
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn insight into the sodium-ion and lithium-ion storage properties of CuS/graphitic carbon nitride nanocomposite†
Dimple P. Dutta *ab,
Dipa D. Pathaka,
Sebin Abrahamc and
Balaji R. Ravurid
*ab,
Dipa D. Pathaka,
Sebin Abrahamc and
Balaji R. Ravurid
aChemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: dimpled@barc.gov.in; Tel: +91-25592308
bHomi Bhabha National Institute, Mumbai 400094, India
cDepartment of Chemistry, Indian Institute of Science Education and Research, Bhopal 462066, India
dDepartment of Physics, School of Science, GITAM Deemed to be University, Hyderabad 502329, India
First published on 25th April 2022
Abstract
Metal sulfides are gaining prominence as conversion anode materials for lithium/sodium ion batteries due to their higher specific capacities but suffers from low stability and reversibility issues. In this work, the electrochemical properties of CuS anode material has been successfully enhanced by its composite formation using graphitic carbon nitride (g-C3N4). The CuS nanoparticles are distributed evenly in the exfoliated g-C3N4 matrix rendering higher electronic conductivity and space for volume alterations during the repeated discharge/charge cycles. The 0.8CuS:0.2g-C3N4 composite when used as an anode for lithium ion coin cell exhibits a reversible capacity of 478.4 mA h g−1 at a current rate of 2.0 A g−1 after a run of 1000 cycles which is better than that reported for CuS composites with any other carbon-based matrix. The performance is equally impressive when 0.8CuS:0.2g-C3N4 composite is used as an anode in a sodium ion coin cell and a reversible capacity of 408 mA h g−1 is obtained at a current rate of 2.0 A g−1 after a run of 800 cycles. A sodium ion full cell with NVP cathode and 0.8CuS:0.2g-C3N4 composite anode has been fabricated and cycled for 100 runs at a current rate of 0.1 A g−1. It can be inferred that the g-C3N4 matrix improves the ion transfer properties, alleviates the volume alteration happening in the anode during the discharge/charge process and also helps in preventing the leaching of polysulfides generated during the electrochemical process.
Introduction
Research on electrochemical energy storage particularly rechargeable batteries has gained tremendous momentum in the last two decades primarily for its seminal role in the field of portable electronics.1 Among various rechargeable battery technologies, the advancement in lithium ion batteries (LIBs) has been the most dramatic.2 Further impetus in LIB technology is being brought about by its application in electric vehicles and renewable energy storage.3 However, the rise in demand for lithium which is scantily available (∼20 ppm) and geopolitically inequitably distributed in the earth's crust is escalating its price and leading to an impending crisis.4 Consequently, there has been a surge of exploration in alternative technologies like those using the more abundant element sodium which can lead to less expensive sodium ion batteries (SIBs).5 However, the cost advantage of SIBs over LIBs is somewhat thwarted by the sluggish kinetics of the comparatively larger and heavier Na+ ions and its higher standard electrode potential (−2.71 V vs. SHE) compared to Li+ ions.6 Nonetheless, it has been predicted that SIBs are poised to play a major role in the renewable energy storage sector where some laxity in gravimetric energy density is tolerable.7 The capacity of both LIBs and SIBs is majorly dependent on the properties of the materials used as electrode (cathode and anode). For currently available commercial LIBs, the standard anode material is graphite which has a theoretical capacity of ∼372 mA h g−1.3 However, for its application in electric vehicles, LIBs need to improve its energy density for which alternative anode materials needs to be identified. In case of SIB, the anode material of choice is hard carbon since it has higher Na+ transport capacity and its chemical potential almost matches with that of Na metal.8 However, the drawback of hard carbon anode is its low initial coulombic efficiency (ICE) and poor rate performance.9 Hence, there is a need to explore better anode materials with higher specific capacity, extended cyclability and adequate rate performance, for both LIBs and SIBs.The conversion type anodes generally have higher capacity compared to insertion type materials like graphite and hard carbon. Transition metal oxides, sulfides, phosphides and nitrides have been explored as conversion type anodes for both LIBs and SIBs.10 The conversion reaction leads to the formation of the corresponding Li/Na salt (oxide, sulphide, phosphide, nitride) with concomitant release of the transition metal during the lithiation/sodiation process. The transition metal sulfides have lower ionicity which helps in smoother insertion/de-insertion of the Li+/Na+ ions and reduces the overpotential linked to ion storage.11 The smaller band gap in transition metal sulfides compared to their oxide counterparts, enhances the electronic conductivity of the former which is also advantageous in the context of electrochemical energy storage.12 Among the various transition metal sulfides, inexpensive and abundantly available CuS gains advantage as an anode material due to its moderately high theoretical capacity (560 mA h g−1) and electronic conductivity (10−3 S cm−1).13,14 However, its performance is marred by the volume expansion/contraction which occurs during the cycling process and leads to pulverization of the anode material and its consequent stripping off from the current collector strip.15 Also, an added point of concern is that the polysulfides formed during the conversion reaction dissolves in the electrolyte leading to loss of the active material.16 Both these factors result in poor cycling performance of CuS and hence various strategies has been adopted over the years to circumvent these problems. Different types of nanoforms of CuS have been studied to compensate for the pulverization effect and suitable coating with conducting materials have been attempted to alleviate the deleterious effect of volume expansion and also improve the electronic conductivity of the resultant anode.15 There are reports on the electrochemical properties of CuS nano-particles/sheets/plates, as well as porous and hollow CuS, when used as anodes for LIBs and SIBs.13–15,17 To recompense for the volume expansion effect and further improve the electronic conductivity of CuS, composites with graphene, graphene-oxide, reduced graphene-oxide, MOF-derived carbon, carbon nanotubes, have also been explored as anode materials.14,18–23 However, synthesis of all of these conductive additives is complex and not cost effective.
g-C3N4 has a layered structure similar to graphene and can be synthesized from inexpensive precursors like melamine, urea, etc. by simple thermal treatment.24 The higher concentration of nitrogen (∼57%) in g-C3N4 results in its lower electronic conductivity compared to that of carbon which hinders its application for Li+/Na+ ion storage.25 However, modified g-C3N4 anodes with greater proportion of terminal pyridinic nitrogen bonds compared to quaternary nitrogen and also its composite with carbon and reduced graphene oxide has been explored as anode material for LIB and SIB and has shown promising results.24,26,27 Improvement in specific capacity of mesoporous CuO has been reported on its composite formation with O-doped g-C3N4 nanospheres with a concomitant increase in capacity retention from ∼47% to ∼75.3% in the latter.28 However, corresponding studies on the electrochemical properties of transition metal sulfide composites with g-C3N4 is relatively unexplored.12,29
CuS composites with g-C3N4 has been studied mainly for their application as visible light active photocatalysts for degradation of organic pollutants and for water splitting.30–32 In this manuscript, the electrochemical properties of CuS/g-C3N4 composite synthesized using simple and scalable precipitation technique has been investigated for Li+/Na+ ion storage and has been compared with that reported for their pristine counterpart. The reason behind the enhanced electrochemical properties of CuS/g-C3N4 composite has been analysed critically and the pivotal role played by the g-C3N4 matrix has been explained through adequate experimental techniques.
Experimental
Material synthesis
All the chemicals used for synthesis of the anode material and fabrication of electrodes were of Analytical Reagent grade and was used without any additional purification process.Synthesis of CuS
In a typical synthesis procedure, aqueous solution (35 ml) of copper(II) nitrate trihydrate (2 g, 8.28 mmol) was taken in a RB flask under N2 and aqueous solution of sodium thiosulphate (1.31 g, 8.28 mmol) was added to it dropwise. Deionized (DI) water was used to make the solutions. The blend was stirred vigorously at 90 °C for 7 h. The black precipitate attained was separated by centrifugation, then washed repeatedly with deionised water and dried under vacuum.Synthesis of g-C3N4
Urea (6 g) was used as a precursor for the synthesis of g-C3N4. It was heated in an alumina crucible in a muffle furnace at 500 °C for 2 h. The product obtained was faint yellow in color. It was sonicated with 20 ml nitric acid for exfoliation and then washed repeatedly with water/ethanol mixture and finally dehydrated in an oven at 70 °C for 6 h.Synthesis of 0.8CuS:0.2g-C3N4 composite
g-C3N4 (0.19 g, 2.06 mmol) was dispersed in 30 ml deionized water. The dispersed mixture was poured in a N2 flushed RB flask and aqueous solution of copper(II) nitrate trihydrate (2 g, 8.28 mmol) was added to it. An aqueous solution of sodium thiosulphate (1.26 g, 8.28 mmol) was added dropwise to the mixture and stirred at 90 °C for 7 h. The precipitate obtained was separated out by centrifugation, cleaned with DI water thrice and then vacuum dried.Physical characterization
The powder X-ray diffraction (XRD) diffraction patterns were obtained on a Rigaku Smart Lab XRD device (10° ≤ 2θ ≤ 80°, scan rate: 0.02° s−1) using Cu-Kα radiation (λ = 1.5406 Å). SEM (scanning electron microscopy) images were recorded on Philips XL 300 ESEM using 30 kV accelerating voltage. Sapphire EDAX attachment of the SEM was used to perform the EDS analysis. Conventional transmission electron microscopy (TEM) images of the samples spread on carbon coated Cu grids were recorded using Carl Zeiss instrument (120 kV). High resolution TEM was done on Jeol-2100F instrument. NETZSCH STA 409 PC instrument was used for the thermogravimetric (TG) studies. The temperature range was between 30–800 °C and the heating was done under air at a rate of 3 °C min−1. The specific surface area has been calculated using the BET (Brunauer–Emmett–Teller) method and the N2 adsorption data was recorded in a Bel Japan Inc., Belsorp II surface area analyzer. HOBIBA LabRAM HR 800 spectrometer with λexc = 632.8 nm laser source was used to record the Raman spectra of the samples. XPS (X-ray photoelectron spectra) was obtained using Al Kα excitation source in AXIS Supra, Kratos Analytical, SHIMADZU instrument.Electrochemical measurements
The working anodes for LIB and SIB were prepared in the following manner. The active material, carbon black (conducting agent) and sodium alginate (binder) was mixed in de-ionized (DI) water in 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio according to their weight%. The paste obtained was smeared on a Cu foil current collector using doctor blade. The area of each electrode was 1 cm−2 and they were dried for 8 h in a hot air oven maintained at 50 °C. The electrolyte used was 1 M LiPF6 in a 50:50 (v/v) mixture of dimethyl carbonate (DMC) and ethylene carbonate (EC) for Li-ion coin cell and 1 M NaClO4 in a 50:50 (v/v) mixture of propylene carbonate (PC) and EC having 5 wt% fluoroethylene carbonate (FEC) added to it in case of Na-ion coin cell. The CR2032 coin cells were assembled in an Argon flushed glove box (Mbraun, Unilab, Germany) using the corresponding anode, Whatman glass microfiber filters GF/D separator soaked in electrolyte and Li/Na foil as counter/reference electrode. Full cell was constructed using Na3V2(PO4)3 (NVP) cathode and the 0.8CuS:0.2g-C3N4 composite as anode using procedure as reported earlier.33 Pre-sodiated anode (5 cycles at 0.2 Ag−1) was used to compensate for the initial coulombic efficiency loss. The mass ratio of cathode: anode was fixed at 4:1. The electrolyte was same as that used for assembling the Na ion half-cell. Neware Battery Test System from Shenzhen Neware Electronic Co, China has been used to record the galvanostatic charge/discharge measurements at room temperature within a voltage window of 0.1–3 V and at various current densities. The specific capacity reported for all the anodes is referenced with respect to the active material mass in the electrode. The cyclic voltammetry (CV) studies were done at 0.1 mV s−1 (voltage range of 0.1–3 V vs. Li/Li+ and 0.1–2.7 V vs. Na/Na+) on a Biologic potentiostat/galvanostat (BCS 810) instrument. Electrochemical impedance spectra (EIS) were recorded on a Novocontrol Alpha-A High Frequency Analyzer (frequency range = 1 Hz to 1 MHz).
