DOI:
10.1039/D2RA02028A
(Paper)
RSC Adv., 2022,
12, 17350-17361
Synthesis and photophysical properties of photostable 1,8-naphthalimide dyes incorporating benzotriazole-based UV absorbers†
Received
29th March 2022
, Accepted 5th June 2022
First published on 13th June 2022
Abstract
We developed a series of blue-emitting 1,8-naphthalimide dyes covalently attached to 2-(2-hydroxyphenyl)-2H-benzotriazoles that retard photodegradation of the fluorophore. The dyes displayed weaker fluorescence emissions than the parent 1.8-naphthalimide. Quantum chemical calculations suggested that the decreased fluorescence was caused by the nonradiative deactivation promoted through the excited state intramolecular proton transfer (ESIPT) in benzotriazole components. The dyes' phosphorescences in a degassed solution at 77 K were more efficient than that of the parent 1.8-naphthalimide, indicating a possible deactivation pathway through intersystem crossing. PMMA films doped with these dyes showed higher resistance against photoaging than the film doped with an equimolar mixture of constituent 1.8-naphthalimide and the benzotriazole derivatives. Thus, the covalently linked benzotriazole units slow fluorophore degradation not only by preferential absorption of harmful UV light, which is found in the film with a simple mixture of two components, but also by the nonradiative deactivation involved in benzotriazole units.
Introduction
In response to increasing interest in the environment and climate change, the effective utilization of solar energy is receiving much attention. Organic compounds and metal complexes that absorb ultraviolet (UV) radiation and emit visible light should be key materials for the effective utilization of sunlight. These dyes can work as wavelength conversion materials to reduce harmful UV light and increase useful visible light through the photophysical process.1–3 They can contribute to the improvement of the power conversion efficiency of photovoltaics due to the poor spectral response in the UV region.1 They also can be utilized to cultivate plants because UV light range is not effective in photosynthesis.4,5 Blue fluorescent dyes are also in demand for existing applications, such as paints and fluorescent brightening agents, as well as for emerging applications. as typified by OLED-emitting materials.6 These dyes have large transition energies comparative to energy close to ultraviolet (UV) light, and therefore the energy of the excited state related to the fluorescence emission is relatively high. During long-term use under incident UV radiation, these classes of dyes are susceptible to photochemical decomposition due to their high-energy excited states. The major challenge in developing efficient blue-emitting dyes relies on their high photostability required for their long-term use. To overcome the stability issue, much effort has been made with regard to molecular design, including the design and arrangement of auxochromes, the substitution of heavy atoms such as sulfur and phosphorus, and the use of coordination bonds.7–9 The method of utilizing energy transfer from organic dyes to inorganic compounds was also investigated for stabilizing dyes.10,11
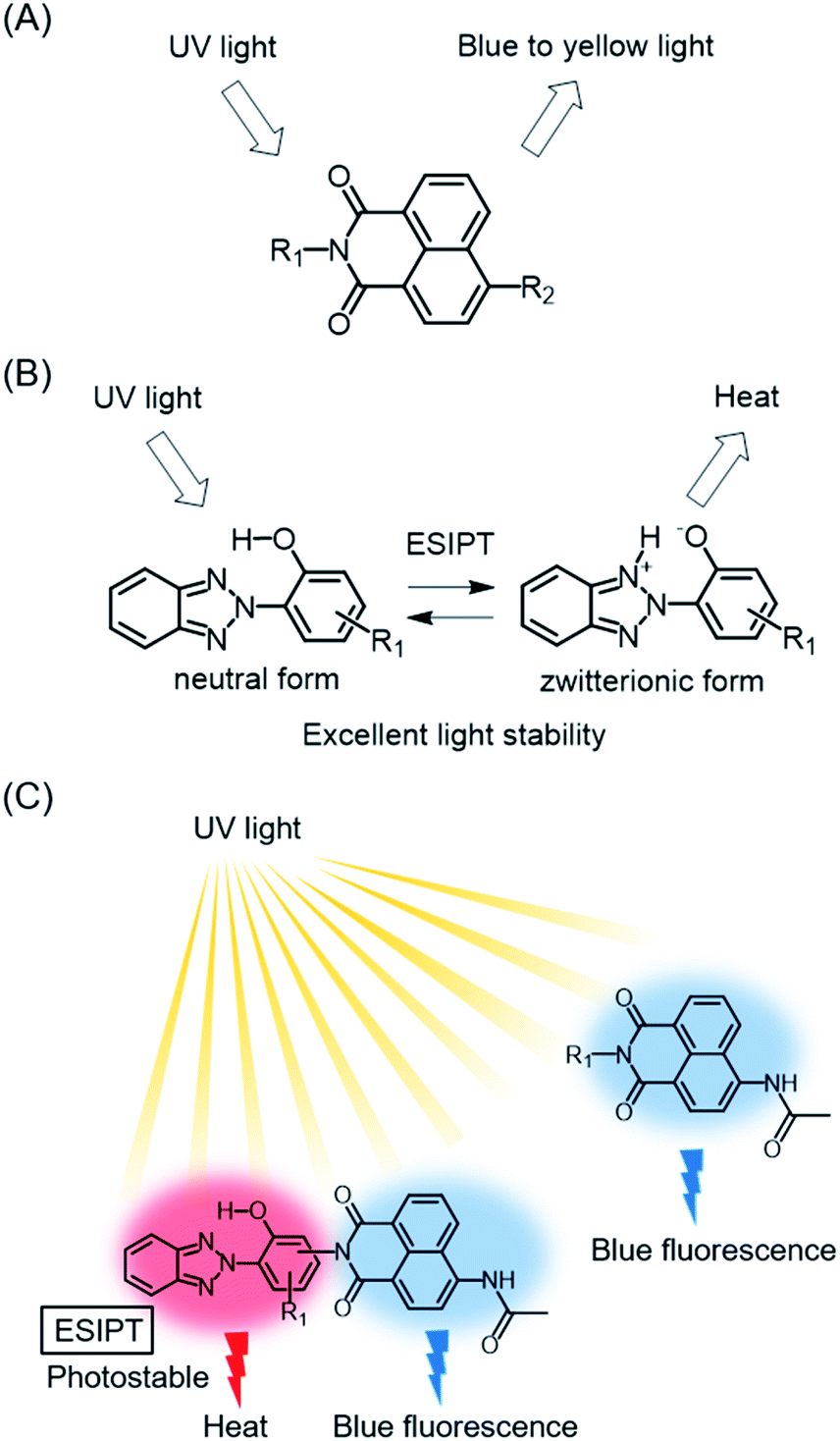
1,8-Naphthalimide derivatives are known as fluorescent dyes with strong blue to yellow fluorescence emission (Fig. 1A). The transition energy and intensity of their fluorescence depend on the substituents as well as on the external environment, and they are known as substantial blue fluorescent dyes used as sensors and bioprobes.12–15 In particular, the substituents attached to the nitrogen atom of the imide group and 4-position, as well as the polarity and pH of the solvent, affect a dye's fluorescent properties through intramolecular charge transfer (ICT) or photoinduced electron transfer (PET).16–22 4-Acetylamino-1,8-naphthalimide derivatives show blue fluorescence that is enhanced especially in polar solvents.16,23,24 However, they have poor stabilities under UV irradiation, possibly due to their wide band gaps. A general strategy to improve their light stability is needed in order to achieve a wide range of applications of these dyes, including wavelength conversion materials working under sunlight.
