DOI:
10.1039/D2RA01968J
(Paper)
RSC Adv., 2022,
12, 14865-14882
Discovery of pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives as novel CDK2 inhibitors: synthesis, biological and molecular modeling investigations†
Received
27th March 2022
, Accepted 28th April 2022
First published on 17th May 2022
Abstract
CDK2 inhibition is an appealing target for cancer treatment that targets tumor cells in a selective manner. A new set of small molecules featuring the privileged pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine scaffolds (4–13) as well as the thioglycoside derivatives (14, 15) were designed, and synthesized as novel CDK2 targeting compounds. The growth of the three examined cell lines was significantly inhibited by most of the prepared compounds. Results revealed that most of the compounds showed superior cytotoxic activities against MCF-7 and HCT-116 with IC50 range (45–97 nM) and (6–99 nM), respectively, and moderate activity against HepG-2 with IC50 range of (48–90 nM) compared to sorafenib (IC50: 144, 176 and 19 nM, respectively). Of these compounds, 14 & 15 showed the best cytotoxic activities against the three cell lines with IC50 values of 45, 6, and 48 nM and 46, 7, and 48 nM against MCF-7, HCT-116 and HepG-2, respectively. Enzymatic inhibitory activity against CDK2/cyclin A2 was achieved for the most potent anti-proliferative compounds. Compounds 14, 13 and 15 revealed the most significant inhibitory activity with IC50 values of 0.057 ± 0.003, 0.081 ± 0.004 and 0.119 ± 0.007 μM, respectively compared to sorafenib (0.184 ± 0.01 μM). Compound 14 displayed potent dual activity against the examined cell lines and CDK2, and was thus selected for further investigations. It exerted a significance alteration in cell cycle progression, in addition to apoptosis induction within HCT cells. Molecular docking simulation of the designed compounds confirmed the good fit into the CDK2 active site through the essential hydrogen bonding with Leu83. In silico ADMET studies and drug-likeness studies using a Boiled Egg chart showed suitable pharmacokinetic properties which helped in structure requirement prediction for the observed antitumor activity.
1. Introduction
Protein kinases are essential in cellular processes and play an important role in the development and progression of many diseases, including cancer,1–3 where kinase mutations may trigger oncogenesis and hence could be key contributors to cancer progression.1 Cyclin dependent kinases (CDK) are responsible for phosphorylation of key components for cell proliferation. They are essential during the cell cycle and become corrupted in tumor cells.4–6 CDK2 plays an important role during cell cycle progress,7,8 and has a catalytic effect in cyclin-dependent protein kinase complexes.7,8 That's why kinase inhibition is becoming one of the most effective approaches for the treatment of cancer nowadays.9,10
The pyrazolopyrimidine moiety represents a common heterocycle nucleus used in the design of many pharmaceutical compounds,11,12 that have a variety of medicinal applications including antimicrobial, antitumor,13 antidiabetic, anti-Alzheimer's disease, anti-inflammatory14,15 and antioxidant applications.16–18
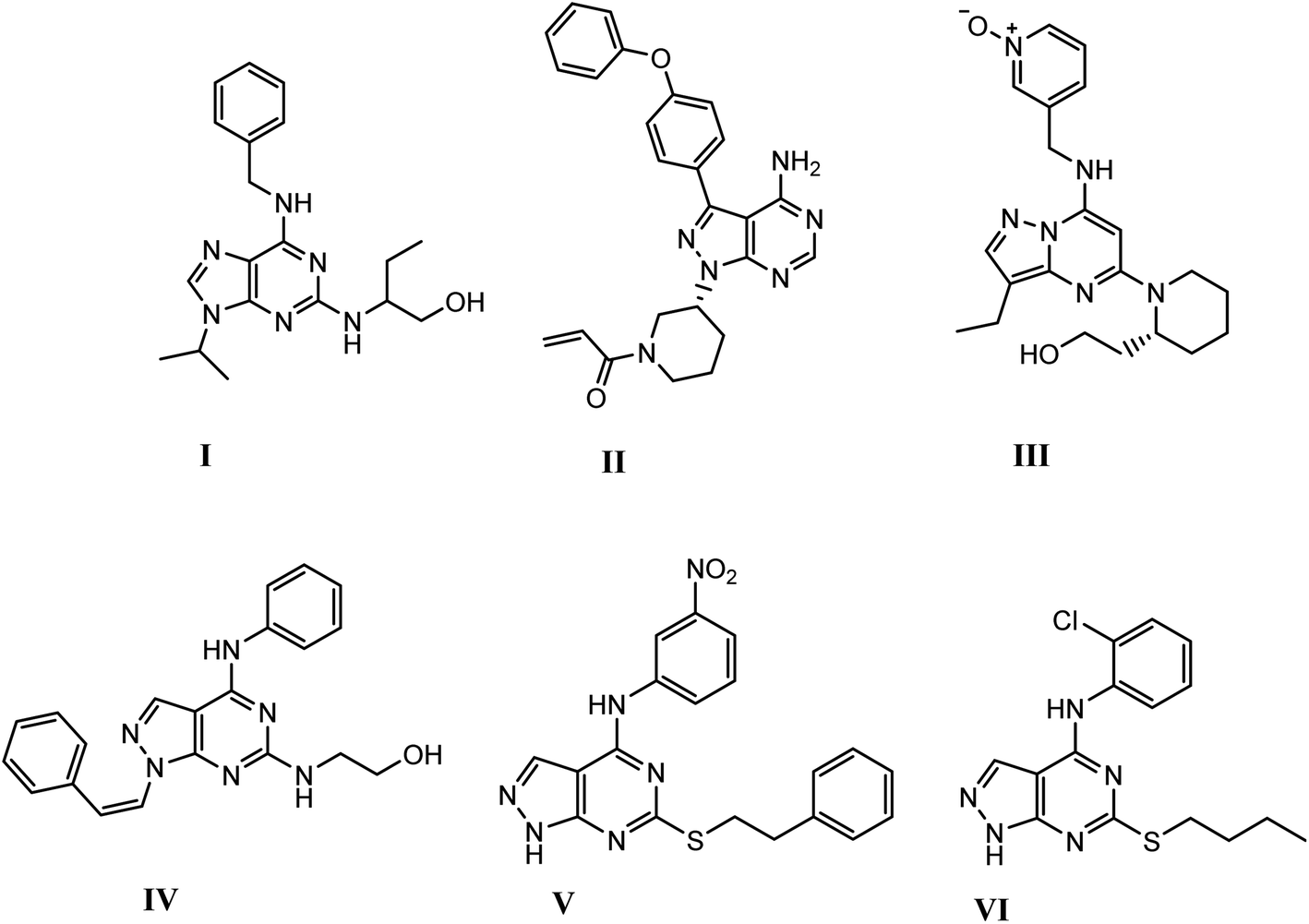
Recently, pyrazolopyrimidine is considered an appealing scaffold for pharmacologically active agents development with antitumor potential.19–24 This scaffold acts as a bioisostere of adenine and retains the main interactions of ATP at the kinase domain.25–28 The pyrazolo[3,4-d]pyrimidine bicycle is of the most employed scaffolds in drug discovery for its great potential as CDK inhibition. It was also considered as a bioisostere of purine ring with potent growth inhibitory activity,29,30 via CDK inhibition as CDK1,31 CDK2 (ref. 32) and 5-lipoxygenase enzymes.33 They also possess significant and specific pharmacological activity in CDK2/cyclin A inhibition.11 Dinaciclib, ibrutinib and roscovitine are known drugs with potent CDK2 inhibition9,12,25,34 (Fig. 1).
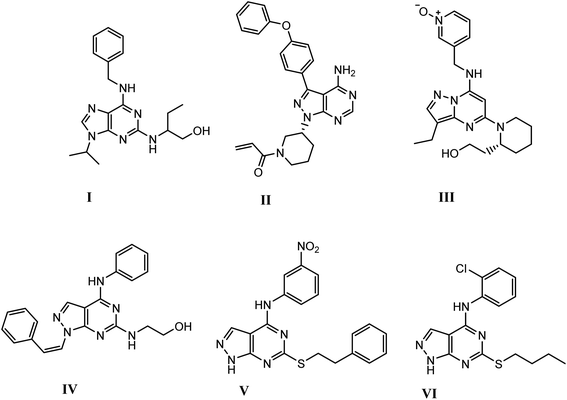 |
| | Fig. 1 Structures of active drugs containing fused pyrimidine (I) roscovitine, (II) ibrutinib & (III) dinaciclib and reported pyrazolo[3,4-d]pyrimidines derivatives (IV), (V) & (VI) as CDK2 inhibitors [9, 12, 25, 34]. | |
Based on the previous work and in continuation of anticancer drug discovery research, the aim of this research was to design and synthesize new pyrazolo[3,4-d]pyrimidine and its glycosyl amino derivatives. As well as the thioxo-pyrazolo[4,3-e][1,2,4]triazolo [1,5-c]pyrimidine compounds and the thioglycoside derivatives, based on pyrazolo[3,4-d]pyrimidine scaffold to develop novel CDK2 inhibitors.34,35 These derivatives were also evaluated for their in vitro anti-proliferative activity against breast cancer (MCF-7), hepatocellular carcinoma (HepG-2) and colorectal carcinoma (HCT-116) cell lines. The potential inhibition of the most potent promising compounds on CDK2/cyclin A was further tested. Also, molecular docking studies were applied to investigate the binding mode of the promising compounds by calculating their binding energies and visualizing their orientations with respect to the active site of CDK-2 protein compared to roscovitine ligand. The computational results were in agreement with the reported observations.
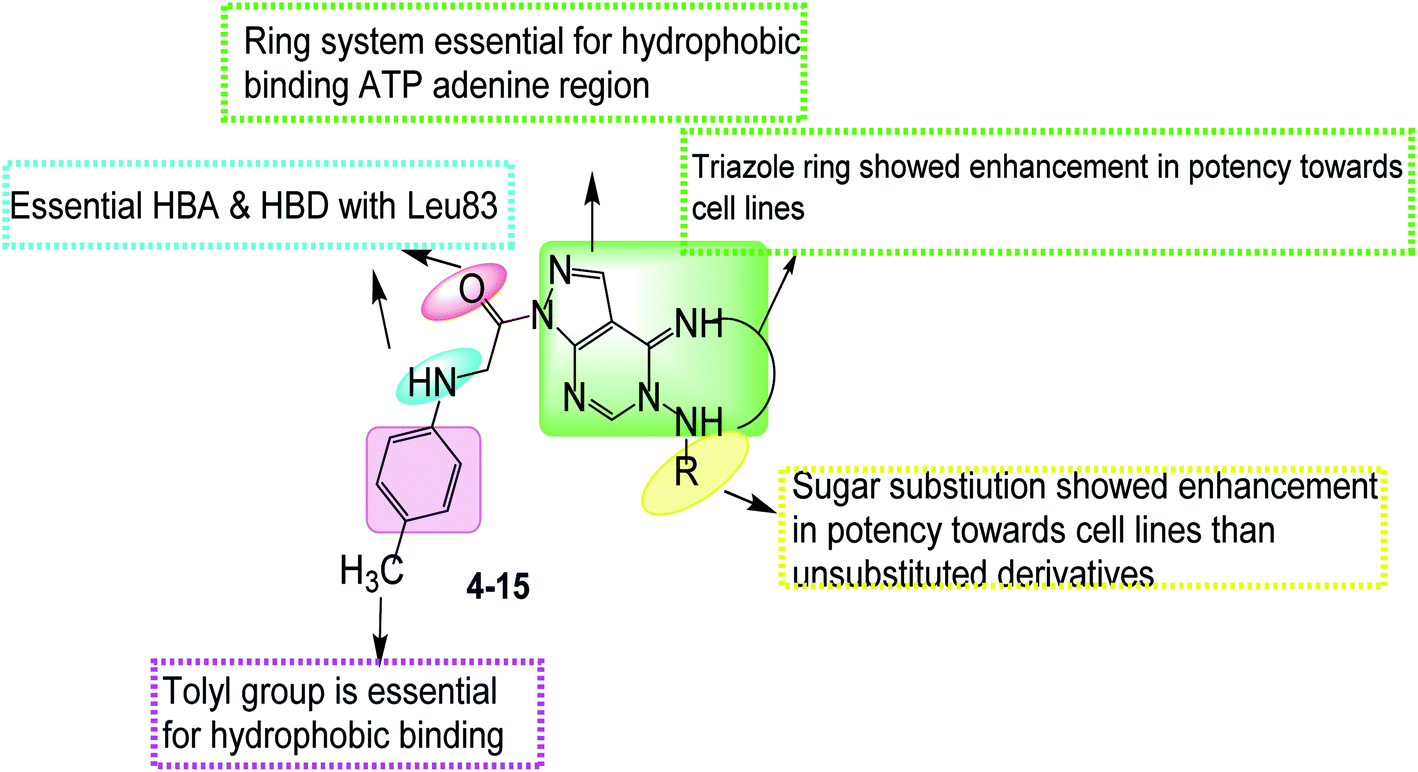
1.1 Rationale design
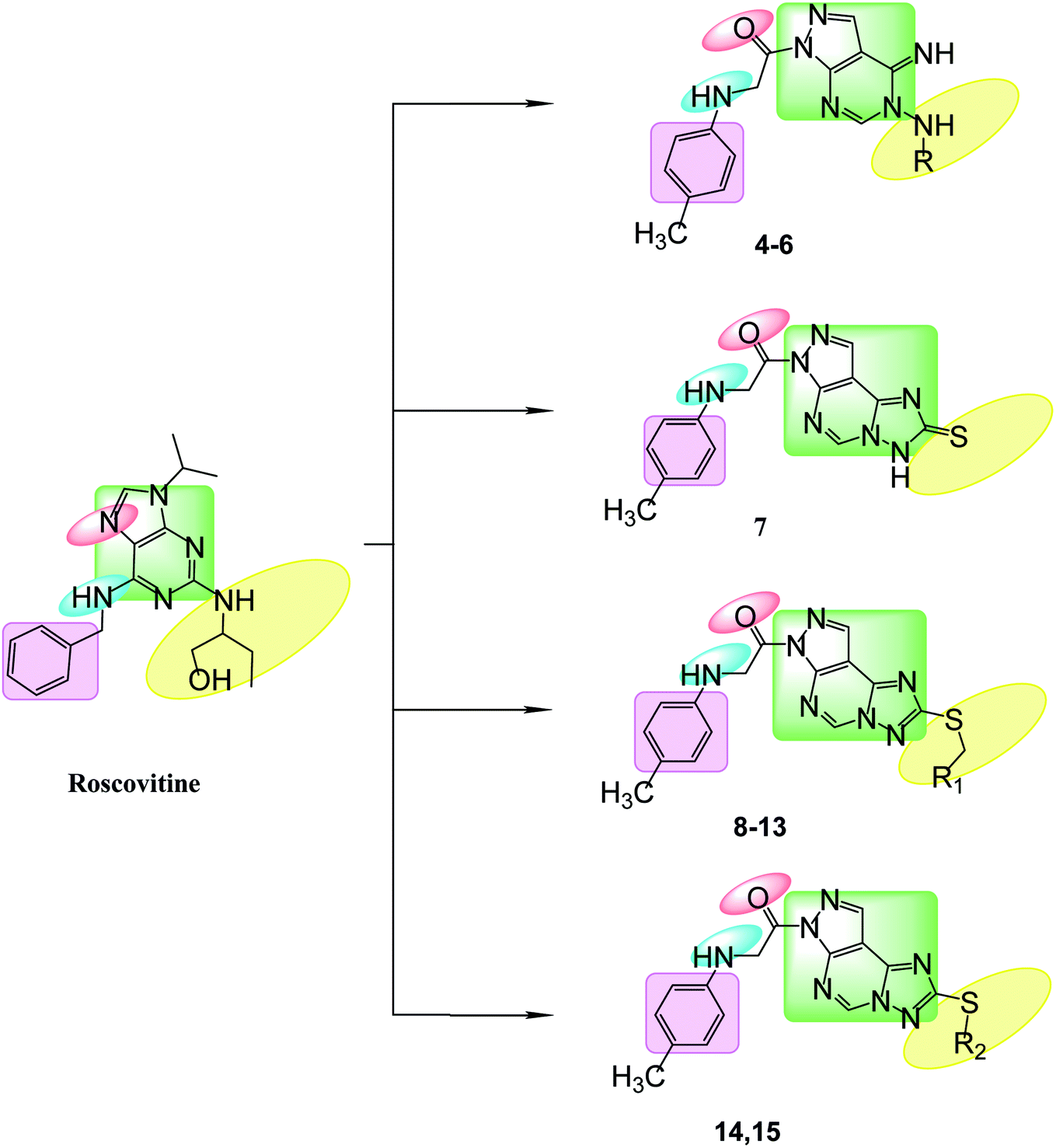
Bioisosteric replacements of purine scaffold of the ligand roscovitine by different ring system as pyrazolo[3,4-d]pyrimidine in compounds 4–6 or pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin in compounds 7–15 to place it in the ATP adenine region as in roscovitine with the required hydrophobic bindings was achieved. The phosphate binding region occupied by –NH of the 6-substituted amino moiety together with N7 in roscovitine was replaced by N arylglycyl moiety in all the proposed compounds, this glycyl group is responsible for the 2 essential hydrogen bonds with Leu83 in most of the compounds as reported by roscovitine.4,25–28 The N butanol at position 2 of roscovitine was replaced by amino group (in compound 4) or N-glycosyl (in compounds 5 and 6) at N5 of pyrazolopyrimidine. Compounds 7–13 possess thio group in position 2 of triazole ring as thioxo in compound 7 or thio alkyl substitution in compounds 8–13. Where an additional hydrogen binding by the thio group was achieved in compounds 11 & 13. The thioglycoside group at position 2 in compounds 14 & 15 showed also additional hydrogen binding via the acetyloxy substitutions. The hydrophobic binding by the benzyl group in roscovitine was maintained by the isosteric replacement by the tolyl group (Fig. 2).
