DOI:
10.1039/D2RA01969H
(Paper)
RSC Adv., 2022,
12, 22108-22118
Anti-multiple myeloma potential of resynthesized belinostat derivatives: an experimental study on cytotoxic activity, drug combination, and docking studies†
Received
27th March 2022
, Accepted 18th July 2022
First published on 10th August 2022
Abstract
Multiple myeloma is a deadly cancer that is a complex and multifactorial disease. In the present study, 12 belinostat derivatives (four resynthesized and eight new), HDAC inhibitors, were resynthesized via either Knoevenagel condensation, or Wittig reaction, or Heck reaction. Then an evaluation of the antiproliferative activities against myeloma cells MOPC-315 was carried out. Amongst them, compound 7f was the most bioactive compound with an IC50 of 0.090 ± 0.016 μM, being 3.5-fold more potent than the reference belinostat (IC50 = 0.318 ± 0.049 μM). Furthermore, we also confirmed the inhibitory activity of 7f in a cellular model. Additionally, we found that the inhibitory activity of 7f against histone deacetylase 6 catalytic activity (HDAC6) is more potent than that of belinostat. Finally, we observed the strong synergistic interaction between the derivative 7f and the proteasome bortezomib inhibitor (CI = 0.26), while belinostat and bortezomib showed synergism with a CI value of 0.36. Taken together, the above results suggest that 7f is a promising HDAC inhibitor deserving further investigation.
1. Introduction
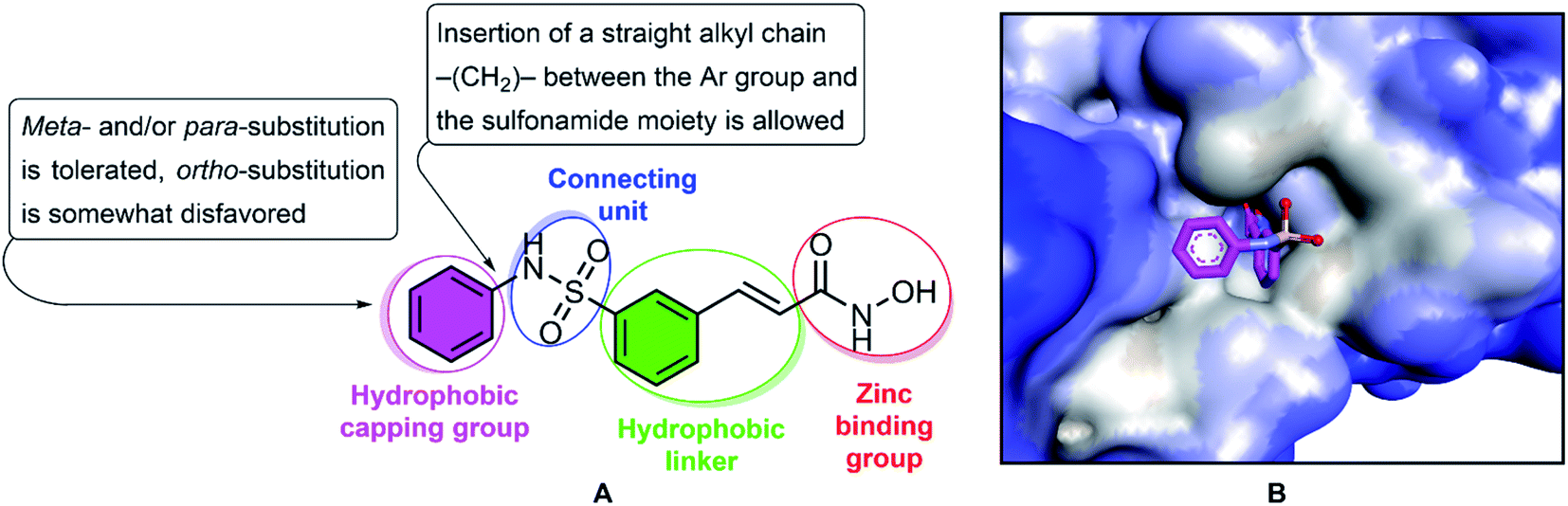
Multiple myeloma is a hematologic malignancy that occurs mainly in elderly patients, and is often relapsed and refractory, and the median survival of each patient is less than a year.1–3 Chemotherapy is one of the major strategies for cancer treatment, and functions by targeting the physiological characteristics of cancer cells, including proliferation, angiogenesis, apoptosis, invasion and migration.4,5 Histone acetylation and deacetylation specifically play an important role in regulating gene expression, influencing the transcription of many genes.6–8 Acetylation is a major form of protein post-translational modification and is responsible for regulating various cellular processes, including cell proliferation and cell survival.9 Acetylation is catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs transfer acetyl groups to lysine residues, whereas HDACs remove acetyl groups from lysine residues.10 Deregulation of histone results in abnormal gene expression profiles involved in controlling cell proliferation, differentiation, and apoptosis of cancer cells and is basically associated with malignancies, including multiple myeloma. HDACs are overexpressed in many types of hematologic malignancies (acute myelogenous leukemia, myelofibrosis, cutaneous T-cell lymphoma, and Hodgkin lymphoma), which inhibition of HDACs can result in the inhibition of cell proliferation and induction of apoptosis.2,11,12 Thus, histone deacetylase inhibitors might represent a novel therapeutic for anticancer drugs that actually alter gene expression. Several HDAC inhibitors have been studied for the treatment of multiple myeloma, including trichostatin A, vorinostat, belinostat, depsipeptide, KD5170, NVP-LAQ824, VPA, ricolinostat and panobinostat.2,13 In 2014, belinostat passed all preclinical and clinical trials with results exceeding expectations and has received FDA market approval for the treatment of patients with relapsed and kind of refractory peripheral T-cell lymphomas (PTCL).14,15 Typically, HDAC inhibitors, such as belinostat, share the same pharmacophore model, consisting of a zinc-binding group (ZBG) to bind the zinc ion at the active site of HDACs, a surface recognition domain (cap group), and a linker that enters the hydrophobic tunnel connecting ZBG to the cap group (Fig. 1).16–18 Various studies have been reported to focus on altering the cap, connective unit (CU), linker, and zinc binding portions of HDAC inhibitors in recent years.19–25 In the structure of belinostat, N-hydroxycinnamamide serves as both the ZBG and the linker group. The hydroxamic acid functional group and the E double bond are essential for activity, and exchange of the E double bond to the Z double bond or the triple bond leads to inactive compounds. The CU of belinostat is a sulfonamide, which has cap–NHSO2–linker is more active than cap–SO2NH–linker and amide. Therefore, replacement is not considered feasible.18 In the present study, we describe the resynthesis of belinostat analogs HDAC inhibitors by keeping the carbon bridge part, CU, and the hydroxamic function group intact while the phenyl frame of belinostat is replaced by ortho- and/or meta- and/or para-substituted phenyl or benzyl. The compounds were evaluated for their antiproliferative potency towards MOPC-315 multiple myeloma cells (in vitro) and the most potent derivative was tested for synergistic interaction with the proteasome bortezomib inhibitor. The derivatives were considered for potential pan-HDAC and HDAC6 inhibition activity based on in vitro and in silico study. This study provided the underlying mechanisms for the use of HDAC inhibitors in the treatment of multiple myeloma and suggested potential therapeutic compounds for future preclinical implications.
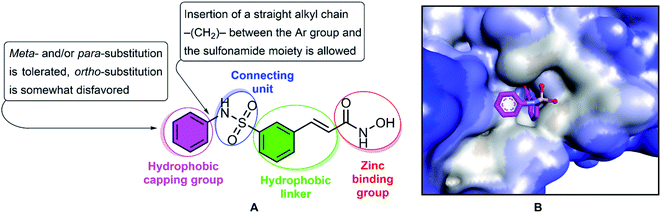 |
| | Fig. 1 The structure and pharmacophoric model of belinostat. HDAC inhibitory SAR (A). Crystal structures of HDAC6-belinostat complexe (PDB ID: 5EEN) (B). | |
2. Results
2.1. Organic synthesis of belinostat analogs
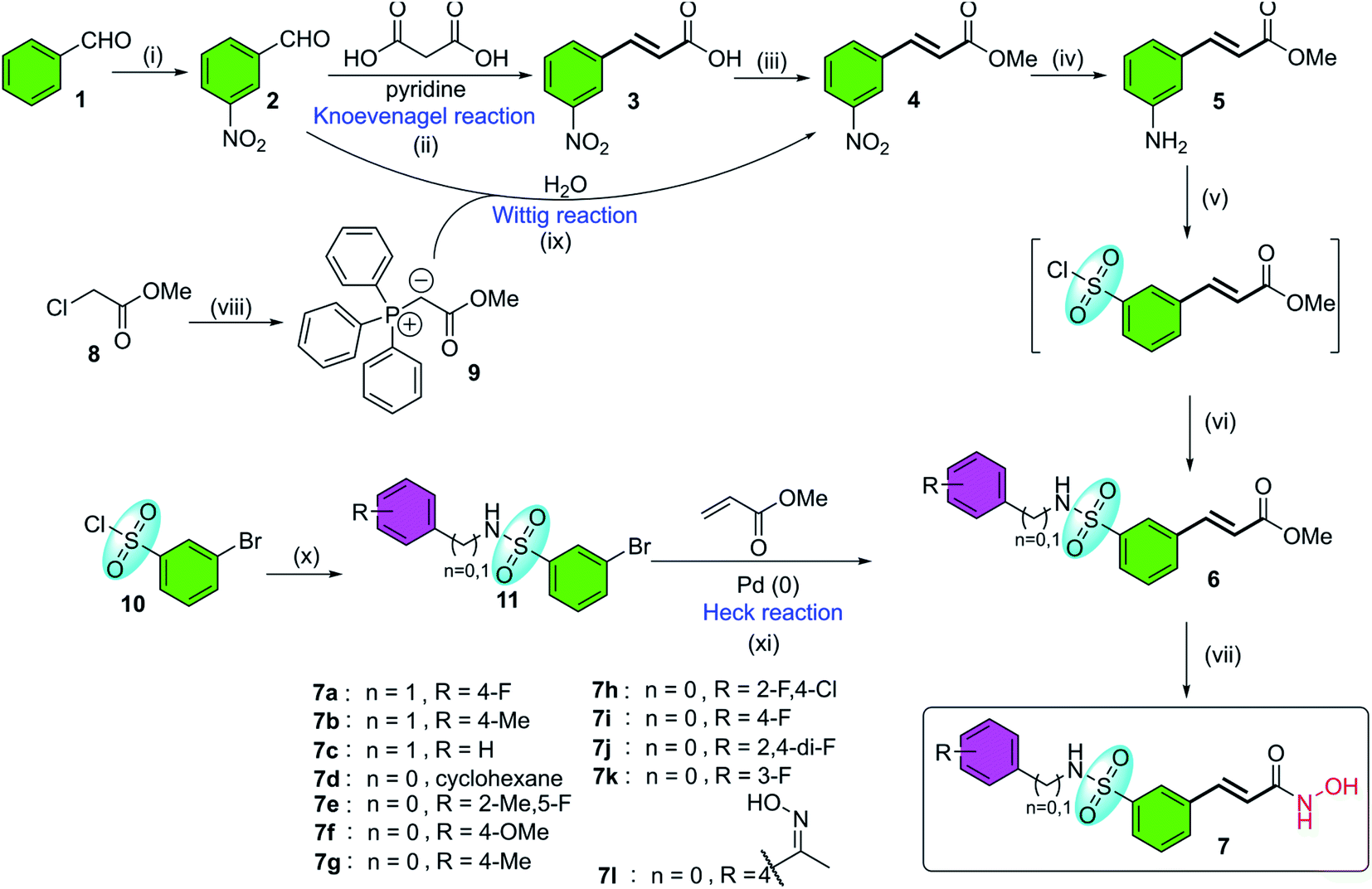
Belinostat analogs were successfully resynthesized through a simple and effective process suited to the laboratory scale that included three to seven steps (Fig. 2).