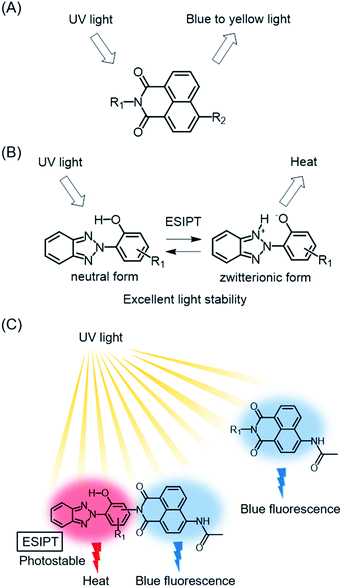 |
| | Fig. 1 (A) Fluorescent 1,8-naphthalimide dye, (B) 2-(2-hydroxyphenyl)-2H-benzotriazole UV absorber, and (C) the hybrid structure and arrangement of 1,8-naphthalimide and benzotriazole UV absorber. | |
In the field of polymer science, polymer degradation is inhibited by blocking or screening out the incident UV radiation through the addition of UV absorbers.25 2-(2-Hydroxyphenyl)-2H-benzotriazoles are widely used as UV absorbers due to their UV absorptivity and remarkable photostability. The derivatives cause excited-state intramolecular proton transfer (ESIPT) by UV irradiation, and form zwitterionic structures that promote nonradiative deactivation (Fig. 1B).26 The internal conversion after ESIPT is very fast due to the presence of a deactivation pathway through the conical intersection, making it difficult to decompose these derivatives within the short lifetimes of the excited states.27,28
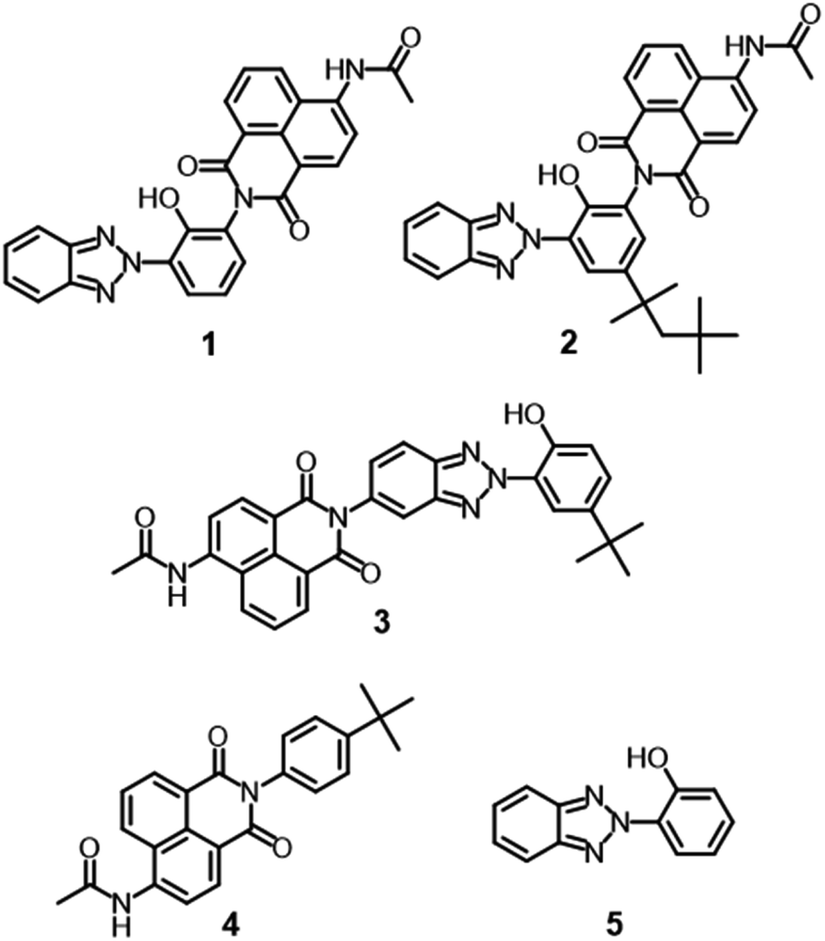
Benzotriazole-based UV absorbers preferentially absorb damaging UV radiation and dissipate it as thermal energy through nonradiative deactivation involving ESIPT. The concept of protecting and stabilizing macromolecules by using UV absorbers can be applicable to metal-free organic dyes. In this context, a hybrid molecule consisting of dyes and UV absorbers should pave the way for the development of durable fluorescent dyes. Apart from our concept, hybrid compounds of 2-(2-hydroxyphenyl)-2H-benzotriazole derivatives and 4-allyloxy-1,8-naphthalimide have been reported as fluorescent brightening agents.29 Since each hybrid compound was used as a pendant group of polymethacrylate, the light stabilities of the hybrid compounds and their detailed optical properties were not clear. For the development of durable blue fluorescent dyes that can be used as wavelength conversion materials for sunlight, it is important to elucidate the photophysical properties and photostability of benzotriazole-naphthalimide hybrids (Fig. 1C). In this study, we designed and synthesized hybrid compounds in which ESIPT-active 2-(2-hydroxyphenyl)-2H-benzotriazole derivatives were incorporated to blue fluorescent 4-acetylamino-1,8-naphthalimide, with the aim of enhancing the photostability of blue fluorescent dyes. We evaluated the effects of the benzotriazole units on the photophysical and photostability properties of dyes in comparison with constituent 4-acetylamino-1,8-naphthalimide (4) and 2-(2-hydroxyphenyl)-2H-benzotriazole (5) (Fig. 2). Hybrid compound 1, its analogue having 1,1,3,3-tetramethylbutyl groups on the 2-hydroxyphenyl unit (2), and a hybrid dye in which the naphthalimide unit was connected at the benzotriazole ring (3) were designed to examine the effects of the hybrid structure on optical properties and durability.
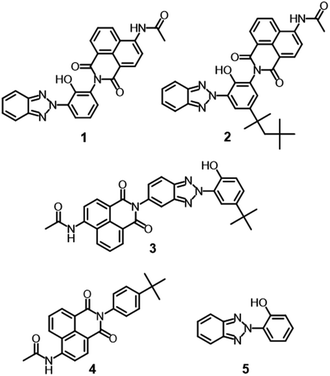 |
| | Fig. 2 Hybrid compounds in the present study. | |
Experimental
General
All starting materials, catalysts, and solvents were purchased from Tokyo Chemical Industry (Tokyo, Japan) and FUJIFILM Wako Pure Chemical (Osaka, Japan). The 1H-NMR spectra were obtained using ECA600 spectrometers (JEOL, Tokyo) operating at 600 MHz or ECX500 spectrometers (JEOL) operating at 500 MHz. The 13C-NMR spectra were obtained using ECA600 spectrometers (JEOL) operating at 150 MHz or ECX500 spectrometers (JEOL) operating at 125 MHz. DMSO-d6 was used as the solvent, and chemical shifts were based on the peak of DMSO. The electrospray ionization mass spectra (ESI-MS) were recorded on an Amazon SL spectrometer (Bruker, Billerica, MA, USA) or a JMS-T100CS spectrometer (JEOL). The elemental analyses were performed on a Yanaco CHN Corder JM-10 analyzer (Yanagimoto, Tokyo). The FT-IR spectra were recorded using a Spectrum Two FT-IR spectrometer (PerkinElmer, Waltham, MA, USA). The absorption spectra were measured in a 1.0 cm quartz cell or 4 μm PMMA film on a UV-1850 spectrophotometer (Shimadzu, Kyoto, Japan) or a U-3900H spectrophotometer (Hitachi, Tokyo). The fluorescence excitation and emission spectra, the phosphorescence spectra, and the fluorescence quantum yields were measured in a 1.0 cm quartz cell for solution at room temperature, in a 5 mm quartz cell for solution at 77 K, and in a 4 μm PMMA film on a FP-8500 spectrofluorometer (Jasco, Tokyo). The phosphorescence spectra were measured with a delay time of 10 ms to remove fluorescence and a sampling time of 40 ms. Fluorescence lifetimes were measured using a FluoroCube spectroanalyzer (Horiba Jobin Yvon, Edison, NJ, USA) with a 390 nm LED light source for excitation. A colloidal silica suspension in water was used as a scatterer to determine the instrumental response. The photostability tests were conducted using a Super Xenon Weather Meter SX75 (Suga, Tokyo) equipped with a xenon lamp in the UV region (300–400 nm) for 24 hours under conditions of 63 °C BPT, 50% humidity, and 60 W m−2 irradiation intensity. Compounds 1,30 3,31 4,31 5,32 and 2-amino-6-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetramethylbutyl)phenol (6),33 were prepared according to the literature procedure. The characterization data of 1, 3–5 and 6 were shown in ESI.†
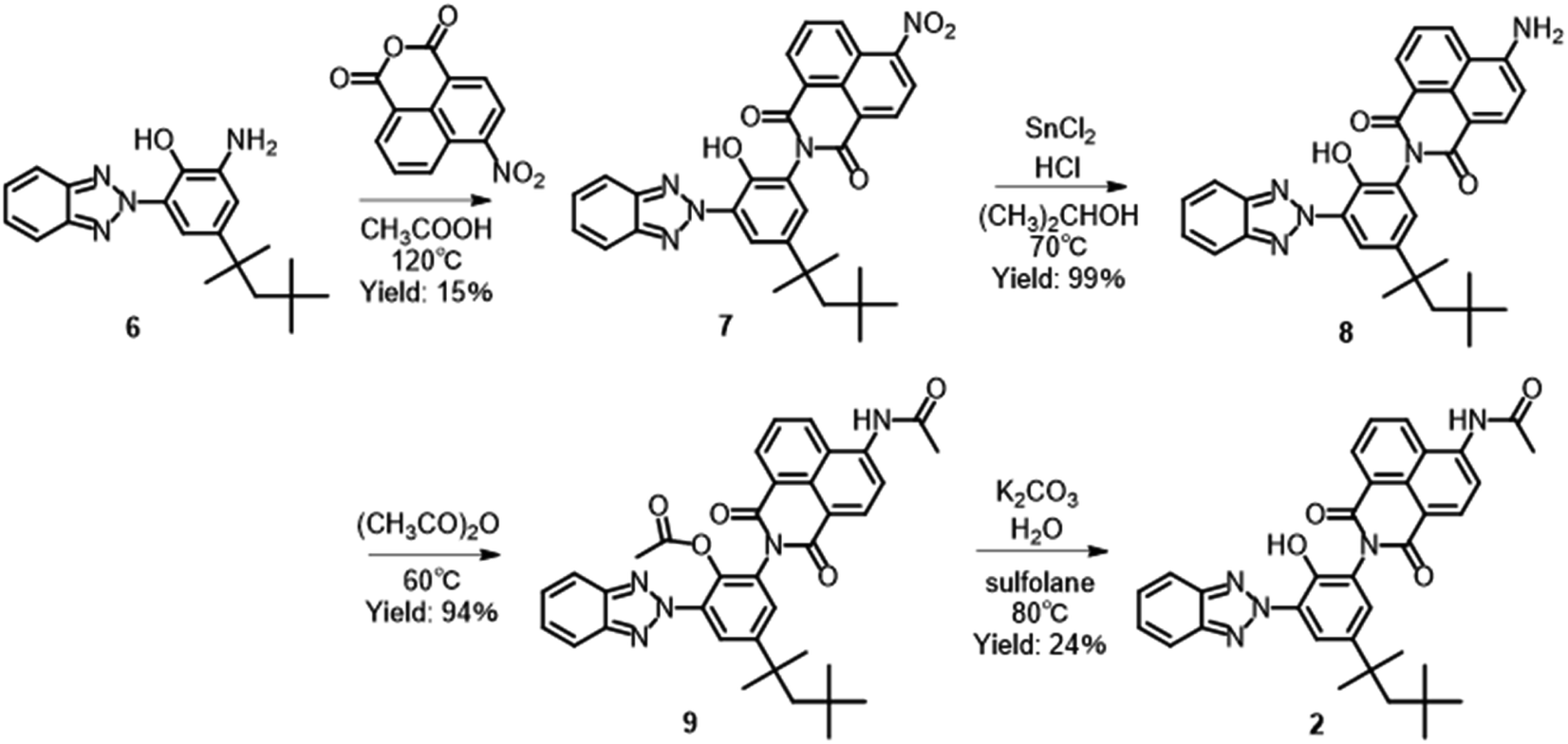
Synthesis of 7
Compound 6 (9.51 g, 0.0281 mol), 4-nitro-1,8-naphthalic anhydride (6.10 g, 0.0251 mol), and acetic acid (75 mL) were added to a 500 mL flask, and the mixture was refluxed at 120 °C for 20 h. After the addition of water (200 mL), the formed precipitate was filtered at 20 °C, washed with isopropyl alcohol and water, and dried to give the crude product of 7 (7.60 g). The crude product of 7 (7.60 g) and acetic acid (70 mL) were added to a 200 mL flask, and the mixture was refluxed at 115 °C for 1 h. The precipitate was filtered at 25 °C, washed with acetic acid, and dried to give compound 7 as a light yellow powder (2.40 g, 4.26 mmol, 15%) with a melting point of 217 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.23 (s, 1H), 8.78–8.80 (d, J = 8.5 Hz, 1H), 8.69–8.70 (d, J = 8.0 Hz, 1H), 8.66–8.68 (d, J = 8.5 Hz, 1H), 8.60–8.61 (d, J = 7.5 Hz, 1H), 8.13–8.16 (dd, J = 8.0, 8.0 Hz, 1H), 8.04–8.06 (m, 2H), 7.83–7.84 (d, J = 1.5 Hz, 1H), 7.66–7.66 (d, J = 2.5 Hz, 1H), 7.52–7.54 (m, 2H), 1.73 (s, 2H), 1.37 (s, 6H), 0.78 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 163.02, 162.24, 149.32, 145.46, 143.74 (2C), 140.81, 131.75, 130.17, 129.98, 129.64, 128.98, 128.94, 128.04, 127.38, 127.33 (2C), 124.34, 123.88, 123.64, 123.52, 122.91, 118.10 (2C), 56.29, 37.91, 32.13, 31.76 (3C), 31.19 (2C). FT-IR (ATR, cm−1): 3073, 2965, 1719, 1678, 1530, 1347, 1243, 782, 747. MS (ESI) m/z: [M–H]− calcd for C32H28N5O5 562.21; Found 561.87. Anal. calcd for C32H29N5O5: C, 68.19; H, 5.19; N, 12.43. Found: 68.25; H, 5.20; N, 12.33.