:1:1 ratio according to their weight%. The paste obtained was smeared on a Cu foil current collector using doctor blade. The area of each electrode was 1 cm−2 and they were dried for 8 h in a hot air oven maintained at 50 °C. The electrolyte used was 1 M LiPF6 in a 50:50 (v/v) mixture of dimethyl carbonate (DMC) and ethylene carbonate (EC) for Li-ion coin cell and 1 M NaClO4 in a 50:50 (v/v) mixture of propylene carbonate (PC) and EC having 5 wt% fluoroethylene carbonate (FEC) added to it in case of Na-ion coin cell. The CR2032 coin cells were assembled in an Argon flushed glove box (Mbraun, Unilab, Germany) using the corresponding anode, Whatman glass microfiber filters GF/D separator soaked in electrolyte and Li/Na foil as counter/reference electrode. Full cell was constructed using Na3V2(PO4)3 (NVP) cathode and the 0.8CuS:0.2g-C3N4 composite as anode using procedure as reported earlier.33 Pre-sodiated anode (5 cycles at 0.2 Ag−1) was used to compensate for the initial coulombic efficiency loss. The mass ratio of cathode: anode was fixed at 4:1. The electrolyte was same as that used for assembling the Na ion half-cell. Neware Battery Test System from Shenzhen Neware Electronic Co, China has been used to record the galvanostatic charge/discharge measurements at room temperature within a voltage window of 0.1–3 V and at various current densities. The specific capacity reported for all the anodes is referenced with respect to the active material mass in the electrode. The cyclic voltammetry (CV) studies were done at 0.1 mV s−1 (voltage range of 0.1–3 V vs. Li/Li+ and 0.1–2.7 V vs. Na/Na+) on a Biologic potentiostat/galvanostat (BCS 810) instrument. Electrochemical impedance spectra (EIS) were recorded on a Novocontrol Alpha-A High Frequency Analyzer (frequency range = 1 Hz to 1 MHz).
Results and discussion
CuS was synthesized via precipitation method using copper(II) nitrate and sodium thiosulphate as precursors which was stirred under N2 cover at 90 °C for 7 h. The reactions leading to the synthesis is given below (eqn (1) and 2):| Cu(NO3)2·3H2O + 2Na2S2O3 = Na2[Cu(S2O3)2] + 2NaNO3 + 3H2O | (1) |
| Na2[Cu(S2O3)2] + H2O → CuS↓ + H2SO4 + Na2SO4 | (2) |
The synthesis procedure of the CuS/g-C3N4 composite samples is schematically represented in Fig. 1. The powder XRD of CuS, g-C3N4 and CuS/g-C3N4 composite is shown in Fig. 2a. The diffraction pattern corresponds to the hexagonal phase of covellite CuS (PCPDF# 782121). g-C3N4 exhibits a sharp strong peak at 27.3° and a weak broad one at 13.2° pertaining to the (002) and (110) diffraction planes, respectively. The strong peak is due to in-plane periodic arrangement of tris-triazine units of g-C3N4 and the weak peak arises due to the inter-planar arrangement of the aromatic rings.30 The CuS/g-C3N4 composite displays peaks pertaining to both CuS and g-C3N4 and a narrower peak at 13.2° is observed for the composite compared to pristine g-C3N4. This indicates that the interplanar arrangement of the aromatic rings in g-C3N4 gets more pronounced in the composite which can be ascribed to the anchoring of the CuS particles on them. The morphology of CuS and CuS/g-C3N4 composite is visible from the SEM images shown in Fig. 2b and c, respectively. CuS shows spherical ball like formation whereas in case of the composite, the CuS particles gets engulfed in the g-C3N4 nanosheets. The presence of Cu, S, C and N along with a minor fraction of O element is seen in the EDS spectrum of the composite (Fig. 2d). The presence of oxygen is probably due to exposure of the sample to air and moisture. Cu and S are present in the composite in 1:1.15 atomic ratio. In contrast, the EDS of CuS confirms presence of only Cu and S elements in a 1:1.1 atomic ratio (Fig. 2e). The TEM images of CuS gives a clearer picture of its actual morphology and it is seen that the particles are spherical in shape with diameter ranging between ∼10–25 nm (Fig. 2f). In case of the CuS/g-C3N4 composite, the TEM image confirms the presence of CuS nanoparticles within the g-C3N4 sheets (Fig. 2g). The diameter of the CuS nanoparticles in the composite range between ∼10–15 nm and the smaller size compared to pristine CuS is attributed to the presence of g-C3N4 nanosheets which arrests further growth of the nanoparticles. The HRTEM image of the CuS/g-C3N4 composite is shown in Fig. 2h. The lattice spacing of 0.28 nm corresponding to the (103) plane of CuS nanoparticle is clearly discernible in the g-C3N4 matrix. Further confirmation for the inclusion of CuS nanoparticles in g-C3N4 matrix in the composite is obtained from Raman spectroscopy. Fig. 2i shows the Raman plots of CuS, g-C3N4 and the 0.8CuS:0.2g-C3N4 composite. A peak at ∼468.6 cm−1 is seen in the Raman plot of CuS which is the optical phonon mode corresponding to S–S stretching.34 The sharpness of the peak indicates the crystalline nature of the sample. The Cu–S vibrational stretching occur at ∼118 cm−1 and 266.2 cm−1.35 These low frequency vibration modes are generally observed in case of nano sized CuS.36 For pristine g-C3N4, the bending vibration corresponding to ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(sp2) is observed at ∼1241 cm−1. The blue shifting of this peak with respect to that seen in the bulk g-C3N4 (1234 cm−1), indicates phonon confinement pertaining to the formation of nanosheet-like structure.37 Other peaks corresponding to various C–N vibrations are observed at 479.1, 542.9, 702.2 and 748.8 cm−1. The signature peaks of both CuS and g-C3N4 are observed in the composite sample. The intensity of the CuS peaks are stronger which can be ascribed to its higher relative concentration in the CuS/g-C3N4 composite. To quantify the proportion of g-C3N4 in the composite, thermogravimetric studies was done under air and the corresponding TG plot is shown in Fig. S1a.† CuS shows a slight mass loss at ∼275 °C followed by an increase in mass which is ascribed to loss of sulfur from CuS leading to formation of Cu2S and then conversion of CuS/Cu2S to CuSO4, respectively.38 The final mass loss between 610–827 °C is due to decomposition of CuSO4 to CuO and the total weight loss is found to be ∼16.6%. The TG curve of g-C3N4 show 100% weight loss between 580–750 °C and is due to its decomposition. For the CuS/g-C3N4 composite, the total weight loss observed at 800 °C is ∼33.2% which indicates that the sample has approximately 20% of g-C3N4 by weight and this is in accordance with the nominal composition.
C(sp2) is observed at ∼1241 cm−1. The blue shifting of this peak with respect to that seen in the bulk g-C3N4 (1234 cm−1), indicates phonon confinement pertaining to the formation of nanosheet-like structure.37 Other peaks corresponding to various C–N vibrations are observed at 479.1, 542.9, 702.2 and 748.8 cm−1. The signature peaks of both CuS and g-C3N4 are observed in the composite sample. The intensity of the CuS peaks are stronger which can be ascribed to its higher relative concentration in the CuS/g-C3N4 composite. To quantify the proportion of g-C3N4 in the composite, thermogravimetric studies was done under air and the corresponding TG plot is shown in Fig. S1a.† CuS shows a slight mass loss at ∼275 °C followed by an increase in mass which is ascribed to loss of sulfur from CuS leading to formation of Cu2S and then conversion of CuS/Cu2S to CuSO4, respectively.38 The final mass loss between 610–827 °C is due to decomposition of CuSO4 to CuO and the total weight loss is found to be ∼16.6%. The TG curve of g-C3N4 show 100% weight loss between 580–750 °C and is due to its decomposition. For the CuS/g-C3N4 composite, the total weight loss observed at 800 °C is ∼33.2% which indicates that the sample has approximately 20% of g-C3N4 by weight and this is in accordance with the nominal composition.