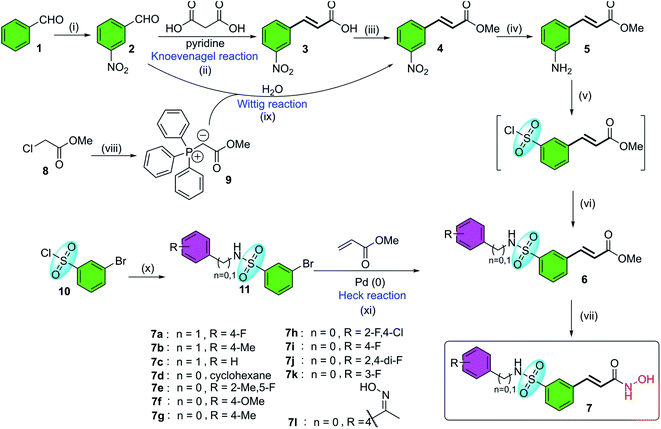 |
| | Fig. 2 The general synthetic procedures of belinostat analogs. (i) KNO3, H2SO4, 0 °C; (ii) reflux, 110 °C; (iii) MeOH, H2SO4, 80 °C; (iv) SnCl2, EtOH, 90 °C; (v(a)) NaNO2, HCl, HOAc, 0 °C; (b) SO2 gas, CuCl, HOAc, H2O, 0 °C; (c) phenyl or benzyl-amine, 1,4-dioxane, rt; (vi) NH2OH·HCl, EtOH, 0 °C; (vii(a)) Ph3P, H2O, 70 °C; (b) NaOH, rt; (viii) H2O, 90 °C; (ix) phenyl or benzyl-amine, DMAP, toluene, 50 °C; (x) triethylamine, toluene, 45–90 °C. | |
The first reaction of the procedure is the nitration reaction employing available commercial materials, including benzaldehyde and the mixture of potassium nitrate and concentrated sulfuric acid. The nitration reaction is followed by Knoevenagel condensation between 2, and malonic acid as a nucleophile. Pyridine is also added as a basic reagent. One advantage of Knoevenagel's employment is that it is highly stereoscopic selective. Before a chain of reactions occurs on the nitro group, the esterification reaction is applied to protect the carboxylic acid group of the Knoevenagel adduct. Alternatively, compound 4 can be prepared through the Wittig reaction generated from starting compound 2. Soon after, a reduction of the –NO2 group was carried out to convert it to an amine functional group. The next step is the transformation from the amine group to the sulfonamide group (it should be noted that the introduction of the sulfonyl chloride group into the aromatic ring is considered the most challenging but vital task of the whole procedure). Finally, preparation of hydroxamates was completed by reacting with hydroxylamine hydrochloride. Furthermore, the synthesis of belinostat derivatives is possible by a 3-step process derived from 3-bromobenzenesulfonyl chloride. However, the starting agent was not readily available and the reaction conditions were harsh. The structures of the final products were confirmed by 1H-NMR, 13C-NMR and HRMS spectra (see ESI†). When using 4′-aminoacetophenone as the starting amine material, a 7l hydroxamate containing a ketoxime moiety is formed when the carbonyl compound 6l is treated with hydroxyl amine (see reaction mechanisms in the ESI†).
2.2. Cytotoxicity screening
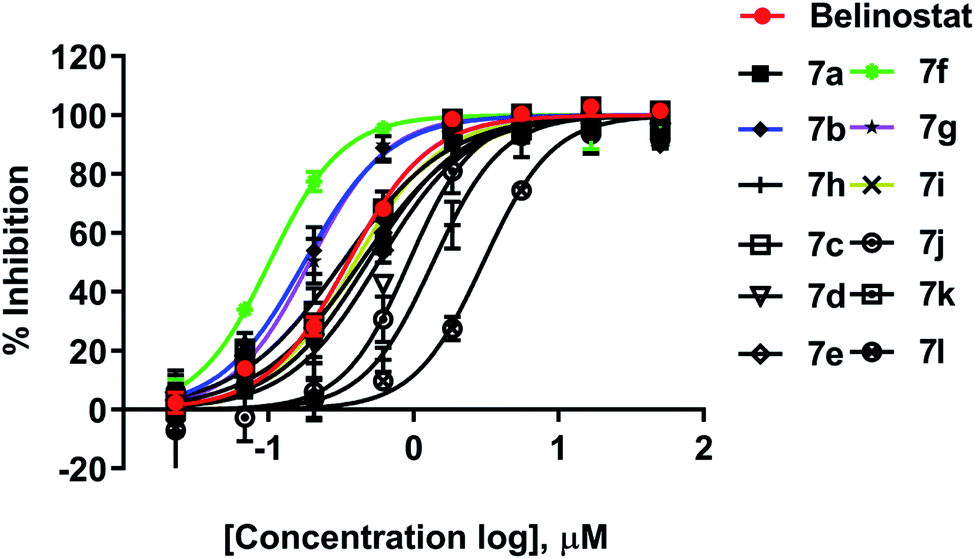
All synthesized compounds were evaluated for their antiproliferative potency toward multiple myeloma cells. Dose–response curve analysis of the selected belinostat is shown in Fig. 3. Among twelve compounds tested, 7b, 7f, and 7g showed significant cytotoxic activity (IC50 = 0.167, 0.090 and 0.159 μM, respectively) with a range from 1.9 to 3.5-fold more toxic than the reference belinostat (IC50 = 0.318 μM) (Table 1), in particular, compound 7f was found to be the most bioactive molecule in this study. In addition, obtained results indicated that compounds 7c and 7i exhibit comparable activity to belinostat, the rests were found to exert the least inhibitory potency on cell proliferation.