Synthesis of 8
Compound 7 (2.40 g, 4.26 mmol), isopropyl alcohol (24 mL), tin(II) chloride (4.04 g, 0.0213 mol), and hydrochloric acid (36%, 4.43 g, 0.0437 mol) were added to a 200 mL flask, and the mixture was stirred at 70 °C for 1 h. The formed precipitate was filtered at 20 °C, washed with water, and dried to give 8 as a yellow powder (2.24 g, 4.20 mmol, 99%) with a melting point of 297 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.24 (s, 1H), 8.65–8.67 (d, J = 8.5 Hz, 1H), 8.44–8.45 (d, J = 7.0 Hz, 1H), 8.20–8.22 (d, J = 8.5 Hz, 1H), 8.04–8.06 (m, 2H), 7.89 (s, 1H), 7.67–7.70 (dd, J = 7.75, 7.75 Hz, 1H), 7.53–7.55 (m, 2H), 7.48 (m, 3H), 6.87–6.89 (d, J = 8.5 Hz, 1H), 1.73 (s, 2H), 1.37 (s, 6H), 0.78 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 163.83, 162.92, 152.74, 145.01, 143.49 (2C), 140.79, 133.91, 131.01, 130.45, 130.16, 129.39, 127.48 (2C), 127.22, 125.66, 124.00, 122.57, 121.79, 119.55, 118.02 (2C), 108.17, 108.11, 56.25, 37.89, 32.14, 31.78 (3C), 31.20 (2C). FT-IR (ATR, cm−1): 3474, 3355, 3255, 2951, 1692, 1658, 1578, 1366, 1346, 1239, 749, 687. MS (ESI) m/z: [M–H]− calcd for C32H30N5O3 532.23; Found 531.77. Anal. calcd for C32H31N5O3: C, 72.03; H, 5.86; N, 13.12. Found: C, 71.82; H, 5.53; N, 12.99.
Synthesis of 2
Compound 8 (1.50 g, 2.81 mmol), acetic anhydride (40.0 g, 0.392 mol), and concentrated sulfuric acid (0.1 mL) were added to a 300 mL flask, and the mixture was stirred at 60 °C for 20 m. After the addition of water (100 mL), the formed precipitate was filtered at 25 °C, washed with water, and dried to give 9 (1.63 g, 2.64 mmol). Compound 9 (1.63 g, 2.64 mmol), potassium carbonate (2.40 g, 0.0174 mol), sulfolane (70 mL), and water (15 mL) were added to a 500 mL flask, and the mixture was stirred at 80 °C for 4 h. After the addition of water (180 mL) and acetic acid (6 mL), the formed precipitate was filtered at 25 °C, washed with methyl alcohol, and dried to give the crude product of 2 (1.86 g). After the crude product of 2 (1.86 g) was dissolved in toluene (30 mL), the mixture was filtered and the insoluble materials were removed. The solvent was removed in reduced pressure, and isopropyl alcohol (30 mL) was added. The formed precipitate was filtered at 20 °C, washed with isopropyl alcohol, and dried to give compound 2 as a yellow powder (0.37 g, 0.643 mmol, 23% from 9) with a sublimation point of 191 °C. 1H NMR (600 MHz, DMSO-d6): δ 10.46 (s, 1H), 10.26 (s, 1H), 8.76–8.77 (d, J = 8.4 Hz, 1H), 8.56–8.57 (d, J = 7.2 Hz, 1H), 8.51–8.52 (d, J = 8.4 Hz, 1H), 8.34–8.36 (d, J = 8.4 Hz, 1H), 8.04–8.06 (m, 2H), 7.91–7.94 (dd, J = 8.1, 8.1 Hz, 1H), 7.87–7.88 (d, J = 1.8 Hz, 1H), 7.60 (d, J = 1.8 Hz, 1H), 7.53–7.54 (m, 2H), 2.30 (s, 3H), 1.73 (s, 2H), 1.37 (s, 6H), 0.79 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 170.18, 164.07, 163.49, 145.75, 144.13 (2C), 141.34, 140.95, 132.12, 131.36, 130.66, 129.90, 129.55, 128.13, 127.94 (2C), 126.91, 125.32, 124.74, 123.61, 123.13, 119.92, 118.75, 118.57 (2C), 56.82, 38.44, 32.65, 32.30 (3C), 31.73 (2C), 24.64. FT-IR (ATR, cm−1): 2954, 1709, 1661, 1588, 1371, 1350, 1240, 778, 746, 688.
MS (ESI) m/z: [M–H]− calcd for C34H32N5O4 574.25; Found 573.81. Anal. calcd for C34H33N5O4: C, 70.94; H, 5.78; N, 12.17. Found: C, 70.77; H, 5.94; N, 12.24.
General method for the fabrication of PMMA films
Each compound (0.05 g) and PMMA pellet (1.95 g) were dissolved in a mixture of methyl ethyl ketone (4 g) and toluene (4 g). Each solution was coated on a glass 2 mm thick with a bar coater no. 20. After drying under reduced pressure, PMMA films with a thickness of 4 μm coated on a glass plate were fabricated.