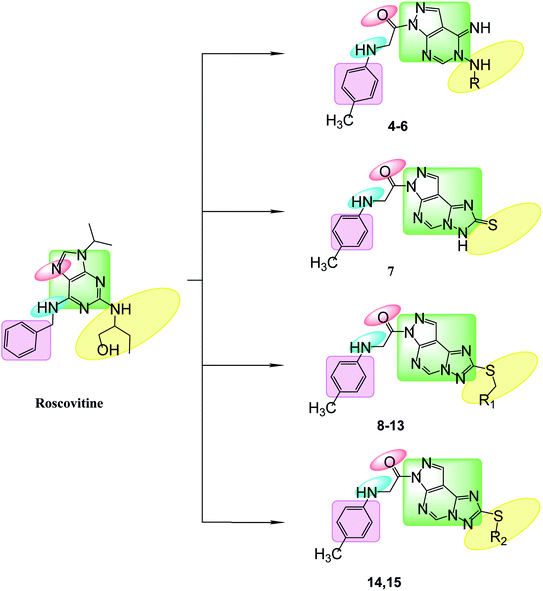 |
| | Fig. 2 Features' similarities for roscovitine ligand and the newly designed compounds as potent CDk2 inhibitors. | |
2. Results and discussion
2.1. Chemistry
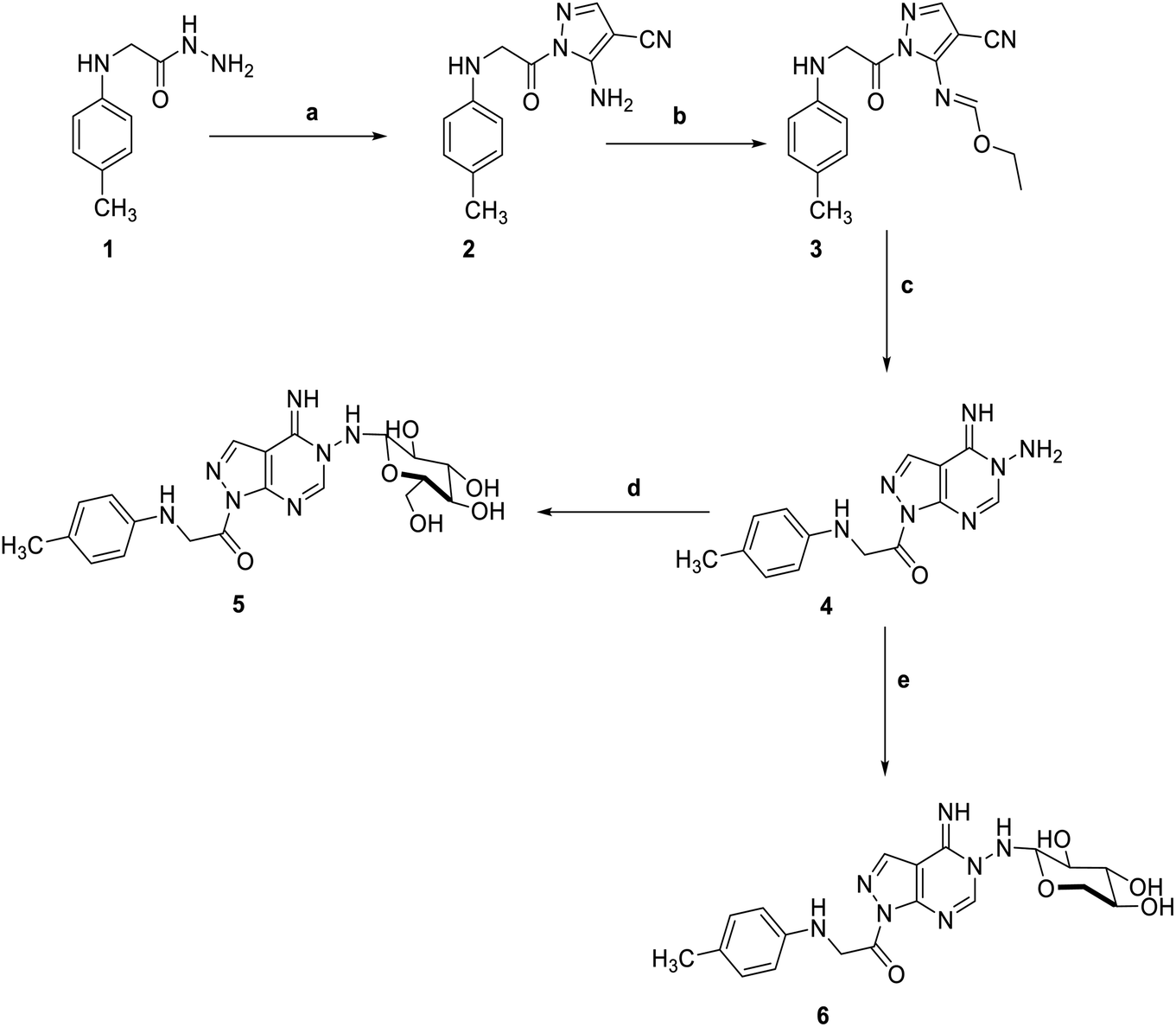
5-Amino-1-(p-tolylglycyl)-1H-pyrazole-4-carbonitrile 2, was prepared by the reaction of 2-(p-tolylamino)acetohydrazide 1 with ethoxymethylene malononitrile in ethanol under reflux. The isolated red solid 2 was then treated with triethoxyorthoformate in acetic anhydride to afford ethyl N-(4-cyano-1-(-tolylglycyl)-1H-pyrazol-5-yl)formimidate 3. The key starting material pyrazolopyrimidine 4 was obtained by treatment of the formimidate derivative 3 with hydrazine hydrate in ethanol. Subsequent reaction of pyrazolopyrimidine 4 with sugar aldoses namely; D-glucose and D-xylose, in the presence of a catalytic amount of acetic acid, afforded the amino-sugar products 5 and 6 respectively (Scheme 1). The structures of this set of novel amino sugars were confirmed by their spectral and elemental analyses data (see Experimental section). 1H NMR spectra of the amino-sugar compounds 5 and 6 showed a signal attributed to H-1 at 5.13 and 5.10 ppm confirming the sp3 hybridization of its corresponding carbon (C-1) and the cyclic form of the sugar moiety which agrees well with the reported results for such type of compounds following such mode of preparation.36
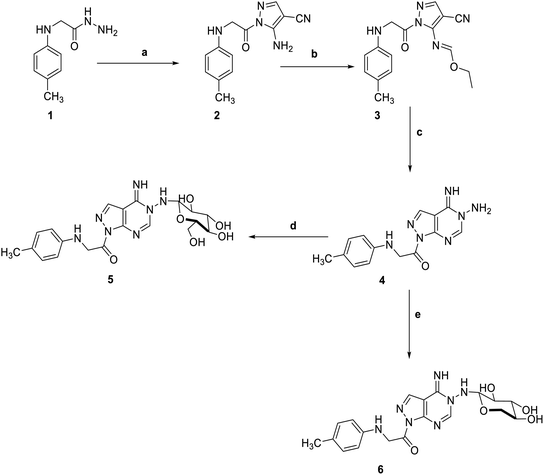 |
| | Scheme 1 Reagents and conditions; (a) 2-(ethoxymethylene)-malononitrile, EtOH, reflux, 6 h. (b) triethyl orthoformate, acetic anhydride, refulx, 6 h. (c) hydrazine hydrate, EtOH, reflux, 6 h. (d) D-Glucose, EtOH, glacial acetic acid, reflux, 3 h. (e) D-Xylose, EtOH, glacial acetic acid, reflux, 3 h. | |
Adding a carbon disulfide to pyrazolopyrimidine 4 in the presence of potassium hydroxide in ethanol afforded the corresponding triazolomercapto derivative 7. The 1H NMR spectra of compound 7 revealed the disappearance of the NH2 and NH signals and instead another broad singlet at 11.66 ppm for the NH in the thione–thiol system appeared. The later was utilized for the preparation of a series of acyclic nucleoside analogs via reaction with acyclic oxygenated halides. Thus, reaction with chloroacetonitrile, 2-chloro-1,1-dimethoxyethane, 2-chloropropane-1,2-diol, and 2-chloroethan-2-ol by stirring or by reflux in ethanol afforded the thioderivatives 8–10 and 12, respectively in good yields. In addition, the hydroxyl compounds 10 and 12 were acetylated with acetic anhydride to yield the corresponding O-acetylated acyclic analogs 11 and 13, respectively (Scheme 2). The 1H NMR spectra of the resulted acetylated derivatives showed the signals of the acetyl-methyl groups in addition to the disappearance of the hydroxyl signals which also confirmed by their IR spectra. The latter showed the carbonyl bands of the ester groups and disappearance of the characteristic bands of the hydroxyl groups (see Experimental section).
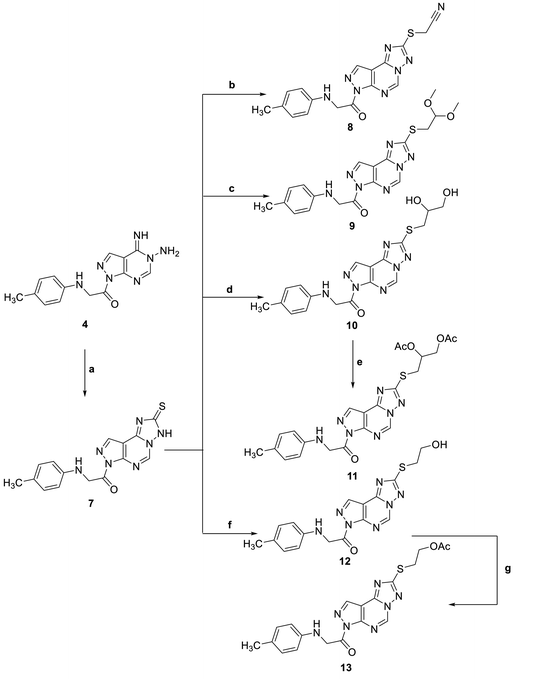 |
| | Scheme 2 Reagents and condition; (a) CS2, KOH, EtOH, reflux, 6 h. (b) Chloroacetonitrile, K2CO3, DMF, 25 C, 8 h. (c) Chloro acetaldhyde dimethyl acetal, K2CO3, DMF, 25 C, 8 h. (d) 2-Chloro-1,2-propandiol, KOH, EtOH, reflux, 3 h. (e) Acetic anhydride, pyridine, reflux, 2 h. (f) 2-Chloroethanol, KOH, EtOH, reflux, 3 h. (g) Acetic anhydride, pyridine, reflux, 2 h. | |
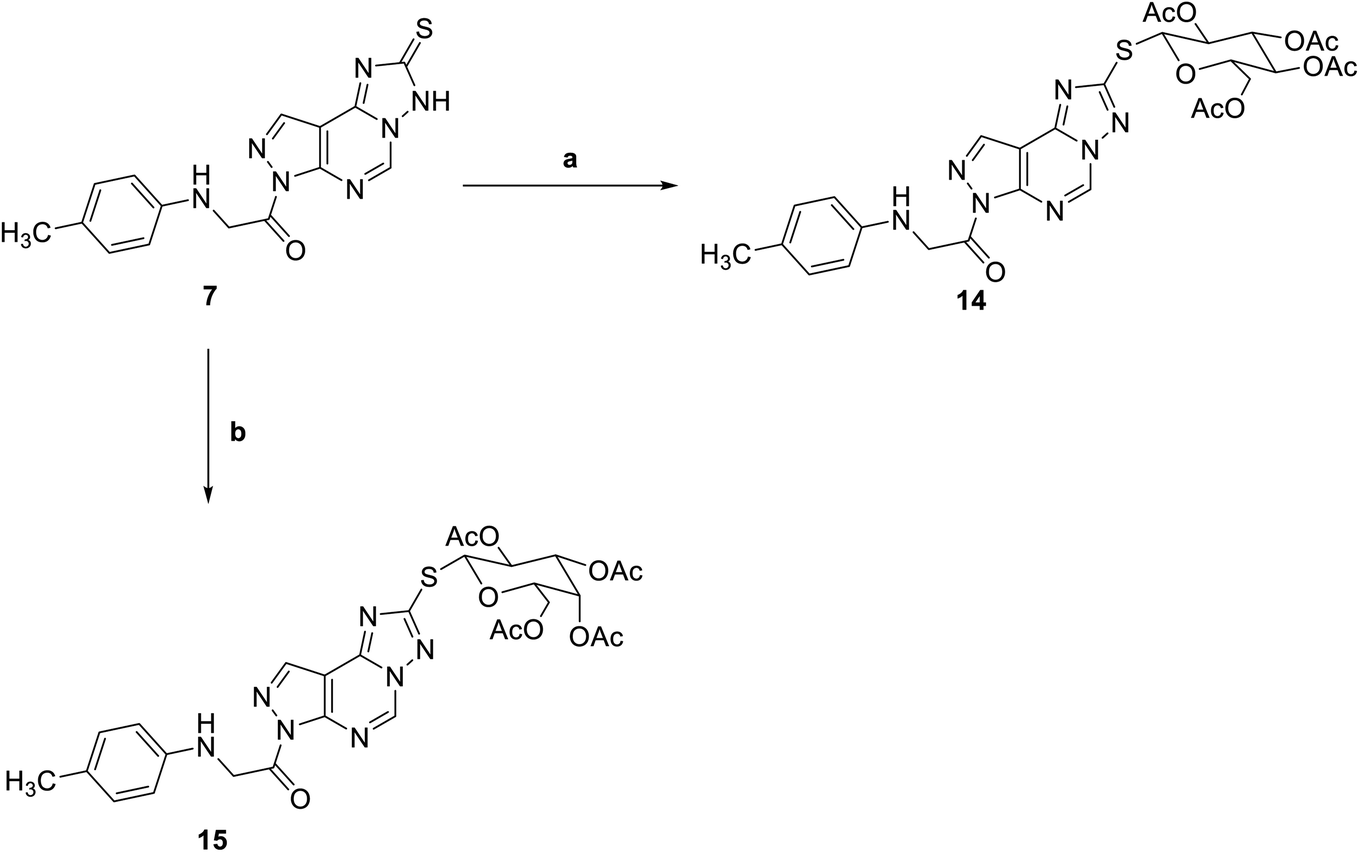
Reaction of triazolo pyrazolo pyridine derivatives 7 with two acetylated glycosyl halides, tetra-O-acetyl-α-D-gluco- and galactopyranosyl bromide, afforded the thioglycoside derivatives 14 and 15 as nucleoside analogs (Scheme 3). The 1H NMR of compound 14 revealed an increase in the integration of aliphatic region at 2.23–5.76 ppm, while the 13C NMR spectrum showed the characteristic signals for the five carbons of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups at δ 165 and 175 ppm.
O groups at δ 165 and 175 ppm.
