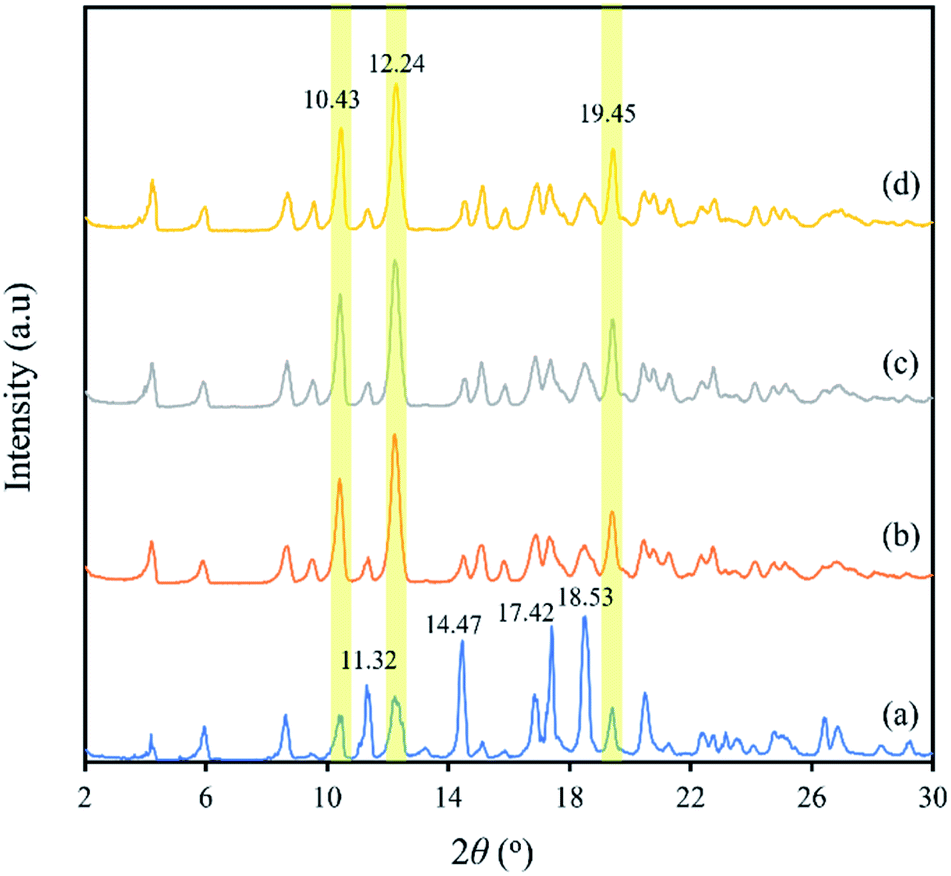 | ||
| Fig. 5 XRD patterns of plain β-CD (a), blank β-CD capsules (b), β-CD-eeHCP(SEEP) capsules (c) and β-CD-GA-eeHCP(SEEP) capsules (d). | ||
3.5. FT-IR analysis
FT-IR was used to demonstrate the interaction between β-CD and eeHCP(SEEP) in the process of forming capsules in terms of peak shape, position and intensity. Fig. 6 shows the FT-IR spectra of eeHCP(SEEP) (Fig. 6a), plain β-CD (Fig. 6b), blank β-CD capsules (Fig. 6c), β-CD-eeHCP(SEEP) capsules (Fig. 6d) and β-CD-GA-eeHCP(SEEP) capsules (Fig. 6e). Typically, eeHCP(SEEP) exhibited three characteristic peaks at 3355.5 cm−1 (for O–H stretching vibrations), 2922.6 cm−1 (for C–H stretching vibrations), and a prominent absorption peak at 1630.5 cm−1 (for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of amides). The absorption bands of plain β-CD were observed at 3369.0, 2924.5, 1649.9, 1157.1 and 1028.8 cm−1, corresponding to the vibration of symmetrical and asymmetrical stretching of the O–H, C–H stretching vibrations, H–O–H bending, C–O stretching vibration, and symmetric stretching link C–O–C, respectively.55 Obviously, the FT-IR spectra of blank β-CD capsules, β-CD-eeHCP(SEEP) capsules as well as β-CD-GA-eeHCP(SEEP) capsules revealed a high degree of similarity to the spectra of plain β-CD, except for the position of O–H absorption peaks which shifted towards lower frequencies. Kayaci et al.56 and Del Toro-Sánchez et al.57 explained these shifts after encapsulation indicated an interaction had occurred between the embedded materials and β-CD via hydrogen bonding, however, a slight shift in the position of O–H absorption peak of blank β-CD capsules was also observed, possibly suggesting that the remaining water molecules after drying were likewise hydrogen-bonded to the β-CD. In addition, it is worth noting that the highly intense peak of eeHCP(SEEP) located at 1630.5 cm−1 was not detected in the spectra of β-CD-eeHCP(SEEP) and β-CD-GA-eeHCP(SEEP) capsules in consistent with the findings reported by Wang et al.55 and Munhuweyi et al.54 who considered this was due to the successful encapsulation by β-CD, however, it should be evidenced in conjunction with a subsequent antibacterial assay. Moreover, combined with the results of SEM, XRD and FT-IR, with the addition of GA