Results and discussion
Synthesis
Compound 2 was synthesized according to Scheme 1. Compound 6 was synthesized by nitration of 2-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetramethylbutyl)phenol, followed by reduction of the nitro group.33 The imidization of 6 with 4-nitro-1,8-naphthalic anhydride gave corresponding 1,8-naphthalimide derivative 7, which was reduced with tin(II) chloride and hydrochloric acid to give amino-substituted 8.34,35 The acetylation of 8 with acetic anhydride followed by hydrolysis of the acetoxy group afforded 2 in 3% yield from 6. The yield of imidization was low at 15% due to the production of N-[3-(2H-benzotriazol-2-yl)-2-hydroxy-5-(1,1,3,3-tetramethyl butyl)phenyl]acetamide as a by-product. The by-product can be recovered and hydrolyzed to obtain 6, that can be used again for the synthesis. Although the number of synthesis processes is increased, it is possible to scale up the synthesis and collect a large amount of the dye. Similarly, compound 1 was prepared from an analogue of 6 without 1,1,3,3-tetramethylbutyl groups.30 A previously reported method was used to successfully synthesize 3.31 Characterization data for 1, 3, 4, and 5 are shown in ESI.†
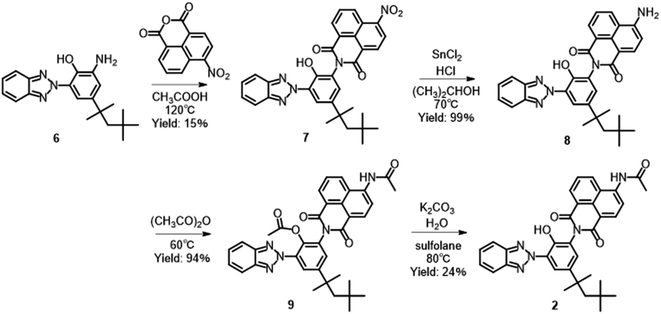 |
| | Scheme 1 Synthesis of compound 2. | |
Electronic absorption and fluorescence properties in solution
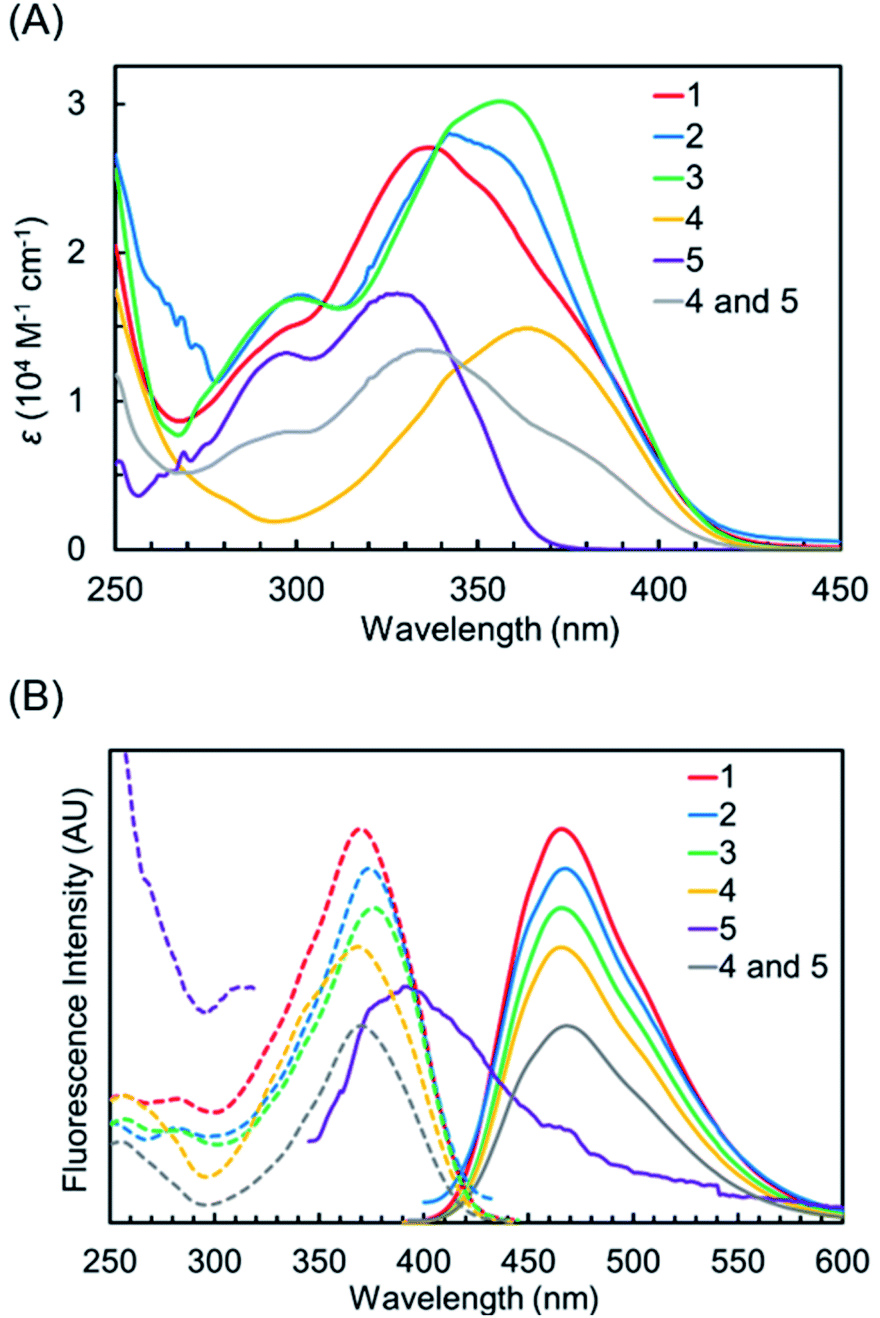
The UV-vis absorption spectra of 1–5 and the equimolar mixture of 4 and 5 in CH3OH are shown in Fig. 3A, and the absorption properties are shown in Table 1. Generally, 2-(2- hydroxyphenyl)-2H-benzotriazole derivatives having intramolecular N–H–O hydrogen bonds between their benzotriazole and phenol groups showed strong absorption at 330–360 nm and weak absorption around 300 nm, which originated from nonhydrogen bonding species.26,36 Benzotriazole derivative 5 showed strong absorption, with a maximum at 328 nm and weak absorption around 300 nm. The electronic absorption of 1,8-naphthalimide derivative 4 was observed in a lower energy region compared to 5. Hybrid compounds 1–3 showed broad absorption bands in the UV region, and their spectral shapes differed slightly from each other. The absorption maxima of 1, 2, and 3 were observed at 337, 342, and 356 nm. The absorption band of 2 was slightly longer than that of 1 due to the electron-donation property of the 1,1,3,3-tetramethylbutyl group on the phenol unit 3, having a different manner of linkage between benzotriazole and naphthalimide components, exhibited absorption in a lower energy region compared to 1 and 2. Thus, both components of hybrid dyes interact with each other to show slightly distinct electronic absorptions. We also examined their absorption properties in CHCl3 and DMSO (Table S1, Fig. S1†). Although the UV absorptions of 1–3 and 5 were hypsochromically shifted in DMSO by breaking down the intramolecular hydrogen bond, their absorptions in CHCl3 were similar to those in CH3OH. For constituent naphthalimide 4, the distinguished solvent effect was not observed.
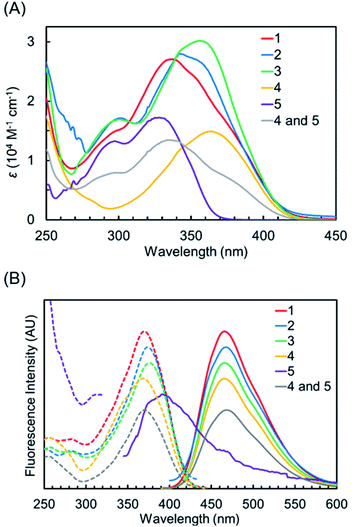 |
| | Fig. 3 (A) UV-vis absorption spectra of 1–5 and the equimolar mixture of 4 and 5 in CH3OH. (B) Fluorescence (solid-line) and excitation (dot-line) spectra of 1–5 and the equimolar mixture of 4 and 5 in CH3OH. | |
Table 1 Optical properties of 1–5 and the equimolar mixture of 4 and 5 in CH3OHa
| Dye |
λabs (nm) |
ε (104 M−1 cm−1) |
λF (nm) |
λEX (nm) |
Stokes shift (103 cm−1) |
ΦF (%) |
| Conc. 20 μM. Conc. 30 μM. Conc. 50 μM. |
| 1 |
337 |
2.70 |
466 |
370 |
5.57 |
56.2 |
| 2 |
342 |
2.80 |
468 |
373 |
5.44 |
7.5 |
| 3 |
356 |
2.02 |
466 |
376 |
5.14 |
41.7 |
| 4b |
364 |
1.49 |
465 |
368 |
5.67 |
82.6 |
| 5c |
328 |
1.73 |
392 |
313 |
6.44 |
0.8 |
| 4 + 5b |
335 |
— |
468 |
370 |
5.66 |
87.1 |
The fluorescence emission and excitation spectra of 1–5 and the equimolar mixture of 4 and 5 in CH3OH are shown in Fig. 3B, and the results are summarized in Table 1. The constituent benzotriazole 5 showed almost no emission due to dominant nonradiative deactivation from the excited state formed after ESIPT.37 In contrast, naphthalimide 4 exhibited fluorescence emission with a maximum at 465 nm and with a large Stokes shift (5.67 × 103). The fluorescence quantum yield of 4 is observed to be high (ΦF = 0.83). The spectral shape of the emission spectrum is a mirror image of its excitation spectrum, in agreement with previous reports.38–40 Hybrid compounds 1–3 exhibited fluorescence emission, and the maxima of their excitation spectra were observed at almost the same range (370 nm for 1, 373 nm for 2, 376 nm for 3, and 368 nm for 4). This indicated that the fluorescence of 1–3 originated from naphthalimide fluorophores. It should be noted that the quantum yields of 1–3 were lower than that of 4. The order of ΦF is 4 > 1 (ΦF = 0.56) > 3 (ΦF = 0.42) > 2 (ΦF = 0.08). This clearly indicated that the benzotriazole components in 1–3 reduced the fluorescence quantum yields of 1,8-naphthalimide fluorophores, although their emissions were not completely quenched. The quantum yield of the simple mixture of 4 and 5 (ΦF = 0.87) is slightly larger than that of 4. The fluorescence of 4 was possibly enhanced by the fluorescence resonance energy transfer from 5 to 4, since weak fluorescence of 5 was overlapped with absorbance of 4.
The fluorescence emissions of 1–3 and 4 shifted hypsochromically with decreasing polarity of the solvents, consistent with previous studies regarding 1,8-naphthalimide derivatives (Table S1, Fig. S2†). The solvatochromic behavior indicated their polar characteristics in the ground state.41 To learn the excited state kinetics of 1–3, we measured the fluorescence lifetime by the time-correlated single-photon counting (TCSPC) method. Then, we calculated the radiative deactivation rate (kr) and the nonradiative deactivation rate (knr) according to the following equations:
where
ΦF and
τ are the fluorescence quantum yield and the lifetime, respectively (Table S1
†). Although the
knr values of
1 and
3 were higher in CHCl
3 and DMSO than in CH
3OH, the
knr values of
2 were higher in CH
3OH than in CHCl
3. The effect of the solvent on the
knr of compound
2 is not well understood at this time. The 1,8-naphthalimide
4 displayed the same tendency. The analysis indicated that the nonradiative deactivation for 1,8-napthalimide components was dominant in CHCl
3 and DMSO. The
knr values of
1–3 are much larger than that of
4. Notably,
2 has a large
knr value in any solvent. Thus, the decreased fluorescence emission of
1–3 appears to stem from the promotion of nonradiative deactivation.