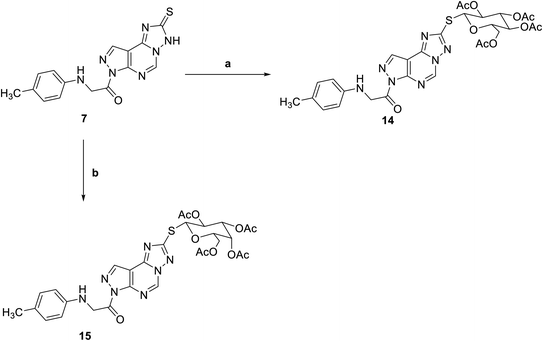 |
| | Scheme 3 Reagents and conditions; (a) 2,3,4,6-tetra-O-acetyl-D-glucopyranosylbromide, KOH, acetone, 25 C, 6 h. (b) 2,3,4,6-Tetra-O-acetyl-D-glactopyranosylbromide, KOH, acetone, 25 C, 6 h. | |
2.2. Biological evaluation
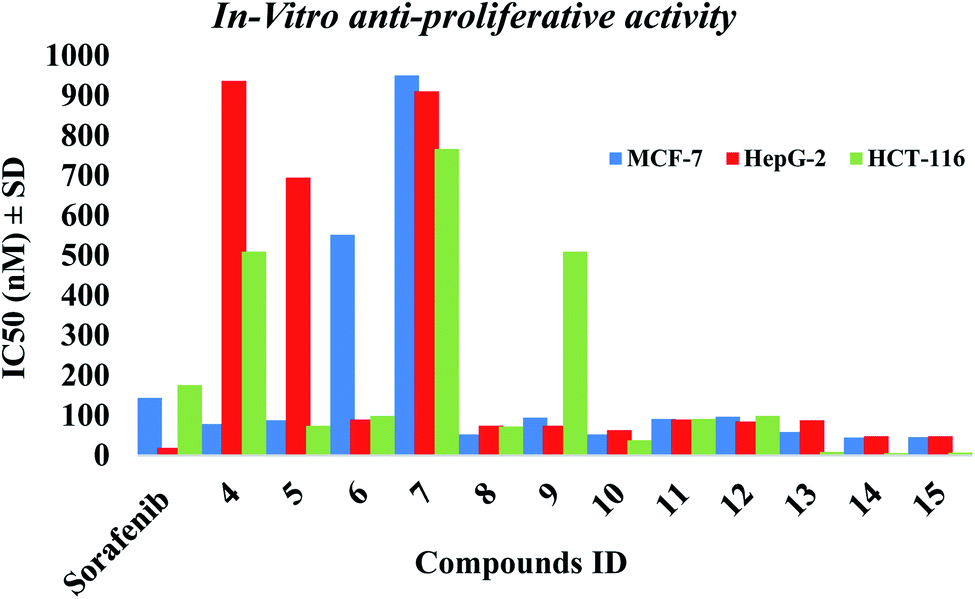
2.2.1. In vitro anti-proliferative activity. The in vitro anti-proliferative activities of all the synthesized compounds using MTT method,37,38 against breast cancer (MCF-7), hepatocellular carcinoma (HepG-2) and colorectal carcinoma (HCT-116) was carried out and compared to reference sorafenib (Table 1). Most of the compounds 5, 8, 10, 11, 12, 13, 14 & 15 showed superior cytotoxic activities against MCF-7 and HCT-116 with IC50 range (45–97 nM) and (7–99 nM), respectively compared to reference (IC50: 144 and 176 nM, respectively). While compounds 5, 8, 10, 11, 12 & 13 showed moderate activity against HepG-2 with IC50 range of (63–90 nM) compared to reference (IC50: 19 nM). Compounds 14 & 15 showed the most potent cytotoxic activities against the three cell lines with IC50 values of 45, 6, 48 nM and 46, 7, 48 nM against MCF-7, HCT-116 and HepG-2, respectively (Fig. 3).
Table 1 The IC50 values of tested compounds against MCF-7, HepG-2 and HCT-116 cancer cell linesa
| Compound ID |
In vitro IC50 (nM) Mean ± SD |
| MCF-7 |
HepG-2 |
HCT-116 |
| Data expressed as mean ± SD from 3 independent repeats (n = 3). Significance from sorafenib at p < 0.0001. |
| Sorafenib |
144 ± 0.47 |
19 ± 0.29 |
176 ± 0.69 |
| 4 |
79 ± 1.4 |
937 ± 12 |
510 ± 33 |
| 5 |
88 ± 0.99 |
695 ± 20 |
74 ± 3.2 |
| 6 |
552 ± 23 |
90 ± 2 |
99 ± 3.2 |
| 7 |
951 ± 33 |
911 ± 23 |
767 ± 32 |
| 8 |
53 ± 2.7 |
74 ± 2.4 |
73 ± 2.7 |
| 9 |
95 ± 4 |
74 ± 1.7 |
510 ± 22 |
| 10 |
53 ± 2.1 |
63 ± 2.1 |
38 ± 0.96 |
| 11 |
91 ± 4.2 |
90 ± 2.7 |
91 ± 1.7 |
| 12 |
97 ± 1.7 |
85 ± 1.8 |
99 ± 2.9 |
| 13 |
59 ± 0.94 |
88 ± 1.8 |
9 ± 0.36 |
| 14 |
45 ± 1.6 |
48 ± 0.69 |
6 ± 0.24 |
| 15 |
46 ± 1.2 |
48 ± 1.1 |
7 ± 0.46 |
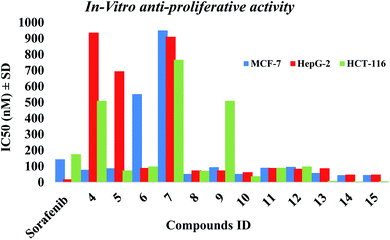 |
| | Fig. 3 The IC50 of tested compounds against MCF-7, HepG-2 and HCT-116 cancer cell lines. Data expressed as mean ± SD from 3 independent repeats (n = 3). | |
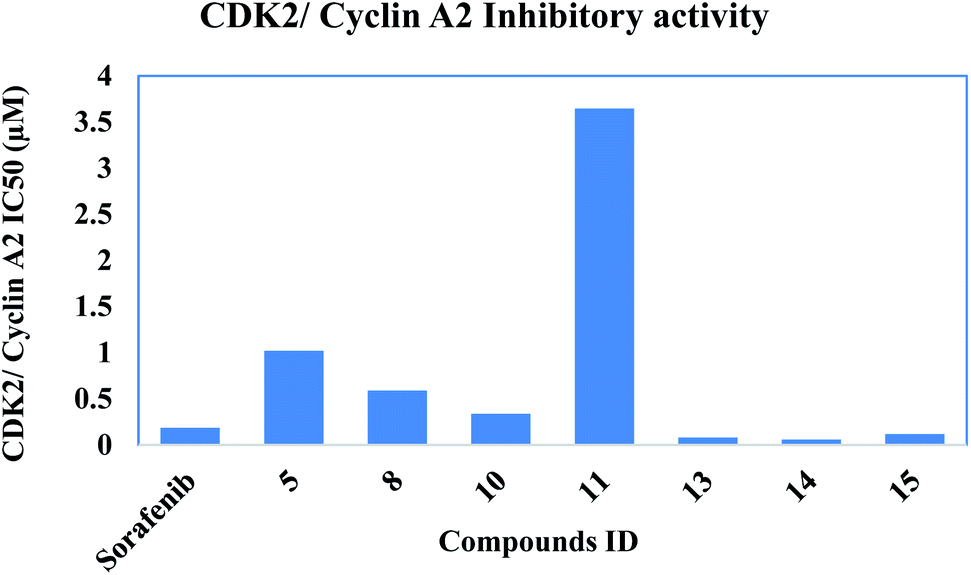
2.2.2. CDK2/cyclin A2 activity. The In vitro CDK2/cyclin A2 assays of the most potent synthesized compounds 5, 8, 10, 11, 13, 14 & 15 that showed high anti-proliferative activity, were carried out applying Promega Kinase-Glo Plus luminescence kinase assay.39 The test depends on ADP measurement after a kinase reaction, where ADP is transferred to ATP and is further converted into light. Here, the produced luminescent signal is in direct correlation to amount of ATP and is inversely correlated to the activity of kinase. The enzymatic inhibitory activity against CDK2/cyclin A2 results was presented in Table 2. All the tested novel compounds showed good inhibitory effect with an IC50 values ranging 0.057 ± 0.003–3.646 ± 0.203 μM compared to sorafenib IC50: 0.184 ± 0.01 μM. Results revealed that compounds 14, 13 and 15 showed significant inhibitory activity with IC50 values of 0.057 ± 0.003, 0.081 ± 0.004 and 0.119 ± 0.007 μM, respectively, compared to control drug sorafenib (Fig. 4).
Table 2 CDK2/cyclin A2 inhibitory activity results of the most potent compounds
| Compound ID |
CDK2/cyclinA2 IC50 (μM) ± SD |
| Sorafenib |
0.184 ± 0.01 |
| 5 |
1.021 ± 0.057 |
| 8 |
0.589 ± 0.033 |
| 10 |
0.336 ± 0.019 |
| 11 |
3.646 ± 0.203 |
| 13 |
0.081 ± 0.004 |
| 14 |
0.057 ± 0.003 |
| 15 |
0.119 ± 0.007 |
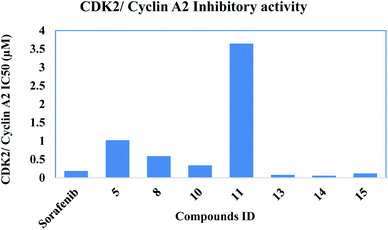 |
| | Fig. 4 Inhibitory activity of the most potent compounds on CDK2/cyclin A2. | |
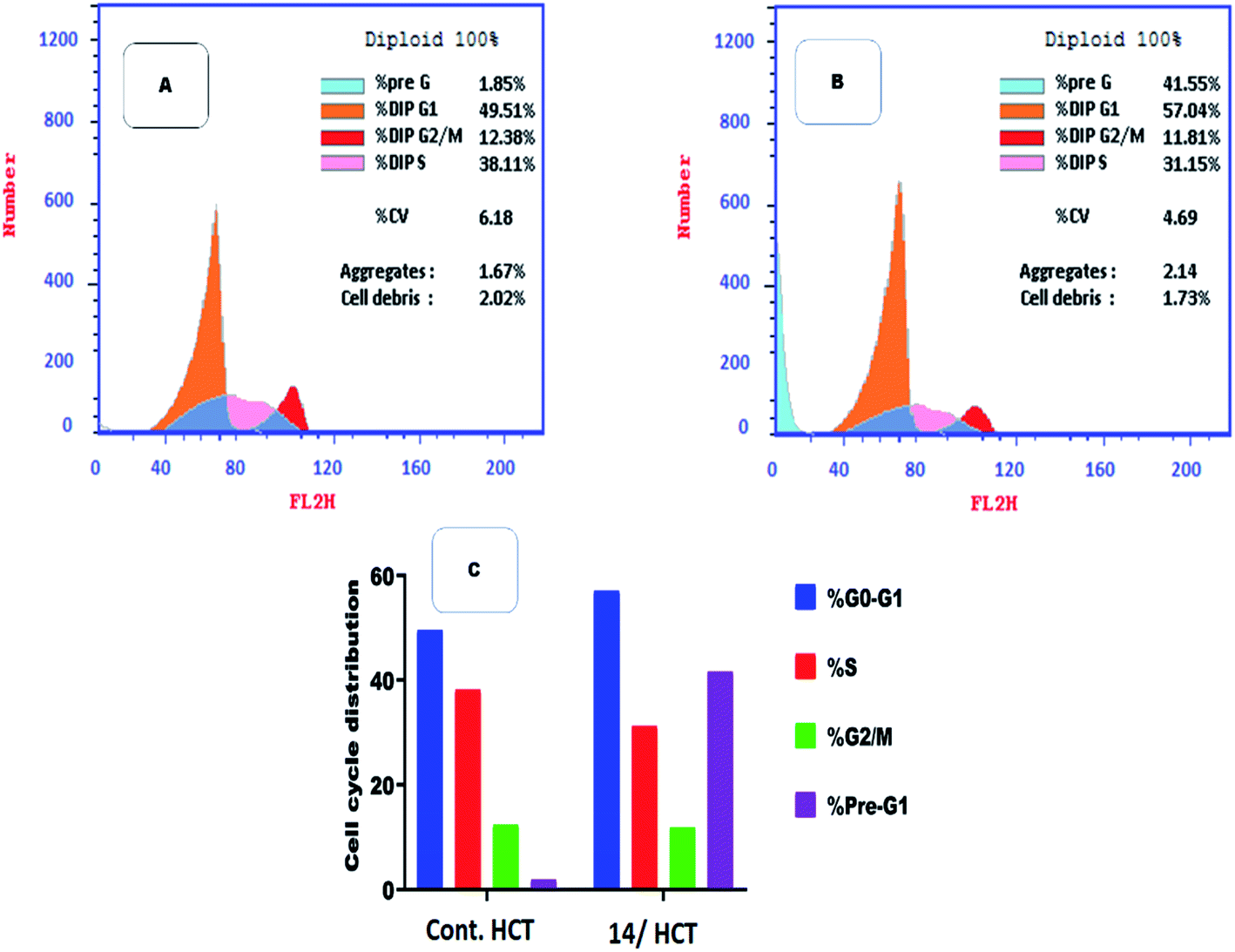
2.2.3. Flow cytometry cell cycle analysis. Among the most potent anti-proliferative screened compounds against the three selected cancer cell lines, compound 14 was selected for cell cycle analysis using flow cytometry assay against HCT cells. The cell cycle analysis is done to investigate the mechanism and the mode of action of the newly synthesized compounds.40 Table 3 presents the results showing Pre-G1 and cell growth arrest at G0-G1 stage. Results were compared to normal control cells, in Dip Pre-G1 with an increase from 1.85% to 41.55% and an increase in Dip G0-G1 phase from 49.51% to 57.04% for compound 14 (Fig. 5). It is worth noting that CDK2 is associated to G1 transition in cell cycle while CDK1 is more associated to G2-M phase. So selectivity to CDK2 was confirmed via the mentioned results.24
Table 3 Flow cytometric analysis for cell cycle distribution of compound 14 on HCT cells
| Compound |
Results DNA content |
| %G0-G1 |
%S |
%G2/M |
%Pre-G1 |
Comment |
| Control/HCT |
49.51 |
38.11 |
12.38 |
1.85 |
— |
| Compound 14/HCT |
57.04 |
31.15 |
11.81 |
41.55 |
Cell growth arrest at G1 |
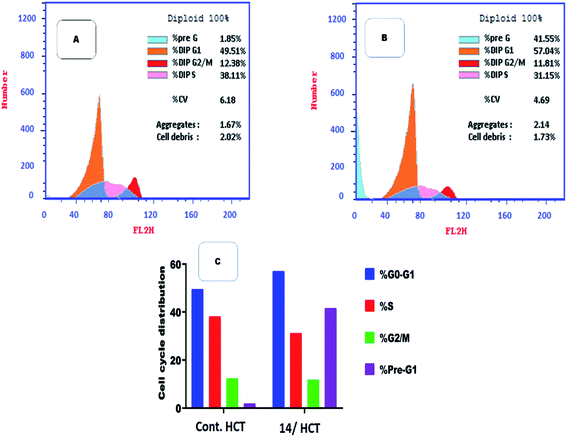 |
| | Fig. 5 Flow cytometric analysis for cell cycle distribution. (A) Control HCT, (B) compound 14, and (C) graphical representation for cell cycle distribution analysis among different treated cells. | |
2.2.4. Flow cytometric analysis of apoptosis
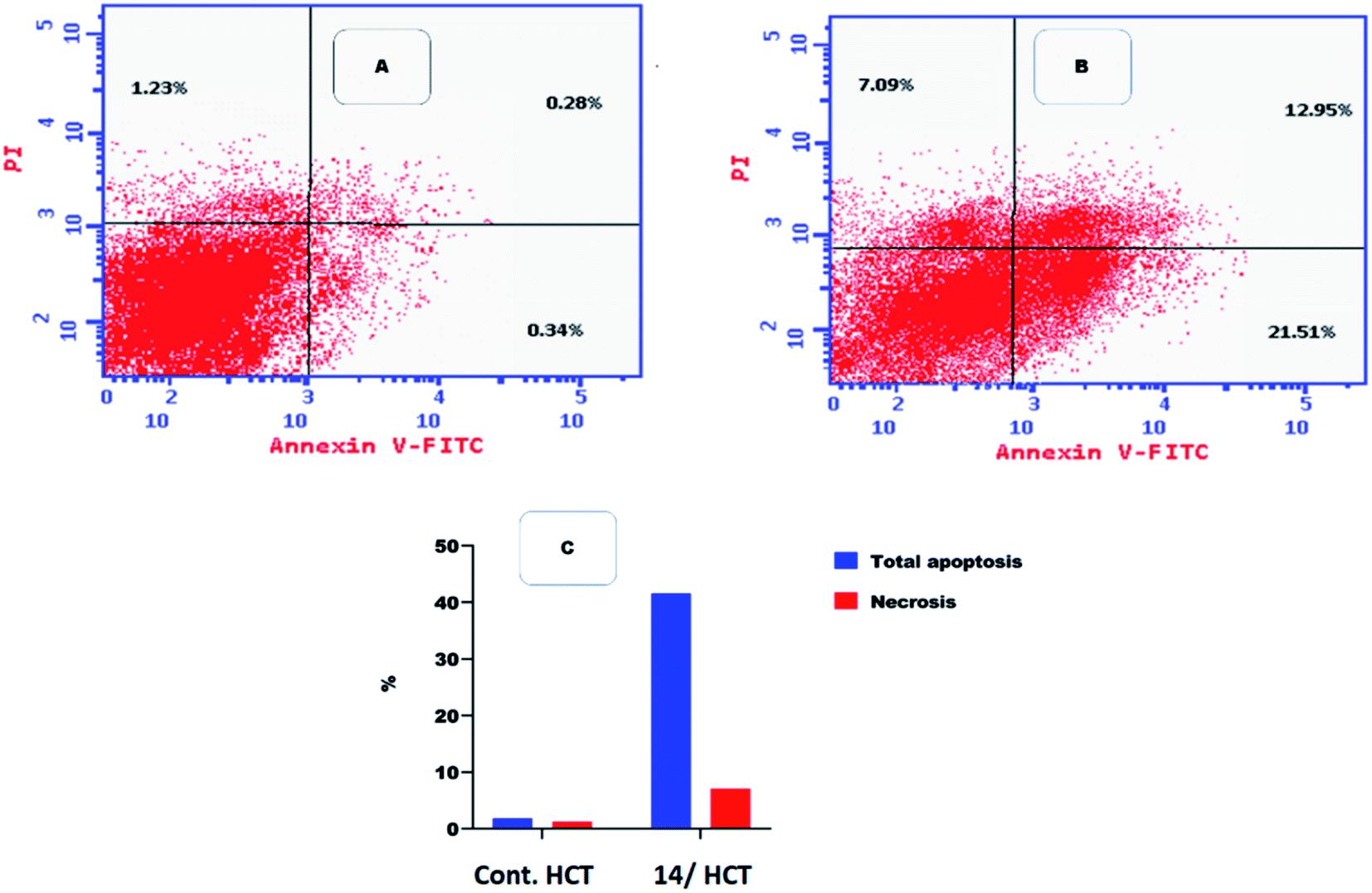
Flow cytometric analysis of apoptosis was performed to examine the potentiality of compound 14 in apoptosis induction against HCT cell line.41
Table 4 and Fig. 6 showed that compound 14 induced apoptosis by 41.55% (21.51 and 12.95 at early and late apoptosis, respectively), which was 22 times more than the standard control (1.85%).