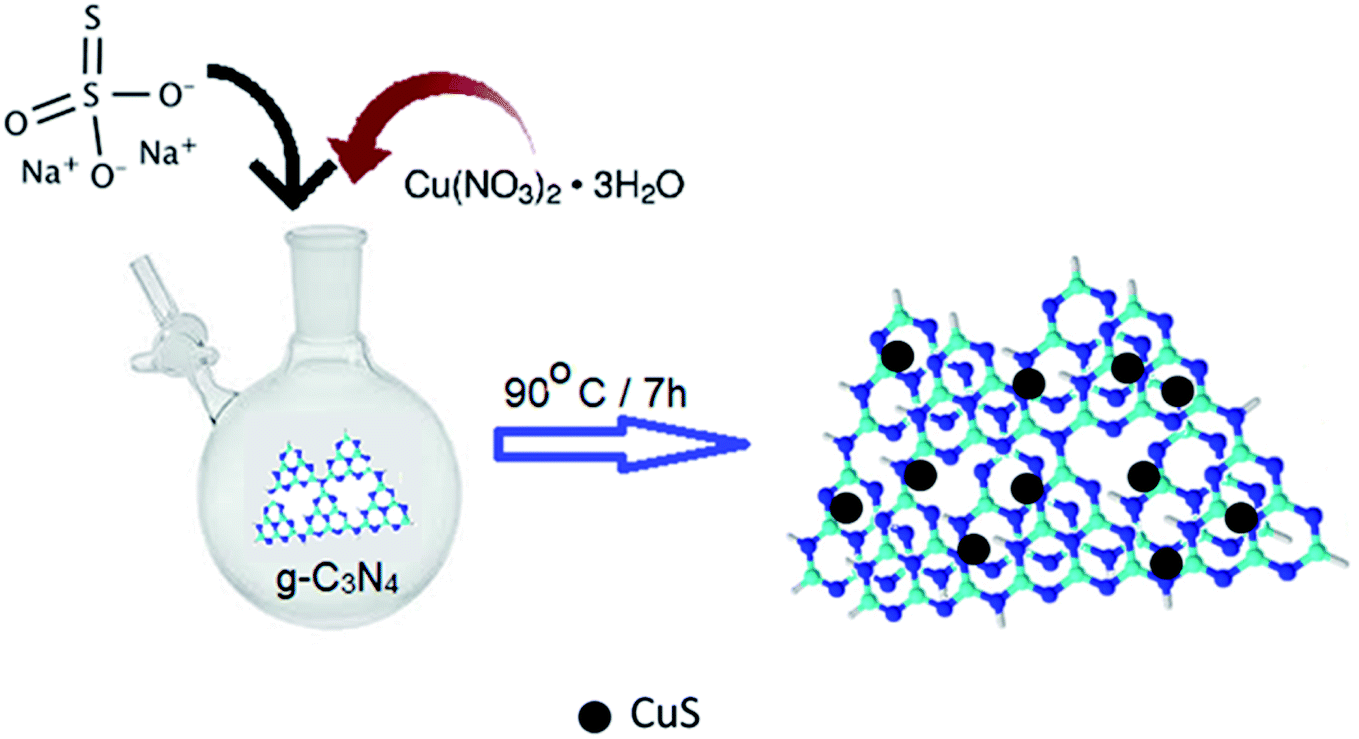 | ||
| Fig. 1 Schematic representation of the synthesis of CuS/g-C3N4 composite. | ||
 | ||
| Fig. 2 (a) Powder XRD of g-C3N4, CuS, 0.8CuS:0.2g-C3N4; (b) SEM of CuS; (c) SEM of 0.8CuS:0.2g-C3N4; (d) EDS of CuS; (e) EDS of 0.8CuS:0.2g-C3N4; (f) TEM of CuS; (g) TEM of 0.8CuS:0.2g-C3N4; (h) HRTEM of 0.8CuS:0.2g-C3N4; (i) Raman spectra of g-C3N4, CuS, 0.8CuS:0.2g-C3N4. | ||
X-ray photoelectron spectroscopy (XPS) has been used to study the chemical and electronic state of the elements present in the CuS/g-C3N4 composite. The survey spectra of CuS, g-C3N4 and 0.8CuS:0.2g-C3N4 is shown in Fig. S1b† and it shows presence of oxygen in all the samples apart from the expected Cu and S in CuS, N and C in g-C3N4 and C, N, Cu, S in the 0.8CuS:0.2g-C3N4 composite. The O 1s peak at ∼532.1 eV is due to the adsorption of atmospheric oxygen on the sample surface.30 The C 1s peak seen in the pristine CuS sample at ∼284.5 eV is attributed to the internal standard used for reference. The survey spectra indicate the absence of any other elemental impurities in the samples. The high resolution XPS of Cu2p in CuS and 0.8CuS:0.2g-C3N4 shows the presence of Cu2p3/2 and Cu2p1/2 peaks (Fig. S1c†). For CuS, the Cu2p3/2 and Cu2p1/2 peaks are observed at 932.4 eV and 952.5 eV, respectively, which indicates presence of Cu in predominantly +1 oxidation state.39 In the 0.8CuS:0.2g-C3N4 composite, broader peaks pertaining to Cu2p3/2 and Cu2p1/2 are observed at 932.1 eV and 951.9 eV, respectively along with a weak satellite peak at 945.4 eV which indicates the presence of Cu in both +1 and +2 oxidation states.40 The presence of Cu2+ can be attributed to surface oxidation. The shift in the Cu 2p3/2 and Cu/S 2p1/2 peaks to lower binding energy in the composite sample and its broadness compared to pristine CuS has been reported in literature and indicates slight chemical interaction between CuS and g-C3N4.30,31 Fig. S1d† depicts the high resolution S2p XPS of CuS. The spectrum obtained could be fitted to S2− 2p3/2 and 2p1/2 peaks at 160.9 and 161.5 eV and (S2)2− 2p3/2 and 2p1/2 peaks at 161.8 and 162.9 eV, respectively. The ratio of area of the sulfide to disulfide peaks is 1:1.9 which is close to that expected for covellite CuS. Another broad peak at a binding energy of 163.5 eV is also seen which has been assigned to elemental sulfur. The results obtained are similar to that reported in earlier literature.39 In case of the 0.8CuS:0.2g-C3N4composite, S2− 2p3/2 and 2p1/2 peaks are seen at 160.7 and 161.4 eV and (S2)2− 2p3/2 and 2p1/2 peaks at 161.6 and 162.7 eV, respectively, and this shift to lower binding energy has been previously reported for CuS/g-C3N4 composites (Fig. S1e†).30 The high resolution N 1s peaks for g-C3N4 and 0.8CuS:0.2g-C3N4 could be fitted to two peaks at ∼398.5 eV and ∼400.4 eV pertaining to sp2 N atoms of (C–NC) moiety and sp3 N of the N-(C)3 group, respectively (Fig. S1f†). The higher intensity of the former peak indicates presence of higher proportion of pyridinic N in g-C3N4 which is beneficial for Li+ ion transport and also leads to better electronic conductivity in both the samples.27 The relative concentration of N-(C)3 group is further reduced in the composite sample and hence it is expected to be a better anode material compared to pristine g-C3N4. The high resolution C 1s spectra of g-C3N4 and 0.8CuS:0.2g-C3N4 shows major peak at ∼288.3 eV and ∼288.7 eV, respectively and they are ascribed to N–CN carbon (Fig. S1g†).41 Hence, both C 1s and N 1s high resolution XPS of 0.8CuS:0.2g-C3N4 indicates presence of higher proportion of pyridinic N bonds which is desirable for its Li+/Na+ ion storage application.
The specific surface area of CuS, g-C3N4 and 0.8CuS:0.2g-C3N4 has been calculated via BET method from the Type IV N2 adsorption/desorption isotherms with H4 pattern of hysteresis loop obtained in case of all the samples (Fig. S2a†). CuS has a specific surface area of ∼30.21 m2 g−1 which is higher than that reported for CuS obtained via precipitation technique (9.56 m2 g−1) and via hydrothermal route (∼14.5 m2 g−1) but less than that obtained using acid mediated dealloying reaction of Ti60Cu40 (48.2–67.9 m2 g−1).17,30,31,42 The slow and controlled addition of reactants with high speed stirring leads to smaller size of CuS nanoparticles and increases the surface area in our sample.43 The specific surface area of the 0.8CuS:0.2g-C3N4 composite sample is 46.63 m2 g−1 which is due to the dispersion of CuS nanoparticles on the g-C3N4 nanosheets. This results in an increase in the surface area of the composite compared to pristine CuS but a decrease compared to that calculated for g-C3N4 (72.15 m2 g−1) since CuS blocks the pores present in g-C3N4. The larger surface area of the composite improves its ability to provide adequate migration channels for Li+/Na+ ions and leads to its better wettability by the electrolyte and can reduce electrode resistance.23 The total pore volume of the samples has been obtained via Barrett–Joyner–Halenda (BJH) method from the corresponding pore size distribution plots (Fig. S2b†). The pore size distribution varies mostly between 5–70 nm for all the samples confirming the presence of mesopores and the total pore volume is highest for g-C3N4 (1.405 cm3 g−1). Pristine CuS has a pore volume of 0.695 cm3 g−1 which can be attributed to the interparticle space. The composite has a higher total pore volume of 0.923 cm3 g−1 compared to CuS but it is less than that of g-C3N4 and this corroborates with the relative specific surface area obtained for all the three samples. The increase in total pore volume and specific surface area in the composite is beneficial for accommodating the volume expansion observed in CuS anodes during cycling.23 This can lead to better cycling performance of the composite by preserving the electrode structure.