Phosphorescence properties in degassed toluene at 77 K
To gain insight into the deactivation pathway of 1–3, we examined their emission properties in low-temperature conditions. The phosphorescence spectra in degassed toluene at 77 K are shown in Fig. 4, prompt fluorescence spectra are shown in Fig. S3,† phosphorescence decay curves are shown in Fig. S4,† and the phosphorescence and fluorescence properties are shown in Table 2. In the measurement condition for phosphorescence (delay time: 10 ms), the parent 1,8-naphthalimide 4 exhibited bimodal emission with maxima at 595 nm and 460 nm. The shape and transition energy of the emission at 460 nm were consistent with its prompt fluorescence (Fig. S3†). This indicated that the emissions at 595 nm and 460 nm are attributable to phosphorescence and delayed fluorescence emission. In contrast, the benzotriazole 5 shows quite weak phosphorescence emission in the higher energy region compared to 4 (4: λp = 595 nm; 5: λp = 567 nm). As shown in Fig. 4, the hybrid dyes 1–3 showed phosphorescence emission together with negligible delayed fluorescence (1: λp = 577 nm; 2: λp = 577 nm; 3: λp = 589 nm), suggesting that 1–3 have a possible deactivation pathway via their triplet excited states formed through intersystem crossing. The phosphorescence intensities of 1–3 and 4 in the same measurement condition increase in the order 4 < 1 < 3 < 2, and these phosphorescence lifetimes are in the order of milliseconds. In light of these phosphorescence intensities, the deactivation via triplet states was promoted in hybrid dyes 1–3. Moreover, the intensity ratios of phosphorescence and delayed fluorescence for 1–3 are much lower than that of 4, suggesting that the reverse intersystem crossing was suppressed in 1–3. Thus, the deactivation pathway of 1–3 was distinct from that of the parent 1,8-naphthalimide 4.
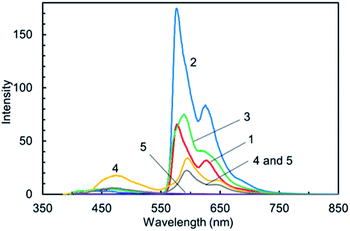 |
| | Fig. 4 Phosphorescence spectra showing relative intensity of 1–5 and the equimolar mixture of 4 and 5 in toluene at 77 K. | |
Table 2 Phosphorescence and fluorescence properties of 1–5 and the equimolar mixture of 4 and 5 in toluene at 77 K
| Compd |
λPa (nm) |
λF (nm) |
λEX (nm) |
Stokes shift (103 cm−1) |
| λP |
λF |
| Measured with delay time: 10 ms. Conc. 20 μM. Conc. 30 μM. Conc. 50 μM. |
| 1b |
577 |
437 |
374 |
9.41 |
3.86 |
| 2b |
577 |
438 |
377 |
9.19 |
3.69 |
| 3b |
589 |
438 |
377 |
9.55 |
3.69 |
| 4c |
595 |
457 |
351 |
11.7 |
6.61 |
| 5d |
567 |
410 |
376 |
8.96 |
2.21 |
| 4 + 5c |
594 |
446 |
377 |
9.69 |
4.10 |
Theoretical calculations
To understand the structural and electronic features of hybrid dyes 1–3 in the ground state (S0), quantum chemical calculations were carried out using density functional theory (DFT) at the CAM-B3LYP/6-31G+(d) level using a polarized continuum model (PCM) of CH3OH to take into account the potential solvent effect. The geometry-optimized structures and the geometrical parameters of the linkage between the two components are shown in Fig. 5 and Table S2.† In hybrid dyes, 1,8-naphthalimide and 2-(2-hydroxyphenyl)-2H-benzotriazole are not placed on the same plane but rather are twisted. The dihedral angles (φ) between the two components are 87.4° for 1, 87.6° for 2, and 87.3° for 3. In contrast, the benzotriazole units are coplanar with the 2-hydroxyphenyl units, forming N–H–O intramolecular hydrogen bonding that may facilitate ESIPT. The bond lengths of the linkage of two components are in the range of the C–N single bond (1.44 Å). The frontier molecular orbitals (FMOs) of their ground states are illustrated in Fig. 6. For 1, both HOMO and LUMO were distributed over the naphthalimide skeletons, whereas the HOMO−1 and LUMO+1 were located on the benzotriazole skeletons. On the other hand, 2 had distinct distributions of FMOs, where the HOMO and HOMO−1 were positioned on both naphthalimide and benzotriazole units. The energy gap between HOMO and HOMO−1 in 2 (0.05 eV) is smaller than that in 1 (0.17 eV). Thus, the alkyl substitution on the 2-hydroxyphenyl unit in 2 caused the elevation of the HOMO−1 energy, and two orbitals may be hybridized. In the case of 3, the HOMO−1 and LUMO were distributed over the naphthalimide skeleton whereas the HOMO and LUMO+1 were distributed over the benzotriazole skeleton. The distributions of HOMO and HOMO-1 might be opposite to 1 due to the addition of 1,8-naphthalimide to the benzotriazole group. Moreover, we analyzed the electronic transitions of 1–3 by time-dependent DFT at the CAM-B3LYP/6-31G+(d) using a PCM of CH3OH. The results of the calculated electronic transition are summarized in Table 3. The S0–S1 transition of 1 and 3 consisted solely of HOMO → LUMO and HOMO−1 → LUMO transitions, respectively. These MOs were distributed over the naphthalimide skeletons; thus the S0–S1 transition can be assigned to π–π* transitions at the naphthalimide units. In 2, two components, HOMO−1 → LUMO (41%) and HOMO → LUMO (55%), contributed to the S0–S1 transition. This indicated that the transition is assigned not to the simple π–π* transition at naphthalimides but rather to the transition having an intramolecular charge transfer feature.
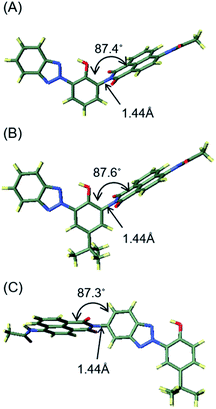 |
| | Fig. 5 Optimized structures of S0 for 1 (A), 2 (B), and 3 (C) calculated at the CAM-B3LYP/6-31G+(d) (solvent: CH3OH). The calculations of 2 were performed on the models in which 1,1,3,3-tetramethylbutyl groups were replaced with tert-butyl groups. | |
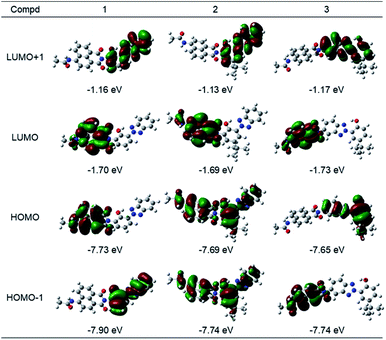 |
| | Fig. 6 Schematic diagram of the frontier molecular orbitals of optimized structures for S0 of 1–3 calculated at the CAM-B3LYP/6-31G+(d) (solvent: CH3OH). The calculations of 2 were performed on the models in which 1,1,3,3-tetramethylbutyl groups were replaced with tert-butyl groups. | |
Table 3 Calculated lowest excited energies (E), oscillator strengths (f), and composition in terms of molecular orbital contributions of optimized structures for S0, neutral S1, and zwitterionic S1 of 1–3 calculated at the CAM-B3LYP/6-31G+(d) (solvent: CH3OH). The calculations of 2 were performed on the models in which 1,1,3,3-tetramethylbutyl groups were replaced with tert-butyl groups
| Compd |
Optimized structures |
na |
E (eV) |
f |
Compositionb |
| n is the ordering number of the calculated excited state. H = HOMO, L = LUMO. |
| 1 |
S0 |
1 |
3.62 |
0.64 |
H → L (95%) |
| |
S0 |
2 |
4.15 |
0.68 |
H-1 → L+1 (89%), H → L+1 (2%) |
| |
Neutral S1 |
1 |
2.98 |
0.69 |
H → L (98%) |
| |
Zwitterionic S1 |
1 |
2.03 |
0.45 |
H → L (97%) |
| 2 |
S0 |
1 |
3.62 |
0.66 |
H-1 → L (41%), H → L (55%) |
| |
S0 |
2 |
4.07 |
0.62 |
H−1 → L+1 (46%), H → L+1 (44%) |
| |
Neutral S1 |
1 |
2.98 |
0.70 |
H → L (98%) |
| |
Zwitterionic S1 |
1 |
1.87 |
0.39 |
H → L (97%) |
| 3 |
S0 |
1 |
3.62 |
0.73 |
H−1 → L (94%) |
| |
S0 |
2 |
4.01 |
0.53 |
H → L+1 (88%), H → L+4 (3%) |
| |
Neutral S1 |
1 |
2.97 |
0.72 |
H → L (98%) |
| |
Zwitterionic S1 |
1 |
1.70 |
0.31 |
H → L (97%) |
As described above, the fluorescence quantum yields of hybrid dyes 1–3 are lower than that of the 1,8-naphthalimide derivative 4. We attribute this to the fluorescence quenching found in 1–3. In the 1,8-naphthalimide dyes, fluorescence was influenced by peripheral substituents through the fluorescence resonance energy transfer (FRET) and photoinduced electron transfer (PeT).42,43 In the former case, the spectral overlap between the emission and the absorbance, which is a fundamental requirement of FRET, was negligible in 1–3, and thus quenching by FRET was excluded as a cause of their decreased fluorescence. In the latter case, the energy level of frontier molecular orbitals is the determining factor. DFT calculations indicated that the PeT from fluorescent naphthalimide to nonfluorescent benzotriazole components in the excited state was thermodynamically unfavorable because the LUMO was located on naphthalimides and LUMO+1 was located on benzotriazoles in 1–3. Therefore, the FRET and PeT should be ruled out as factors in the decrease of the fluorescence intensities of 1–3. For further insight into the quenching mechanism in 1–3, we characterized the ESIPT in 1–3 by the computational analysis of first excited state geometries. We optimized S1 state structures having neutral forms and zwitterionic forms obtained by ESIPT (Table S2, Fig. S5†). Although the dihedral angles between benzotriazole and naphthalimide components were slightly changed in S1 zwitterionic forms, no significant conformational changes were observed for the excited states of 1–3. The FMOs in neutral S1 and zwitterionic S1 forms are illustrated in Fig. 7. For all hybrid dyes, HOMOs and LUMOs are distributed over the naphthalimide components in neutral forms of S1 states. It is noteworthy that these distributions were switched in the S1 zwitterionic structures, where HOMOs and LUMOs were placed on the nonfluorescent benzotriazole components. This suggested the possibility that hybrid dyes cause nonradiative deactivation from the zwitterionic forms due to the contribution of benzotriazole components. Then, to confirm the possibility of ESIPT in these dyes, the potential energy curves were represented by changing the N-H bond length by 0.075 Å for 12 steps using TD-CAM-B3LYP/6-31G+(d) with PCM (CH3OH) in the S1 state and CAM-B3LYP/6-31G+(d) with PCM (CH3OH) in the S0 state (Fig. 8A–C).44–46 Table S2† shows the geometrical parameters and total energies of the optimized structures of the S0, the neutral S1, and the zwitterionic S1 of 1–3. As shown in Fig. 8, the S0 states of 1–3 were destabilized along with the decrease in the N-H bond length. In the excited state, however, the energies of S1 were highest at 1.25 Å of the N-H bond lengths and had local minimum values at 1.05 Å of the N–H bond length, indicating that zwitterionic structures formed by proton transfer are stable and thus that the ESIPT is thermodynamically allowed for 1–3. From these calculated results, we suggested the scenario of the fluorescence quenching found in 1–3 (Fig. 8D). After the photoexcitation, ESIPT occurred and then formed thermodynamically allowed zwitterionic forms in which nonradiative deactivation was dominant due to the contribution of benzotriazole components. Additionally, the deactivation pathway involved in the triplet states, which are indicated by phosphorescence spectra, should also be taken into account. Thus, the incorporation of benzotriazole components on the naphthalimide fluorophore has an impact on the components' photophysical properties and leads to the reduction of fluorescence emissions.