Table 4 Effect of compound 14 on apoptosis in HCT cells
| Compound |
Apoptosis |
Necrosis |
| Total |
Early |
Late |
| Control HCT |
1.85 |
0.34 |
0.28 |
1.23 |
| Compound 14/HCT |
41.55 |
21.51 |
12.95 |
7.09 |
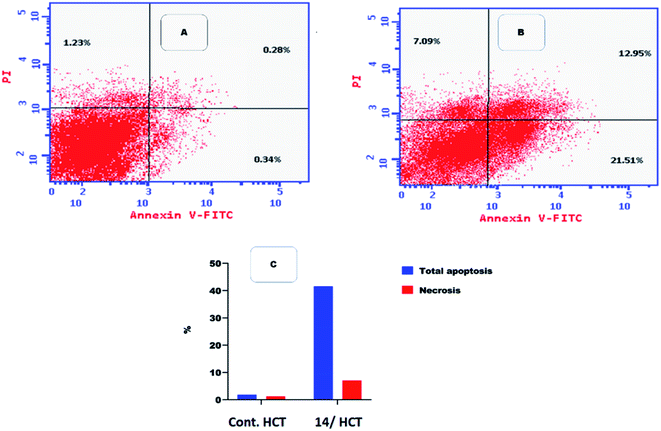 |
| | Fig. 6 Flow cytometric analysis of apoptosis among treated cells. (A) Control HCT, (B) compound 14, and (C) graphical illustration of apoptosis% among different treated cells. | |
2.3. In silico studies
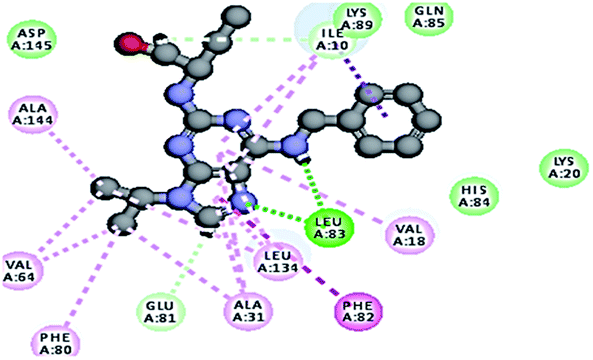
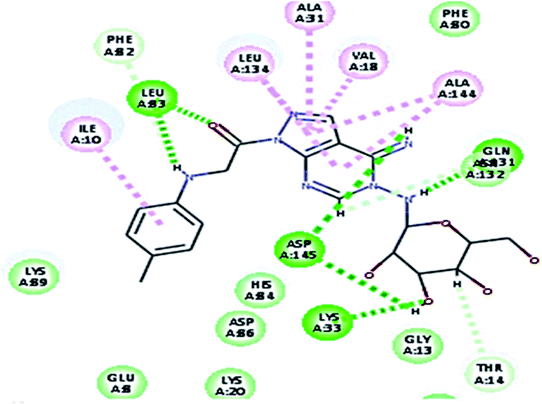
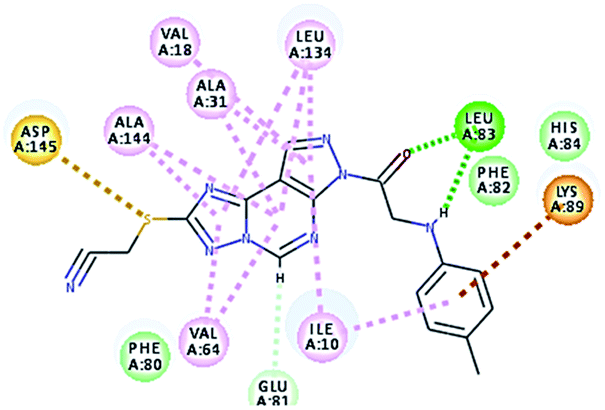
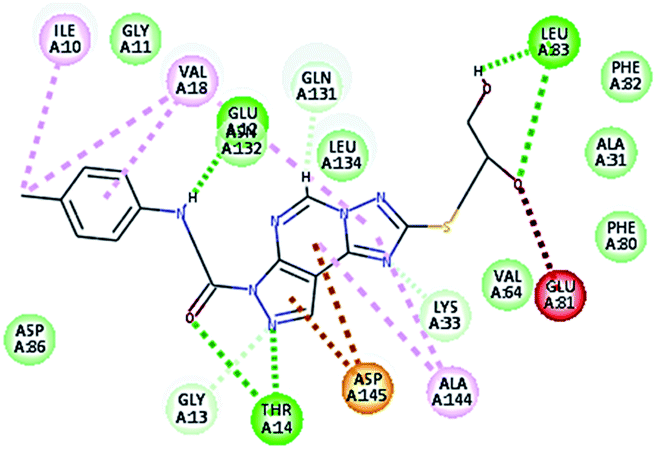
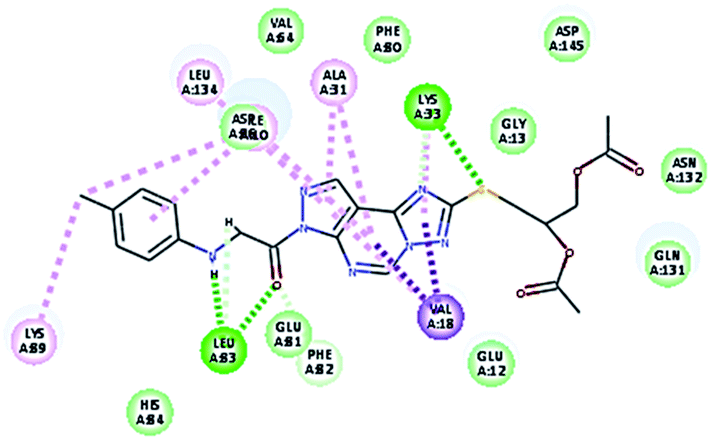
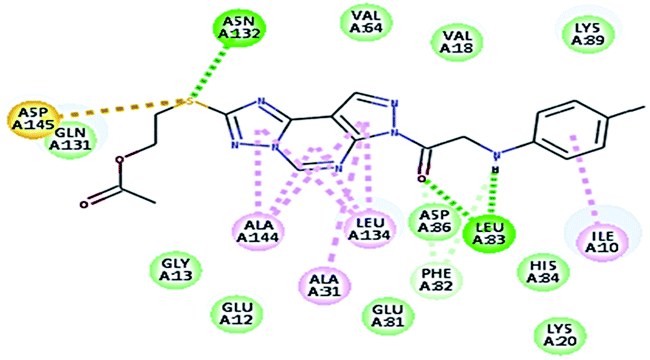
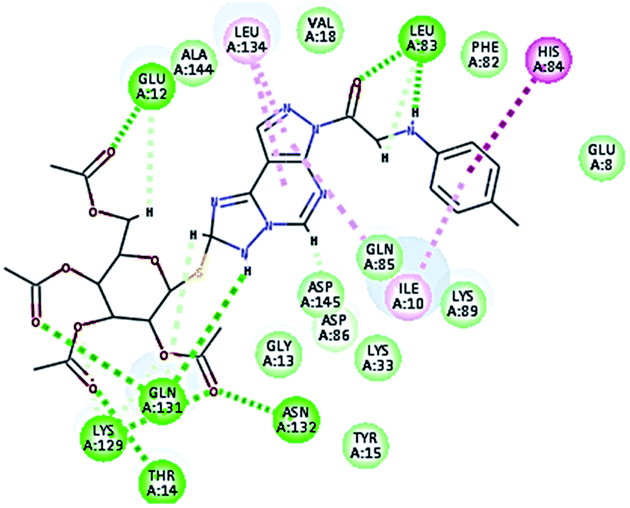
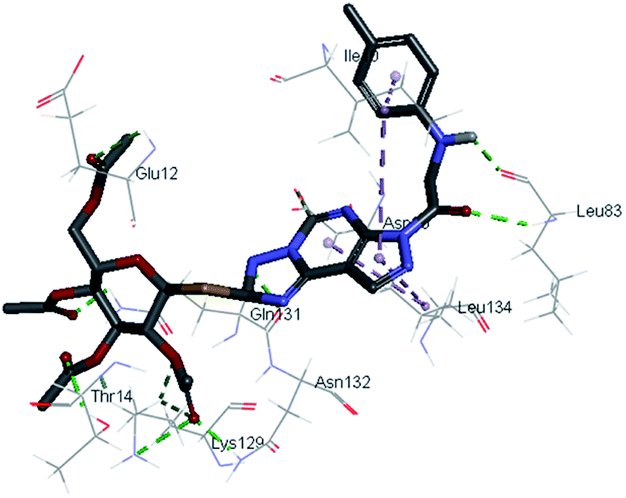
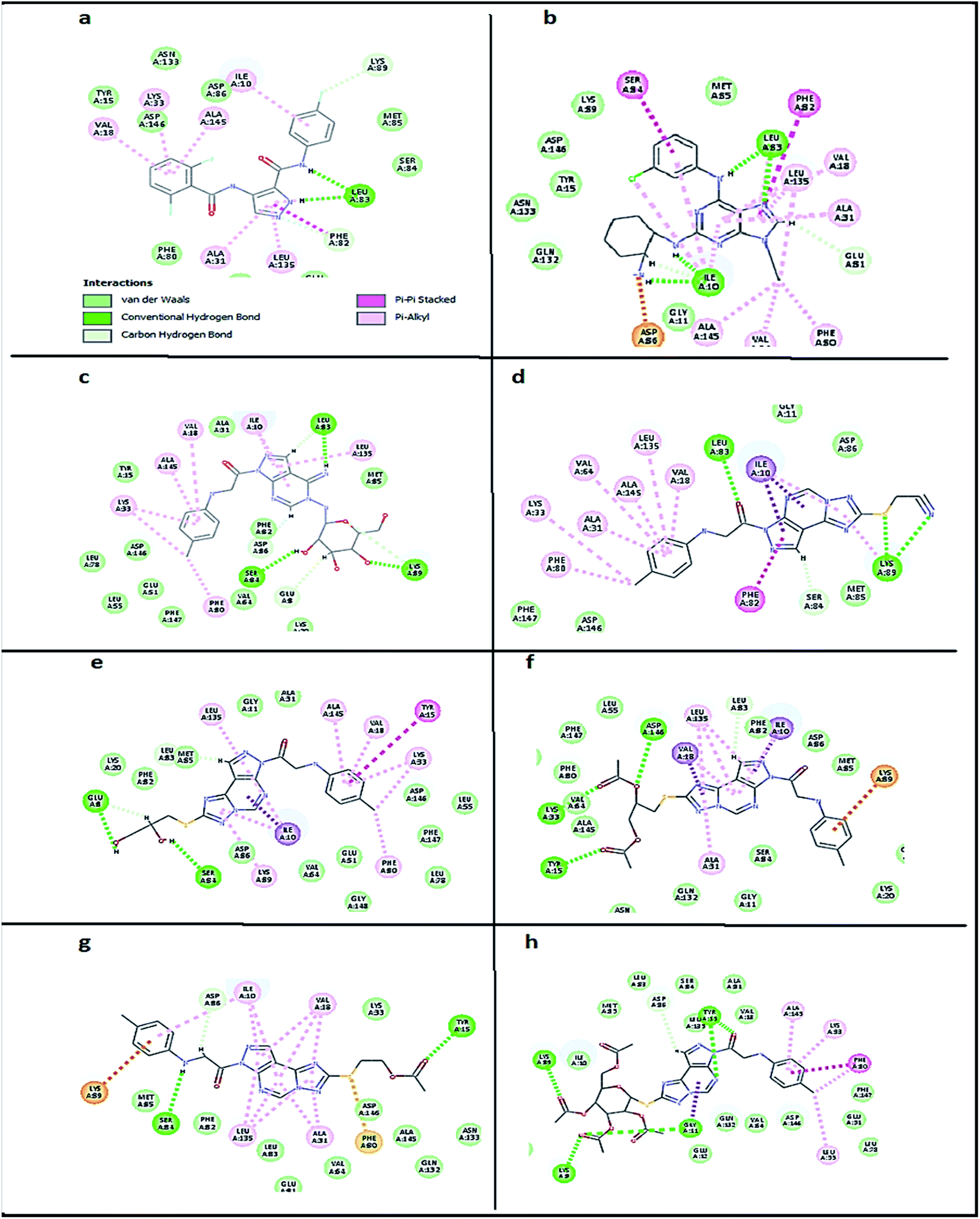
2.3.1. Molecular docking. Molecular docking study was applied using C-Docker protocol in Discovery Studio 4.0 Software. The compounds that showed CDk2 inhibitory activity (compounds 5, 8, 10, 11, 13, 14 & 15) were prepared and docked into the binding site of CDK2 enzyme. The analysis study of the binding modes of the designed compounds was used to interpret the biological results and to obtain more explanation about the binding poses and interactions to the key amino acids in the binding site. The X-ray crystallographic structure of CDk2 complexed with roscovitine (PDB ID: 2A4L) revealed the two essential hydrogen bonds with Leu83.9,12 Validation was confirmed via re-docking of roscovitine in the active site of CDk2 with RMSD value = 0.5 Å°. The selected pose out of ten for each compound that showed maximum similarity to the binding mode of the ligand is considered the best pose. The presented docking study showed comparable binding modes between the lead compound and the docked molecules. The binding mode with the essential amino acids and the interaction energy of the most biologically active synthesized compounds are summarized in Table 5.