as an emulsifier to the co-solvent, no significant change was observed, possibly because the presence of ethanol in the co-solvent was sufficient to achieve a good dispersibility of EOs, just as discussed in the Section 3.3. Therefore, it could be considered that GA was not required to the co-solvent in this study and only the β-CD-eeHCP(SEEP) capsules were used for the antibacterial assay.
O stretching vibration of amides). The absorption bands of plain β-CD were observed at 3369.0, 2924.5, 1649.9, 1157.1 and 1028.8 cm−1, corresponding to the vibration of symmetrical and asymmetrical stretching of the O–H, C–H stretching vibrations, H–O–H bending, C–O stretching vibration, and symmetric stretching link C–O–C, respectively.55 Obviously, the FT-IR spectra of blank β-CD capsules, β-CD-eeHCP(SEEP) capsules as well as β-CD-GA-eeHCP(SEEP) capsules revealed a high degree of similarity to the spectra of plain β-CD, except for the position of O–H absorption peaks which shifted towards lower frequencies. Kayaci et al.56 and Del Toro-Sánchez et al.57 explained these shifts after encapsulation indicated an interaction had occurred between the embedded materials and β-CD via hydrogen bonding, however, a slight shift in the position of O–H absorption peak of blank β-CD capsules was also observed, possibly suggesting that the remaining water molecules after drying were likewise hydrogen-bonded to the β-CD. In addition, it is worth noting that the highly intense peak of eeHCP(SEEP) located at 1630.5 cm−1 was not detected in the spectra of β-CD-eeHCP(SEEP) and β-CD-GA-eeHCP(SEEP) capsules in consistent with the findings reported by Wang et al.55 and Munhuweyi et al.54 who considered this was due to the successful encapsulation by β-CD, however, it should be evidenced in conjunction with a subsequent antibacterial assay. Moreover, combined with the results of SEM, XRD and FT-IR, with the addition of GA as an emulsifier to the co-solvent, no significant change was observed, possibly because the presence of ethanol in the co-solvent was sufficient to achieve a good dispersibility of EOs, just as discussed in the Section 3.3. Therefore, it could be considered that GA was not required to the co-solvent in this study and only the β-CD-eeHCP(SEEP) capsules were used for the antibacterial assay.