The CV plots of pristine CuS and 0.8CuS:0.2g-C3N4 composite at scan rate of 0.1 mV s−1 while functioning as anode in LIB configuration is shown in Fig. 3a and b, respectively. Reduction peaks are seen at 2.11 eV and 1.62 eV during 1st cycle discharge which is due to formation of Cu2S and its subsequent conversion to yield Li2S and Cu, respectively.44 Apart from this, a broad peak between 0.3–0.9 eV with weak intensity is seen which disappears in the consecutive cycles and is assigned to the formation of SEI layer.18 The oxidation peaks are seen at 1.91 eV and 2.37 eV which is attributed to formation of Cu2−xS and the overpotential pertaining to Li2S, respectively.44 From second cycle onwards, the cathodic peak at 2.11 eV disappears and this is attributed to the irreversible nature of the reaction leading to the formation of Cu2S:
| 2CuS + 2Li → Cu2S + Li2S | (3) |
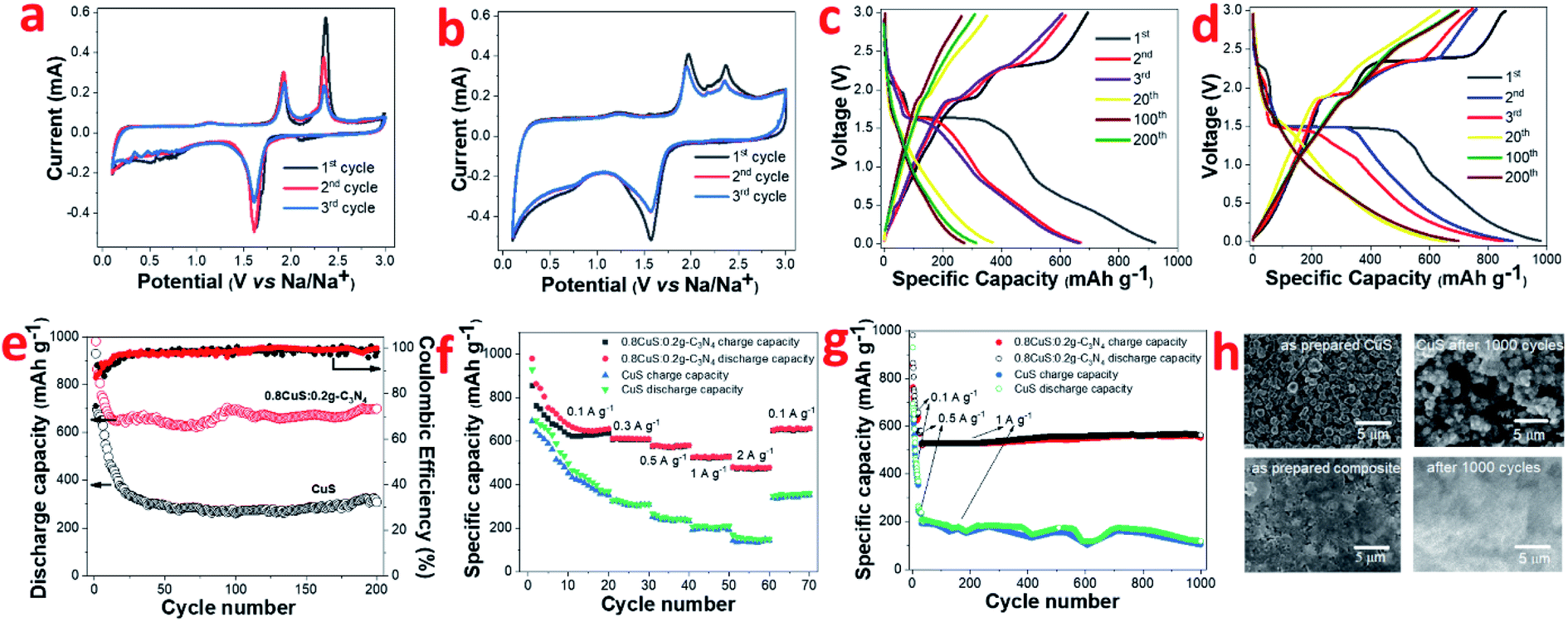 | ||
| Fig. 3 In LIB half cell: (a) CV plot of CuS; (b) CV plot of 0.8CuS:0.2g-C3N4; (c) galvanostatic discharge charge of CuS; (d) galvanostatic discharge charge of 0.8CuS:0.2g-C3N4; (e) discharge capacity and coulombic efficiency plot, (f) rate capability, (g) long term cyclability of CuS and 0.8CuS:0.2g-C3N4; (h) SEM of as prepared and after 1000 cycle run of CuS and 0.8CuS:0.2g-C3N4 electrode. | ||
The anodic peak at 2.37 eV pertaining to Li2S overpotential exhibits a sharp decrease in intensity with cycling and this is similar to that observed in earlier reports.44 For the 0.8CuS:0.2g-C3N4 composite, the area covered under the CV plot is larger than that observed in case of pristine CuS and this indicates an increase in the capacity of the composite anode (Fig. 3b).28 The broad and very weak cathodic peak at ∼2.31 eV pertaining to Cu2S formation disappears from the second cycle and the strong cathodic peak at 1.56 eV denoting further reduction to form Cu and Li2S persists but shift to slightly higher value of 1.57 eV. The anodic peaks are seen at 1.96 eV and 2.37 eV and is ascribed to the reactions leading to formation of Cu2S. There is almost no change in CV plot of the 0.8CuS:0.2g-C3N4 composite from second cycle onwards which indicate its better electrochemical stability compared to the pristine CuS electrode.
The galvanostatic discharge/charge (GDC) plots corresponding to few representative cycles of the CuS and 0.8CuS:0.2g-C3N4 composite anode in LIB mode within the voltage range of 0.01–3.0 V is shown in Fig. 3c and d, respectively. The GDC experiments were performed under current density of 0.1 A g−1. CuS exhibits a first discharge/charge capacity of ∼930/693 mA h g−1 which decreases to ∼369/353 mA h g−1 by the 20th cycle. The discharge plot exhibits plateaus at ∼2.1 and 1.6 eV during the first cycle but the former plateau region is not seen in the consecutive cycles. In case of the charge plots, the plateau region is seen at ∼1.9 and 2.4 eV in the first cycle with considerable reduction in the stretch of plateau region in the consecutive cycles. The results obtained match with the cathodic and anodic peak positions observed in the CV plot of pristine CuS. The plateau region in GDC plots becomes almost indiscernible by the 20th cycle and considerable loss in specific capacity is observed by the 200th run. The first discharge plot of the 0.8CuS:0.2g-C3N4 composite exhibits two voltage plateaus at ∼2.3 and 1.5 eV whereas the discharge plots for the second and third cycle shows only one voltage plateau at ∼1.5 eV. The charge plots of the composite exhibit voltage plateaus at ∼1.9 and 2.37 eV but the length of the plateau region diminishes with cycling. The results agree with that observed in the CV plots of the composite sample. The first cycle discharge/charge renders a specific capacity of ∼981/855 mA h g−1 yielding an initial coulombic efficiency (ICE) of 87.15%. The ICE is higher in case of the composite sample compared to that observed in pristine CuS (74.5%). The irreversible loss in the first cycle is caused by formation of the SEI layer with concomitant electrolyte breakdown and the irreversible nature of reaction between Li and CuS due to formation of polysulfides.19 Higher ICE value in the composite indicates better stability of SEI which can be attributed to the presence of g-C3N4 which restricts the polysulfide intermediates from diffusing out into the electrolyte. The presence of the pyridinic N atoms in g-C3N4 binds to the lithium ions of the polar polysulfide species and restricts its diffusion.45 The coulombic efficiency (CE) of both CuS and the 0.8CuS:0.2g-C3N4 composite increases to ∼94.8% and ∼96.2%, respectively, by the 20th run (Fig. 3e). However, compared to the first cycle charge capacity, the capacity retention after a run of 200 cycles is ∼81.4% for the composite which is much higher than obtained in case of pristine CuS anode (∼44.5%) and is a signature of its better electrochemical property (Fig. 3e). The anode with g-C3N4 nanosheets as active material exhibits a specific capacity of ∼202 mA h g−1 (current density = 0.1 A g−1, 200 cycles) in LIB configuration with a CE of ∼99.56% by the 3rd cycle which matches with earlier reports and establishes its suitability as a composite host for CuS nanoparticles (Fig. S3†).12 The specific charge capacity of the 0.8CuS:0.2g-C3N4 anode after 200 cycles is ∼699.3 mA h g−1 which is enhanced drastically compared to ∼308.6 mA h g−1 delivered by the pristine CuS anode (Fig. 3e). The reversible capacity of the pristine CuS anode is less than that observed for anodes with CuS nano-rods/wires but more than that with CuS microflowers, which highlights the role of nanostructures in the final reversible capacity delivered (Table S1†). The higher surface area and enhanced pore volume of the 0.8CuS:0.2g-C3N4 composite shortens the diffusion path of the Li+ ions and ensures its better wettability by the electrolyte which accounts for its higher specific capacity compared to pristine CuS.
The rate capability of pristine CuS and the 0.8CuS:0.2g-C3N4 anode has been tested at 0.1, 0.3, 0.5, 1 and 2 A g−1 current density via GDC within 0.01–3 V range (Fig. 3f). The first 20 cycles of discharge/charge were at 0.1 A g−1 and for all other current density, the anodes were cycled for 10 runs. At 2 A g−1 current density, the 0.8CuS:0.2g-C3N4 anode displayed a reversible capacity of ∼477.5 mA h g−1 and maintained a CE of ∼99.6%. On immediate reduction of current density to 0.1 A g−1, the reversible capacity increased to ∼657.5 mA h g−1 by the 70th cycle run for the 0.8CuS:0.2g-C3N4 anode which is slightly higher than that observed in case of its 20th cycle run at similar current density. This indicates activation of the electrode and also its stability inspite of repeated volume alteration during the lithiation/de-lithiation process. For the pristine CuS anode, increasing the current rate to 2 A g−1 yielded a meagre reversible capacity of ∼142.9 mA h g−1 with CE of ∼96.2%. The value of both the parameters are reduced compared to that of the composite anode. On decreasing current to 0.1 A g−1, the reversible capacity increases to ∼352.9 mA h g−1 which is slightly lower than that obtained in the 20th cycle run of the anode (∼357.9 mA h g−1). The 0.8CuS:0.2g-C3N4 composite anode shows ∼25% loss in capacity on increase in current rate from 0.1 to 2 A g−1 as opposed to ∼60% capacity loss exhibited by pristine CuS under similar variation in current rate. Approximately 60% loss in specific capacity with similar current rate variation has been reported in case of small CuS nanorods.46 Hence, it can be deduced that addition of g-C3N4 support to CuS, improves its rate capability.
The cycling ability of both pristine CuS and 0.8CuS:0.2g-C3N4 composite anode at a moderately high current rate of 1 A g−1 has been tested for 1000 cycles and the matching plot is shown in Fig. 3g. The first 25 cycle run is at a current of 0.1 A g−1 followed by 10 cycles at 0.5 A g−1 and this was done to activate the electrodes. The 0.8CuS:0.2g-C3N4 composite anode shows appreciable stability and delivers a reversible capacity of ∼551 mA h g−1 at the end of the 1000th cycle. Compared to it, the capacity delivered by the pristine CuS anode keeps fluctuating and decreases to ∼104 mA h g−1 after 1000 cycles. The post mortem analysis of the electrodes after 1000 runs has been done via SEM and has been compared with the as prepared electrodes (Fig. 3h). In case of the CuS electrode, cracks and fissures are seen after 1000 cycle run which peels off the active material from the Cu contact and cause fluctuation as well as reduction in its capacity. In case of the 0.8CuS:0.2g-C3N4 composite electrode, extensive pulverization is seen after 1000 cycles but no obvious discontinuity or gaps is visible in the electrode. Hence, addition of g-C3N4 helps in withstanding the structural stress brought about by the volume alteration during the lithiation/de-lithiation process in CuS.