Table 5 The C-Docker interaction energy and the binding mode of the most potent newly synthesized compounds compared to roscovitine ligand
| Compound name |
Binding mode |
C-DOCKER interaction energy (kcal mol−1) |
Key amino acids/interaction |
Distance Å in hydrogen bonds |
| Lead compound roscovitine |
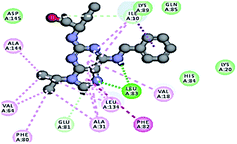 |
55.75 |
1 HBA with NH group of Leu83 |
2.33 |
| 1 HBD with oxygen atom of the carbonyl group of Leu83, |
2.87 |
| Hydrophobic binding with Leu134, Val18, Val64, Ala31, Ala144, Ile10, Phe80, Lys89 |
|
| 5 |
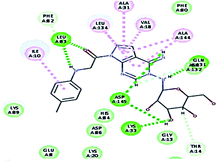 |
51.35 |
1 HBA through NH group with Leu83 |
2.40 |
| 1 HBD through oxygen atom of the carbonyl group of Leu83, |
2.01 |
| 1 HBA with Lys33, |
2.44 |
| 2 HBD with Asp145, hydrophobic binding with Leu134, Val18, Ala31, Ala144, Ile10. |
2.31–2.05 |
| 8 |
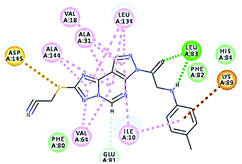 |
52.75 |
1 HBA through NH group with Leu83 |
2.40 |
| 1 HBD through oxygen atom of the carbonyl group of Leu83. |
2.04 |
| Hydrophobic binding with Leu134, Val18,Val64, Ala31, Ala144, Ile10. |
|
| 10 |
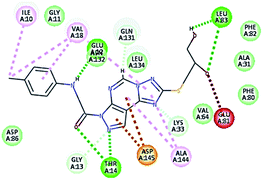 |
53.05 |
1 HBA, 1HBD through –OH groups of the dihydroxypropyl thio group with Leu83, |
2.41 |
| 2.01 |
| 1 HBD Glu132, |
2.43 |
| 2 HBA Thr14, hydrophobic binding with Val18, Ala144, Ile10, aromatic interaction with Asp145. Unfavorable interaction with Glu81. |
2.07–2.31 |
| 11 |
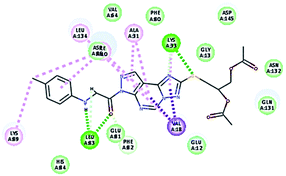 |
49.88 |
1 HBA through NH group with Leu83 |
2.02 |
| 1 HBD through oxygen atom of the carbonyl group of Leu83, |
2.13 |
| 1 HBA with Lys33, hydrophobic with Val18, Ala31, Leu134 and Lys89. |
2.22 |
| 13 |
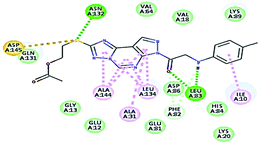 |
61.55 |
1 HBA through NH group with Leu83 |
2.04 |
| 1 HBD through oxygen atom of the carbonyl group of Leu83, |
1.79 |
| 1 HBA with Asn132. Hydrophobic with Ile10, Ala144, Ala31, Leu134. Sulphur–hydrogen bonding interaction with Asp145. |
2.43 |
| 14 |
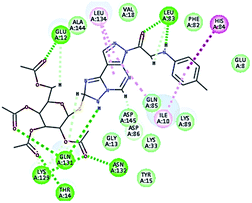 |
62.79 |
1 HBA through NH group with Leu83 |
2.44 |
| 1 HBD through an oxygen atom of the carbonyl group of Leu83, |
2.03 |
| 1 HBD, 1 HBA with Gln131, |
2.33–2.41 |
| 1 HBA with Glu12, Asn132, Lys129, Thr14. Hydrophobic with Leu134 and Ile10, His84 |
2.40-2.13–2.33-2.12 |
| 15 |
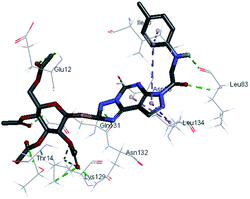 |
62.79 |
1 HBA through NH group with Leu83 |
2.44 |
| 1 HBD through an oxygen atom of the carbonyl group of Leu83, |
2.03 |
| 1 HBD with HBA with Gln131, |
2.33–2.41 |
| 1 HBA with Glu12, Asn132, Lys129, Thr14. hydrophobic with Leu134 and Ile10, His84 |
2.40-2.13–2.33-2.12 |
It was observed that all the docked molecules revealed the two essential hydrogen bonds with LEU83 as HBD via –NH of glycyl group with binding distance range of (1.79–2.13 Å) and HBA via the oxygen atom of the carbonyl group of the glycyl with binding distance range of (2.02–2.41) Å (Table 5). Compound 10 (the dihydroxy propyl thio derivative) showed a different binding mode where the two essential hydrogen bonds with Leu83 was performed via the –OH groups of the dihydroxypropyl thio group. While compounds 11 & 13 showed an extra hydrogen bond via the thio group. On the other side compound 14 & 15 showed 5 extra hydrogen bonds with the carbonyl groups of the 4 acetoxy thioglycoside groups and 1 extra hydrogen bond via the –NH of the triazole ring, that may explain the superior biological results.
To confirm the selectivity of the novel potent compounds on CDK2 enzyme rather than CDK1, docking study was performed on human CDK1/CyclinB1/CKS2 with inhibitor {[(2,6-difluorophenyl)carbonyl]amino}-N-(4-fluorophenyl)-1H-pyrazole-3-carboxamide (PDB ID: 4Y72). Results showed different binding mode of the targeted compounds compared to co-crystallized ligand and lead compound CGP74514A. Although both lead compounds showed 2 hydrogen binding with Leu83 in addition to the hydrophobic binding to both Ala31 and Leu135, it was observed that all the tested compounds had different binding mode than the reported one,42 with interaction energy of range of (E = − 61.22 to −45.33 kcal mol−1) compared to lead compounds (E = − 59.60, −58.65 kcal mol−1). Where compounds 5 and 8 showed only 1 hydrogen bond with Leu83, while the other compounds missed the 2 essential hydrogen bonds with Leu83 (Fig. 7). Also compounds 5 and 8 missed one hydrophobic binding with Ala31, while the other compounds missed the two reported hydrophobic binding with either Ala31 or Leu135 (Fig. 7).
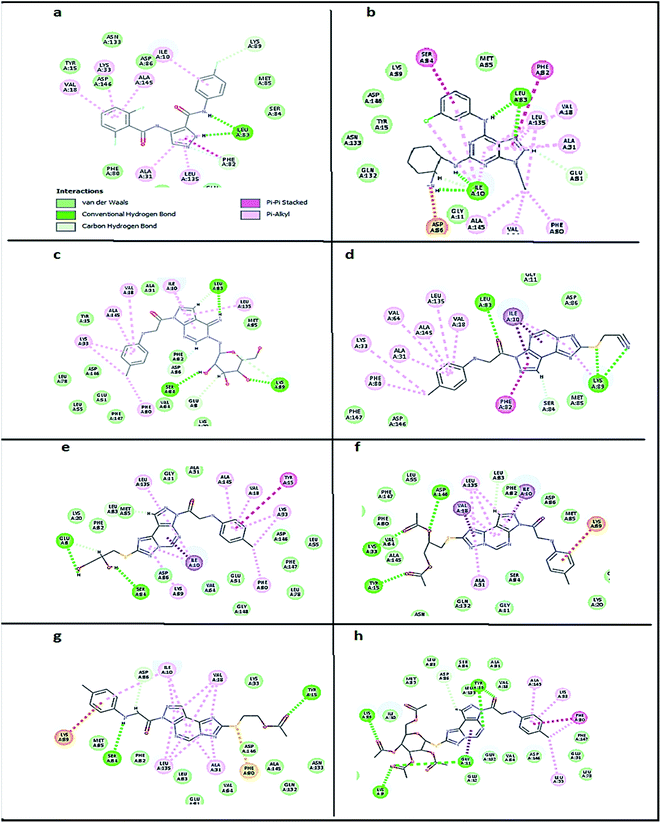 |
| | Fig. 7 2D diagram showing docking results on CDK1(PDB ID: 4Y72) of: (a) co-crystallized ligand (E = − 59.60 kcal mol−1), (b) lead compound CGP74514A (E = − 58.65 kcal mol−1), (c) compound 5 (E = − 55.10 kcal mol−1), (d) compound 8 (E = − 45.33 kcal mol−1), (e) compound 10 (E = − 50.13 kcal mol−1), (f) compound 11 (E = − 56.23 kcal mol−1), (g) compound 13 (E = − 50.55 kcal mol−1) and (h) compound 14 (E = − 61.22 kcal mol−1). | |
2.3.2. In silico predictive ADMET study. The ADMET study is mainly concerned with the chemical structure of the molecule, it includes several parameters calculation using Discovery Studio 4.0 Software; blood–brain barrier level, absorption level, atom based log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P98 (A logP98), 2D polar surface area (ADMET 2D PSA), Cytochrome P450 2D6 (CYP2D6), hepatotoxicity probability, aqueous solubility level and Plasma protein binding logarithmic level (PPB Level).
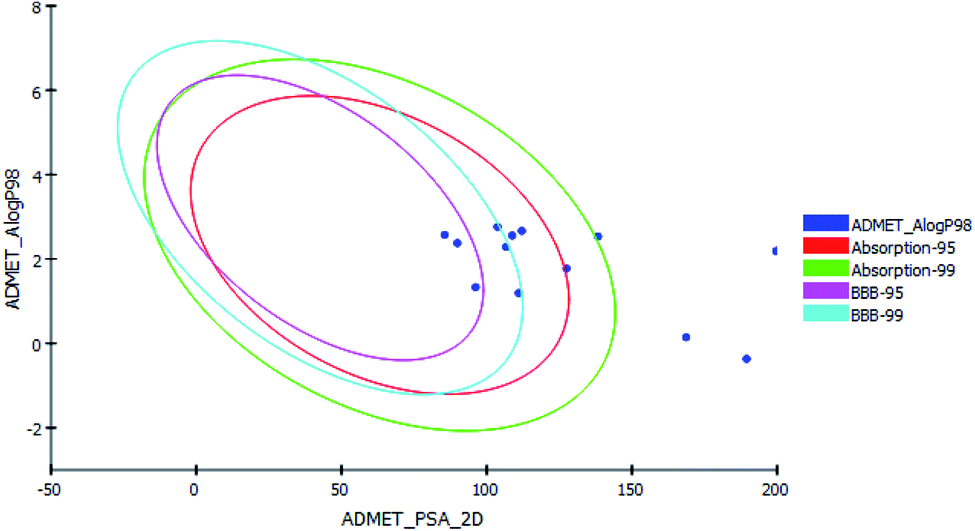
P98 (A logP98), 2D polar surface area (ADMET 2D PSA), Cytochrome P450 2D6 (CYP2D6), hepatotoxicity probability, aqueous solubility level and Plasma protein binding logarithmic level (PPB Level).In ADMET plot (Fig. 8), all the compounds had BBB level of 3 and 4, hence they are not able to pass the blood–brain barrier. Most of the compounds had absorption level = 0 or 1, thus estimated to have good to moderate human intestinal absorption, whilst only compounds 4, 5, 11, 14 and 15 showed low absorption. Most of the compounds showed ADME aqueous solubility level between 2 and 3 which indicates good aqueous solubility except compounds 7, 8, 9 & 13. The key property (PSA) was linked to drug bioavailability. Therefore, molecules which are passively absorbed and PSA < 140 are thought to have lower bioavailability. Most of the synthesized compounds were predicted to present good passive oral absorption except compounds 4, 5, 14 & 15 which showed good bioavailability results with PSA range: 168.67–199.705. Compounds 3, 4, 5, 11, 14 and 15 showed no hepatotoxicity. Also, all the compounds are considered non inhibitor to Cytochrome P450 2D6 (CYP2D6) (Table 6).
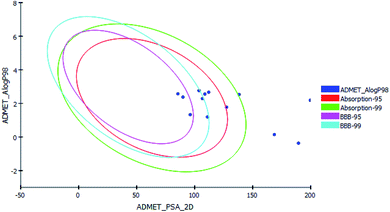 |
| | Fig. 8 The ADMET plot using calculated PSA_2D and A logP98 properties. | |
Table 6 ADMET predictions of the newly synthesized compounds
| Name |
BBB LEVEL |
ADMET Absorption level |
ADMET A logP98 |
ADMET PSA 2D |
Cytochrome P450 2D6 (CYP2D6) (non-inhibitor) |
Hepato-toxicity |
ADMET Aq solubility level |
PPB-level (highly bounded) |
| 2 |
3 (Low) |
0 (Good) |
1.331 |
96.195 |
−8.00592 |
True |
3 (Good) |
13.1974 |
| 3 |
3 (Low) |
0 (Good) |
2.376 |
89.908 |
−5.56285 |
False (non-toxic) |
3 (Good) |
12.5838 |
| 4 |
4 |
3 (v. Low) |
0.143 |
168.67 |
−6.93254 |
False (non-toxic) |
3 (Good) |
17.2665 |
| 5 |
4 |
3 (v. Low) |
−0.368 |
189.486 |
−4.50282 |
False (non-toxic) |
3 (Good) |
16.8921 |
| 6 |
3 (Low) |
0 (Good) |
1.193 |
111.023 |
−6.96286 |
True |
3 (Good) |
14.0489 |
| 7 |
3 (Low) |
0 (Good) |
2.572 |
85.529 |
−6.84541 |
True |
2 (Low) |
14.0495 |
| 8 |
4 |
0 (Good) |
2.556 |
108.786 |
−5.40681 |
True |
2 (Low) |
15.4385 |
| 9 |
3 (Low) |
0 (Good) |
2.763 |
103.711 |
−5.6183 |
True |
2 (Low) |
22.1754 |
| 10 |
4 |
1 (Moderate) |
1.775 |
127.482 |
−5.15483 |
True |
3 (Good) |
17.524 |
| 11 |
4 |
2 (Low) |
2.533 |
138.313 |
−7.01132 |
False (non-toxic) |
3 (Good) |
17.9553 |
| 12 |
3 (Low) |
0 (Good) |
2.286 |
106.667 |
−4.74756 |
True |
3 (Good) |
15.9934 |
| 13 |
4 |
0 (Good) |
2.665 |
112.082 |
−5.66217 |
True |
2 (Low) |
16.1493 |
| 14 |
4 |
3 (v. Low) |
2.186 |
199.705 |
−8.90741 |
False (non-toxic) |
3 (Good) |
18.2753 |
| 15 |
4 |
3 (v. Low) |
2.186 |
199.705 |
−8.90741 |
False (non-toxic) |
3 (Good) |
18.2753 |
Since gastrointestinal absorption and blood barrier penetration are considered the two essential pharmacokinetic behaviors to be estimated during the drug discovery processes, the Brain Or IntestinaL EstimateD permeation method (BOILED-egg) was used as a predictive model computing the lipophilicity and polarity of tested molecules. The obtained results from the two physicochemical descriptors were translated into molecular design, conserving simplicity, accuracy and speed within a graphical output of the model. The BOILED-egg can be applied starting from the early steps of drug discovery till the evaluation of drug candidates for development.43