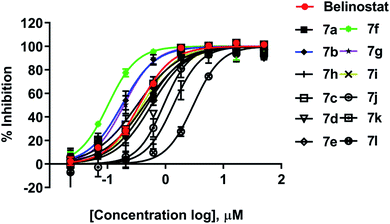 |
| | Fig. 3 Dose–response curve analysis of belinostat for anti-cancer activity against MOPC-315. | |
Table 1 Cytotoxicity of belinostat derivatives against MOPC-315 cell line
|
| Compd |
A position |
IC50 (μM) |
Fold change |
| Compound 7f was chosen for further studies. Belinostat was used as a positive control. |
| 7a |
4-Fluorobenzyl |
0.457 ± 0.033 |
0.695 |
| 7b |
4-Methylbenzyl |
0.167 ± 0.014 |
1.907 |
| 7c |
Benzyl |
0.291 ± 0.044 |
1.093 |
| 7d |
Cyclohexyl |
0.444 ± 0.181 |
0.714 |
| 7e |
5-Fluoro-2-methylphenyl |
0.380 ± 0.073 |
0.835 |
| 7fa |
4-Methoxyphenyl |
0.090 ± 0.016 |
3.522 |
| 7g |
4-Methylphenyl |
0.159 ± 0.046 |
1.993 |
| 7h |
4-Chloro-2-fluorophenyl |
1.029 ± 0.469 |
0.309 |
| 7i |
4-Fluorophenyl |
0.312 ± 0.114 |
1.019 |
| 7j |
2,4-Difluorophenyl |
0.784 ± 0.198 |
0.405 |
| 7k |
3-Fluorophenyl |
0.365 ± 0.019 |
0.869 |
| 7l |
(Z)-4-(1-(Hydroxyimino)ethyl)phenyl |
2.141 ± 1.246 |
0.148 |
| Belinostatb |
Phenyl |
0.318 ± 0.049 |
1.000 |
2.3. Combination study
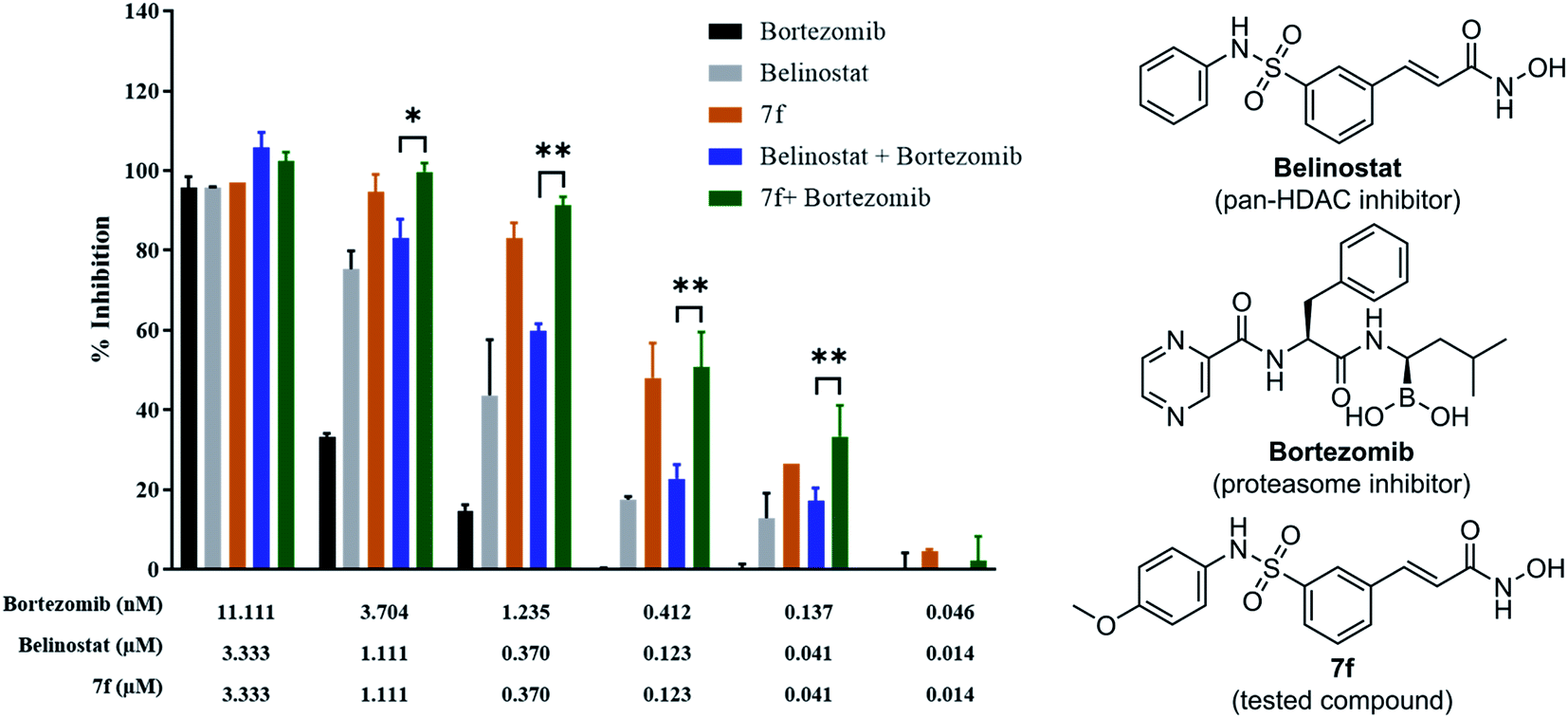
To determine whether synergism between 7f and bortezomib occurred in multiple myeloma cells, as previously observed between belinostat and bortezomib,26 MOPC-315 cells were exposed (24 h) from micro-molar to sub-micromolar concentrations in the presence or absence of low concentrations (0.046 to 11.11 nM) of 7f and belinostat as a control, after which cell proliferation was monitored by cytotoxicity test. In all cases, single-agent treatment was shown with minimal to modest toxicity, whereas combined exposure resulted in a pronounced increase in cell death, indicating a synergistic interaction (Fig. 4 and Table 2). The combination index (CI) values were calculated by Chou's method.27 The combined effect of 7f and bortezomib (strong synergism with CI value = 0.26) is significantly higher than that of belinostat and bortezomib (synergism with CI value = 0.36). The result suggests that the combination of 7f and bortezomib is more potent than that of belinostat and bortezomib.
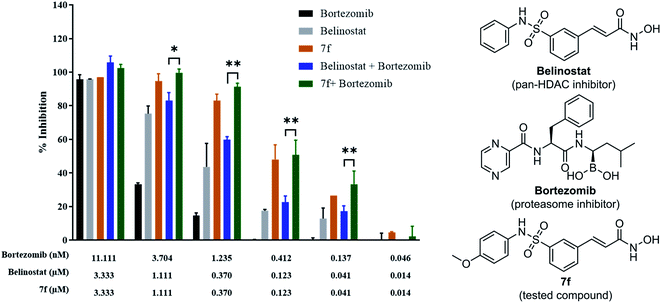 |
| | Fig. 4 Synergistic inhibition effect of 7f and bortezomib on proliferation of multiple myeloma cells. | |
Table 2 Drug–drug combinations
| Drug combination |
CI valuesa at inhibition of |
Weighted average CI valuesb |
Assigned symbolc |
Description |
| 50% |
75% |
90% |
95% |
| CI values are based on the combination index isobologram equations: CI = [(D)1/(Dx)1] + [(D)2/(Dx)2], where Dx = Dm[fa/(1 − fa)]1/m. Because the high degrees of effects are more important than the low degrees of effects, the weighted CI value was designed as: CIwt = [CI50 + 2CI75 + 3CI90 + 4CI95]/10. Degree of synergism or antagonism are based on the ranges of CI values as described in Pharmacological Reviews.27 |
| Belinostat + bortezomib |
0.488 |
0.410![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
0.344 |
0.305 |
0.36 |
+++ |
Synergism |
| 7f + bortezomib |
0.572 |
0.359 |
0.225 |
0.164 |
0.26 |
++++ |
Strong synergism |
2.4. HDAC activity analysis
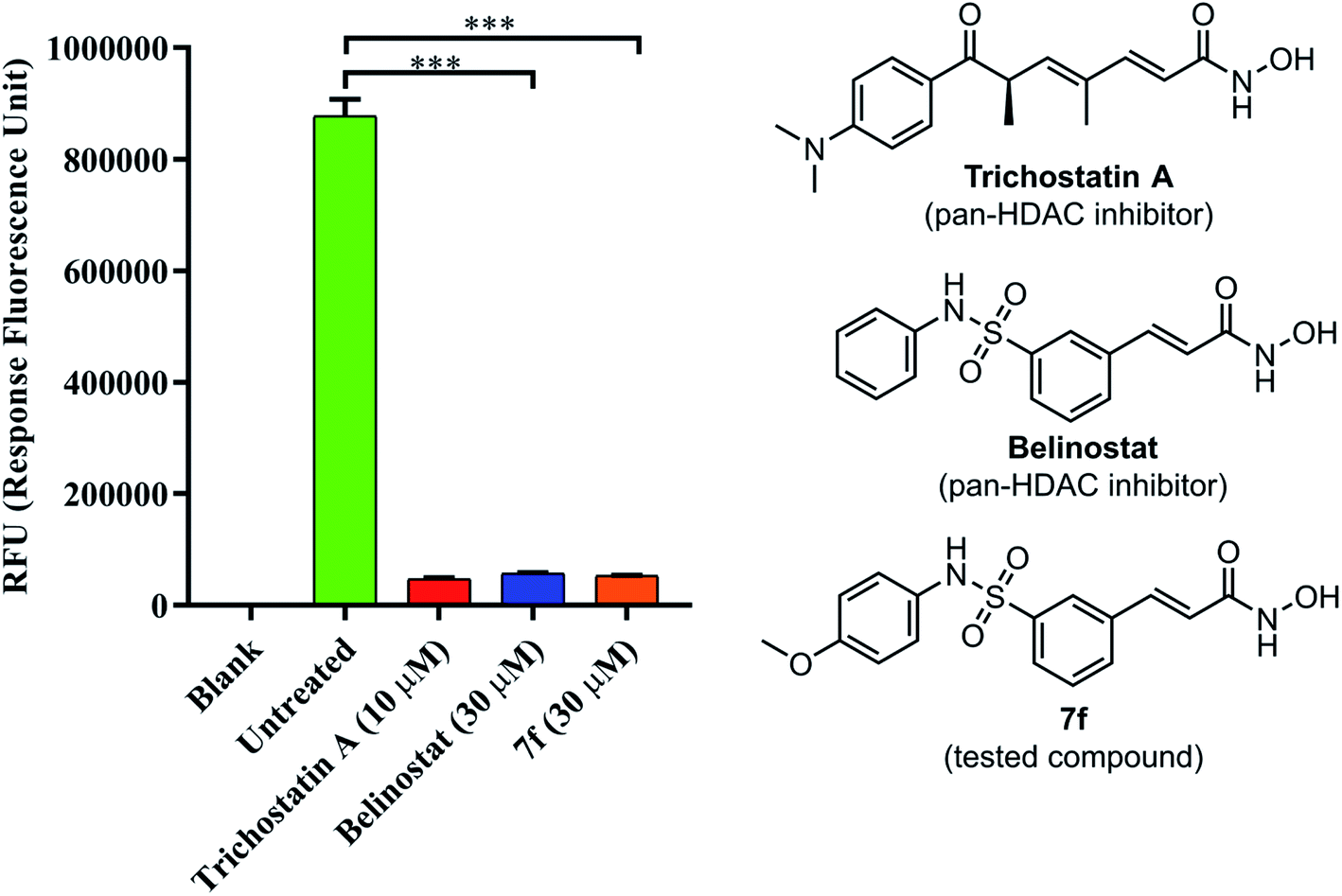
To determine if 7f could inhibit a global histone deacetylase (HDAC) activity, a fluorescence-based assay for HDACs was employed. As shown in Fig. 5, HDAC activity corresponded to the high fluorescence signal obtained in the control group; however, the activity was decreased in presence of belinostat or 7f. The assay showed the ability of compound 7f to inhibit HDACs that can deacetylate histone.