The evaluation of reaction kinetics in the CuS and 0.8CuS:0.2g-C3N4 composite anode has been done via electrochemical impedance studies. The electrochemical impedance spectra (EIS) for both anodes (as-prepared and after a run of 100 cycles) have been recorded within a frequency spread of 0.1–1 MHz and the Nyquist plots is shown in Fig. S4.† The inset of Fig. S4† depicts the corresponding equivalent circuit where Rs, Rcf and Rct denotes the resistance arising due to the electrode and electrolyte, the impedance due to SEI formation and the charge transfer resistance arising due to Li+ diffusion between electrolyte and electrode, respectively. The straight line obtained in the low frequency part of the Nyquist plot is due to Warburg impedance (W) pertaining to Li+ ion diffusion. The equivalent circuit includes a constant phase element (CPE) pertaining to the formation of SEI layer, and also double layer capacitance (C). The fitting of the equivalent circuit to the Nyquist plot has been done using EIS Spectrum Analyser software and the corresponding values of Rs, Rcf and Rct are given in Table 1. Both anodes show lower Rcf and Rct values after 100 runs compared to the fresh as-prepared electrodes suggesting lower interface and charge transfer impedance which is due to subtle changes in the SEI layer and better kinetics of the conversion reaction with repeated cycling, respectively. Similar results have been documented in case of heterostructured FeS2/CuS anodes.47 The pristine CuS anode has higher Rct and Rs compared to the composite both before and after cycling which suggests that the presence of g-C3N4 reduces the charge transfer impedance by providing better electrode/electrolyte interaction, enhances the electronic conductivity and promotes Li+ diffusion in the sample. Eqn (4) has been used to estimate the Li-ion diffusion coefficient (DLi+) in CuS and 0.8CuS:0.2g-C3N4 composite anode both before and after a run of 100 cycles:
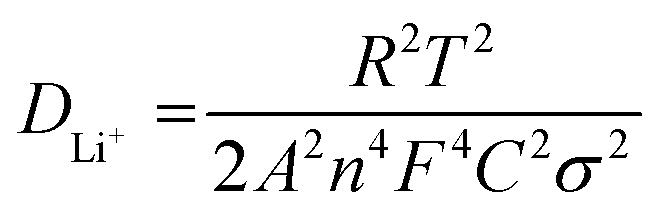 | (4) |
| Electrode | Rs (Ω) | Rcf (Ω) | Rct (Ω) | σ (Ω s−0.5) | DLi+ (cm2 s−1) |
|---|---|---|---|---|---|
| CuS (as prepared) | 8.6822 | 15.5213 | 81.3672 | 63.67 | 0.87 × 10−11 |
| CuS (after 100 cycles) | 10.5710 | 9.9952 | 75.1959 | 60.39 | 0.97 × 10−11 |
| 0.8CuS:0.2g-C3N4 (as prepared) | 3.1212 | 12.8843 | 78.6288 | 37.56 | 2.51 × 10−11 |
| 0.8CuS:0.2g-C3N4 (after 100 cycles) | 2.0903 | 9.6578 | 71.5132 | 26.15 | 5.18 × 10−11 |
In the above equation, T, R, A, C and F represents absolute temperature, gas constant, area of anode covered with active material (1 cm2), Li-ion molar density in electrode and Faraday constant, respectively. n denotes number of electrons involved in the half reaction during redox process, and σ is Warburg factor which is the slope of the Z′ vs. ω−1/2 plot as given in eqn (5)
| Z′ = RD + RL + σω−1/2 | (5) |
The σ and DLi+ values are listed in Table 1 and the DLi+ values are higher in case of the 0.8CuS:0.2g-C3N4 composite anode both before and after cycling compared to the CuS anode. This confirms the faster Li+ ion diffusion in the composite which supports the results obtained from the Nyquist plots.
The electrochemical performance of 0.8CuS:0.2g-C3N4 composite anode has been compared with other CuS anodes reported in literature (Table S2†). It can be deduced that the 0.8CuS:0.2g-C3N4 composite has much enhanced electrochemical properties compared to that reported for most of the graphene and reduced graphene oxide based composites of CuS. The initial coulombic efficiency (ICE) of ∼87.2% obtained in the 0.8CuS:0.2g-C3N4 composite anode is the highest among all the CuS composites reported and that exhibited by the pure CuS anode (74.5%). This indicates that the dispersion of CuS nanoparticles on g-C3N4 nanosheets leads to an improvement in its electrochemical performance.
Conversion type anode materials like CuS have better applicability in SIBs compared to intercalation type materials like graphite due to the larger size of Na+ ions compared to Li+ ions which negatively impacts its movement. Hence it was of interest to study the electrochemical performance of both CuS and the 0.8CuS:0.2g-C3N4 composite as anode materials for SIB application. The CV of CuS exhibits multiple cathodic and anodic peaks which are not reversible for the first three cycles (Fig. 4a) suggesting multistep process for the electrochemical reaction.13 During the first scan, cathodic peaks are seen at 0.55 V, 1.44 V and 1.87 V. During the subsequent scans, the peak at 0.55 V is not seen which indicates that it is due to the irreversible SEI layer formation. The other two peaks are due to the insertion of Na+ ions into CuS to form Na2S and Cu. The first anodic scan exhibits a broad peak at ∼1.72 V and two more peaks at 2.13 V and 2.21 V which are different from that observed in subsequent scans. The CV plots clearly indicate irreversibility and some structure change in the material. The anodic peaks seen at 1.65 V and 2.19 V during the third scan pertains to formation of Cu2S from Na2S and Cu.48 The corresponding discharge/charge curves for CuS for the first three cycles is shown in Fig. 4c. The discharge and charge plateaus match with the peaks observed in CV. The ICE obtained for this CuS anode is ∼86% which is comparable to that stated in earlier reports (Table S3†). The coincidence of CV plots for the 0.8CuS:0.2g-C3N4 composite is better than that observed for the pristine CuS anode suggesting decreased irreversible loss and better structure stability (Fig. 4b). The broad cathodic peak observed between ∼0.25–0.35 V observed in the composite in the first cycle is due to SEI layer formation. The slight loss in intensity of the anodic peaks during second and third cycle indicate specific capacity fading. The cathodic peaks at 1.24 V and 1.89 V are attributed to Na+ insertion into the CuS/g-C3N4 composite structure with the concomitant formation of Na2S whereas the broad anodic peaks at ∼1.60 V and 2.20 V is indicative of Cu2S formation.48 The galvanostatic discharge/charge plots exhibit multiple plateau region which corroborates with the results obtained from CV studies (Fig. 4d). Overlapping of the plateau regions in the second and third cycle indicate enhanced reversibility in the 0.8CuS:0.2g-C3N4 composite compared to that seen in case of pristine CuS. The cycling performance of CuS and 0.8CuS:0.2g-C3N4 at 0.1 A g−1 is shown in Fig. 4e. After 200 cycles, the specific capacity retention is ∼40.6% and ∼63.4% compared to the first cycle for CuS and 0.8CuS:0.2g-C3N4, respectively. A rapid decrease in charge capacity is observed in the first 25 cycles for CuS electrode which then increases unevenly up to 170 cycles and drops again and is a signature of electrochemical activation resulting from constant disintegration and reformation of active material to generate fresh sites for Na+ ion storage.12 The deleterious effect of volume expansion leading to rapidly changing specific capacity during cycling is buffered by the presence of g-C3N4 support in the 0.8CuS:0.2g-C3N4 composite. Consequently, the cycling performance is quite steady in case of the composite and the enhanced specific capacity (479.5 mA h g−1 after 200 cycles at 0.1 A g−1) is attributed to higher surface area and pore volume which ensures higher number of Na+ ions storage sites and better electrode/electrolyte contact. The 0.8CuS:0.2g-C3N4 composite also exhibits better rate performance compared to the CuS electrode (Fig. 4f). In case of CuS, the specific reversible capacity (mA h g−1)/current rate (A g−1) obtained varies as 410/0.1, 312/0.3, 276/0.5, 246/1, 190/2 and reverts to 316 mA h g−1 on reducing the current rate to 0.1 A g−1. For the composite, the specific reversible capacity decreases from ∼508 mA h g−1 at current rate of 0.1 A g−1 to ∼408 mA h g−1 at current rate of 2 A g−1. On decreasing current rate back to 0.1 A g−1, the reversible capacity obtained is 531 mA h g−1 which is slightly higher than the initially obtained value and can be attributed to the better electrochemical activation of the cycled electrode. Long term cycling of both electrodes has been studied at a higher current rate of 1 A g−1 after initial activation of the electrode at 0.1 A g−1 for 10 cycles and 0.8CuS:0.2g-C3N4 composite exhibits stable performance with a reversible capacity of ∼424 mA h g−1 after a total run of 800 cycles (Fig. 4g). The CuS electrode runs for 800 cycles but the specific capacity is lower than the composite and exhibit considerable fluctuation. The post mortem SEM of both electrodes after 800 cycles is shown in Fig. S5a and b.† Deep fissures and cracks are evident in the cycled CuS electrode which is not conducive for its electrochemical performance. In case of the 0.8CuS:0.2g-C3N4 composite, the surface of the electrode turns uneven but the absence of any major gaps ensures better connectivity and stability in the structure which augments the capacity and rate performance. The Nyquist plots for as prepared CuS and 0.8CuS:0.2g-C3N4 composite and after 100 cycles is shown is Fig. 4h. The semicircle observed at high and intermediate frequencies correspond to the resistance due to SEI layer formation and charge transfer and the straight line at low frequency is attributed to the diffusion of Na+ ions in the half cell. The corresponding equivalent circuit shown in Fig. S6† has been obtained by fitting the Nyquist plots by means of EIS Spectrum Analyser software and the values of Rs, Rcf and Rct is listed in Table 2. The Rct value of the composite is lower than that of pristine CuS both in as prepared and after 100 cycles which indicates an improvement in the conductivity of the former. Also, the steeper gradient of the oblique line of the composite compared to pristine CuS at low frequency before the testing indicate better diffusion of sodium ions in the former which is due to the even spreading of the CuS particles in the g-C3N4 matrix leading to an open structure. The remarkable decrease in Rct values on cycling of the CuS as well as 0.8CuS:0.2g-C3N4 composite electrode has been reported earlier and has been attributed to the formation of Cu2S which has higher electrical conductivity.49 The calculated DNa+ values are also given in Table 2 and the improvement in Na+ ion diffusion coefficient in the composite is indicative of the support extended by g-C3N4 in the ion transfer process.