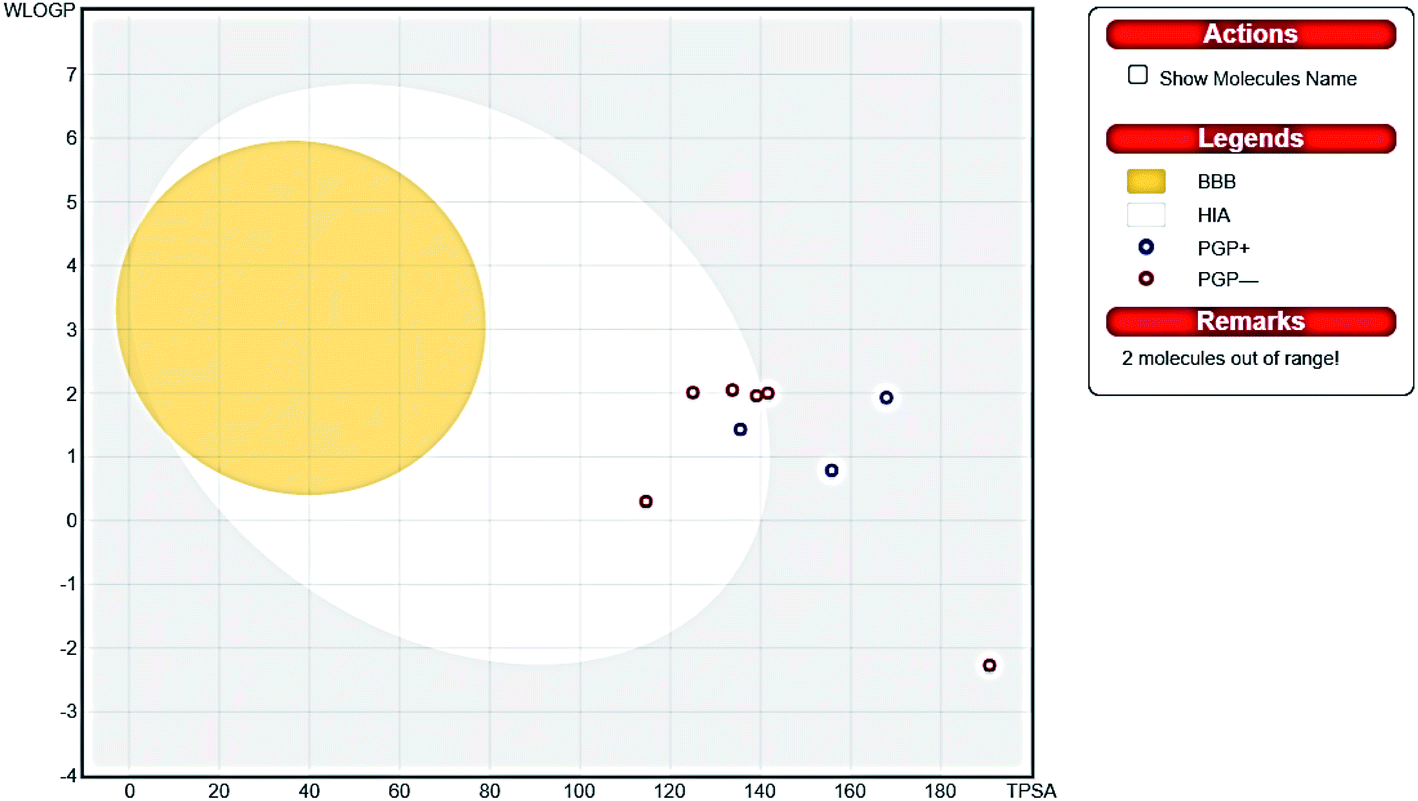
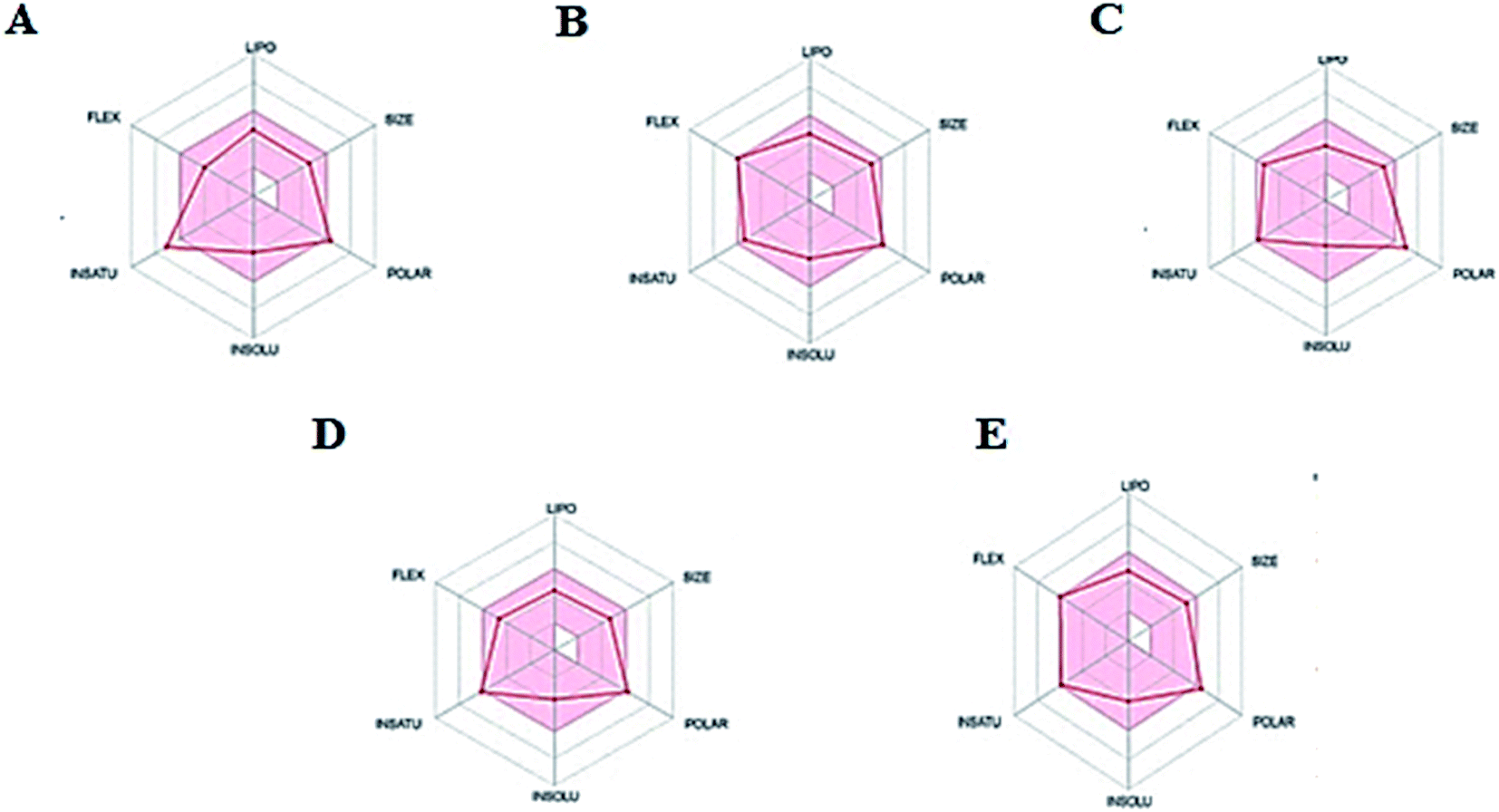
Thus, the pharmacokinetic properties of the tested compounds were predicted using SwissADME online tool.44 And results were aligned with ADMET study by Discovery Studio showing that compounds 7, 8, 9 & 12 expected to have high GIT absorption since they were in the white section of the Boiled egg chart for human intestinal absorption,43 while compounds 11, 14 & 15 were located in the grey area, showing low GIT solubility (Fig. 9). Regarding BBB penetration all the tested compounds were located away from the BBB penetration yellow region, so they probably have no capacity to penetrate the BBB and therefore can be safely used without CNS effect. Moreover, the bioavailability radar chart showed that all the tested compounds are in the desired range (pink region) of five parameters from the six parameters used for oral absorption prediction: FLEX (Flexibility), LIPO (Lipophilicity), INSOLU (Solubility), SIZE and POLAR (Polarity) (Fig. 10). Compounds 8, 9, 10, 12 & 13 showed good oral bioavailability. Also, all the compounds were predicted to be non-inhibitors to the cytochrome P450 2D6.
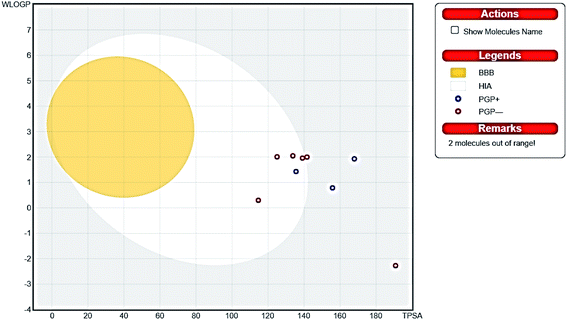 |
| | Fig. 9 Boiled egg chart showing the oral absorption and BBB penetration ability of the tested compounds. | |
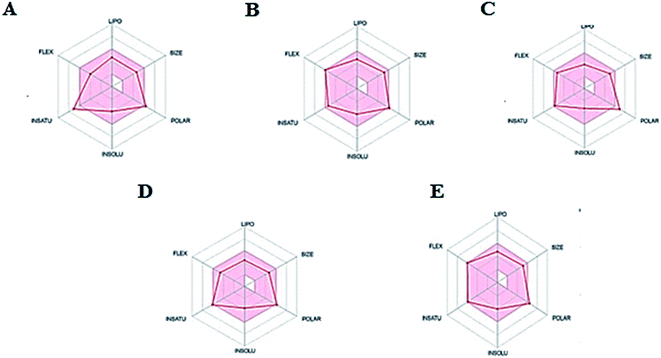 |
| | Fig. 10 The oral bioavailability radar chart of compounds (A) 8, (B) 9, (C) 10, (D) 12 & (E) 13. Pink zone describes the range of the optimal oral bioavailability property values of and the red lines expressed the predicted properties. | |
2.3.3 Structure activity relationship. In the pyrazolo[3,4-d]pyrimidine scaffold in compounds (4–6), it was observed that the N5 pyrazolopyrimidine D-glucose substitution showed potent activity against MCF-7 & HCT-116 cell lines, D-xylose substitution showed potent activity against HCT-116 & moderate activity against HepG-2 cell lines. Compared to unsubstituted group that showed potent activity against MCF-7 cell lines only (Fig. 11).
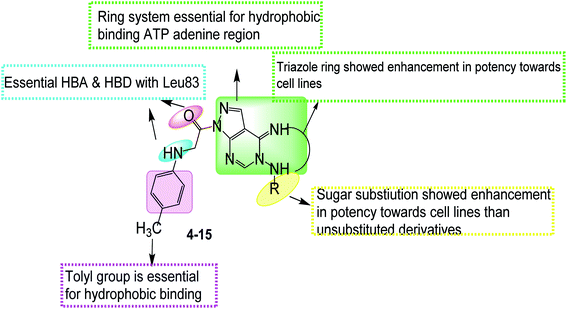 |
| | Fig. 11 Structure activity relationship for compounds (4–15). | |
While by the triazole ring fusion in pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin scaffold (7–15), the in vitro antiproliferative activity results revealed that compounds 14 and 15 showed the best cytotoxic results that might be related to the thioglycosidic substitution on triazole ring. The thioalkyl substituents as the monoacetoxy alkyl group in compound 13 showed more potent cytotoxic results compared to the acetonitrile in 8, diacetoxy alkyl in 11, monohydroxy alkyl in 12 as potent activity against MCF-7 & HCT-116 cell lines & moderate activity against HepG-2 cell lines. While the thioxo derivative in 7 was inactive, dimethoxy alkyl subistitution in 9 lost HCT-116 cell line activity, dihydroxy alkyl in 10 showed different binding mode with Leu83 (Fig. 11).
3. Conclusion
New series of pyrazolo[3,4-d]pyrimidine and pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine compounds (4–13) and the thioglycoside derivatives (14, 15) designed as novel CDK2 targeting compounds were synthesized. Most of the compounds showed superior cytotoxic activities against MCF-7 and HCT-116, and moderate activity against HepG-2 compared to sorafenib. Compounds 14 & 15 showed the best cytotoxic activities against the three cell lines. Also, compounds 14, 13 and 15 revealed the most significant inhibitory activity against CDK2/cyclin A2. From which the best compound, compound 14 was used to apply cell cycle test which showed cell arrest at G1 phase and apoptosis at Pre-G1 phase on HCT cells. and confirmed CDK2 selectivity.
Molecular docking study revealed that all the potent anti-proliferative tested compounds were of comparable docking interaction energy to that of roscovitine CDK2 ligand, compounds 13 (E = −61.55 kcal mol−1),14 and 15 (E = −62.79 kcal mol−1) showed the best docking interaction energy compared to roscovitine (E = −55.75 kcal mol−1) with similar binding mode forming the essential hydrogen bonding with Leu83 with selective CDK2 activity. While different binding mode to CDK1 ligand was observed after docking. In silico ADMET studies and drug-likeness was predicted and Boiled egg chart showed good pharmacokinetic properties. This helped in prediction of structure requirements necessary for the observed antitumor activity.
4. Experimental
4.1. Chemistry
All melting points were measured using a Reichert Thermovar apparatus and are uncorrected. The IR spectra were recorded on a PerkinElmer model 1720 FTIR spectrometer for KBr disc. Routine NMR measurements were made on a JEOL ECA-500 II spectrometer. Chemical shifts were reported in δ scale (ppm) relative to TMS as a reference standard and the coupling constants J values are given in Hz. 13C NMR were recorded at 125 MHz. The progress of the reactions was monitored by TLC using aluminum silica gel plates 60 F245. Spectral measurements and Elemental analyses were performed at the Micro-analytical center at the Faculty of science, Mansoura University, Mansoura. Compound 1 was synthesized according to a reported procedure.45
4.1.1 5-Amino-1-(p-tolylglycyl)-1H-pyrazole-4-carbonitrile (2). A solution of the acid hydrazide 1 (1.7 g, 10 mmol) and 2(ethoxymethylene)malononitrile (1.2 g, 10 mmol) in absolute ethanol (20 mL) was refluxed for 6 hours, then cooled to room temperature. The resulted solid was recrystallized from ethanol to afford the pyrazole compound 2 as red solid, yield 80%, mp 111–112 °C; IR (KBr): ![[small nu, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e154.gif) cm−1: 3340, 3250 (NH, NH2), 2205 (CN), 1660 (CO), 1630 (CN); 1H-NMR (DMSO-d6) (ppm): 8.23 (s, 1H, pyrazolo-H3), 7.43 (brs, 1H, NH, D2O exchangeable), 6.88 (d, J = 7.4 Hz, 2H, Ar–H), 6.48 (d, J = 7.4 Hz, 2H, Ar–H), 6.11 (brs, 2H, NH2, D2O exchangeable), 4.13 (s, 2H, CH2) 2.64 (s, 3H, CH3); MS (m/z): 255; anal. calc. for (C13H13N5O): C, 61.17; H, 5.13; N, 27.43; found C, 61.19; H, 5.17; N, 27.49%.
cm−1: 3340, 3250 (NH, NH2), 2205 (CN), 1660 (CO), 1630 (CN); 1H-NMR (DMSO-d6) (ppm): 8.23 (s, 1H, pyrazolo-H3), 7.43 (brs, 1H, NH, D2O exchangeable), 6.88 (d, J = 7.4 Hz, 2H, Ar–H), 6.48 (d, J = 7.4 Hz, 2H, Ar–H), 6.11 (brs, 2H, NH2, D2O exchangeable), 4.13 (s, 2H, CH2) 2.64 (s, 3H, CH3); MS (m/z): 255; anal. calc. for (C13H13N5O): C, 61.17; H, 5.13; N, 27.43; found C, 61.19; H, 5.17; N, 27.49%.
4.1.2 Ethyl-N-(4-cyano-1-(p-tolylglycyl)-1H-pyrazol-5-yl)formimidate (3). A mixture of the substituted pyrazole derivative 2 (2.5 g, 10 mmol) and triethyl orthoformate (1.4 g, 10 mmol) in acetic anhydride (20 mL) was heated under reflux for 6 hours, then cooled to room temperature. The precipitated formed was filtered and recrystallized by ethanol to afford the ester derivative 3 as Brown solid, yield 78%, mp 146–147 °C; IR (KBr): cm−1: 3320 (NH), 2205 (CN), 1650 (CO), 1618 (CN); 1H-NMR (DMSO-d6) (ppm): 8.24 (s, 1H, pyrazolo-H3), 8.17 (s, 1H, NCH), 7.40 (brs, 1H, NH, D2O exchangeable), 6.88 (d, J = 7.4 Hz, 2H, Ar–H), 6.48 (d, J = 7.4 Hz, 2H, Ar–H), 4.12 (s, 2H, CH2), 3.60 (q, J = 5.2 Hz, 2H, CH2), 2.67 (s, 3H, CH3), 1.2 (t, J = 5.2 Hz, 3H, CH3); MS (m/z): 311; anal. calc. for (C16H17N5O2): C, 61.72; H, 5.50; N, 22.49; found C, 61.76; H, 5.55; N, 22.54%.
4.1.3 1-(5-Amino-4-imino-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-(p-tolylamino)ethan-1-one (4). To solution of the ethyl ester derivative 3a (3.1 g, 10 mmol) in absolute ethanol (20 mL), hydrazine hydrate (99%, 1 mL, 20 mmol) was added dropwise. The reaction mixture was heated at reflux for 6 h then allowed to cool down to room temperature. The formed solid was separated by filtration and crystallized from methanol to provide the desired product as brown crystals (78%) mp = 169–170 °C; IR (KBr) cm−1: 3340–3210 (NH2, NH), 1660 (CO), 1615 (CN); 1H-NMR (DMSO-d6) (ppm): 11.42 (brs, 1H, NH, D2O exchangeable), 8.77 (brs, 1H, NH, D2O exchangeable), 8.51 (s, 1H, pyrimidine-H2), 7.98 (s, 1H, pyrazolo-H3), 7.56 (d, J = 12 Hz, 1H, phenyl-H2, H6), 7.07 (d, J = 12 Hz, 1H, phenyl-H3, H5), 5.51 (brs, 2H, NH2, D2O exchangeable), 4.12 (s, 2H, CH2), 2.70 (s, 3H); 13C NMR (DMSO-d6) δ: 168.7, 161.4, 153.5, 146.8, 143.0, 133.0, 129.4, 127.1, 122.2, 116.6, 55.9, 23.6; MS (m/z) 297; anal. calc. for: (C14H15N7O): C, 56.56; H, 5.09; N, 32.98%; found: C, 56.62; H, 5.14; N, 33.05%.