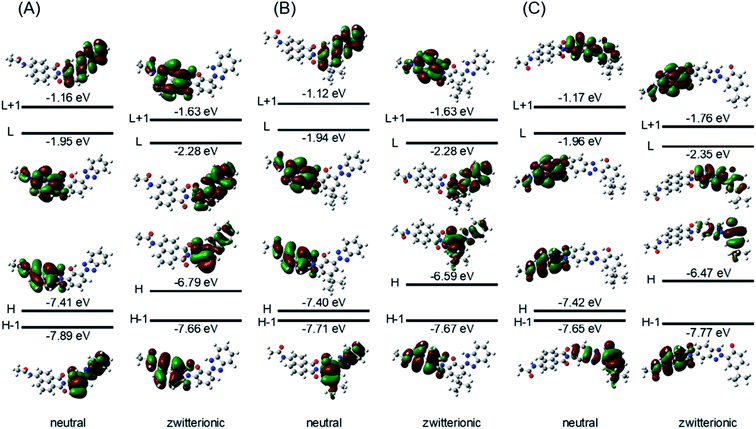 |
| | Fig. 7 Schematic diagram of the frontier molecular orbitals of optimized structures for neutral S1 and zwitterionic S1 of 1 (A), 2 (B), and 3 (C) calculated at the CAM-B3LYP/6-31G+(d) (solvent: CH3OH). The calculations of 2 were performed on the models in which 1,1,3,3-tetramethylbutyl groups were replaced with tert-butyl groups. H means HOMO and L means LUMO. | |
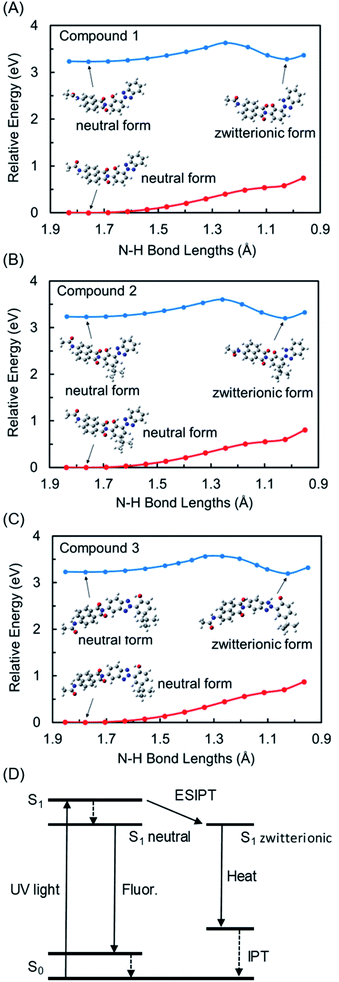 |
| | Fig. 8 Optimized structures and potential energy curves of S0 (red) and S1 (blue) for 1 (A), 2 (B), and 3 (C) calculated at the CAM-B3LYP/6-31G+(d) (solvent: CH3OH). The calculations of 2 were performed on the models in which 1,1,3,3-tetramethylbutyl groups were replaced with tert-butyl groups. (D) Assumed energy diagram for 1–3. | |
Photostability of polymer films doped with hybrid dyes
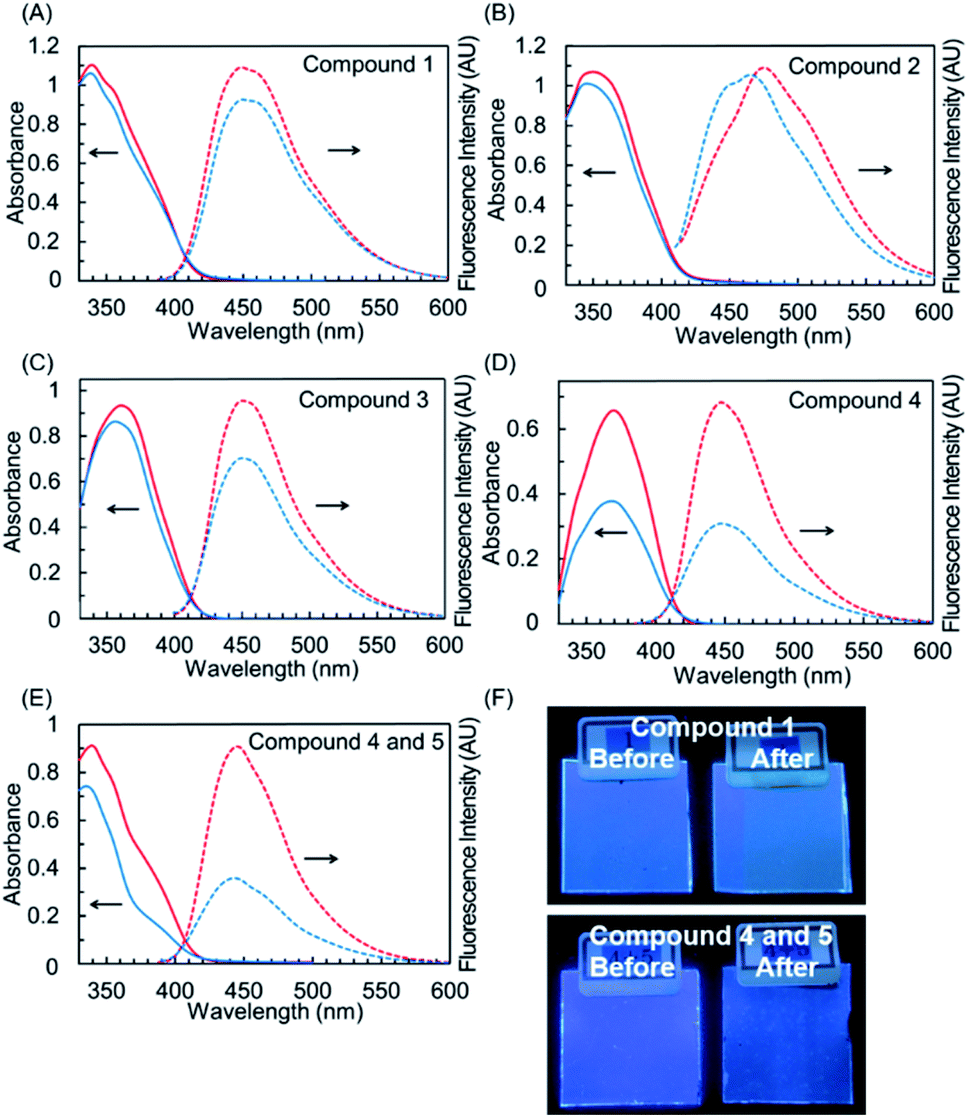
To characterize the wavelength conversion properties as well as their photostable features of hybrid dyes, we fabricated PMMA films doped with hybrid dyes 1–3. PMMA films with 4, 5, and an equimolar mixture of 4 and 5 were also fabricated for comparison. The normalized absorption and fluorescence spectra of these films are shown in Fig. 9A–E, and the results are summarized in Tables 4 and S3.† These films showed UV absorptions comparable to their absorptions in solution, indicating that the dyes were not aggregated in PMMA. Films doped with 1–3 emitted in the blue region, and their fluorescence quantum yields (ΦF) were 0.33 for 1, 0.18 for 2, and 0.20 for 3. Similar to the case with quantum yields in solution, the fluorescence quantum yields were lower than that of the parent naphthalimide 4 (ΦF = 0.51). The PMMA films with 1–3 could convert UV light to visible light, although the quantum yields were impaired, probably due to the nonradiative deactivation stemming from the benzotriazole components found in the photophysical study in solution. The fluorescence property of all PMMA films doped with the dyes was maintained under natural light over a period of over a year. We further examined these films from the aspect of durability against photoaging. Photostability was evaluated by the accelerated photoaging condition, which is comparable to 6 days of exposure to sunlight (irradiance: 60 W m−2 at 300 to 400 nm; temperature: 60 °C at black panel temp.; humidity: 50%; time: 24 h; integrated irradiance: 5.18 MJ m−2). The film doped with 4 showed a significant drop in absorbance (−43% at λmax) after the photoaging, indicating severe degradation of 4 in this condition.