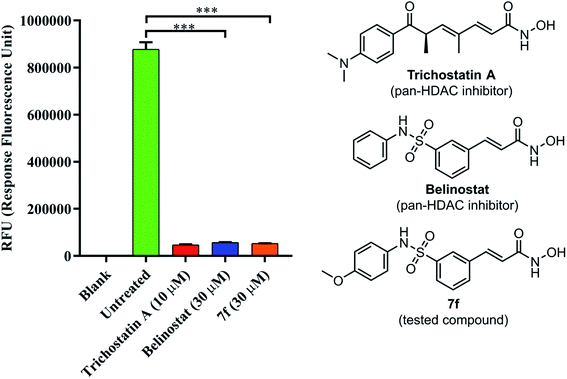 |
| | Fig. 5 The pan-HDAC inhibitory activity of belinostat and 7f. Blank: cell only; untreated: cell and developer mix; trichostatin A as a positive control. | |
2.5. In vitro inhibitory activity against HDAC6 enzyme
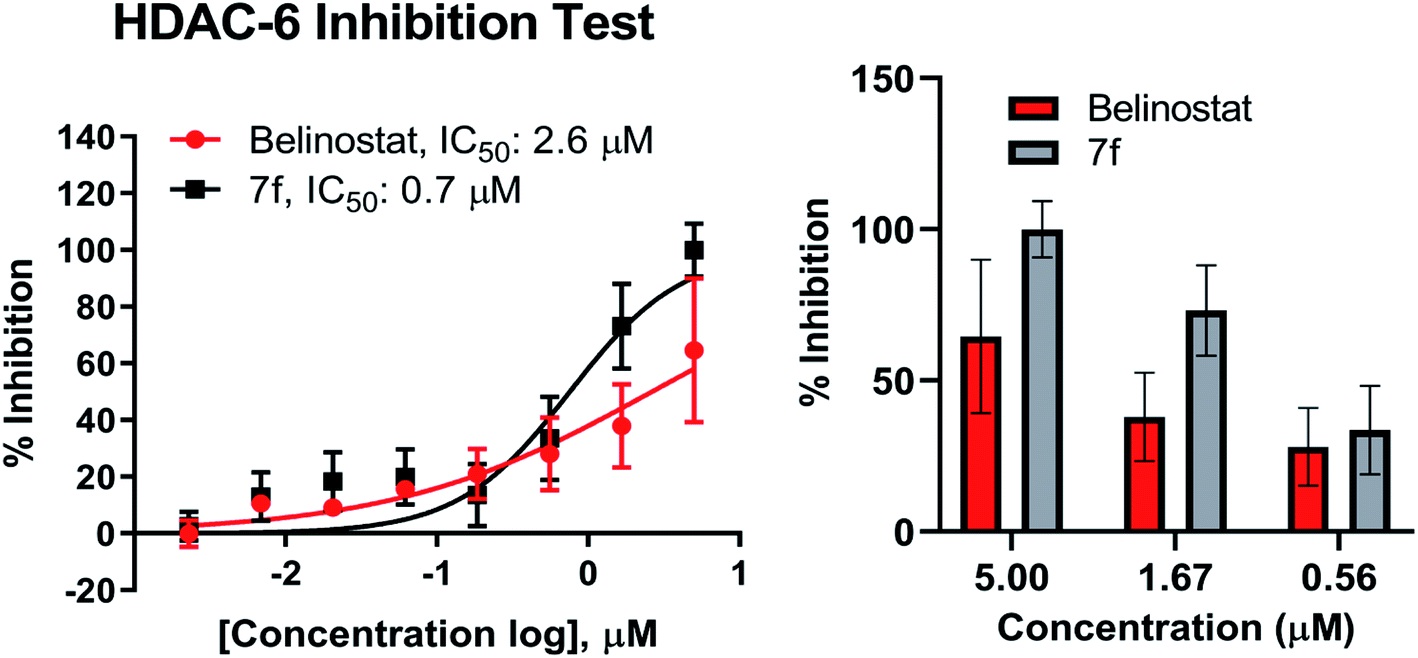
Compound 7f was evaluated for its inhibitory activity against HDAC6 isoform, in which belinostat was utilized as the positive control. Compound 7f exhibited inhibitory activity against HDAC6 in a dose-dependent manner in the concentration range of 2 nM to 5 μM (Fig. 6). Compound 7f reached an IC50 value of 0.7 μM while belinostat did a higher IC50 value of 2.6 μM.
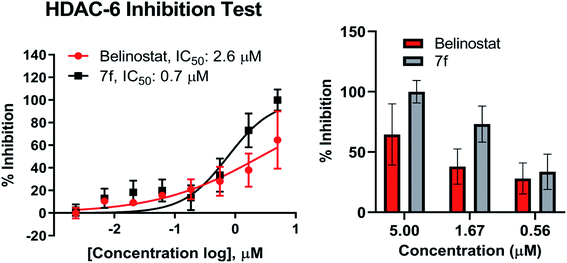 |
| | Fig. 6 Dose–response curve analysis of 7f for inhibitory activity against HDAC6. IC50 values were calculated by 8-point DRC analysis and experiments were repeated at least twice independently. | |
2.6. Docking protocol
Abnormal HDAC6 enzyme activity is associated with multiple myeloma manifestations; therefore, inhibition of HDAC6 is considered a therapeutic target for multiple myeloma.13,28,29
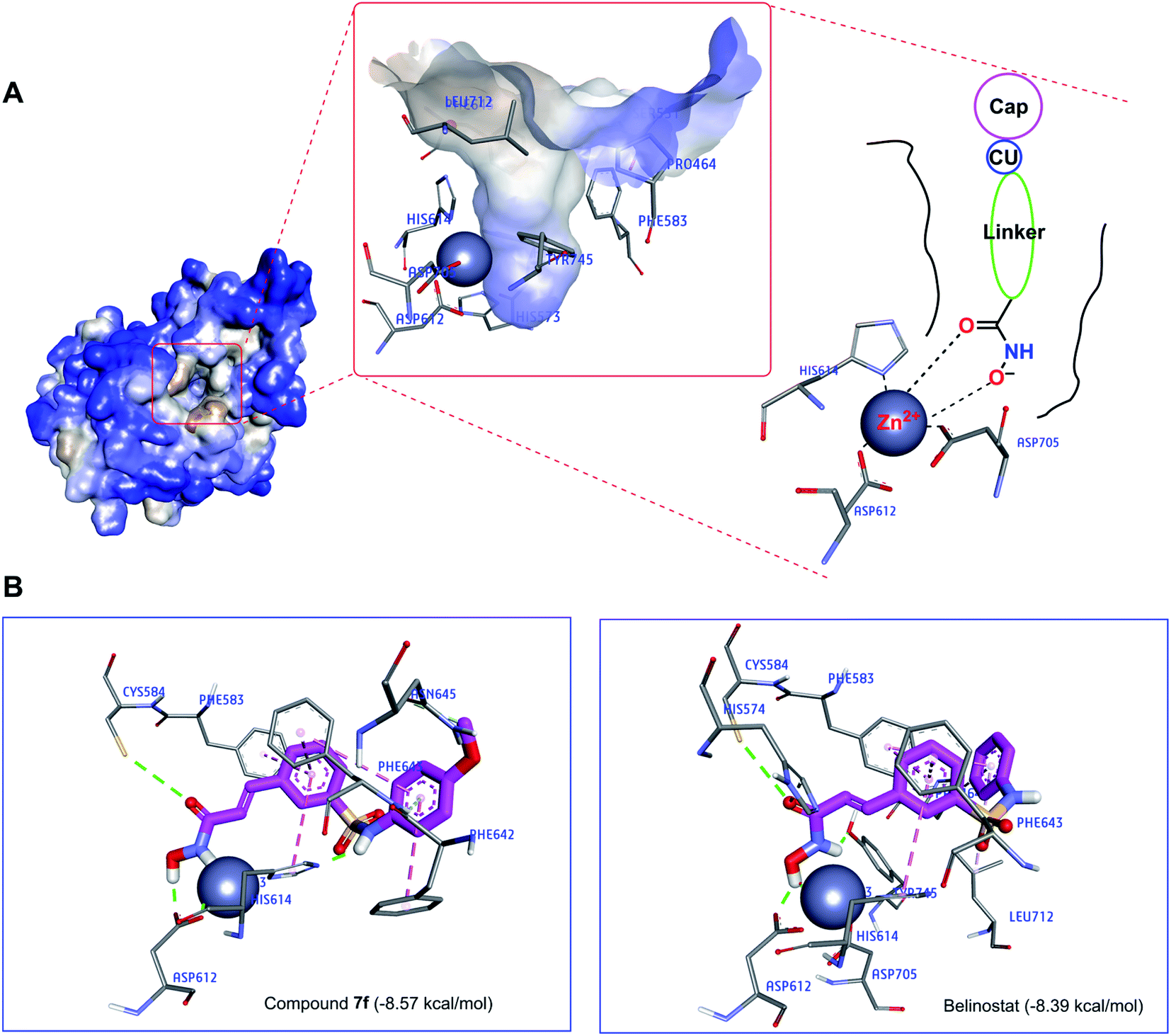
Recently, the well-known antineoplastic compound ricolinostat, a selective HDAC6 inhibitor with an IC50 value of 5 nM, has shown to be a novel potential candidate for the treatment of multiple myeloma.28,30 The compound 7f displayed HDAC6 inhibitor from fluorescence-based assay (Fig. 6). For a better understanding of the molecular interaction between 7f and enzyme HDAC6, which were docked to the active site of HDAC6 using Autodock4Zn. The three-dimensional structures have been reported by Gohlke et al. that ligands calculated partial charge using PM6 basic function could significantly increase docking accuracy and cluster population of the most accurate docking.31 According to the ranking criteria of Autodock, the more negative value of docking energy, the better binding affinity of the compound towards targeted receptor.32,33 Obtained dock score for belinostat was −8.39 kcal mol−1, thus, any molecules whose docking energies are close or more negative than this threshold would be considered to have a high binding affinity toward HDAC6 and potential to inhibit the function of this enzyme. Panobinostat was approved by the FDA for the treatment of recurrent multiple myeloma, it was also considered the second control.34 Table 3 presents dock results of 7f in combination with the ligand efficiency value (LE). Potential lead compounds when the LE value was greater than 0.3.35 In addition, the redocking results of the control are considered reliable when the RMSD value does not exceed 2.0 (ESI†). In the docked result, compound 7f exhibited the dock score of −8.57 kcal mol−1, suggesting that it has the high binding affinity to the HDAC6 enzyme. The binding mode analysis of the reference inhibitor, belinostat, revealed that Pro464, His574, Phe583, Cys584, Asp612, His614, Phe643, Asp705, Leu712, Tyr745 are key residues of HDAC6 that participated in the formation of interaction with this ligand. Previous studies have indicated that key interactions for HDAC inhibitor binding, in addition to those involving ZBG, appear to be pi–pi interactions between the central aromatic scaffold and both phenyllalaninine (Phe583, Phe643). The HDACs inhibitors are embedded in a lipophilic channel whose walls consist of Pro464, Ser531, His573, His574, Phe583, Asp612, His614, Phe643, Asp705, Leu712, Tyr745, which are assumed as important for inhibitory activities (Fig. 7A).36 HDAC inhibition depends on ZBG, the two oxygen atoms of ZBG coordinate with zinc, which participates in hydrogen bonds with His573, His574, Cys584, Asp612, Asp705 and Tyr745 by van der Waals forces. Some studies reported that two contiguous histidines, one aspartate and one tyrosine were observed to form hydrogen bonds with the ZBG.28,36,37 Compounds that bind HDAC6 at the active site were displayed relatively similar characteristics. Dock pose analysis of selected molecules showed that, in general, the pi–pi stacking establishment between the centre aromatic group and Phe583/Phe643 helps to stabilize the linker in the pocket. Another bond which is the pi–pi stacked interaction with His614 also contributes to the stabilization of the complex. Additionally, substituted cap group presents significant interactions including pi-donor and pi–pi stacked interaction with Asn645, Phe643, and Phe642 (Fig. 7B). These distinct characteristics may be the key to the lead HDAC6 inhibitors designed for the treatment of multiple myeloma. By combining both the docking score and the analysis criteria of the binding mode, the compound 7f was assumed to have the high binding affinity towards the targeted enzyme, which formed H-bond interactions with essential residues at the active site. An array of hydrophobic interaction was observed as contributed that further stabilizing the interaction in the binding site.