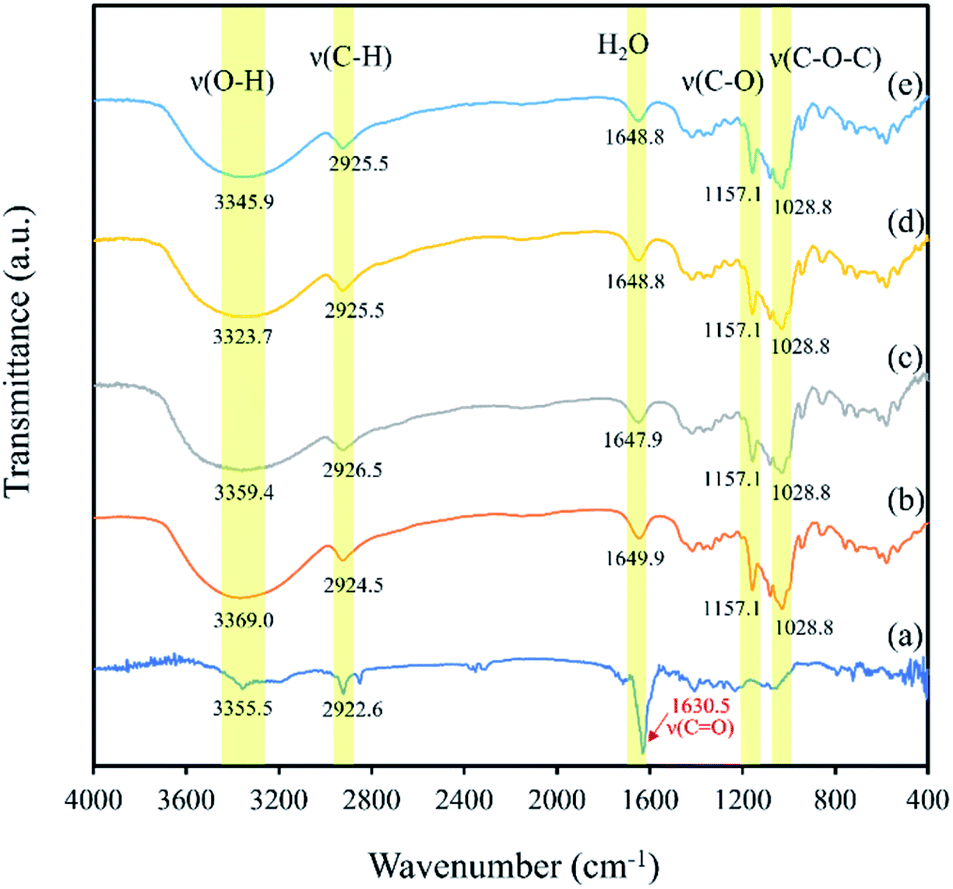 | ||
| Fig. 6 FT-IR spectra of eeHCP(SEEP) (a), plain β-CD (b), blank β-CD capsules (c), β-CD-eeHCP(SEEP) capsules (d) and β-CD-GA-eeHCP(SEEP) capsules (e). | ||
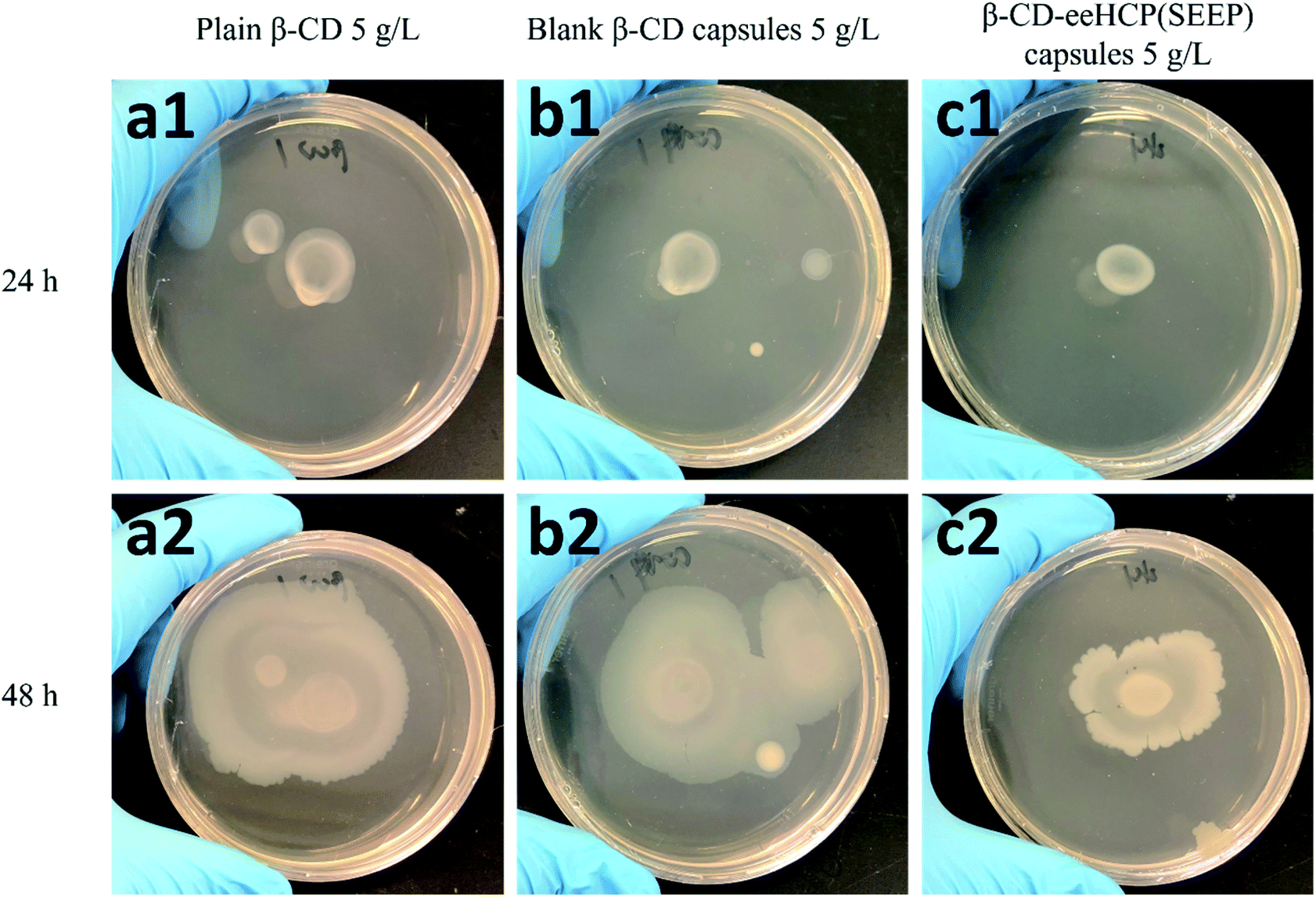
3.6. Antibacterial activity of capsules
To investigate the antibacterial activity of the fabricated capsules, an antibacterial assay using B. subtilis as a study object was performed. The growth and breeding status of B. subtilis in TSA medium with the addition of plain β-CD (a), blank β-CD capsules (b) and β-CD-eeHCP(SEEP) capsules (c) was observed after an addition of 10 μL diluted B. subtilis at two different concentrations, as shown in Fig. 7 (6 × 104 CFU mL−1) and Fig. 8 (6 × 106 CFU mL−1), respectively. Plain β-CD and blank β-CD capsules resulted in a rapid growth and breeding of B. subtilis due to the fact that β-CD is a polysaccharide that can provide a carbohydrate source and blank β-CD capsules were merely the precipitated product of dissolved β-CD. On the contrary, the addition of capsules containing eeHCP(SEEP) showed a potent inhibitory ability. There appeared to be a slight difference after incubation for 24 h in the growth of B. subtilis among the addition of plain β-CD, blank β-CD capsules and β-CD-eeHCP(SEEP) capsules; however, when the incubation was continued to 48 h, a distinctive difference was reflected. Also, remarkably similar results were exhibited whether the concentration of B. subtilis added was 6 × 104 or 6 × 106 CFU mL−1. This fact indirectly revealed the eeHCP(SEEP) was successfully encapsulated within hollow cavities of β-CD, as estimated from the FT-IR spectra. Most importantly, this satisfactory antibacterial activity of the β-CD-eeHCP(SEEP) capsules fabricated by the co-precipitation method in this study provides a promising direction for the use of eco-friendly antibacterial agents for food packaging industry. | ||