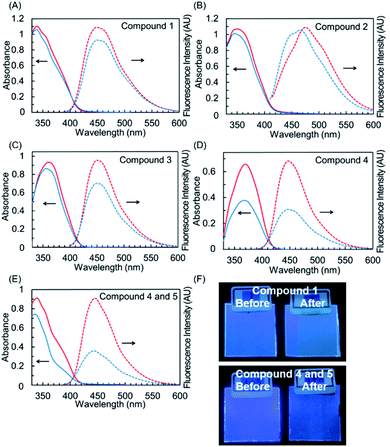 |
| | Fig. 9 UV-vis absorption (solid-line) and fluorescence (dot-line) spectra of 1 (A), 2 (B), 3 (C), 4 (D), and the equimolar mixture of 4 and 5 (E) in PMMA matrix before simulated solar light irradiation (red) and after simulated solar light irradiation (blue). (F) The photographs under UV light (365 nm) of PMMA films doped with 1 (upper) and the equimolar mixture of 4 and 5 (lower) before and after simulated solar light irradiation. | |
Table 4 Photostability test for PMMA films doped with 1–5 and the equimolar mixture of 4 and 5a
| Compd |
λabs (nm) |
Absorbance before irradiation |
Absorbance after irradiation |
Decrease (%) |
| PMMA films were irradiated with simulated solar light for 24 h in a super xenon weather meter under conditions of 63 °C BPT, 50% humidity, and 60W m−2 irradiation intensity. |
| 1 |
340 |
1.103 |
1.055 |
4.3 |
| 2 |
349 |
1.070 |
1.007 |
5.9 |
| 3 |
361 |
0.935 |
0.853 |
8.8 |
| 4 |
370 |
0.657 |
0.377 |
42.7 |
| 5 |
333 |
1.958 |
1.935 |
1.1 |
| 4 and 5 |
339 |
0.912 |
0.730 |
20.0 |
In the film including the equimolar mixture of 4 and benzotriazole 5, the decrease in absorbance after aging was suppressed (−20%) in comparison to that of the 4-doped film. This indicated that the degradation of 4 was retarded through the shielding of UV light by the agency of 5 with photostability provided by ultrafast nonradiative deactivation involving ESIPT. In contrast to the PMMA films doped with 4 and 5, films 1–3 showed no drop in absorbance. They almost maintained absorbance after aging, with quite small decreases (4% for 1, 6% for 2, 9% for 3). Thus, hybrid dyes 1–3 have high photostability in PMMA matrix. Considering the 20% decrease in absorbance in the film with the equimolar mixture of 4 and 5, the photostabilities of 1–3 did not originate only from the simple UV shielding effect enhanced by the benzotriazole components. As seen in the study of photophysical properties in solution, the deactivation pathways of the hybrid dyes are different from that of 4 or 5. Their inherent properties in the excited state should affect the photochemical reaction under continued irradiation with UV light. After the harsh photo aging, the polymer films with 1–3 maintained blue emission, as shown in the pictures of films under UV light (Fig. 9F, S6†). These results indicated the potential of these films for application to wavelength converters for sunlight.
Conclusion
To show the possibility and limitations of the concept of protecting and stabilizing blue fluorescent dyes by utilizing UV absorbers, we synthesized hybrid compounds in which ESIPT-active 2-(2-hydroxyphenyl)-2H-benzotriazole derivatives were incorporated into blue fluorescent 4-acetylamino-1,8-naphthalimide (1–3). Both components in hybrid dyes interacted with one another and showed electronic absorptions slightly different from those of constituent 1,8-naphthalimide derivative 4 and benzotriazole derivative 5. The hybrid dyes 1–3 displayed fluorescence emissions in the blue region, but their fluorescence quantum yields were lower than that of constituent 1,8-naphthalimide 4. Moreover, their phosphorescence emissions in toluene at 77 K were stronger than that of 4. Thus, the photophysical properties of 1–3 were not consistent with the simple mixing of constituent 1,8-naphthalimide 4 and benzotriazole 5. From the DFT calculations, the decrease in fluorescence found in the hybrid dyes may have been involved in the ESIPT of 2-(2-hydroxyphenyl)-2H-benzotriazole components. Although the incorporation of benzotriazole components promoted the nonradiative deactivation, the PMMA films doped with hybrid dyes emitted blue in moderate strength. The hybrid dye-doped blue-emitting films showed higher photostability under the harsh photoaging condition, in which the film doped with the simple mixture of 4 and 5 showed severe decomposition of the fluorophore. The high durability against photoaging should come from their photophysical properties, including dominant nonradiative deactivation provided by benzotriazoles. Thus, the photostability of hybrid dyes can be enhanced substantially by sacrificing fluorescence emission. The hybrid dye 1, its analogue bearing alkyl substituents 2, and the analogue with a different arrangement of the two components 3 displayed fluorescence with respective quantum efficiency as well as respective photostability. These results suggested that the balance between fluorescence intensity and photostability in polymer matrices can be tuned by modifying the structure of the hybrid dye. The hybridization of fluorescent dyes with UV absorbers can expand the method of stabilizing fluorophores and pave the way for the development of wavelength conversion materials for sunlight. These photostable hybrid compounds can be prepared by simple organic reactions without rare-earth metals as well as heavy metals, therefore they can be prepared in low cost and have low toxicity. Due to their low cost production and low toxicity, we believe they are suitable for use as wavelength conversion materials especially in the agricultural sector.
Conflicts of interest
There are no conflicts to declare.
References
- H. Kataoka, T. Kitano, T. Takizawa, Y. Hirai, T. Nakanishi and Y. Hasegawa, Photo- and thermo-stable luminescent beads composed of Eu(III) complexes and PMMA for enhancement of silicon solar cell efficiency, J. Alloys Compd., 2014, 601, 293–297 CrossRef CAS.
- T. Jin, S. Inoue, K. Machida and G. Adachi, Photovoltaic cell characteristics of hybrid silicon devices with lanthanide complex phosphor-coating film, J. Electrochem. Soc., 1997, 144, 4054–4058 CrossRef CAS.
- H. Nakamura, Y. Shirakawa, H. Kitamura, N. Sato, O. Shinji, K. Saito and S. Takahashi, Mechanism of wavelength conversion in polystyrene doped with benzoxanthene: emergence of a complex, Sci. Rep., 2013, 3, 2502 CrossRef PubMed.
- I. Vass, E. Turcsányi, E. Touloupakis, D. Ghanotakis and V. Petrouleas, The mechanism of UV-A radiation-induced inhibition of photosystem II electron transport studied by EPR and chlorophyll fluorescence, Biochemistry, 2002, 41, 10200–10208 CrossRef CAS PubMed.
- R. V. Grondelle and E. Boeker, Limits on natural photosynthesis, J. Phys. Chem. B, 2017, 121, 7229–7234 CrossRef PubMed.
- J. Tagare and S. Vaidyanathan, Recent development of phenanthroimidazole-based fluorophores for blue organic light-emitting diodes (OLEDs): an overview, J. Mater. Chem. C, 2018, 6, 10138–10173 RSC.
- M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguchi, A highly photostable near-infrared labeling agent based on a phospha-rhodamine for long-term and deep imaging, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS PubMed.
- N. Ando, H. Soutome and S. Yamaguchi, Near-infrared fluorescein dyes containing a tricoordinate boron atom, Chem. Sci., 2019, 10, 7816–7821 RSC.
- S. Zhang, J. Fan, Z. Li, N. Hao, J. Cao, T. Wu, J. Wang and X. Peng, A bright red fluorescent cyanine dye for live-cell nucleic acid imaging, with high photostability and a large Stokes shift, J. Mater. Chem. B, 2014, 2, 2688–2693 RSC.
- G. Yeroslavsky, M. Umezawa, K. Okubo, K. Nigoghossian, D. T. K. Dung, K. Miyata, M. Kamimura and K. Soga, Stabilization of indocyanine green dye in polymeric micelles for NIR-II fluorescence imaging and cancer treatment, Biomater. Sci., 2020, 8, 2245–2254 RSC.
- J. Li, J. Lin, Y. Huang, X. Xu, Z. Liu, Y. Xue, X. Ding, H. Luo, P. Jin, J. Zhang, J. Zou and C. Tang, Organic Fluorescent Dyes Supported on Activated Boron Nitride: A Promising Blue Light Excited Phosphors for High-Performance White Light-Emitting Diodes, Sci. Rep., 2015, 5, 8492 CrossRef CAS PubMed.