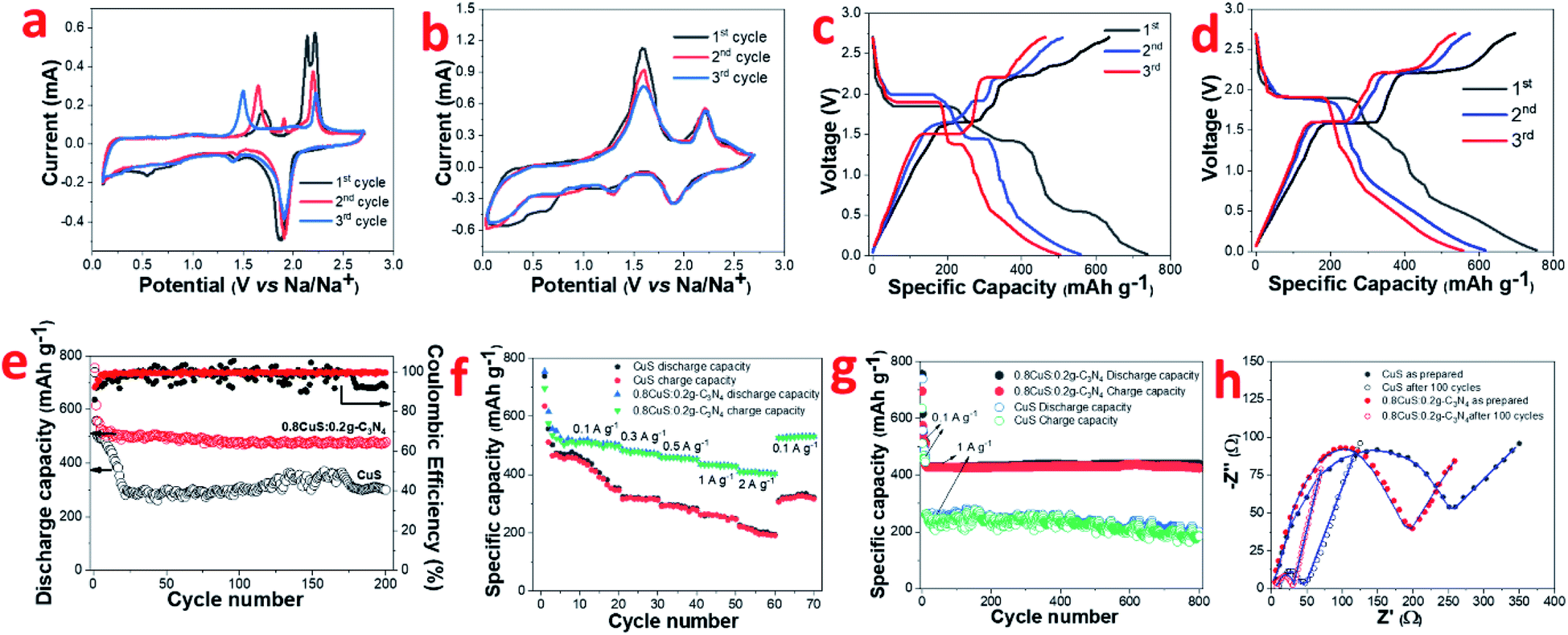 | ||
| Fig. 4 In SIB half cell: (a) CV plot of CuS; (b) CV plot of 0.8CuS:0.2g-C3N4; (c) galvanostatic discharge charge of CuS; (d) galvanostatic discharge charge of 0.8CuS:0.2g-C3N4; (e) discharge capacity and coulombic efficiency plot, (f) rate capability, (g) long term cyclability of CuS and 0.8CuS:0.2g-C3N4; (h) Nyquist plots of as prepared and after 100 cycle run of CuS and 0.8CuS:0.2g-C3N4 electrode. | ||
| Electrode | Rs (Ω) | Rcf (Ω) | Rct (Ω) | σ (Ω s−0.5) | DNa+ (cm2 s−1) |
|---|---|---|---|---|---|
| CuS (as prepared) | 6.6135 | 27.3062 | 218.1103 | 3.87 | 1.02 × 10−13 |
| CuS (after 100 cycles) | 10.7270 | 18.4956 | 31.0023 | 12.57 | 0.97 × 10−12 |
| 0.8CuS:0.2g-C3N4 (as prepared) | 4.7282 | 22.7626 | 168.3922 | 2.59 | 2.27 × 10−13 |
| 0.8CuS:0.2g-C3N4 (after 100 cycles) | 3.2610 | 12.0883 | 16.7219 | 9.85 | 1.58 × 10−12 |
The electrochemical kinetics in the 0.8CuS:0.2g-C3N4 composite electrode during the cycling process has been assessed via recording CV plots at various scan rates (Fig. 5a). With scan rate increase, the current intensity is enhanced and there is a slight variation in the peak positions suggesting that diffusion is not the only mechanism driving the electrochemical reaction. The relation between scan rate (ν) and current (i) varies as follows:
|
logi = blogν + loga
| (6) |
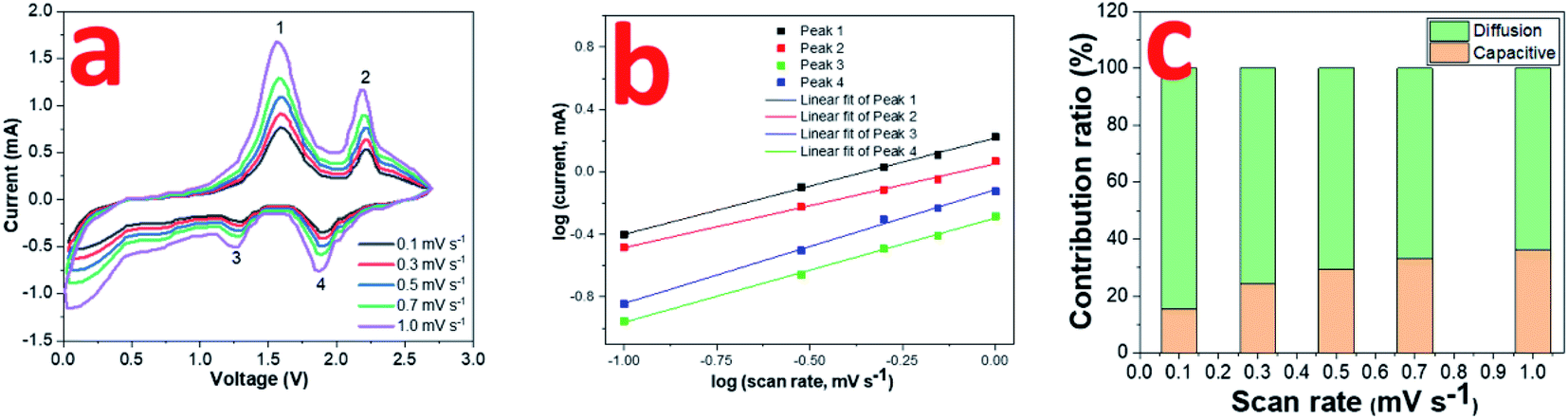 | ||
| Fig. 5 (a) CV curves of 0.8CuS:0.2g-C3N4 composite for SIBs at scan rates ranging from 0.1 to 1 mV s −1; (b) log(i) versus log(ν) plots; (c) contribution ratio of capacities at different scan rates. | ||
The value of b varies between 0.5 and 1 with b = 0.5 denoting diffusion controlled and b = 1 indicating capacitive process.12 The b values obtained from the slope of the log(i) and log(ν) plots are found to be 0.62, 0.54, 0.67 and 0.73 for peaks 1, 2, 3 and 4, respectively, which indicates both diffusion and capacitive contribution in the electrochemical process (Fig. 5b). The capacitive contribution results in the improved rate capability exhibited by the 0.8CuS:0.2g-C3N4 composite electrode whereas the dominant diffusion contribution yields higher specific capacity to it.50 The current contribution by the diffusion and capacitive process at any scan rate (ν) is given by the following equation:
| i = k1ν + k2ν1/2 | (7) |
The actual process of sodium storage in the composite has been explored by ex situ XPS studies. The high resolution Cu2p and S2p XPS spectra was recorded for CuS and 0.8CuS:0.2g-C3N4 composite cells at various potentials (1 V, 0.1 V, 2.7 V) during the first discharge and charge process (Fig. 6a–d). Compared to the before cycling plot, the Cu2p spectrum at 1.0 V discharge exhibits a broad weak satellite peak at ∼944 eV indicating the existence of Cu2+ in the sample (Fig. 6a). The Cu2p3/2 and Cu2p1/2 peaks also bifurcate confirming presence of both Cu+ and Cu2+. On further discharge up to 0.1 V, the satellite peak disappears and the Cu2p3/2 and Cu2p1/2 peaks observed at 932.9 and 953.3 eV, respectively, is due to the formation of Cu(0). Charging up to 2.7 V again shows a satellite peak at ∼944.5 eV and the Cu2p3/2 and Cu2p1/2 peaks indicate presence of both Cu+ and Cu2+. More clarity regarding the underlying process is obtained from the S2p XPS spectra (Fig. 6b). The broad peak at 169.2 eV seen in all the post-mortem analysis spectra is due to presence of sulfate which arises due to oxidation of the Na2S formed during the conversion reaction.51 Compared to the before cycling spectrum, on discharge up to 1.0 V, the broad peak in the region between ∼161–164 eV has been ascribed to the formation of polysulfides Na2Sn where n values can range from 2 to 8.42 On further discharge to 0.1 V, the peaks seen at 161.7 eV and 162.6 eV are due to Na2S and lower polysulfides, respectively.42 When charged to 2.7 V, the distinct peaks at 162.0 and 162.9 eV pertains to S2p3/2 and S2p1/2 of Cu2S.51 The broad tail of the peak up to ∼164 eV persists which indicates that the presence of polysulfides cannot be ruled out. At state of full discharge and charge, the small peak at ∼168 eV indicates the presence of Na2S in the sample.42 In case of the 0.8CuS:0.2g-C3N4 composite electrode, the fitted high resolution Cu2p XPS spectrum before cycling shows peaks pertaining to Cu+ and Cu2+, the latter being ascribed to surface oxidation (Fig. 6c). On discharge to 1.0 V, the bifurcation of the peak pertaining to Cu2p3/2 gets more pronounced while that of Cu2p1/2 gets broader. Further discharge up to 0.1 V shows presence of Cu2p3/2 and Cu2p1/2 peaks at 931.4 eV and 951.8 eV, respectively, which is on the lower binding energy side compared to that observed for the material before cycling and pertains to formation of Cu(0). Though the satellite peak at ∼944 eV is not seen, the fitted spectra shows presence of very low intensity peaks at 932.7 eV and 953.2 eV, which might be originating from some residual Cu+ or Cu2+ present in the electrode. Charging of the electrode to 2.7 V results in the formation of predominantly Cu+ with the Cu2p3/2 and Cu2p1/2 peaks at 932.1 eV and 952.1 eV, respectively. On comparing Fig. 6a and c, it can be inferred, that at state of full discharge at 0.1 V, the 0.8CuS:0.2g-C3N4 composite electrode has better conversion of Cu+ to Cu(0) compared to the pristine CuS electrode. Similar to the pristine CuS electrode, the post mortem S2p XPS spectra of the 0.8CuS:0.2g-C3N4 composite exhibits a broad peak at ∼169.2 eV indicating the presence of sulfate due to the oxidation of Na2S formed in the electrode during the discharge/charge process (Fig. 6d).51 Formation of polysulfide (Na2Sn with n = 2 to 8) is indicated due to the presence of broad peaks in the ∼161–164 eV range when the electrode is discharged at 1.0 V.41 Full discharge of the electrode at 0.1 V exhibit peaks at 161.7 and 162.5 eV pertaining to Na2S and lower polysulfides, respectively.52 On charging to 2.7 V, binding energy peaks pertaining to presence of sulfide is clearly discernible along with broad peak at ∼169.2 eV indicating presence of sulfate group. However, the Na2S peak at ∼168 eV is very weak and the main peak of Na2S at 161.7 eV merges with the 2p3/2 S2− peaks at 161.4 eV. The other peak at 162.5 eV correspond to S2p1/2 of S2−. The broad tail peak at ∼163 to 164 eV pertaining to polysulfides seen in the charged pristine CuS electrode is less conspicuous in the composite electrode. This indicates that in the 0.8CuS:0.2g-C3N4 composite, the g-C3N4 matrix helps in reducing the polysulfide leaching and similar observation has been reported in case of g-C3N4 reinforced Li–S batteries.53