4.1.4 General procedure a. A mixture of aminopyrazolopyrimidine compound 4 (2.9 g, 10 mmol), the aldose sugar namely; D-glucose or D-xylose [10 mmol suspended in water (1 mL)] and glacial acetic acid (0.5 mL) in ethanol (25 mL) was allowed to be refluxed for 6 hours. Half of the amount of the solvent was evaporated under reduced pressure and the resulting solution was cooled to room temperature then left to stand in a refrigerator at 5–8 °C overnight. The resulting precipitated solid was filtered, dried and recrystallized by methanol to afford the N-glycosyl compounds 5 or 6, respectively.
4.1.5 1-(4-Imino-5-((β-D-glucopyranosyl)amino)-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-(p-tolylamino)ethan-1-one (5). Brown solid, yield 57%, mp 191–192 °C; IR (KBr): cm−1: 3425–3400 (OH), 3325 (NH), 1660 (CO), 1620 (CN); 1H-NMR (DMSO-d6) (ppm): 11.25 (brs, 1H, NH, D2O exchangeable), 8.74 (brs, 1H, NH, D2O exchangeable), 8.51 (s, 1H, pyrimidine-H2), 7.79 (s, 1H, pyrazolo-H3), 7.60 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.99 (d, J = 8 Hz, 1H, phenyl-H3, H5), 6.71 (brs, 1H, NH, D2O exchangeable), 5.10–5.13 (m, 1H), 4.97 (brs, 1H, OH, D2O exchangeable), 4.66 (brs, 1H, OH, D2O exchangeable), 4.63–4.62 (m, 2H), 4.43 (brs, 2H, 2OH, D2O exchangeable), 4.13 (s, 2H, CH2), 3.87–3.93 (m, 3H), 3.59–3.65 (m, 1H), 2.70 (s, 3H); 13C NMR (DMSO-d6) δ: 169.0, 161.4, 153.4, 146.8, 143.0, 134.3, 128.8, 127.1, 122.5, 116.3, 93.0, 83.6, 75.7, 72.2, 71.8, 62.1, 55.5, 23.9; MS (m/z) 459; anal. calc. for: (C20H25N7O6): C, 52.28; H, 5.48; N, 21.34%; found: C, 52.35; H, 5.56; N, 21.40%.
4.1.6 1-(4-Imino-5-((β-D-xylopyranosyl)amino)-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-2-(p-tolylamino)ethan-1-one (6). Brown solid, yield 66%, mp 183–144 °C; IR (KBr): cm−1: 3455–3440 (OH), 3325 (NH), 1668 (CO), 1615 (CN); 1H-NMR (DMSO-d6) (ppm): 11.25 (brs, 1H, NH, D2O exchangeable), 8.72 (brs, 1H, NH, D2O exchangeable), 8.51 (s, 1H, pyrimidine-H2), 8.02 (s, 1H, pyrazolo-H3), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.96 (d, J = 8 Hz, 1H, phenyl-H3, H5), 6.68 (brs, 1H, NH, D2O exchangeable), 5.08–5.10 (m, 1H), 4.83 (brs, 1H, OH, D2O exchangeable), 4.43 (brs, 2H, 2OH, D2O exchangeable), 4.12 (s, 2H, CH2), 3.90–3.96 (m, 2H), 3.71–3.76 (m, 3H), 2.68 (s, 3H); 13C NMR (DMSO-d6) δ: 168.7, 161.4, 153.4, 146.8, 143.0, 134.3, 128.8, 127.1, 122.5, 116.3, 93.0, 83.6, 75.7, 70.5, 62.1, 55.5, 23.9; MS (m/z) 429; anal. calc. for: (C19H23N7O5): C, 53.14; H, 5.40; N, 22.83%; found: C, 53.23; H, 5.47; N, 22.88%.
4.1.7 1-(2-Thioxo-2,3-dihydro-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-2-(p-tolylamino)ethan-1-one (7). To a well stirred mixture of compound 4 (2.9 g, 10 mmol), potassium hydroxide (0.67 g, 12 mmol) in ethanol 25 (mL), carbon disulfide (0.4 mL, 12 mmol) was added dropwise over 30 min. The reaction mixture was heated on water bath at 70 °C for 1 hour then refluxed for 10 hours. The solvent was evaporated under vacuum, then poured onto ice-cold water (50 mL) and the solution was acidified, the precipitate formed was filtered, washed with water and dried. Crystallization from ethanol gave the thione derivative 7 as brown solid, yield 73%, m p 17′–172 °C; IR (KBr): cm−1: 3328–3310 (NH), 1666 (CO), 1615 (CN); 1H-NMR (DMSO-d6) (ppm): 11.66 (brs, 1H, NH, D2O exchangeable), 8.96 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 8.13 (s, 1H, pyrazolo-H3), 7.59 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.86 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.12 (s, 2H, CH2), 2.64 (s, 3H); 13C NMR (DMSO-d6) δ: 176.4, 170.5, 160.7, 154.2, 146.5, 143.0, 131.2, 129.1, 127.1, 122.2, 115.3, 56.2, 23.9; MS (m/z) 339; anal. calc. for: (C15H13N7OS): C, 53.09; H, 3.86; N, 28.89%; found: C, 53.13; H, 3.90; N, 28.93%.
4.1.8 2-((7-(p-Tolylglycyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)thio)acetonitrile (8). A mixture of 7 (3.3 g, 10 mmol), chloroacetonitrile (0.7 g, 11 mmol) and anhydrous potassium carbonate (1.4 g, 10 mmol) in dimethyl formamide (20 mL) was stirred for 8 hours at room temperature. The reaction mixture was poured into ice-cold water (50 mL) with vigorous stirring for 30 minutes and the formed precipitate was filtered, dried and recrystallized from ethanol to give the nitrile derivative 8 as brown powder, yield 56%; mp > 300 °C; IR (KBr): cm−1: 3340 (NH), 2220 (CN), 1660 (CO), 1620 (CN); 1H-NMR (DMSO-d6) (ppm): 8.74 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.96 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.54 (s, 2H), 4.12 (s, 2H, CH2), 2.64 (s, 3H); 13C NMR (DMSO-d6) δ: 169.8, 166.3, 159.4, 154.2, 146.8, 142.7, 131.2, 129.1, 127.1, 122.5, 118.0, 116.3, 54.5, 23.6, 17.0; MS (m/z) 378; anal. calc. for: (C17H14N8OS): C, 53.96; H, 3.73; N, 29.61%; found: C, 54.02; H, 3.80; N, 29.66%.
4.1.9 General procedure b. A solution of pyrazolopyrimidine derivative 7 (1.4 g, 10 mmol), chloro-acetaldhyde dimethyl acetal (10 mmol) or 3-chloropropane-1,2-diol, and anhydrous potassium hydroxide (0.67 g, 10 mmol) in ethanol (25 mL) was heated under reflux with stirring for 10 hours. The solvent was evaporated under reduced pressure and ice-cold water was added to the remaining residue with continuous stirring to afford a precipitate which was filtered, dried and recrystallized from ethanol–water mixture 1:1 to afford the dimethoxy product 9 and 10, respectively.
4.1.10 1-(2-((2,2-Dimethoxyethyl)thio)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-2-(p-tolylamino)ethan-1-one (9). Gray solid, yield 74%, mp 197–198 °C; IR (KBr): cm−1: 3325 (NH), 1670 (CO), 1615 (CN); 1H-NMR (DMSO-d6) (ppm): 8.74 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.60 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.98 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.87–4.90 (m, 1H, OCH), 4.12 (s, 2H), 3.65 (s, 6H), 3.09–3.12 (m, 2H, SCH2), 2.68 (s, 3H); 13C NMR (DMSO-d6) δ: 169.8, 166.3, 159.7, 156.2, 146.8, 142.7, 131.9, 129.4, 127.1, 122.9, 115.9, 96.8, 67.3, 55.9, 35.3, 23.9; MS (m/z) 427; anal. calc. for: (C19H21N7O3S): C, 53.38; H, 4.95; N, 22.94%; found: C, 53.42; H, 4.99; N, 22.98%.
4.1.11 1-{2-[(2,3-Dihydroxypropyl)thio]-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl}-2-(p-tolylamino)ethan-1-one (10). Brownish solid, yield 66%, mp > 300 °C; IR (KBr): cm−1: 3420 (OH), 3315 (NH), 1670 (CO), 1620 (CN); 1H-NMR (DMSO-d6) (ppm): 8.72 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.90 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.76 (brs, 1H, OH, D2O exchangeable), 4.57 (brs, 1H, OH, D2O exchangeable), 4.12 (s, 2H), 3.70–3.73 (m, 1H), 3.45–3.48 (m, 2H), 3.15–3.17 (m, 2H), 2.68 (s, 3H); 13C NMR (DMSO-d6) δ: 170.1, 166.3, 159.3, 154.2, 146.8, 142.7, 131.6, 129.4, 127.4, 122.2, 115.3, 71.2, 65.6, 55.5, 35.3, 22.5; MS (m/z) 413; anal. calc. for: (C18H19N7O3S): C, 52.29; H, 4.63; N, 23.71%; found: C, 52.36; H, 4.71; N, 23.77%.
4.1.12 3-((7-(p-Tolylglycyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)thio)propane-1,2-diyl diacetate (11). A mixture of compound 10 (4.1 g, 10 mmol) and acetic anhydride (5 mL) in pyridine (15 mL) was stirred at room temperature for 8 hours then allowed to cool down to room temperature and poured on cold water (30 mL) with vigorous stirring for 2 hours. The formed precipitate was filtered, washed with water then with potassium hydrogen carbonate and dried. Crystallization form methanol afford the acetylated product as Brown solid, yield 71%, mp. 280–282 °C; IR (KBr): cm−1: 3305 (NH), 1666 (CO); 1H-NMR (DMSO-d6) (ppm): 8.80 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.60 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.98 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.32–4.37 (m, 1H), 4.12 (s, 2H), 3.42–3.48 (m, 2H), 3.12–3.22 (m, 2H), 2.64 (s, 3H), 2.20 (s, 6H); 13C NMR (DMSO-d6) δ: 172.2, 171.8, 169.7, 165.6, 159.0, 153.8, 146.8, 142.7, 131.2, 129.4, 127.4, 121.8, 115.2, 74.2, 67.7, 55.2, 37.1, 23.6, 17.3, 17.0; MS (m/z) 497; anal. calc. for: (C22H23N7O5S): C, 53.11; H, 4.66; N, 19.71%; found: C, 53.19; H, 4.73; N, 19.78%.
4.1.13 1-(2-((2-Hydroxyethyl)thio)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-2-(p-tolylamino)ethan-1-one (12). A solution of pyrazolopyrimidine derivative 7 (1.4 g, 10 mmol), 1-chloroethanol (0.8 g, 10 mmol) and anhydrous potassium hydroxide (0.67 g, 10 mmol) in ethanol (25 mL) was heated under reflux for 12 hours. The solvent was evaporated under reduced pressure and ice-cold water was added to the remaining residue with continuous stirring to afford a precipitate which was filtered, dried and recrystallized from methanol–water mixture 1:1 to provide the hydroxyethoxy compound 12 as Brown solid, yield 65%, mp 179–180 °C; IR (KBr): cm−1: 3380 (OH), 3340 (NH), 1668 (CO), 1615 (CN); 1H-NMR (DMSO-d6) (ppm): 8.72 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 8.03 (s, 1H, pyrazolo-H3), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 7.04 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.81 (brs, 1H, OH, D2O exchangeable), 4.12 (s, 2H), 3.68–3.71 (m, 2H), 3.48–3.51 (m, 2H), 2.68 (s, 3H); 13C NMR (DMSO-d6) δ: 170.1, 165.9, 159.4, 153.8, 146.5, 142.7, 131.2, 129.4, 127.1, 122.2, 115.3, 66.3, 55.2, 32.6, 23.9; MS (m/z) 497; anal. calc. for: (C17H17N7O2S): C, 53.25; H, 4.47; N, 25.57%; found: C, 53.33; H, 4.52; N, 25.61%.
4.1.14 2-((7-(p-Tolylglycyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)thio)ethyl acetate (13). To a mixture of compound 12 (4.9 g, 10 mmol) and acetic anhydride (5 mL) in pyridine (15 mL) was stirred at room temperature for 14 hours then cooled to room temperature. Ice-cold water (35 mL) was added with vigorous stirring for 3 hours. The formed precipitate was filtered, washed with water then with potassium hydrogen carbonate and dried. Crystallization form methanol afford the acetylated products as brown powder, yield 67%, mp. 162–163 °C; IR (KBr): cm−1: 3318 (NH), 1675 (CO), 1618 (CN); 1H-NMR (DMSO-d6) (ppm): 8.80 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 7.04 (d, J = 8 Hz, 1H, phenyl-H3, H5), 4.12 (s, 2H), 3.93–3.96 (m, 2H), 3.51–3.54 (m, 2H), 2.68 (s, 3H), 2.32 (s, 3H); 13C NMR (DMSO-d6) δ: 171.5, 169.4, 165.9, 159.7, 153.4, 146.1, 142.7, 131.2, 129.1, 127.1, 122.5, 115.3, 67.3, 55.5, 23.6, 17.3; MS (m/z) 425; anal. calc. for: (C19H19N7O3S): C, 53.64; H, 4.50; N, 23.05%; found: C, 53.69; H, 4.55; N, 23.11%.
4.1.15 General procedure c. To a suspension of potassium hydroxide (0.67 g, 10 mmol) in water (1 mL), a solution of pyrazolopyrimidine derivative 7 (1.4 g, 10 mmol) in acetone (20 mL) was added with continuous stirring for about 15 minutes, then a solution of the glycosyl bromide namely; 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide or 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide (12 mmol) in acetone (10 mL) was added portion-wise and the resulting mixture was stirred at room temperature for 5 hours. The solvent was evaporated under reduced pressure and the residue was washed with cold water (15 mL) to remove any remained inorganic substance. Extraction of the product with ethyl acetate (50 mL) followed by removal of the solvent afforded a solid material which was treated with petroleum ether/diethyl ether mixture 3:1 (30 mL) with stirring for 15 minutes for further purification. The formed solid product was filtered and dried to give compounds 15a or 15b, respectively.
4.1.16 1-(2-((Tetra-O-acetyl-D-glucopyranosyl)thio)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-2-(p-tolylamino)ethan-1-one (14). Pale yellow solid, yield 77%, mp 160–161 °C; IR (KBr): cm−1: 3325 (NH), 1670 (CO), 1738 (CO), 1618 (CN); 1H-NMR (DMSO-d6) (ppm): 8.66 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.98 (s, 1H, pyrazolo-H3), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.86 (d, J = 8 Hz, 1H, phenyl-H3, H5), 5.71–5.76 (m, 1H), 5.01–5.07 (m, 2H), 4.29–4.34 (m, 3H), 4.12 (s, 2H), 3.68–3.73 (m, 1H), 2.68 (s, 3H), 2.23 (s, 12H); 13C NMR (DMSO-d6) δ: 173.6, 173.2, 171.8, 171.5, 169.4, 165.3, 159.4, 153.8, 146.1, 142.7, 131.6, 129.1, 127.1, 122.2, 115.6, 84.3, 80.8, 76.7, 71.5, 67.3, 63.1, 55.5, 23.6, 18.7, 18.3, 17.7, 17.3; MS (m/z) 669; anal. calc. for: (C29H31N7O10S): C, 52.01; H, 4.67; N, 14.64%; found: C, 52.09; H, 4.74; N, 14.71%.