| Fig. 7 Growth of 10 μL 6 × 104 CFU mL−1 B. subtilis with plain β-CD (a), blank β-CD capsules (b) and β-CD-eeHCP(SEEP) capsules (c) after incubation for 24 h and 48 h. | ||
 | ||
| Fig. 8 Growth of 10 μL 6 × 106 CFU mL−1 B. subtilis with plain β-CD (a), blank β-CD capsules (b) and β-CD-eeHCP(SEEP) capsules (c) after incubation for 24 h and 48 h. | ||
4 Conclusions
This study developed a promising antibacterial agent containing an HC extract for food packaging. Two extraction methods: MCE method and SEEP method, were attempted to extract EOs from the HC. Although the eeeHC(MCE) exhibited the greatest anti-B. subtilis activity, the eeHCP(SEEP) was chosen for the capsule synthesis, taking into account the operational and economical aspects.The results of GC-MS showed the content of decanal which plays a definitely crucial role in the antibacterial activity in eeHCP(SEEP) was higher than that in eeHC(SEEP), explaining a higher antibacterial activity of eeHCP(SEEP). Afterwards, the co-precipitation method using β-CD was employed to produce capsules to protect the EOs of eeHCP(SEEP). Consequently, the fabricated capsules were in powder form with the dimensions of approx. 80 μm in diameter. The SEM and XRD results showed the β-CD crystallites in form of thinner plates became oriented upon precipitation. From the comprehensive results of SEM, XRD and FT-IR, GA added with the aim of increasing the dispersibility of EOs in the co-solvent was not required in this study due to the presence of ethanol in the co-solvent was sufficient to achieve a good dispersibility of EOs. Combining the results of FT-IR analysis and an antibacterial assay, it is revealed that the eeHCP(SEEP) was successfully encapsulated within hollow cavities of β-CD. Moreover, the antibacterial assay demonstrated a satisfactory antibacterial activity of the fabricated β-CD-eeHCP(SEEP) capsules, making them prospective for food packaging as an eco-friendly antibacterial agent.