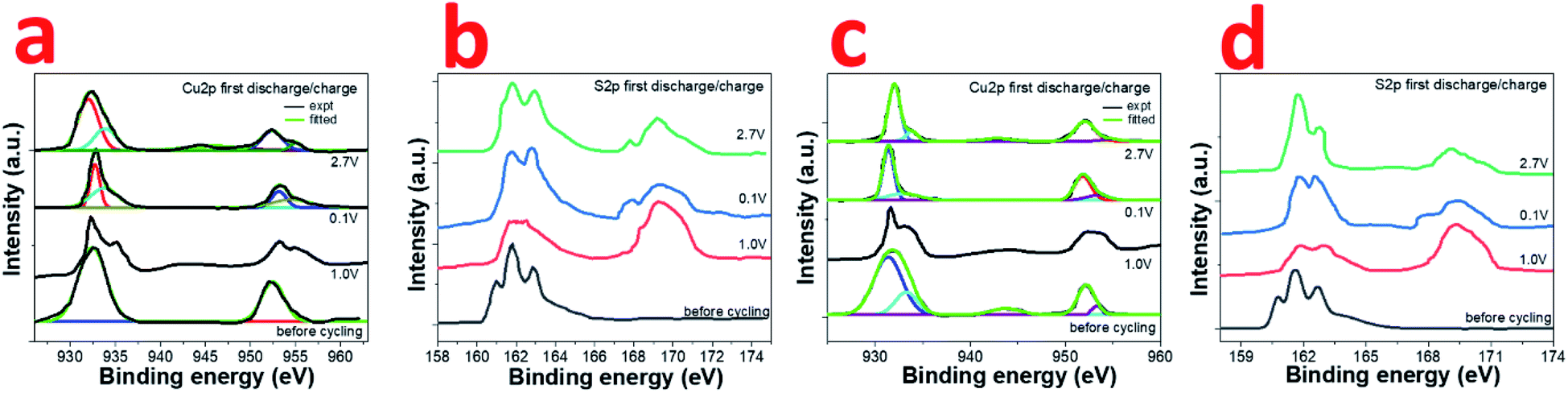 | ||
| Fig. 6 Ex situ high resolution (a) Cu2p; (b) S2p XPS of CuS electrode; (c) Cu2p; (d) S2p XPS of 0.8CuS:0.2g-C3N4 composite electrode at as prepared, discharge at 1.0 V, discharge at 0.1 V and charge at 2.7 V state. | ||
From the above ex situ XPS data, certain inferences can be drawn. The presence of Cu2+ in the post mortem samples cannot be unambiguously assigned to its formation in the electrochemical reaction process. Since the XPS experiment has been done ex situ, there is a possibility that partial oxidation of Cu+ to Cu2+ occurred during the loading of the sample. In the pristine CuS and 0.8CuS:0.2g-C3N4 composite electrode, the S2p spectrum after discharge at 0.1 V, shows presence of Cu(0) metal, Na2S and other polysulfides (Na2Sn). This indicates that the discharge process can be summed up by the following reaction:
| CuS + 2Na → Na2S + Cu |
However, the reaction occurs via intermediate steps of Na intercalation in CuS, followed by formation of Na2S and Cu(0). The presence of other polysulfides can be accounted for as Na reacts with the excess sulfur present in the sample as indicated by EDS and also by the S2p XPS spectra of it before cycling. On charging to 2.7 V, the S2p XPS of pristine CuS electrode indicates the presence of Cu2S in the sample along with Na2S and other polysulfides. The presence of Cu2S indicates that only one Na+ ion of Na2S could desodiate in the electrode during charging and this contributes to the lowering of specific capacity on cycling. However, the presence of Cu2S improves the Rct of the electrode on cycling which corroborates with the results obtained from EIS data. For the composite electrode, the S2p XPS obtained on charging to 2.7 V shows presence of sulfide groups indicating formation of Cu2S which denotes some irreversibility in the reaction. However, the specific capacity remains high and is maintained throughout the repeated cycling and this cannot be explained if only one Na+ ion is transported in the system. It is to be noted that the presence of g-C3N4 ensures that there is less loss of sulfur through polysulfide leaching and hence the capacity loss is minimized. To understand the origin of Cu2S in the sample, the composite electrode was stripped off from the current collector after 1st charge and the ratio of Cu to S was analysed using EDS. The atomic ratio obtained was 1.89:1 and the excess copper most probably gets extracted from the Cu collector as the reaction between CuS and Cu is thermodynamically feasible.15 SEM of the stripped Cu current collector has a pitted appearance exhibiting loss of Cu from the surface (Fig. S7†). Similar observation has been reported in case of CuS electrodes synthesized by direct heating of S on copper current collector strips.15 Hence, it can be inferred that in the 0.8CuS:0.2g-C3N4 composite, g-C3N4 helps in mitigating the volume expansion of CuS during the sodiation/desodiation cycles and also reduces the polysulfide leaching which enhance its electrochemical properties compared to other CuS composites reported in literature (Table S4†). The sulfiphilic nature of Cu leads to the formation of Cu2S,54 but the specific capacity is not compromised since the total amount of sulfur remains mostly unaltered in the composite electrode.
A sodium ion full cell has been assembled using NVP cathode and pre-sodiated 0.8CuS:0.2g-C3N4 anode. The construction and electrochemical performance of the NVP cathode is discussed in supplementary section (Fig. S8†). The insertion of sodium ion in the composite anode was done to reduce the loss of sodium ions during the initial cycles which impacts the full cell performance.55 The full cell was cycled at 0.1 A g−1 between the voltage range 0.2–3.0 V and the cycle performance and GDC plots are shown in Fig. 7a and b, respectively. A discharge capacity of ∼266 mA h g−1 was obtained after a 100-cycle run and a capacity retention of ∼84.9% was observed compared to the first cycle.
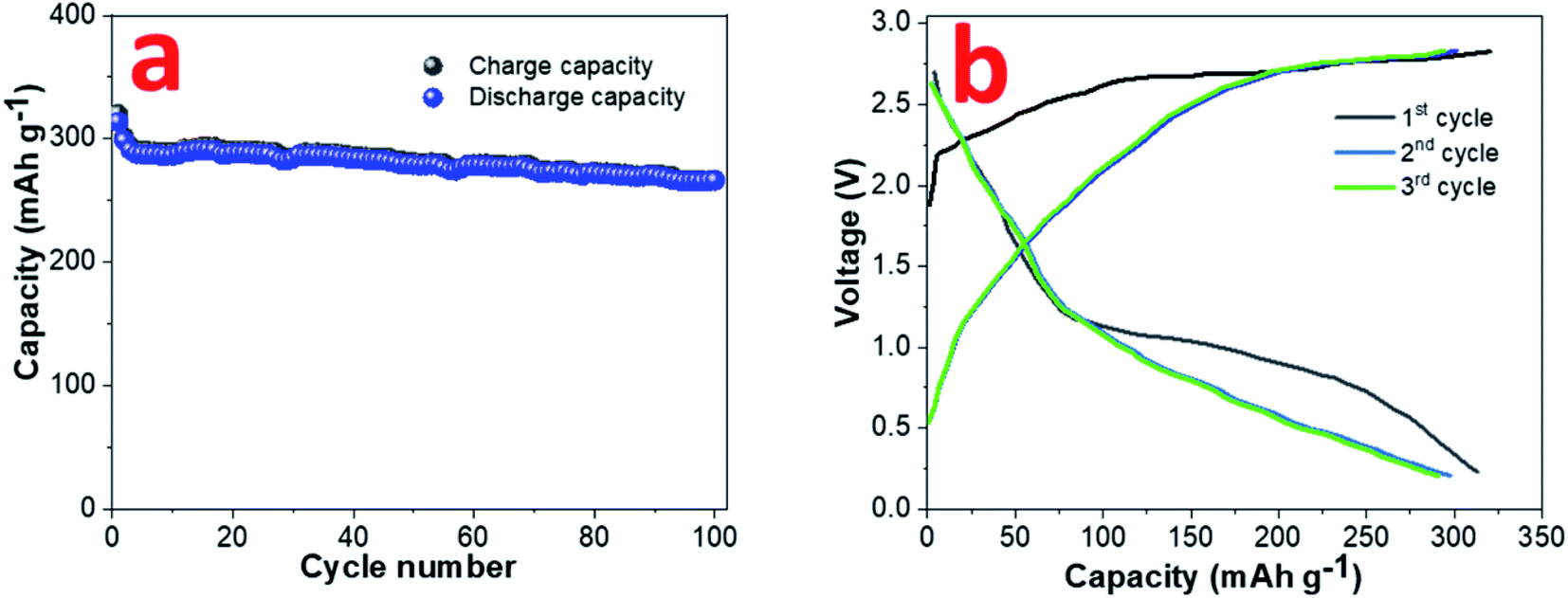 | ||
| Fig. 7 (a) Cyclability and (b) GDC plots at 0.1 A g−1 current rate for sodium ion full cell with NVP cathode and pre-sodiated 0.8CuS:0.2g-C3N4 anode. | ||
Conclusion
The synthesis of 0.8CuS:0.2g-C3N4 composite has been done via precipitation method using exfoliated g-C3N4 nanosheets as matrix for in situ dispersion of the CuS nanoparticles. The pristine CuS nanoparticles exhibits a reversible capacity of 304 mA h g−1 at 0.1 A g−1 current rate after a run of 200 cycles which improves to 478.4 mA h g−1 at 2.0 A g−1 current rate after 1000 cycles run in the 0.8CuS:0.2g-C3N4 composite when used as anode in lithium ion half-cell. In sodium ion half-cell, the composite delivers a reversible capacity of 408 mA h g−1 at 2.0 A g−1 current rate after a run of 800 cycles which is superior to that exhibited by pristine CuS (212 mA h g−1, 1.0 A g−1, 800 cycles). The presence of pyridinic N enriched exfoliated g-C3N4 improves the Li+/Na+ ion conductivity and the higher surface area and pore volume of the composite compared to pristine CuS improves the penetration of electrolyte in the anode. The even dispersion of CuS nanoparticles in g-C3N4 prevents agglomeration and shortens the diffusion length of the ions and also provides space for their movement during the discharge/charge process thereby reducing the detrimental effect of volume alteration associated with these conversion anodes. The formation of Cu2S from CuS during the electrochemical reaction has been established via ex situ XPS studies and the reduction in reversible capacity with cycling has been accounted for in case of pristine CuS anode. However, in the 0.8CuS:0.2g-C3N4 composite, the presence of g-C3N4 prevents leaching of the polysulfides but ex situ XPS indicates transformation of CuS to Cu2S. The enhanced reversible capacity observed in the composite is attributed to the prevention of polysulfide leaching and formation of Cu2S via addition of Cu in the anode from the Cu current collector. Finally, a sodium ion full cell with NVP cathode and 0.8CuS:0.2g-C3N4 anode has been constructed which yields a capacity of 266 mA h g−1 at 0.1 A g−1 after a run of 100 cycles.Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- Y. Liang, C. -Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. -Q. Huang, D. Yu, Y. Liu, M. -M. Titirici, Y. -L. Chueh, H. Yu and Q. Zhang, InfoMat., 2019, 1, 6–32 CrossRef CAS.