Table 3 Interaction residues of compound 7f obtained from molecular docking simulation
| Compound |
BEa (kcal mol−1) |
Interacting residuesb |
ZBG⋯Zn2+ distancec (Å) |
LEd |
Binding energy. The results were analyzed using Discovery Studio Visualizer. Choose the shortest distance between HO⋯Zn2+ or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Zn2+ interactions. Ligand efficiency; compounds with LE > 0.3 as potential lead compounds. Control. O⋯Zn2+ interactions. Ligand efficiency; compounds with LE > 0.3 as potential lead compounds. Control. |
| 7f |
−8.57 |
His574, Phe583, Cys584, Asp612, His614, Phe642, Phe643, Asn645 |
1.35 |
0.36 |
| Belinostate |
−8.39 |
Pro464, His574, Phe583, Cys584, Asp612, His614, Phe643, Asp705, Leu712, Tyr745 |
2.25 |
0.38 |
| Panobinostate |
−8.97 |
His463, Pro464, His573, Phe583, Cys584, Asp612, His614, Phe643, Asp705, Tyr745 |
1.71 |
0.35 |
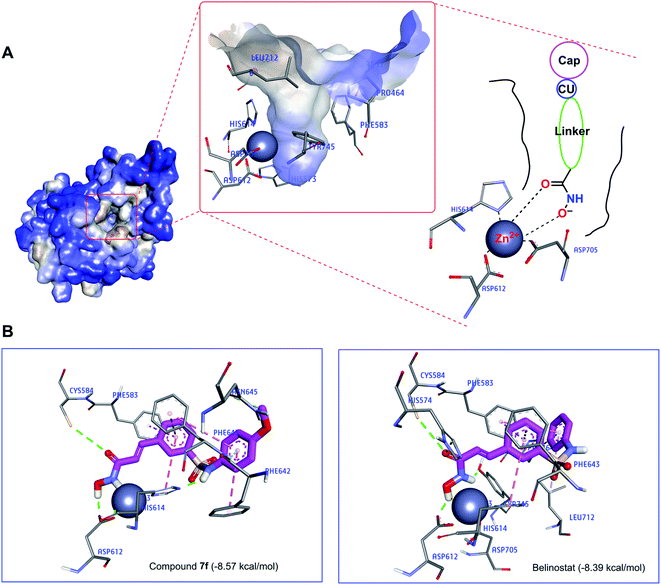 |
| | Fig. 7 (A) The active site of HDAC6 enzyme (blue light for hydrophilic to brown light for hydrophobic). (B) Interaction of compounds (7f and belinostat) in the HDAC6 binding site suggested by molecular docking studies. | |
3. Discussion
Multiple myeloma is a hematological B cell malignancy and is still considered as an incurable disease. In previous studies, HDAC-mediated inhibition of non-histone targets was found to cause anti-myeloma effects and HDAC inhibitors also have emerged as a novel therapy in the treatment of such hematological cancers.38 Belinostat is a potent HDAC inhibitor, tested as monotherapy for the treatment of multiple myeloma.39 Our study provided evidence that belinostat derivatives are effective against multiple myeloma cell line (MOPC-315). Twelve derivatives were selected and resynthesized to use as starting materials for cytotoxicity and drug combination assay of multiple myeloma cells. Four compounds (7c, 7f, 7g, 7k) were selected from a previous study of Finn et al.,40 which displayed as potent HDAC enzyme inhibitors at low inhibitory nanomolars. Based on cytotoxicity screening results and different substituents at the cap position of belinostat, compounds were classified into three groups. Group I consists of three strong active compounds, which ranged from 1.907 to 3.522-fold including 7b, 7f and 7g (IC50 = 0.167, 0.090, and 0.159 μM, respectively). The cap of this compounds bearing only an electron-donating group (methoxyl or methyl), the presence of the –OCH3 group at the para-phenyl position (7f) resulted in a 3.5-fold increase in inhibition compared with the belinostat compound (IC50 = 0.318 μM). Compounds 7c (IC50 = 0.291 μM) and 7i (IC50 = 0.312 μM) have almost the same activity as belinostat (1.019 to 1.093-fold), which is classified in group II. The remaining compounds that show less activity than belinostat are classified in group III, however, the IC50 value of the compounds still reached the micromolar value. Substituted phenyl cap group of belinostat including simple benzyl (7c) or 4-fluorophenyl (7i) did not reduce the inhibition of the parent drug. However, substituted 4-fluorobenzyl (7a, IC50 = 0.450 μM) reduces the potency of the generic drug significantly. Surprisingly, the compound 7j (IC50 = 0.784 μM) with the 2,4-difluorophenyl showed poor active as compared to its analog 7i (IC50 = 0.312 μM) without the substituted ortho-fluoro atom. The ketoxime group present in the structure of 7l showed a 6.5-fold decrease in activity (IC50 = 2.141 μM). In summary, the structure–activity relationship (SAR) of belinostat derivatives against multiple myeloma cell lines was considered as follows: para-substitution patterns across the aromatic ring by electron-donating group of cap lead to highly potent compounds, the meta or ortho or both-substitution leads to poor active compounds. Insertion of a straight alkyl chain –(CH2)– between the Ar group and the sulfonamide moiety is allowed if the Ar group has electron-donating group in para-substitution.
The development of medicine and the combination of multiple functions of drugs to treat diseases is getting good results. Specifically, belinostat is used in combination with 17-AAG as therapy for MDA-MB-231 breast cancer cells.41 Belinostat was combined with carboplatin and paclitaxel for good results in metastatic lung cancer.42 The human acute myeloid leukaemia, multiple myeloma, and lymphoblastic leukaemia, belinostat was synergized with proteasome inhibitor bortezomib, eicosanoid biosynthesis inhibitor dexamethasone.26,39 Therefore, compound 7f was also evaluated for its synergistic effect with the protease inhibitor bortezomib against the MOPC-315 multiple myeloma cells (Table 2). Interestingly, additive effects between 7f and bortezomib were shown to be stronger than the combination of belinostat and bortezomib. This finding also suggests a further animal study on this combination of bortezomib and 7f in the future.
Belinostat is known as a hydroxamate-based pan-HDAC inhibitor. It, thus, prompted us to examine 7f and belinostat in the same assay against a pan-HDAC. To verify the pan-HDAC inhibition of 7f, we have tested 7f using a fluorometric HDAC activity assay kit. As described by the manufacturer, using an assay kit provided a fast and fluorescence-based method that eliminates radioactivity, extractions, or chromatography, as used in traditional assays, which is well suited for high throughput screening applications. The results showed that 7f strongly inhibited HDACs, almost even more potent than belinostat.
Recent studies have demonstrated that aberrant HDAC6 enzyme activity may be responsible for multiple myeloma.28 The belinostat and panobinostat (pan-HDAC inhibitor) showed high efficiency in the treatment of multiple myeloma.28,43,44 Recently, ricolinostat, a selective inhibitor of HDAC6 (IC50 = 5 nM) has been considered as a new candidate for the treatment of multiple myeloma (currently in clinical trials), which also combined with bortezomib and dexamethasone for refractory multiple myeloma in 2017.45 Compound 7f were evaluated in vitro for inhibitory activity against purified HDAC6 enzyme using a fluorescence-based activity assay. As a result, 7f displayed as the strongest inhibitor of HDAC6 enzyme. At 5 μM, this substance caused inhibition of 100% while belinostat was 64.5% (Fig. 6). Compound 7f inhibited HDAC6 with an IC50 of 0.7 μM (Fig. 6), which was 3.71-fold more potent than belinostat (IC50 = 2.6 μM) in the same assay.