Author contributions
P. K. performed most of the experimental work and wrote the manuscript. J. P. A., A. N. and M. K. guided and assisted in part of the experiments. T. E. supervised the whole study and revised the manuscript. All authors have given final approval to publish the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Open Facility Centre, University of Tsukuba for allowing us to use their facilities. The authors are also thankful to the Academic Computing & Communications Centre, University of Tsukuba for providing the SPSS software.References
- Y. Ma, P. Liu, C. Si and Z. Liu, J. Macromol. Sci., Part B: Phys., 2010, 49, 994–1001 CrossRef CAS.
- T.-T. Tsai, T.-H. Huang, C.-J. Chang, N. Yi-Ju Ho, Y.-T. Tseng and C.-F. Chen, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- P. J. P. Espitia, N. d. F. F. Soares, R. F. Teófilo, J. S. dos Reis Coimbra, D. M. Vitor, R. A. Batista, S. O. Ferreira, N. J. de Andrade and E. A. A. Medeiros, Carbohydr. Polym., 2013, 94, 199–208 CrossRef CAS PubMed.
- M. Carbone, D. T. Donia, G. Sabbatella and R. Antiochia, J. King Saud Univ., Sci., 2016, 28, 273–279 CrossRef.
- N. C. Martins, C. S. Freire, C. P. Neto, A. J. Silvestre, J. Causio, G. Baldi, P. Sadocco and T. Trindade, Colloids Surf., A, 2013, 417, 111–119 CrossRef CAS.
- N. Omerović, M. Djisalov, K. Živojević, M. Mladenović, J. Vunduk, I. Milenković, N. Ž. Knežević, I. Gadjanski and J. Vidić, Compr. Rev. Food Sci. Food Saf., 2021, 20, 2428–2454 CrossRef PubMed.
- Z. Wu, X. Huang, Y. C. Li, H. Xiao and X. Wang, Carbohydr. Polym., 2018, 199, 210–218 CrossRef CAS PubMed.
- T.-T. Tsai, T.-H. Huang, C.-J. Chang, N. Yi-Ju Ho, Y.-T. Tseng and C.-F. Chen, Sci. Rep., 2017, 7, 3155 CrossRef PubMed.
- P. Kanmani and J.-W. Rhim, Food Chem., 2014, 148, 162–169 CrossRef CAS PubMed.
- A. Ben Arfa, L. Preziosi-Belloy, P. Chalier and N. Gontard, J. Agric. Food Chem., 2007, 55, 2155–2162 CrossRef CAS PubMed.