- J. B. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef.
- S. Choi and G. Wang, Adv. Mater. Technol., 2018, 3, 1700376 CrossRef.
- B. Jones, R. J. R. Elliott and V. Nguyen-Tien, Appl. Energy, 2020, 280, 115072 CrossRef CAS PubMed.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS.
- I. E. Moctar, Q. Ni, Y. Bai, F. Wu and C. Wu, Funct. Mater. Lett., 2018, 11, 1830003 CrossRef.
- K. Jiang, Z. Chen and X. Meng, ChemElectroChem, 2019, 6, 2825–2840 CrossRef CAS.
- Y. Xiao, J. Y. Hwang, I. Belharouak and Y. K. Sun, Nano Energy, 2017, 32, 320–328 CrossRef CAS.
- D. D. Pathak, D. P. Dutta, B. R. Ravuri, A. Ballal, A. C. Joshi and A. K. Tyagi, Electrochim. Acta, 2021, 370, 137715 CrossRef CAS.
- L. Wu, J. Gao, Z. Qin, Y. Sun, R. Tian, Q. Zhang and Y. Gao, J. Power Sources, 2020, 479, 228518 CrossRef CAS.
- L.-H. Wang, Y.-K. Dai, Y.-F. Qin, J. Chen, E.-L. Zhou, Q. Li and K. Wang, Materials, 2020, 13, 3797 CrossRef CAS.
- H. Kim, M. K. Sadan, C. Kim, S.-H. Choe, K.-K. Cho, K.-W. Kim, J.-H. Ahn and H.-J. Ahn, J. Mater. Chem. A, 2019, 7, 16239–16248 RSC.
- J. Zhang, Y. Zhao, Y. Zhang, J. Li, M.-R. Babaa, N. Liu and Z. Bakenov, Nanotechnology, 2020, 31(6pp), 095405 CrossRef CAS PubMed.
- C. An, Y. Ni, Z. Wang, X. Li and X. Liu, Inorg. Chem. Front., 2018, 5, 1045 RSC.
- H.-C. Tao, X.-L. Yang, L.-L. Zhang and S.-B. Ni, J. Phys. Chem. Solids, 2014, 75, 1205–1209 CrossRef CAS.
- C. Feng, L. Zhang, M. Yang, X. Song, H. Zhao, Z. Jia, K. Sun and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15726–15734 CrossRef CAS PubMed.
- S. Iqbal, A. Bahadur, A. Saeed, K. Zhou, M. Shoaib and M. Waqas, J. Colloid Interface Sci., 2017, 502, 16–23 CrossRef CAS PubMed.
- H. Liu, L. Zhang and H. Ruan, Int. J. Electrochem. Sci., 2018, 13, 4775–4781 CrossRef CAS.
- N. R. Kim, J. Choi, H. J. Yoon, M. E. Lee, S. U. Son, H.-J. Jin and Y. S. Yun, ACS Sustainable Chem. Eng., 2017, 5, 9802–9808 CrossRef CAS.
- J. Li, D. Yan, T. Lu, W. Qin, Y. Yao and L. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 2309–2316 CrossRef CAS PubMed.
- G. M. Veith, L. Baggetto, L. A. Adamczyk, B. Guo, S. S. Brown, X.-G. Sun, A. A. Albert, J. R. Humble, C. E. Barnes and M. J. Bojdys, Chem. Mater., 2013, 25, 503–508 CrossRef CAS.
- M. Hankel, D. Ye, L. Wang and D. J. Searles, J. Phys. Chem. C, 2015, 119, 21921–21927 CrossRef CAS.
- G.-M. Weng, Y. Xie, H. Wang, C. Karpovich, J. Lipton, J. Zhu, J. Kong, L. D. Pfefferle and A. D. Taylor, Angew. Chem., Int. Ed., 2019, 58, 13727–13733 CrossRef CAS PubMed.
- S. Wang, Y. Shi, C. Fan, J. Liu, Y. Li, X.-L. Wu, H. Xie, J. Zhang and H. Sun, ACS Appl. Mater. Interfaces, 2018, 36(10), 30330–30336 CrossRef PubMed.
- H. S. H. Mohamed, L. Wu, C.-F. Li, Z.-Y. Hu, J. Liu, Z. Deng, L.-H. Chen, Y. Li and B.-L. Su, ACS Appl. Mater. Interfaces, 2019, 36(11), 32957–32968 CrossRef PubMed.
- H. T. Huu, X. D. N. Thi, K. N. Van, S. J. Kim and V. Vo, Materials, 2019, 12, 1730 CrossRef CAS PubMed.
- X. Chen, H. Li, Y. Wu, H. Wu, L. Wu, P. Tan, J. Pan and X. Xiong, J. Colloid Interface Sci., 2016, 476, 132–143 CrossRef CAS PubMed.
- A. Khan, U. Alam, W. Raza, D. Bahnemann and M. Muneer, J. Phys. Chem. Solids, 2018, 115, 59–68 CrossRef CAS.
- Z. Xu, B. Xu, K. Qian, Z. Li, F. Ding, M. Fan, Y. Sun and Y. Gao, RSC Adv., 2019, 9, 25638 RSC.
- R. K. Anish, M. R. Panda, D. P. Dutta and S. Mitra, Carbon, 2019, 143, 402–412 CrossRef.
- S. Yadav, K. Shrivas and P. K. Bajpai, J. Alloys Compd., 2019, 772, 579–592 CrossRef CAS.
- P. Kumar and R. Nagarajan, Inorg. Chem., 2011, 50, 9204–9206 CrossRef CAS PubMed.
- T. Hurma and S. Kose, Optik, 2016, 127, 6000–6006 CrossRef CAS.
- J. Jiang, L. Ou-yang, L. Zhu, A. Zheng, J. Zou, X. Yi and H. Tang, Carbon, 2014, 80, 213–221 CrossRef CAS.
- M. Nafees, S. Ali, K. Rasheed and S. Idrees, Appl. Nanosci., 2012, 2, 157–162 CrossRef CAS.
- Y. Xie, A. Riedinger, M. Prato, A. Casu, A. Genovese, P. Guardia, S. Sottini, C. Sangregorio, K. Miszta, S. Ghosh, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2013, 135, 17630–17637 CrossRef CAS PubMed.
- W. Shuang, H. Huang, M. Liu, T. Bai, J. Zhang and D. Wang, J. Sol. State Chem., 2021, 302, 122348 CrossRef CAS.
- P. Jiménez-Calvo, C. Marchal, T. Cottineau, V. Caps and V. Kelle, J. Mater. Chem. A, 2019, 7, 14849–14863 RSC.
- Y. Xiao, D. Su, X. Wang, S. Wu, L. Zhou, Y. Shi, S. Fang, H.-M. Cheng and F. Li, Adv. Energy Mater., 2018, 8, 1800930 CrossRef.
- R. Valenzuela, M. C. Fuentes, C. Parra, J. Baeza, N. Duran, S. K. Sharma, M. Knobel and J. Freer, J. Alloys Compd., 2009, 488, 227–231 CrossRef CAS.
- H. Li, Y. Wang, J. Huang, Y. Zhang and J. Zhao, Electrochim. Acta, 2017, 225, 443–451 CrossRef CAS.
- S. Majumder, M. Shao, Y. Deng and G. Chen, J. Power Sources, 2019, 431, 93–104 CrossRef CAS.
- M. Zhou, N. Peng, Z. Liu, Y. Xi, H. He, Y. Xia, Z. Liu and S. Okada, J. Power Sources, 2016, 306, 408–412 CrossRef CAS.
- X. Xu, L. Li, H. Chen, X. Guo, Z. Zhang, J. Liu, C. Mao and G. Li, Inorg. Chem. Front., 2020, 7, 1900 RSC.
- Q. Chen, M. Ren, H. Xu, W. Liu, J. Hei, L. Su and L. Wang, ChemElectroChem, 2018, 5, 2135–2141 CrossRef CAS.
- H. Kim, M. K. Sadan, C. Kim, S.-H. Choe, K.-K. Cho, K.-W. Kim, J.-H. Ahn and H.-J. Ahn, J. Mater. Chem. A, 2019, 7, 16239–16248 RSC.
- W. Shuang, H. Huang, M. Liu, T. Bai, J. Zhang and D. Wang, J. Solid State Chem., 2021, 302, 122348 CrossRef CAS.
- Q.-Q. Pan and H.-Q. Peng, Adv. Mater. Sci. Eng., 2018, 4656424 Search PubMed.
- I. Mínguez-Bacho, M. Courté, C. Shi and D. Fichou, Mater. Lett., 2015, 159, 47–50 CrossRef.
- X. Wang, G. Li, M. Li, R. Liu, H. Li, T. Li, M. Sun, Y. Deng, M. Feng and Z. Chen, J. Energy Chem., 2021, 53, 234–240 CrossRef.
- P. Li, L. Ma, T. Wu, H. Ye, J. Zhou, F. Zhao, N. Han, Y. Wang, Y. Wu, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1800624 CrossRef.
- D. Zhao, M. Yin, C. Feng, K. Zhan, Q. Jiao, H. Li and Y. Zhao, ACS Sustainable Chem. Eng., 2020, 8(30), 11317–11327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02014a |
| This journal is © The Royal Society of Chemistry 2022 |