4.1.17 1-(2-((Tetra-O-acetyl-D-galactopyranosyl)thio)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-7-yl)-2-(p-tolylamino)ethan-1-one (15). Pale yellow solid, yield 77%, mp 155–156 °C; IR (KBr): cm−1: 3345 (NH), 1668 (CO), 1740 (CO), 1618 (CN); 1H-NMR (DMSO-d6) (ppm): 8.67 (brs, 1H, NH, D2O exchangeable), 8.50 (s, 1H, pyrimidine-H2), 7.98 (s, 1H, pyrazolo-H3), 7.58 (d, J = 8 Hz, 1H, phenyl-H2, H6), 6.86 (d, J = 8 Hz, 1H, phenyl-H3, H5), 5.82–5.74 (m, 1H), 5.01–5.07 (m, 2H), 4.29–4.38 (m, 3H), 4.12 (s, 2H), 3.68–3.73 (m, 1H), 2.68 (s, 3H), 2.23 (s, 12H); 13C NMR (DMSO-d6) δ: 173.6, 173.2, 171.8, 171.5, 169.4, 165.6, 159.4, 153.8, 146.1, 142.7, 131.6, 129.1, 127.1, 122.2, 115.6, 84.3, 80.8, 76.7, 71.5, 67.3, 63.1, 55.5, 23.6, 18.7, 18.3, 17.7, 17.3; MS (m/z) 669; anal. calc. for: (C29H31N7O10S): C, 52.01; H, 4.67; N, 14.64%; found: C, 52.07; H, 4.72; N, 14.70%.
4.2 Biological assays
4.2.1 In vitro anti-proliferative activity. The In vitro cytotoxicity of all the synthesized compounds against cancer cell lines breast cancer (MCF-7), hepatocellular carcinoma (HepG-2) and colorectal carcinoma (HCT-116) was carried out and compared to reference sorafenib. The assay was performed by applying the MTT assay procedure.37,38 In brief, in MTT assay, 5000–10000 cells per well were plated in a 96-well plate and allowed to grow 24 h, then treated with media that contain increased concentrations of tested compounds as 100 μL complete growth medium were mixed with 100 μL for each compound per well for 48 hours before applying the assay. After that, the media were withdrawn, and 100 μL of MTT was applied to each well, which was then incubated for 4 hours. The formed formazan crystals then solubilized by adding 100 μL of dimethyl sulfoxide (DMSO) solution. The cells viability was determined by measuring the optical density (OD) of each well of the developed purple color spectrophotometrically at 570 nm using an ELISA microplate reader (Epoc-2 C micro-plate reader, Bio Tek, VT, USA). The optical density of produced color was measured at 570 nm. The IC50 values (the concentration required to inhibit cell viability by 50%) were calculated, with the data expressed as a percentage of control cells (100 percent of cell viability).
4.2.2 CDK2/cyclin A2 assay. The In vitro assay of CDK2/cyclin A2 protein kinase was carried out on the most potent synthesized compounds 5, 8, 10, 11, 13, 14 & 15 that showed the highest anti-proliferative activities on human cell lines. The assay was proceeded in Egypt applying Kinase-Glo Plus luminescence kinase Assay kit (Promega).39 The Protocol steps were applied by enzyme and substrate dilution. Where, 1 μL of inhibitor, 5% DMSO, 2 μL of enzyme and 2 μL of substrate/ATP mix were added after ATP and inhibitors being diluted in Kinase Buffer. Incubation for 10 min at room temperature was allowed, then 5 μL of Reagent ADP-Glo™ was added and been incubated for another 40 min. After that, An amount equivalent to 10 μL of Kinase Detection Reagent was added to be incubated at room temperature for another 30 minutes. Luminescence was (where integration time range 0.5–1 second). The luminescent signal is in direct relation to the present quantity of ATP and in inverse relation to the activity of the kinase enzyme.39
4.2.3 Flow cytometry cell cycle analysis. The cell cycle analysis protocol was performed on compound 14 on HCT cells. The test is based on content of DNA measurement via staining using propidium iodide. Cells were first washed in PBS before being kept at 4 °C for 3 min through the dropwise using vortex addition of cold 70% ethanol, this is to avoid cell clumping and to ensure fixation as well. Then 50 μL from a stock of 100 μg mL−1 ribonuclease was added to selectively stain only DNA. Finally, an amount of 200 μL from a stock solution of 50 μg mL−1 of propidium iodide was added.40
4.2.4 Flow cytometric analysis of apoptosis. For the detection of apoptosis in treated cells, Annexin V—FITC—apoptosis detection kit (PN IM3546) was used, followed by flow cytometric analysis according to manufacturer protocol. In this assay, HCT cells were allowed to grow in a 25 cm3 flask until 70–80% confluence. Then HCT cells were treated with compound 14 for 48 h followed by a wash in PBS and suspended in 1× binding buffer. To 100 μL of the cell suspensions, 1 μL of annexin V-FITC solution and 5 μL of dissolved PI were added and incubated for 15 min in the dark. Then 400 μL of ice-cold 1× binding buffer was added and mixed gently. The flow cytometric analysis for the percentage of apoptotic cells was performed on a COULTER® EPICS® XL™ Flow Cytometer (USA).41
4.3. Molecular modeling studies
Molecular docking study using CDOCKER protocol in Discovery Studio 4.0 Software was carried out. The targeted compounds were docked into the CDK2 active site. The X-ray crystallographic structure of CDk2 complexed with roscovitine (PDB ID: 2A4L) was downloaded from PDB.9,12 Human CDK1 complexed with inhibitor (PDB ID: 2Y72) was also downloaded from PDB to confirm CDK2 selectivity of the novel compounds.42 The binding mode of the designed compounds was studied to explain their biological results and to detect the essential hydrogen bonding with Leu83. Where the best pose out of ten for each compound was selected compared to the ligand binding mode. In silico ADMET studies using Discovery Studio 4.0 Software and drug-likeness applying Boiled egg chart using https://www.swissadme.ch/index.php,44 were carried out to predict the pharmacokinetic properties of the targeted compounds which helped in structure requirements prediction for the observed antitumor activity.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contribution
All authors have contributed in all the steps of building up the article and approved the final article.
Conflicts of interest
All the authors declare that they have no conflict of interest.
Acknowledgements
Acknowledgement for Future University in Egypt (FUE) for performing molecular modeling study in Computer Aided Drug Design labs. The cell lines were obtained from American Type Tissue Culture Collection (ATCC, Rockville, MD).
References
- D. J. Baillache and A. Unciti-Broceta, RSC Med. Chem., 2020, 11(10), 1112–1135 RSC . https://pubs.rsc.org/en/content/articlelanding/2020/md/d0md00227e.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298(5600), 1912–1934, DOI:10.1126/science.1075762.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1(4), 309–315, DOI:10.1038/nrd773.
- N. E. Abd El-Sattar, E. H. Badawy, W. H. AbdEl-Hady, M. I. Abo-Alkasem, A. A. Mandour and N. S. Ismail, Chem. Pharm. Bull., 2021, 69(1), 106–117, DOI:10.1248/cpb.c20-00714.
- J. Chen, L. Pang, W. Wang, L. Wang, J. Z. Zhang and T. Zhu, J. Biomol. Struct. Dyn., 2020, 38(4), 985–996, DOI:10.1080/07391102.2019.1591304.
- S. S. Liang, X. G. Liu, Y. X. Cui, S. L. Zhang, Q. G. Zhang and J. Z. Chen, SAR QSAR Environ. Res., 2021, 32(7), 573–594, DOI:10.1080/1062936X.2021.1934896.
- G. Kontopidis, C. McInnes, S. R. Pandalaneni, I. McNae, D. Gibson, M. Mezna, M. Thomas, G. Wood, S. Wang, M. D. Walkinshaw and P. M. Fischer, Chem. Biol., 2006, 13(2), 201–211, DOI:10.1016/j.chembiol.2005.11.011.
- Y. Li, J. Zhang, W. Gao, L. Zhang, Y. Pan, S. Zhang and Y. Wang, Int. J. Mol. Sci., 2015, 16(5), 9314–9340, DOI:10.3390/ijms16059314.
- E. M. Husseiny, Bioorg. Chem., 2020, 102, 104053, DOI:10.1016/j.bioorg.2020.104053.
- S. Wee, D. Dhanak, H. Li, S. A. Armstrong, R. A. Copeland, R. Sims, S. B. Baylin, X. S. Liu and L. Schweizer, Ann. N. Y. Acad. Sci., 2014, 1309(1), 30–36, DOI:10.1111/nyas.12356.
- S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. A. Hampannavar, N. Thapliyal and V. N. Palakollu, Eur. J. Med. Chem., 2017, 126, 298–352, DOI:10.1016/j.ejmech.2016.11.019.
- G. M. Ali, D. A. Ibrahim, A. M. Elmetwali and N. S. Ismail, Bioorg. Chem., 2019, 86, 1–4, DOI:10.1016/j.bioorg.2019.01.008.
- A. M. Farag and A. M. Fahim, Synthesis, biological evaluation and DFT calculation of novel pyrazole and pyrimidine derivatives, J. Mol. Struct., 2019, 1179, 304–314, DOI:10.1016/j.molstruc.2018.11.008.
- N. Gokhan-Kelekc, S. Yabanoglu, E. Kupeli, U. Salgın, O. Ozgen, G. Ucar, E. Yesilada, E. Kendi, A. Yesilada and A. A. Bilgin, Bioorg. Med. Chem., 2007, 15(17), 5775–5786, DOI:10.1016/j.bmc.2007.06.004.
- R. Nithyabalaji, H. Krishnan and R. Sribalan, J. Mol. Struct., 2019, 1186, 1–10, DOI:10.1016/j.molstruc.2019.02.095.
- E. Hernandez-Vazquez, S. Salgado-Barrera, J. J. Ramirez-Espinosa, S. Estrada-Soto and F. Hernandez-Luis, Bioorg. Med. Chem., 2016, 24(10), 2298–2306, DOI:10.1016/j.bmc.2016.04.007.
- I. F. Nassar, A. F. El Farargy, F. M. Abdelrazek and N. S. Ismail, Nucleosides, Nucleotides Nucleic Acids, 2017, 36(4), 275–291, DOI:10.1080/15257770.2016.1276290.
- I. F. Nassar, S. R. Atta-Allah and A. S. Elgazwy, J. Enzyme Inhib. Med. Chem., 2015, 30(3), 396–405, DOI:10.3109/14756366.2014.940936.
- A. E. Rashad, A. E. Mahmoud and M. M. Ali, Eur. J. Med. Chem., 2011, 46(4), 1019–1026, DOI:10.1016/j.ejmech.2011.01.013.
- G. S. Hassan, H. H. Kadry, S. M. Abou-Seri, M. M. Ali and A. E. Mahmoud, Bioorg. Med. Chem., 2011, 19(22), 6808–6817, DOI:10.1016/j.bmc.2011.09.036.
- M. E. El-Naggar, A. S. Hassan, H. M. Awad and M. F. Mady, Molecules, 2018, 23(6), 1249, DOI:10.3390/molecules23061249.
- M. Radi, E. Dreassi, C. Brullo, E. Crespan, C. Tintori, V. Bernardo, M. Valoti, C. Zamperini, H. Daigl, F. Musumeci, F. Carraro, A. Naldini, I. Filippi, G. Maga, S. Schenone and M. Botta, J. Med. Chem., 2011, 54(8), 2610–2626, DOI:10.1021/jm1012819.
- K. J. Curran, J. C. Verheijen, J. Kaplan, D. J. Richard, L. Toral-Barza, I. Hollander, J. Lucas, S. Ayral-Kaloustian, K. Yu and A. Zask, Bioorg. Med. Chem. Lett., 2010, 20(4), 1440–1444, DOI:10.1016/j.bmcl.2009.12.086.
- Y. Li, W. Gao, F. Li, J. Wang, J. Zhang, Y. Yang, S. Zhang and L. Yang, Mol. BioSyst., 2013, 9(9), 2266–2281, 10.1039/c3mb70186g.
- S. J. Almehmadi, A. M. Alsaedi, M. F. Harras and T. A. Farghaly, Bioorg. Chem., 2021, 117, 105431, DOI:10.1016/j.bioorg.2021.105431.
- N. S. Ismail, E. M. Ali, D. A. Ibrahim, R. A. Serya and D. A. Abou El Ella, Future J. Pharm. Sci., 2016, 2(1), 20–30, DOI:10.1016/j.fjps.2016.02.002.
- M. Chauhan and R. Kumar, Bioorg. Med. Chem., 2013, 21(18), 5657–5668, DOI:10.1016/j.bmc.2013.07.027.
- S. Schenone, M. Radi, F. Musumeci, C. Brullo and M. Botta, Chem. Rev., 2014, 114(14), 7189–7238, DOI:10.1021/cr400270z.
- G. Bocci, A. Fioravanti, C. La Motta, P. Orlandi, B. Canu, T. Di Desidero, L. Mugnaini, S. Sartini, S. Cosconati, R. Frati, A. Antonelli, P. Berti, P. Miccoli, F. Da Settimo and R. Danesi, Biochem. Pharmacol., 2011, 81(11), 1309–1316, DOI:10.1016/j.bcp.2011.03.022.
- M. A. Abdelgawad, R. B. Bakr, O. A. Alkhoja and W. R. Mohamed, Bioorg. Chem., 2016, 66, 88–96, DOI:10.1016/j.bioorg.2016.03.011.
- J. Le Brazidec, A. Pasis, B. Tam, C. Boykin, C. Black, D. Wang, G. Claassen, J. Chong, J. Chao, J. Fan, K. Nguyen, L. Silvian, L. Ling, L. Zhang, M. Choi, M. Teng, N. Pathan, S. Zhao, T. Li and A. Taveras, Bioorg. Med. Chem. Lett., 2012, 22(5), 2070–2074, DOI:10.1016/j.bmcl.2012.01.019.
- S. Cherukupalli, B. Chandrasekaran, R. R. Aleti, N. Sayyad, G. A. Hampannavar, S. R. Merugu, H. R. Rachamalla, R. Banerjee and R. Karpoormath, J. Mol. Struct., 2019, 1176, 538–551, DOI:10.1016/j.molstruc.2018.08.104.
- A. Rahmouni, S. Souiei, M. A. Belkacem, A. Romdhane, J. Bouajila and H. B. Jannet, Bioorg. Chem., 2016, 66, 160–168, DOI:10.1016/j.bioorg.2016.05.001.
- S. Cherukupalli, B. Chandrasekaran, V. Kryštof, R. R. Aleti, N. Sayyad, S. R. Merugu, N. D. Kushwaha and R. Karpoormath, Bioorg. Chem., 2018, 79, 46–59, DOI:10.1016/j.bioorg.2018.02.030.
- A. S. Elgazwy, N. S. Ismail and H. S. Elzahabi, Bioorg. Med. Chem., 2010, 18(21), 7639–7650, DOI:10.1016/j.bmc.2010.08.033.
- M. T. Abdel-Aal, W. A. El-Sayed, S. M. El-Kosy and E. S. El-Ashry, Arch. Pharm., 2008, 341(5), 307–313, DOI:10.1002/ardp.200700154.
- A. S. Girgis, J. Stawinski, N. S. Ismail and H. Farag, Eur. J. Med. Chem., 2012, 47, 312–322, DOI:10.1016/J.Ejmech.2011.10.058.
- I. M. Fawzy, K. M. Youssef, D. S. Lasheen, N. S. Ismail and K. A. Abouzid, Future Med. Chem., 2018, 10(12), 1421–1433, DOI:10.4155/fmc-2017-0242.
- J. Hennek, J. Alves, E. Yao, S. A. Goueli and H. Zegzouti, Anal. Biochem., 2016, 495, 9–20, DOI:10.1016/j.ab.2015.11.007.
- P. Pozarowski and Z. Darzynkiewicz, InCheckpoint controls and cancer, Humana Press., 2004, pp. 301–11. doi: DOI:10.1385/1-59259-811-0:301.
- A. Ismail, A. S. Doghish, B. E. Elsadek, S. A. Salama and A. D. Mariee, Steroids, 2020, 160, 108656, DOI:10.1016/j.steroids.2020.108656.
- D. J. Wood, S. Korolchuk, N. J. Tatum, L. Z. Wang, J. A. Endicott, M. E. Noble and M. P. Martin, Cell Chem. Biol., 2019, 26(1), 121–130, DOI:10.1016/j.chembiol.2018.10.015.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11(11), 1117–1121 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717 , PMID:28256516, https://www.swissadme.ch/index.php.
- W. Zou, X. Ma, W. Hua, B. Chen, Y. Huang, D. Wang and G. Cai, Oncol. Lett., 2016, 11(1), 551–558, DOI:10.3892/ol.2015.3963.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mohammed T. Abdel Aalb,
Wael A. El-Sayedcd,
Mahmoud A. E Shahinb,
Elsayed G. E. Elsakkae,
Mahmoud Mohamed Mokhtare,
Maghawry Hegazye,
Mohamed Hagrasf,
Asmaa A. Mandourg and
Nasser S. M. Ismail
*a,
Mohammed T. Abdel Aalb,
Wael A. El-Sayedcd,
Mahmoud A. E Shahinb,
Elsayed G. E. Elsakkae,
Mahmoud Mohamed Mokhtare,
Maghawry Hegazye,
Mohamed Hagrasf,
Asmaa A. Mandourg and
Nasser S. M. Ismail