- L. Motelica, D. Ficai, A. Ficai, R.-D. Truşcă, C.-I. Ilie, O.-C. Oprea and E. Andronescu, Foods, 2020, 9, 1801 CrossRef CAS PubMed.
- L. Motelica, D. Ficai, O.-C. Oprea, A. Ficai, V.-L. Ene, B.-S. Vasile, E. Andronescu and A.-M. Holban, Nanomaterials, 2021, 11, 2377 CrossRef CAS PubMed.
- L. Motelica, D. Ficai, O. Oprea, A. Ficai, R.-D. Trusca, E. Andronescu and A. M. Holban, Pharmaceutics, 2021, 13, 1020 CrossRef CAS PubMed.
- V. G. L. Souza, C. Rodrigues, S. Valente, C. Pimenta, J. R. A. Pires, M. M. Alves, C. F. Santos, I. M. Coelhoso and A. L. Fernando, Coatings, 2020, 10, 110 CrossRef CAS.
- R. Ribeiro-Santos, M. Andrade and A. Sanches-Silva, Curr. Opin. Food Sci., 2017, 14, 78–84 CrossRef.
- P. S. G. R. a. Safe, U.S. Food and Drug Administration, 2022, Title 21-Food and Drugs, https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=182 Search PubMed.
- T. Nakatsu, A. T. Lupo Jr, J. W. Chinn Jr and R. K. Kang, in Studies in natural products chemistry, Elsevier, 2000, vol. 21, pp. 571–631 Search PubMed.
- J. Dai and R. J. Mumper, Molecules, 2010, 15, 7313–7352 CrossRef CAS PubMed.
- J. S. Raut and S. M. Karuppayil, Ind. Crops Prod., 2014, 62, 250–264 CrossRef CAS.
- A. C. Gargano, C. Costa and M. Costa, Tree For. Sci. Biotechnol., 2008, 2, 121–124 Search PubMed.
- E. F. de Matos, B. S. Scopel and A. Dettmer, J. Environ. Chem. Eng., 2018, 6, 1989–1994 CrossRef CAS.
- Z. Xiao, W. Liu, G. Zhu, R. Zhou and Y. Niu, J. Sci. Food Agric., 2014, 94, 1482–1494 CrossRef CAS PubMed.
- D. Poncelet in Surface chemistry in biomedical and environmental science, Springer, 2006, pp. 23–34 Search PubMed.
- W. Li, G. Wu, H. Chen and M. Wang, Colloids Surf., A, 2009, 333, 133–137 CrossRef CAS.
- J. Oxley in Microencapsulation in the food industry, Elsevier, 2014, pp. 35–46 Search PubMed.
- S. Freiberg and X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS PubMed.
- S. Freitas, H. P. Merkle and B. Gander, J. Controlled Release, 2005, 102, 313–332 CrossRef CAS PubMed.
- H. Sawalha, K. Schroen and R. Boom, Chem. Eng. J., 2011, 169, 1–10 CrossRef CAS.
- P. S. Elizei and W. Krasaaekoopt, Agric. Nat. Resour., 2014, 48, 893–907 Search PubMed.
- M. G. Bah, H. M. Bilal and J. Wang, Soft Matter, 2020, 16, 570–590 RSC.
- M. Peanparkdee, S. Iwamoto and R. Yamauchi, Reviews in Agricultural Science, 2016, 4, 56–65 CrossRef.
- X. Pan, H. Li, D. Chen, J. Zheng, L. Yin, J. Zou, Y. Zhang, K. Deng, M. Xiao and L. Meng, Int. J. Anal. Chem., 2021, 2021 Search PubMed.
- L. Yang and J.-G. Jiang, Pharm. Biol., 2009, 47, 1154–1161 CrossRef CAS.
- Z. Wu, X. Deng, Q. Hu, X. Xiao, J. Jiang, X. Ma and M. Wu, Front. Pharmacol., 2021, 12 CAS.
- J. Pharmacopoeia, Pharmaceuticals and Medical Devices Agency, 2021, 18th, https://www.pmda.go.jp/english/rs-sb-std/standards-development/jp/0029.html Search PubMed.