To better understand the molecular mechanisms, the study also performed the molecular docking of compound 7f at the active site of the HDAC6 enzyme. The 7f are embedded in a lipophilic channel whose walls consist of Pro464, Ser531, His573, His574, Phe583, Asp612, His614, Phe643, Asp705, Leu712, Tyr745 and Zn2+ ion. The strong zinc-binding ability of ZBG was observed. Through molecular docking analysis, compound 7f was found to insert into the active site of HDAC6 and was surrounded by some amino acids including His574 (van der Waals), Phe583 (pi–pi stacked), Cys584 (hydrogen bond), Asp612 (hydrogen bond), His614 (hydrogen bond and pi–pi T shaped), Asn645 (hydrogen bond), Phe642 (pi–pi stacked), Phe643 (hydrogen bond and pi–pi T shaped).
In summary, major focus of our research project, is the identification of belinostat derivatives as potential therapeutic compounds with exciting opportunities in multiple myeloma, structural activity relationships, and premise for animal studies to clinical trials in the future.
4. Conclusions
In conclusion, we have successfully synthesized 12 analogues from benzaldehyde with a considerable yield, and the procedure is convenient and has economic advantages. The key step in these processes is the conversion of benzaldehyde to 3-formylbenzenesulfonyl chloride. In the biological activity studies, compound 7f was proved to be the most effective molecule, with 3.5-fold enhanced efficiency than reference drug belinostat against multiple myeloma MOPC-315. The 7f/bortezomib regimen is highly effective in inducing cell death. The inhibitory activity of 7f against pan-HDAC and HDAC6 was remarkably higher than that of belinostat. The molecular mechanism of 7f–HDAC6 complex was studied using the molecular docking method. The docking score and binding mode analysis also demonstrated that compound 7f could be a promising inhibitor of this targeted enzyme. Our findings suggest that compound 7f could be a potent candidate for further pharmacological studies.
5. Experimental
5.1. Chemistry
All reagents were purchased from commercial sources and were used without further purification. Reactions were monitored by thin-layer chromatography (TLC) on 0.2 mm precoated silica gel 60 F254 plates (Merck) and compounds were visualized on TLC with UV-light. Derivatives were synthesized in the laboratory and then purified by flash column chromatography using silica gel 45–63 μm (230–400 mesh), 60 Å pore size. The nuclear magnetic resonance (NMR) spectra were recorded on Bruker Ascend 400 and Bruker Ascend 500. Electrospray ionization mass spectrometry (ESI-MS) analyses were recorded by an Agilent 1100. Chemical shift values (δ) are given in parts per million (ppm) downfield from tetramethylsilane as internal reference, coupling constants are given in hertz (Hz) and spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet) or m (multiplet). Belinostat derivatives were prepared adopting previously reported methods as shown in Fig. 2.40,46–48
5.1.1 Belinostat. 1H-NMR (400 MHz, DMSO-d6, δ ppm): 10.80 (1H, s, –OH), 10.27 (1H, s, –SO2NH–), 9.10 (1H, s, –NH–), 7.90 (s, 1H, ArH), 7.76 (1H, d, J = 7.0 Hz, ArH), 7.69 (1H, d, J = 7.1 Hz, ArH), 7.55 (1H, t, J = 7.5 Hz, ArH), 7.45 (1H, d, J = 15.8 Hz, Ar–CHCH–), 7.22 (2H, t, J = 7.3 Hz, ArH), 7.08 (2H, d, J = 7.5 Hz, ArH), 7.02 (1H, t, J = 6.9 Hz, ArH), 6.49 (1H, d, J = 15.8 Hz, Ar–CHCH–). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 162.54, 140.72, 137.94, 137.02, 136.32, 132.47, 130.48, 129.67, 127.55, 125.20, 124.80, 121.81, 120.82. ESI-MS, m/z (%) 316.8 ((M − H)−, 100).
5.1.2 Compound 7a. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 9.11 (1H, s, –NH–), 8.23 (1H, t, J = 6.5 Hz, –SO2NH–), 7.89 (1H, s, ArH), 7.78 (1H, d, J = 7.5 Hz, ArH), 7.75 (1H, d, J = 8 Hz, ArH), 7.59 (1H, t, J = 7.75 Hz, ArH), 7.50 (1H, d, J = 15.5 Hz, Ar–CHCH–), 7.27–7.24 (2H, m, ArH), 7.08–7.04 (2H, m, ArH), 6.54 (1H, d, J = 16 Hz, Ar–CHCH–), 4.02 (2H, d, J = 6.5 Hz, –CH2–). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 160.57, 140.04, 136.27, 131.94, 130.37, 130.12, 130.04, 125.02, 121.63, 115.49, 115.28, 45.88. IDA-MS m/z (%) 351.0808 ((M + H)+, 100).
5.1.3 Compound 7b. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 9.11 (1H, s, –NH–), 8.14 (1H, t, J = 6.25 Hz, –SO2NH–), 7.87 (1H, s, ArH), 7.76 (2H, t, J = 8.75 Hz, ArH), 7.59 (1H, t, J = 7.75 Hz, ArH), 7.48 (d, J = 16 Hz, 1H), 7.09 (d, J = 8 Hz, 2H), 7.04 (d, J = 8 Hz, 2H), 6.54 (1H, d, J = 16 Hz, Ar–CHCH–), 3.97 (2H, d, J = 6.5 Hz, –CH2–), 2.23 (3H, s, –CH3). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 160.86, 142.10, 136.75, 130.33, 129.19, 128.08, 125.02, 121.55, 117.42, 46.46, 21.10. IDA-MS m/z (%) 347.1060 ((M + H)+, 100).
5.1.4 Compound 7c. 1H NMR (500 MHz, DMSO-d6, δ ppm): 10.78 (1H, s, –OH), 9.13 (1H, s, –NH–), 8.20 (1H, br, –SO2NH–), 7.92 (1H, s, ArH), 7.77 (2H, t, J = 8.25 Hz, ArH). 7.59 (1H, t, J = 7.75 Hz, ArH), 7.49 (1H, d, J = 15.5 Hz, Ar–CHCH–), 7.28–7.18 (5H, m, ArH), 6.55 (1H, d, J = 16 Hz, Ar–CHCH–), 4.02 (2H, s, –CH2–). 13C-NMR (100 MHz, CD3OD, δ ppm): 164.14, 141.89, 138.26, 137.04, 135.95, 130.89, 129.42, 128.02, 127.55, 127.36, 125.47, 119.36, 46.56. ESI-MS m/z (%) 333.0920 ((M + H)+, 100).
5.1.5 Compound 7d. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.80 (1H, s, –OH), 9.12 (1H, s, –NH–), 7.97 (1H, s, –SO2NH–), 7.79 (2H, dd, J1 = 1.5 Hz, J2 = 7.75 Hz, ArH), 7.68 (1H, d, J = 7 Hz, ArH), 7.61 (1H, t, J = 7.75 Hz, ArH), 7.51 (1H, d, J = 15.5 Hz, Ar–CHCH–), 6.56 (1H, d, J = 16 Hz, Ar–CHCH–), 2.96–2.95 (1H, m, cyclohexane), 1.56–1.55 (5H, m, cyclohexane), 1.19–1.08 (5H, m, cyclohexane). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 143.56, 141.16, 136.27, 131.81, 130.39, 124.75, 121.59, 52.59, 33.68, 25.29, 24.77. ESI-MS m/z (%) 325.1228 ((M + H)+, 100).
5.1.6 Compound 7e. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 9.81 (1H, s, –SO2NH–), 7.99 (1H, d, J = 7.5 Hz, ArH), 7.92 (1H, s, ArH), 7.70–7.61 (1H, m, ArH), 7.61 (1H, t, J = 8 Hz, ArH), 7.61 (1H, d, J = 15.5 Hz, Ar–CHCH–), 7.18–7.15 (1H, m, ArH), 6.97–6.93 (1H, m, ArH), 6.82–6.80 (1H, m, ArH), 6.53 (1H, d, J = 16 Hz, Ar–CHCH–), 1.94 (3H, s, –CH3). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 178.98, 162.53, 161.73, 141.51, 137.07, 137.01, 136.38, 132.59, 132.53, 132.43, 130.56, 124.89, 120.93, 17.28. ESI-MS m/z (%) 351.0811 ((M + H)+, 100).
5.1.7 Compound 7f. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 9.94 (1H, s, –SO2NH–), 9.10 (1H, s, –NH–), 7.85 (1H, s, ArH), 7.76 (1H, d, J = 7.5 Hz, ArH), 7.62 (1H, d, J = 7.5 Hz, ArH), 7.56 (1H, t, J = 7.5 Hz, ArH), 7.46 (1H, d, J = 16 Hz, Ar–CHCH–), 6.97 (2H, d, J = 9 Hz, ArH), 6.81–6.79 (2H, m, ArH), 6.50 (1H, d, J = 15.5 Hz, Ar–CHCH–), 3.66 (3H, s, –CH3). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 162.54, 157.16, 140.69, 137.06, 136.22, 132.36, 130.35, 130.32, 127.61, 125.15, 124.15, 121.72, 114.82, 55.62. ESI-MS m/z (%) 349.0862 ((M + H)+, 100).
5.1.8 Compound 7g. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 10.13 (1H, s, –SO2NH–), 9.11 (1H, s, –NH–), 7.88 (1H, s, ArH), 7.77 (1H, d, J = 8 Hz, ArH), 7.67 (1H, d, J = 8 Hz, ArH), 7.56 (1H, t, J = 7.75 Hz, ArH), 7.46 (1H, d, J = 16 Hz, Ar–CHCH–), 7.03 (2H, d, J = 8.5 Hz, ArH), 6.97 (2H, d, J = 8.5 Hz, ArH), 6.50 (1H, d, J = 16 Hz, Ar–CHCH–), 2.18 (3H, s, –CH3). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 140.78, 136.26, 135.31, 134.09, 132.38, 130.41, 130.08, 127.57, 125.18, 121.35, 20.76. ESI-MS m/z (%) 333.0893 ((M + H)+, 100).
5.1.9 Compound 7h. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 10.34 (1H, s, –SO2NH–), 9.11 (1H, br, –NH–), 7.89 (1H, s, ArH), 7.83 (1H, d, J = 7.5 Hz, ArH), 7.67 (1H, d, J = 8 Hz, ArH), 7.60 (1H, t, J = 7.75 Hz, ArH), 7.48 (1H, d, J = 16 Hz, Ar–CHCH–), 7.45–7.40 (1H, m, ArH), 7.27–7.22 (2H, m, ArH), 6.51 (1H, d, J = 16 Hz, Ar–CHCH–). 13C-NMR (100 MHz, CD3OD, δ ppm): 164.11, 156.68, 154.20, 140.65, 138.12, 136.02, 131.53, 129.44, 127.49, 126.98, 125.56, 124.61, 124.56, 123.53, 119.59, 116.22, 115.98. ESI-MS m/z (%) 371.0266 ((M + H)+, 100).
5.1.10 Compound 7i. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.81 (1H, s, –OH), 10.25 (1H, s, –SO2NH–), 9.12 (1H, s, –NH–), 7.88 (1H, s, ArH), 7.78 (1H, d, J = 7.5 Hz, ArH), 7.65 (1H, d, J = 7.5 Hz, ArH), 7.57 (1H, t, J = 7.75 Hz, ArH), 7.46 (1H, d, J = 16 Hz, Ar–CHCH–), 7.08 (4H, d, J = 6.5 Hz, ArH), 6.50 (1H, d, J = 16 Hz, Ar–CHCH–). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 167.57, 160.86, 158.47, 142.32, 141.12, 140.45, 136.36, 134.14, 132.50, 130.49, 127.56, 125.19, 123.63, 123.55, 121.84, 116.52, 116.29, 113.20. ESI-MS m/z (%) 337.0660 ((M + H)+, 100).
5.1.11 Compound 7j. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.80 (1H, s, –OH), 10.17 (1H, s, –SO2NH–), 9.10 (1H, s, –NH–), 7.85 (1H, s, ArH), 7.82 (1H, d, J = 7.5 Hz, ArH), 7.64 (1H, d, J = 8 Hz, ArH), 7.59 (1H, t, J = 7.75 Hz, ArH), 7.48 (1H, d, J = 16 Hz, Ar–CHCH–), 7.25–7.20 (2H, m, ArH), 7.06–7.02 (1H, m, ArH), 6.50 (1H, d, J = 15.5 Hz, Ar–CHCH–). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 162.54, 144.63, 141.12, 137.03, 136.29, 132.62, 130.43, 127.54, 124.96, 121.75, 112.32, 105.38, 105.14, 105.11, 104.88. ESI-MS m/z (%) 355.0566 ((M + H)+, 100).
5.1.12 Compound 7k. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 10.82 (1H, s, –OH), 10.81 (1H, s, –SO2NH–), 9.12 (1H, s, –NH–), 7.95 (1H, s, ArH), 7.80 (1H, d, J = 7.5 Hz, ArH), 7.74 (1H, d, J = 8 Hz, ArH), 7.59 (1H, t, J = 7.75 Hz, ArH), 7.48 (1H, d, J = 16 Hz, Ar–CHCH–), 7.29–7.25 (1H, m, ArH), 6.94–6.83 (3H, m, ArH), 6.52 (1H, d, J = 16 Hz, Ar–CHCH–). 13C-NMR (100 MHz, DMSO-d6, δ ppm): 163.90, 162.52, 161.48, 140.45, 139.95, 139.85, 136.48, 132.62, 131.54, 131.45, 130.64, 127.53, 125.27, 121.94, 116.08, 111.30, 107.17, 106.92. ESI-MS m/z (%) 337.0661 ((M + H)+, 100).
5.1.13 Compound 7l. 1H-NMR (500 MHz, DMSO-d6, δ ppm): 11.06 (1H, d, J = 1 Hz, =N–OH), 10.81 (1H, s, –OH), 10.46 (1H, s, –SO2NH–), 9.11 (1H, s, –NH–), 7.95 (1H, s, ArH), 7.78 (1H, d, J = 7.5 Hz, ArH), 7.72 (1H, d, J = 7.5 Hz, ArH), 7.58 (1H, t, J = 7.75 Hz, ArH), 7.52 (2H, d, J = 8 Hz, ArH), 7.47 (1H, d, J = 15.5 Hz, Ar–CHCH–), 7.11 (2H, d, J = 8 Hz, ArH), 6.52 (1H, d, J = 16 Hz, Ar–CHCH–), 2.05 (3H, d, J = 0.5 Hz, –CH3). ESI-MS m/z (%) 343.0759 ((M + H)+, 100), 358.0869 ((M + H)+, 77), 376.0974 ((M + H)+, 70).
5.2. Biological methods and synergy analysis
MOPC-315 cell line was obtained from ATCC. The cells were cultured in vitro in RPMI 1640 (Invitrogen, Paisley, UK) containing 10% fetal bovine serum (Gibco), and 1% penicillin. For cytotoxicity assays, cells were seeded into 96-well culture plates at 2 × 104 cells per well in 0.1 mL of RPMI 1640 medium, then incubated at 37 °C with 5% CO2 and at 90% humidity. After 4 h, the different concentrations of drugs were treated and further incubated for 24 h. Cell viabilities were measured using CELLOMAXTM Viability Assay kit (Precaregene, Korea). UV absorption at 450 nm was measured using an Infinite M200 micro-plate reader (Tecan, Zürich, Switzerland). Combination index (CI) values were calculated using the Chou–Talalay method.27
5.3. Fluorometric HDAC activity assay
MCF-7 cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (Gibco), and 1% penicillin in an incubator kept at 37 °C and 5% CO2. HDAC activity was assessed using an HDAC Activity Fluorometric Assay Kit (Cat#K330-100, Bio-Vision Incorporated, USA) according to the manufacturer's instructions. Briefly, cells were seeded at 5 × 103 cells per well in a 96-well cell culture plate. After 24 h, the old media was removed, 99 μL of new media, 1 μL of HDAC substrate and 1 μL of belinostat or 7f (the final concentration is 30 μM) or 1 μL of trichostatin A was added and incubated for 2 h. Then, 100 μL of developer mix was added to each well and incubated for 30 minutes at 37 °C. Fluorescence was measured with a multi-well plate reader (excitation at 368 nm and emission at 442 nm).
5.4. In vitro HDAC6 inhibition activity
Activity against HDAC6 was performed similarly to previously reported.49 Summarization of the protocol, the inhibitor diluted to indicated concentrations, which were mixed with HDAC6 enzyme and the substrate for 30 min including 25 μL of HDAC assay buffer, 2.5 μL of 1 mg mL−1 BSA, 2.5 μL of 200 μM HDAC substrate 2, 2.5 μL of HDAC6, and 2.5 μL of belinostat or 7f in DMSO. Stop the reaction by adding 25 μL of HDAC assay developer incubate at room temperature for 15 min. The fluorescence intensity was measured according to the manufacturer's instructions (absorption at 485 nm wavelength and emission at 528 nm). Belinostat was served as a positive control.
5.5. Molecular docking studies
The three-dimensional structures of designed molecules (ligands) were prepared using MarvinSketch version 19.27.0 and PyMOL version 2.2.2.27. The energy minimization was carried out using MM2 force field and quantum chemical calculations were per-formed by PM6 semi-empirical method implemented in Gaussian 09.50 Belinostat has been proved by Buckley et al. in 2007 as a novel inhibitor of histone deacetylase, thus, it was selected as reference ligand for the docking simulation.51 Crystal structure of HDAC6 in complex with belinostat inhibitor (PDB code: 5EEN) was retrieved from the Protein Data Bank archive (PDB Bank).36 The Graphical User Interface program named Autodock Tools 1.5.6 (ADT) was employed to set up input data. Details of molecular docking simulation are given in ESI.† The docking simulation procedure was performed by AutoDock4Zn utilizing Lamarckian genetic algorithm and an empirical binding free energy function.52 Search space was restricted to a grid box size of 66 each in x, y and z dimensions which was centered on the active site of protein and a grid spacing of 0.375 Å. A total of 50 runs were performed for each docking and the rest of the parameters were set to default values.
Author contributions
Cuong Quoc Nguyen, Quang De Tran, Minh Quan Pham, Quang Le Dang, and Hong Phuong Nguyen conceived the idea and designed the works. Quang De Tran, Cuong Quoc Nguyen, Huynh Nhu Thao, Nguyen Trong Tuan, Bui Thi Buu Hue, Nguyen Quoc Chau Thanh and Nguyen Van Ky performed chemical synthesis, purification, and structure identification. Cuong Quoc Nguyen and Pham Minh Quan performed molecular docking studies. Tran Phuong Hoa, Tran Duy Binh, Quang Le Dang, Hong Phuong Nguyen and Su-Geun Yang performed, analyzed and supervised biological assays. Quang De Tran, Cuong Quoc Nguyen, Minh Quan Pham, Hong Phuong Nguyen, Tran Phuong Hoa, Quang Le Dang, Su-Geun Yang and Bui Thi Buu Hue wrote, prepared, and edited the original draft. All authors have approved the final version of the manuscript.
Conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgements
This work was supported by the Korean government (MOE and MSIT) (2020R1A2B5B02002377, 2018R1A6A1A03025523, and 2019M3E5D1A02069623), and the Ministry of Education and Training of Vietnam under grant number B2019-TCT-37 (TCT-05).
References
- R. A. Kyle, Mayo Clin. Proc., 1975, 50, 29–40 CAS.
- J. Moreaux, T. Reme, W. Leonard, J.-L. Veyrune, G. Requirand, H. Goldschmidt, D. Hose and B. Klein, Br. J. Cancer, 2013, 109, 676–685 CrossRef CAS PubMed.
- S. V. Rajkumar, M. A. Dimopoulos, A. Palumbo, J. Blade, G. Merlini, M.-V. Mateos, S. Kumar, J. Hillengass, E. Kastritis, P. Richardson, O. Landgren, B. Paiva, A. Dispenzieri, B. Weiss, X. LeLeu, S. Zweegman, S. Lonial, L. Rosinol, E. Zamagni, S. Jagannath, O. Sezer, S. Y. Kristinsson, J. Caers, S. Z. Usmani, J. J. Lahuerta, H. E. Johnsen, M. Beksac, M. Cavo, H. Goldschmidt, E. Terpos, R. A. Kyle, K. C. Anderson, B. G. M. Durie and J. F. S. Miguel, Lancet Oncol., 2014, 15, e538–e548 CrossRef.
- T. Liu, X. Liu and W. Li, Oncotarget, 2016, 7, 40800–40815 CrossRef PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- P. A. Wade, Hum. Mol. Genet., 2001, 10, 693–698 CrossRef CAS PubMed.
- W. Weichert, A. Röske, V. Gekeler, T. Beckers, M. P. Ebert, M. Pross, M. Dietel, C. Denkert and C. Röcken, Lancet Oncol., 2008, 9, 139–148 CrossRef CAS.
- W. Weichert, Cancer Lett., 2009, 280, 168–176 CrossRef CAS PubMed.
- A. Drazic, L. M. Myklebust, R. Ree and T. Arnesen, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864, 1372–1401 CrossRef CAS PubMed.
- C. Zhang, J. F. Zhong, A. Stucky, X.-L. Chen, M. F. Press and X. Zhang, Clin. Epigenet., 2015, 7, 117 CrossRef PubMed.
- H. Zhang, Y.-P. Shang, H. Chen and J. Li, Hepatol. Res., 2017, 47, 149–159 CrossRef CAS PubMed.
- S. Ropero and M. Esteller, Mol. Oncol., 2007, 1, 19–25 CrossRef CAS PubMed.
- T. Harada, T. Hideshima and K. C. Anderson, Int. J. Hematol., 2016, 104, 300–309 CrossRef CAS PubMed.
- J. A. Plumb, P. W. Finn, R. J. Williams, M. J. Bandara, M. R. Romero, C. J. Watkins, N. B. L. Thangue and R. Brown, Mol. Cancer Ther., 2003, 2, 721–728 CAS.
- R. M. Poole, Drugs, 2014, 74, 1543–1554 CrossRef CAS PubMed.
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, Nature, 1999, 401, 188–193 CrossRef CAS PubMed.
- M. Manal, M. J. N. Chandrasekar, J. Gomathi Priya and M. J. Nanjan, Bioorg. Chem., 2016, 67, 18–42 CrossRef CAS PubMed.
- J. Fischer and W. E. Childers, Successful Drug Discovery, John Wiley & Sons, 2017, vol. 2 Search PubMed.
- C. Wang, T. E. Eessalu, V. N. Barth, C. H. Mitch, F. F. Wagner, Y. Hong, R. Neelamegam, F. A. Schroeder, E. B. Holson, S. J. Haggarty and J. M. Hooker, Am. J. Nucl. Med. Mol. Imaging, 2013, 4, 29–38 Search PubMed.
- J.-H. Zhang, M. Mottamal, H.-S. Jin, S. Guo, Y. Gu, G. Wang and L.-M. Zhao, Future Med. Chem., 2019, 11, 2765–2778 CrossRef CAS PubMed.
- V. Andrianov, V. Gailite, D. Lola, E. Loza, V. Semenikhina, I. Kalvinsh, P. Finn, K. D. Petersen, J. W. A. Ritchie, N. Khan, A. Tumber, L. S. Collins, S. M. Vadlamudi, F. Björkling and M. Sehested, Eur. J. Med. Chem., 2009, 44, 1067–1085 CrossRef CAS PubMed.
- P. Linciano, L. Pinzi, S. Belluti, U. Chianese, R. Benedetti, D. Moi, L. Altucci, S. Franchini, C. Imbriano, C. Sorbi and G. Rastelli, J. Enzyme Inhib. Med. Chem., 2021, 36, 2080–2086 CrossRef CAS PubMed.
- L. Marek, A. Hamacher, F. K. Hansen, K. Kuna, H. Gohlke, M. U. Kassack and T. Kurz, J. Med. Chem., 2013, 56, 427–436 CrossRef CAS PubMed.
- J. Melesina, C. V. Simoben, L. Praetorius, E. F. Bülbül, D. Robaa and W. Sippl, ChemMedChem, 2021, 16, 1336–1359 CrossRef CAS PubMed.
- P. Linciano, R. Benedetti, L. Pinzi, F. Russo, U. Chianese, C. Sorbi, L. Altucci, G. Rastelli, L. Brasili and S. Franchini, Bioorg. Chem., 2021, 106, 104462 CrossRef CAS PubMed.
- Y. Dai, S. Chen, L. Wang, X.-Y. Pei, L. B. Kramer, P. Dent and S. Grant, Br. J. Haematol., 2011, 153, 222–235 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- S. Pulya, Sk. A. Amin, N. Adhikari, S. Biswas, T. Jha and B. Ghosh, Pharmacol. Res., 2021, 163, 105274 CrossRef CAS PubMed.
- M. Cea, A. Cagnetta, M. Gobbi, F. Patrone, P. G. Richardson, T. Hideshima and K. C. Anderson, Curr. Pharm. Des., 2013, 19, 734–744 CrossRef CAS.
- J. Cao, W. Lv, L. Wang, J. Xu, P. Yuan, S. Huang, Z. He and J. Hu, Cell Death Dis., 2018, 9, 1–11 CrossRef PubMed.
- H. Gohlke, M. Hendlich and G. Klebe, J. Mol. Biol., 2000, 295, 337–356 CrossRef CAS PubMed.
- M. Quan Pham, K. B. Vu, T. N. H. Pham, L. T. T. Huong, L. Hoang Tran, N. Thanh Tung, V. V. Vu, T. Hai Nguyen and S. Tung Ngo, RSC Adv., 2020, 10, 31991–31996 RSC.
- N. T. Dan, H. D. Quang, V. Van Truong, D. Huu Nghi, N. M. Cuong, T. D. Cuong, T. Q. Toan, L. G. Bach, N. H. T. Anh, N. T. Mai, N. T. Lan, L. Van Chinh and P. M. Quan, Sci. Rep., 2020, 10, 11429 CrossRef CAS PubMed.
- P. Neri, N. J. Bahlis and S. Lonial, Expert Opin. Invest. Drugs, 2012, 21, 733–747 CrossRef CAS PubMed.
- S. Schultes, C. de Graaf, E. E. J. Haaksma, I. J. P. de Esch, R. Leurs and O. Krämer, Drug Discovery Today: Technol., 2010, 7, e157–e162 CrossRef CAS PubMed.
- Y. Hai and D. W. Christianson, Nat. Chem. Biol., 2016, 12, 741–747 CrossRef CAS PubMed.
- N. J. Porter, A. Mahendran, R. Breslow and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13459–13464 CrossRef CAS PubMed.
- Y. Imai, E. Ohta, S. Takeda, S. Sunamura, M. Ishibashi, H. Tamura, Y. Wang, A. Deguchi, J. Tanaka, Y. Maru and T. Motoji, JCI Insight, 2016, 1, e85061 CrossRef PubMed.
- D. Sullivan, S. Singhal, M. Schuster, J. Berenson, P. Gimsing, F. Wislöff, A. Waage, M. Alsina, R. Gerwien, A. Clarke, K. Moller and C. E. Ooi, Blood, 2006, 108, 3583 CrossRef.
- P. W. Finn, M. Bandara, C. Butcher, A. Finn, R. Hollinshead, N. Khan, N. Law, S. Murthy, R. Romero, C. Watkins, V. Andrianov, R. M. Bokaldere, K. Dikovska, V. Gailite, E. Loza, I. Piskunova, I. Starchenkov, M. Vorona and I. Kalvinsh, Helv. Chim. Acta, 2005, 88, 1630–1657 CrossRef CAS.
- Y. Zuo, H. Xu, Z. Chen, F. Xiong, B. Zhang, K. Chen, H. Jiang, C. Luo and H. Zhang, Oncol. Rep., 2020, 43, 1928–1944 CAS.
- S. Waqar, S. Chawla, B. Mathews, D. Park, T. Song, M. R. Choi, T. Niederman and R. Govindan, J. Thorac. Oncol., 2017, 12, S888–S889 CrossRef.
- Z. Rana, S. Diermeier, M. Hanif and R. J. Rosengren, Biomedicines, 2020, 8, 22 CrossRef CAS PubMed.
- E. Eleutherakis-Papaiakovou, N. Kanellias, E. Kastritis, M. Gavriatopoulou, E. Terpos and M. A. Dimopoulos, J. Oncol., 2020, 2020, e7131802 Search PubMed.
- D. T. Vogl, N. Raje, S. Jagannath, P. Richardson, P. Hari, R. Orlowski, J. G. Supko, D. Tamang, M. Yang, S. S. Jones, C. Wheeler, R. J. Markelewicz and S. Lonial, Clin. Cancer Res., 2017, 23, 3307–3315 CrossRef CAS PubMed.
- L. Yang, X. Xue and Y. Zhang, Synth. Commun., 2010, 40, 2520–2524 CrossRef CAS.
- X. Bao, D. Song, X. Qiao, X. Zhao and G. Chen, Org. Process Res. Dev., 2016, 20, 1482–1488 CrossRef CAS.
- H. A. Reisch, P. Leeming and P. S. Raje, US Pat., US8642809B2, 2014 Search PubMed.
- L. Peng, Z. Yuan and E. Seto, in Chromatin Protocols, ed. S. P. Chellappan, Springer, New York, NY, 2015, pp. 95–108 Search PubMed.
- Z. Bikadi and E. Hazai, J. Cheminf., 2009, 1, 15 Search PubMed.
- M. T. Buckley, J. Yoon, H. Yee, L. Chiriboga, L. Liebes, G. Ara, X. Qian, D. F. Bajorin, T.-T. Sun, X.-R. Wu and I. Osman, J. Transl. Med., 2007, 5, 49 CrossRef PubMed.
- D. Santos-Martins, S. Forli, M. J. Ramos and A. J. Olson, J. Chem. Inf. Model., 2014, 54, 2371–2379 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The NMR spectra of the synthesized compounds. See https://doi.org/10.1039/d2ra01969h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Cuong Quoc Nguyen
*b,
Cuong Quoc Nguyen