- K. N. Hearn, T. D. Nalder, R. P. Cox, H. D. Maynard, T. D. M. Bell, F. M. Pfeffer and T. D. Ashton, Modular synthesis of 4-aminocarbonyl substituted 1,8-naphthalimides and application in single molecule fluorescence detection, Chem. Commun., 2017, 53, 12298–12301 RSC.
- M. V. Refalo, J. C. Spiteri and D. C. Magri, Covalent attachment of a fluorescent ‘Pourbaix sensor’ onto a polymer bead for sensing in water, New J. Chem., 2018, 42, 16474–16477 RSC.
- E. B. Veale, G. M. Tocci, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Demonstration of bidirectional photoinduced electron transfer (PET) sensing in 4-amino-1,8-naphthalimide based thiourea anion sensors, Org. Biomol. Chem., 2009, 7, 3447–3454 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Recent advances
in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- P. A. Panchenko, Y. V. Fedorov, O. A. Fedorova, V. P. Perevalov and G. Jonusauskas, Synthesis and spectral properties of 4-amino- and 4-acetylamino-N-arylnaphthalimides containing electron-donating groups in the N-aryl substituent, Russ. Chem. Bull., Int. Ed., 2009, 58, 1233–1240 CrossRef CAS.
- I. Grabchev, S. Sali, R. Betcheva and V. Gregoriou, New green fluorescent polymer sensors for metal cations and protons, Eur. Polym. J., 2007, 43, 4297–4305 CrossRef CAS.
- J. Gan, K. Chen, C.-P. Chang and H. Tian, Luminescent properties and photo-induced electron transfer of naphthalimides with piperazine substituent, Dyes Pigm., 2003, 57, 21–28 CrossRef CAS.
- E. Martín, J. L. G. Coronado, J. J. Camacho and A. Pardo, Experimental and theoretical study of the intramolecular charge transfer on the derivatives 4-methoxy and 4-acetamide 1,8-naphthalimide N-substituted, J. Photochem. Photobiol., A, 2005, 175, 1–7 CrossRef.
- A. Manna and S. Chakravorti, Charge transfer in 1,8-naphthalimide: A combined theoretical and experimental approach, Photochem. Photobiol., 2010, 86, 47–54 CrossRef CAS PubMed.
- A. Pardo, E. Martin, J. M. L. Poyato, J. J. Camacho, J. M. Guerra, R. Weigand, M. F. Braña and J. M. Castellano, N-substituted 1,8-naphthalimide derivatives as high efficiency laser dyes, J. Photochem. Photobiol., A, 1989, 48, 259–263 CrossRef CAS.
- S. Chatterjee, S. Pramanik, S. U. Hossain, S. Bhattacharya and S. C. Bhattacharya, Synthesis and photoinduced intramolecular charge transfer of N-substituted 1,8-naphthalimide derivatives in homogeneous solvents and in presence of reduced glutathione, J. Photochem. Photobiol., A, 2007, 187, 64–71 CrossRef CAS.
- R. J. Hodgkiss, G. W. Jones, A. Long, R. W. Middleton, J. Parrick, M. R. L. Stratford, P. Wardman and G. D. Wilson, Fluorescent markers for hypoxic cells: A study of nitroaromatic compounds, with fluorescent heterocyclic side chains, that undergo bioreductive binding, J. Med. Chem., 1991, 34, 2268–2274 CrossRef CAS PubMed.
- K. Yasuda, K. Okabe, K. Inukai and K. Itou, Fluorescence spectra of substituted naphthalimides. Solvent effect, Nippon Kagaku Zasshi, 1959, 80, 962–965 CrossRef CAS.
- F. Xie, T. Zhang, P. Bryant, V. Kurusingal, J. M. Colwell and B. Laycock, Degradation and stabilization of polyurethane elastomers, Prog. Polym. Sci., 2019, 90, 211–268 CrossRef CAS.
- M. Wiechmann, H. Port, W. Frey, F. Lärmer and T. Elsässer, Time-Resolved Spectroscopy on Ultrafast Proton Transfer In 2-(2′-Hydroxy-5′-methylphenyl)benzotriazole in Liquid and Polymer Environments, J. Phys. Chem., 1991, 95, 1918–1923 CrossRef CAS.
- S. Pijeau, D. Foster and E. G. Hohenstein, Excited-state dynamics of a benzotriazole photostabilizer: 2-(2′-hydroxy-5′-methylphenyl)benzotriazole, J. Phy. Chem. A, 2017, 121, 6377–6387 CrossRef CAS PubMed.
- G. Woessner, G. Goeller, P. Kollat, J. J. Stezowski, M. Hauser, U. K. A. Klein and H. E. A. Kramer, Photophysical and photochemical deactivation processes of ultraviolet stabilizers of the (2-hydroxyphenyl)benzotriazole class, J. Phys. Chem., 1984, 88, 5544–5550 CrossRef CAS.
- V. Bojinov, I. Panova and I. Grabchev, Novel adducts of a 2-(2-hydroxyphenyl)-benzotriazole and a blue emitting benzo[de]isoquinoline-1,3-dione for ‘‘one-step’’ fluorescent brightening and stabilization of polymers, Polym. Degrad. Stab., 2005, 88, 420–427 CrossRef CAS.
- T. Uesaka, T. Ishitani, T. Fujiwara, S. Yoshida, T. Shimeno, H. Nakazumi and T. Maeda, Japan Patent, JP2018-158912A, 2018 Search PubMed.
- T. Uesaka, T. Ishitani, T. Fujiwara, K. Yamada, T. Shimeno, H. Nakazumi and T. Maeda, Japan Patent, JP2016-169181A, 2016 Search PubMed.
- J. Rosevear and J. F. K. Wilshire, Preparation of some 2-(2′H-benzotriazol-2'-yl)phenol ultraviolet absorbers: application of the transalkylation reaction, Aust. J. Chem., 1985, 38, 1163–1176 CrossRef CAS.
- K. Mizukawa, J. Ueno and K. Fukui, Japan Patent, JP1999-158163A, 1999 Search PubMed.
- V. B. Bojinov and I. K. Grabchev, Novel functionalized 2-(2-hydroxyphenyl)-benzotriazole- benzo[de]isoquinoline-1,3-dione fluorescent UV absorbers synthesis and photostabilizing efficiency, J. Photochem. Photobiol., A, 2005, 172, 308–315 CrossRef CAS.
- V. B. Bojinov, I. P. Panova and J.-M. Chovelon, Novel blue emitting tetra- and pentamethylpiperidin-4-yloxy-1,8-naphthalimides as photoinduced electron transfer based sensors for transition metal ions and protons, Sens. Actuators, B, 2008, 135, 172–180 CrossRef CAS.
- A. L. Huston and G. W. Scott, Spectroscopic and kinetic investigations of internally hydrogen-Bonded (hydroxyphenyl)benzotriazoles, J. Phys. Chem., 1987, 91, 1408–1413 CrossRef CAS.
- T. Uesaka, S. Yagi and T. Maeda, Synthesis and Fluorescence Property of 5-amino-2-(2-hydroxyphenyl)-2H-benzotriazole Derivatives, J. Jpn. Soc. Colour Mater., 2020, 93, 194–201 CrossRef CAS.
- K. Yasuda, K. Inukai and K. Ito, Fluorescence of naphthalimide derivatives. Consideration on the mirror-image relation between fluorescence and absorption spectra, Nippon Kagaku Zasshi, 1958, 79, 897–899 CrossRef CAS.
- C.-G. Niu, P.-Z. Qin, G.-M. Zeng, X.-Q. Gui and A.-L. Guan, Fluorescence sensor for water in organic solvents prepared from covalent immobilization of 4-morpholinyl-1, 8-naphthalimide, Anal. Bioanal. Chem., 2007, 387, 1067–1074 CrossRef CAS PubMed.
- L. Biczók, P. Valat and V. Wintgens, Effect of molecular structure and hydrogen bonding on the fluorescence of hydroxy-substituted naphthalimides, Phys. Chem. Chem. Phys., 1999, 1, 4759–4766 RSC.
- L. Wang, M. Fujii, M. Yamaji and H. Okamoto, Fluorescence behaviour of 2-, 3- and 4-amino-1,8-naphthalimides: effects of the substitution positions of the amino functionality on the photophysical properties, Photochem. Photobiol. Sci., 2018, 17, 1319–1328 CrossRef CAS PubMed.
- P. A. Panchenko, Y. V. Fedorov, O. A. Fedorova and G. Jonusauskas, FRET versus PET: ratiometric chemosensors assembled from naphthalimide dyes and crown ethers, Phys. Chem. Chem. Phys., 2015, 17, 22749–22757 RSC.
- J. S. Sidhu and N. Singh, FRET and PET paired dual mechanistic carbon dots approach for tyrosinase sensing, J. Mater. Chem. B, 2018, 6, 4139–4145 RSC.
- N. S. Tasheh, N. K. Nkungli and J. N. Ghogomu, A DFT and TD-DFT study of ESIPT-mediated NLO switching and UV absorption by 2-(2′-hydroxy-5′-methylphenyl)benzotriazole, Theor. Chem. Acc., 2019, 138, 1–17 Search PubMed.
- N. Zhang, T. Zhang, L. Wen, L. Wan, J. Yan and K. Zheng, Tuning the excited-state intramolecular proton transfer (ESIPT) process of indole–pyrrole systems by π-conjugation and substitution effects: experimental and computational studies, Phys. Chem. Chem. Phys., 2020, 22, 1409–1415 RSC.
- J. S. Chen, P. W. Zhou, L. Zhao and T. S. Chu, A DFT/TDDFT study of the excited state intramolecular proton transfer based sensing mechanism for the aqueous fluoride chemosensor BTTPB, RSC Adv., 2014, 4, 254–259 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Shigeyuki Yagi
b,
Shigeyuki Yagi