- S. K. Das, S. Mahanta, B. Tanti, H. Tag and P. K. Hui, Mol. Diversity, 2021, 1–24 Search PubMed.
- M. Zeng, L. Li and Z. Wu, PLoS One, 2020, 15, e0238828 CrossRef CAS PubMed.
- J. Fu, L. Dai, Z. Lin and H. Lu, Chin. Med., 2013, 4, 101–123 CrossRef.
- M. Kumar, S. K. Prasad and S. Hemalatha, Pharmacogn. Rev., 2014, 8, 22–35 CrossRef PubMed.
- H. D. Kwon, I. H. Cha, W. K. Lee, J. H. Song and I. H. Park, Prev. Nutr. Food Sci., 1996, 1, 208–213 CAS.
- A. Arana-Sánchez, M. Estarrón-Espinosa, E. Obledo-Vázquez, E. Padilla-Camberos, R. Silva-Vázquez and E. Lugo-Cervantes, Lett. Appl. Microbiol., 2010, 50, 585–590 CrossRef PubMed.
- M. Kfoury, L. Auezova, H. Greige-Gerges and S. Fourmentin, Carbohydr. Polym., 2015, 131, 264–272 CrossRef CAS PubMed.
- M. Karaaslan, F. Şengün, Ü. Cansu, B. Başyiğit, H. Sağlam and A. Karaaslan, Food Chem., 2021, 337, 127748 CrossRef CAS PubMed.
- W.-C. Hsieh, C.-P. Chang and Y.-L. Gao, Colloids Surf., B, 2006, 53, 209–214 CrossRef CAS PubMed.
- J. K. Rutz, C. D. Borges, R. C. Zambiazi, M. M. Crizel-Cardozo, L. S. Kuck and C. P. Noreña, Food Chem., 2017, 220, 59–66 CrossRef CAS PubMed.
- S. Songkro, P. Yapong, P. Puechpan, D. Maneenuan and P. Boonme, Songklanakarin J. Sci. Technol., 2018, 40, 767–775 CAS.
- O. Vega, J. J. Araya, M. Chavarría and E. Castellón, J. Sol-Gel Sci. Technol., 2016, 79, 584–595 CrossRef CAS.
- H. Lee, S. Lee, I. Cheong and J. Kim, J. Microencapsulation, 2002, 19, 559–569 CrossRef CAS PubMed.
- C. Sebaaly, A. Jraij, H. Fessi, C. Charcosset and H. Greige-Gerges, Food Chem., 2015, 178, 52–62 CrossRef CAS PubMed.
- Y. Sekita, K. Murakami, H. Yumoto, H. Mizuguchi, T. Amoh, S. Ogino, T. Matsuo, Y. Miyake, H. Fukui and Y. Kashiwada, Biosci., Biotechnol., Biochem., 2016, 80, 1205–1213 CrossRef CAS PubMed.
- J. F. Ayala-Zavala, H. Soto-Valdez, A. González-León, E. Álvarez-Parrilla, O. Martín-Belloso and G. A. González-Aguilar, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 359–368 CrossRef CAS.
- Y. Nakai, K. Yamamoto, K. Terada, A. Kajiyama and I. Sasaki, Chem. Pharm. Bull., 1986, 34, 2178–2182 CrossRef CAS.
- M. Sauceau, E. Rodier and J. Fages, J. Supercrit. Fluids, 2008, 47, 326–332 CrossRef CAS.
- K. Munhuweyi, O. J. Caleb, A. J. van Reenen and U. L. Opara, LWT, 2018, 87, 413–422 CrossRef CAS.
- J. Wang, Y. Cao, B. Sun and C. Wang, Food Chem., 2011, 127, 1680–1685 CrossRef CAS.
- F. Kayaci and T. Uyar, J. Agric. Food Chem., 2011, 59, 11772–11778 CrossRef CAS PubMed.
- C. L. Del Toro-Sánchez, J. F. Ayala-Zavala, L. Machi, H. Santacruz, M. A. Villegas-Ochoa, E. Alvarez-Parrilla and G. A. González-Aguilar, J. Inclusion Phenom. Macrocyclic Chem., 2010, 67, 431–441 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |