
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo statins and cromolyn as possible drugs against the cytotoxicity of Aβ(31–35) and Aβ(25–35) peptides: a comparative study by advanced computer simulation methods†
Fredrik Blomgren,
Alexander Rodin,
Wojciech Chrobak ,
Dawid Wojciech Pacut,
Jan Swenson and
Inna Ermilova*
,
Dawid Wojciech Pacut,
Jan Swenson and
Inna Ermilova*
Department of Physics, Chalmers University of Technology, Fysikgränd 4, Göteborg 41258, Sweden. E-mail: ina.ermilova@gmail.com; inna.ermilova@chalmers.se; Tel: +46-728487773
First published on 4th May 2022
Abstract
In this work, possible effective mechanisms of cromolyn, atorvastatin and lovastatin on the cytotoxicity of Aβ(31–35) and Aβ(25–35) peptides were investigated by classical molecular dynamics and well-tempered metadynamics simulations. The results demonstrate that all the drugs affect the behavior of the peptides, such as their ability to aggregate, and alter their secondary structures and their affinity to a particular drug. Our findings from the computed properties suggest that the best drug candidate is lovastatin. This medicine inhibits peptide aggregation, adsorbs the peptides on the surface of the drug clusters, changes the secondary structure and binds to MET35, which has been seen as the reason for the toxicity of the studied peptide sequences. Moreover, lovastatin is the drug which previously has demonstrated the strongest ability to penetrate the blood–brain barrier and makes lovastatin the most promising medicine among the three investigated drugs. Atorvastatin is also seen as a potential candidate if its penetration through the blood–brain barrier could be improved. Otherwise, its properties are even better than the ones demonstrated by lovastatin. Cromolyn appears to be less interesting as an anti-aggregant from the computational data, in comparison to the two statins.
Introduction
The amyloid-β peptide (Aβ) is formed through atypical intramembranous cleavages of the amyloid precursor protein.1 Through the clustering of these peptides, misfolded and soluble oligomers are formed, which are known for inducing cytotoxicity and, thus, affecting a number of important functions in the human brain.2,3 This has led to a hypothesis that the aggregation of Aβ has a pathological correlation with neurodegenerative diseases, such as Alzheimer’s (AD) and Parkinson’s (PD).4Today, there are no fully developed pharmaceuticals that cure AD or PD; the medications approved for these usages have only mitigating effects on the symptoms caused by the diseases. With an increasingly aging population, the societal costs will become more and more substantial and, therefore, the demand for a solution to this problem is increasing. In the U.S. only, an estimated 6.2 million people aged 65 and above are living with AD in 2021. This is more than one in nine people within this age group.5
There are numbers of possible ways to prevent the amyloid cascade. One could try to inhibit the production of Aβ through the control of BACE1 (β-site amyloid precursor protein cleaving enzyme 1)6 and γ-secretase activity,7 or one could use immunotherapy8 as a way to neutralize the toxicity of Aβ. On the other hand, the aggregation of peptides could also be prevented.4
Clinical testing of drugs can be time consuming and costly. From 2002 through 2012, the AD drug development failure rate was 99.6%.9 A logical choice to circumvent this issue is to investigate drugs that are already approved for other pharmaceutical purposes and of which the side effects are more extensively known.
This study focuses on two types of statins, atorvastatin and lovastatin, and cromolyn (Fig. 1). All the drugs have been clinically tested against other diseases. Statins are mainly used in cholesterol regulation to prevent cardiovascular diseases10–12 and cromolyn is used to prevent the release of histamine and can be found in medications against asthma.13,14 Various studies have been performed on these compounds as a possible drug against the cytotoxicity of Aβs, and they have shown promising effects.15–19
 | ||
| Fig. 1 Simulated molecules. | ||
Statins have been subjects to numerous studies in connections to AD and PD,20 since cholesterol plays an important part in neurotransmitter receptor expression, mitochondrial function, synapse development and other functionalities that are vital to the normal functionality of the brain.21–23 This suggests that an unbalance in cholesterol levels can affect cognition negatively and cause cognitive impairment, which is believed to be reversible.24 However, there are reasons to believe that the use of statins can also affect the brain functionality positively and the research on their effect on AD and PD patients points to this. A clinical study by Shalaby et al.,18 conducted on 108 PD cases and 124 controls, revealed an inverse association between PD and statin use. Another clinical study conducted by Lin et al.25 also showed a beneficial trend of the MDS-UPDRS motor score of PD patients when given a dosage of lovastatin. There are, however, a number of clinical trials that have failed to show beneficial effects of statin use as a treatment against AD. On the other hand, Geifman et al.26 performed a meta-analysis of many of these studies and found that the long term use of statins actually revealed trends of both enhanced cognitive performance and benefits in the treatment of all endotypes of AD, but was most beneficial for ApoE4/ApoE4-genotyped people. The lipophilicity of a statin may also play an important part in the potential use of statins as medication against AD or PD. Shepardson et al.19 pointed out that lipophilic statins, such as lovastatin, can more readily cross the blood–brain barrier than less lipophilic statins, such as atorvastatin, and that this can be a cause of why statins have shown various results in treatment of AD.
Similar to statins, cromolyn has proven to be a good candidate as well.16,27,28 Zhang et al.16 found in their in vivo study on mice that a combination of cromolyn and ibuprofen could be used to promote phagocytosis in microglia that targets the accumulation of Aβ(1–42) and Aβ(1–40), although an increase in Aβ(1–38) was discovered. Another study by Hori et al.,28 conducted both in vitro and in vivo, showed similar results. The study revealed that cromolyn inhibits Aβ(1–42) and Aβ(1–40) aggregation and causes a decrease in soluble Aβ oligomers. Lozupone et al.29 mentioned in their study that a mix of cromolyn and ibuprofen is one of the most eminent emerging drugs that can prevent the amyloid cascade, but stressed the importance of getting a complete understanding of the pathogenesis of AD and PD. They suggested that the pathogenesis of the disease may vary at different stages and that the Aβ cascade may be a response to upstream events, in which the brain attempts to amend neuronal damage.
Nevertheless, the understanding of the pathogenesis of AD and PD may not be complete, but the cytotoxicity of Aβ has been established. Furthermore, the Aβ peptides can have different lengths that differ in their degree of toxicity. Many peptides discovered in the brain of AD and PD patients belong to the sequence Aβ(1–43), but they are not equally toxic.30 The most toxic part of this sequence is (25–35), because it can aggregate within hours and still retain the toxicity of the full length of Aβ(1–43).31,32
However, mechanistic insights of the peptide aggregation processes and their toxicity, depending on the sequence, still need to be understood in order to develop better treatments against AD and PD. For instance, comparative studies of two shorter peptides Aβ(25–35) and Aβ(31–35) (Fig. 1) demonstrated that they invoke different toxic mechanisms.33–35 Misiti et al.35 showed that Aβ(31–35) acts through apoptotic mechanisms while Aβ(25–35) may induce neurotoxicity through the adherent cell count.
Another interesting fact about these short peptides is that the presence of MET35 in the C-terminal plays a big role in their toxicity.36–38 For example, the reverse sequence (35–25) does not appear to be toxic.39
The toxicity of short peptides makes them interesting targets for computational investigations, because such calculations would be less costly to perform than for longer peptides. Nevertheless, there are not many atomistic modeling works carried out with Aβ(25–35) and Aβ(31–35).40–43 For instance, in our other in silico paper by Chrobak et al.,42 we elucidated how cannabidiol interacts with Aβ(31–35) and Aβ(25–35). Furthermore, Ermilova et al.43 investigated Aβ(25–35) in the presence of a lipid membrane and discovered that in the presence of cholesterol, peptides aggregate on the membrane surface.
Atomistic simulations of atorvastatin, lovastatin and cromolyn together with Aβ peptides have not been performed previously. Therefore, in order to understand the possible mechanisms of action of the listed drugs, we carried out in silico studies on them in mixtures with Aβ(31–35) and Aβ(25–35).
The goal of this work is to understand which one of the three drugs would be most suitable in treatments of neurodegenerative diseases out of results from classical molecular dynamics (MD) and well-tempered metadynamics simulations. MD simulations can give an idea about the natural behavior of peptides and drugs on a nanosecond time-scale. For instance, one can obtain a structural understanding about how a small change in concentration can affect the structures and interactions between the peptides and drugs.44–46
Well-tempered metadynamics simulations are used in order to obtain a thermodynamical understanding of the interactions, where the free energy can determine whether aggregation or binding can occur spontaneously at the given temperature or not.47–50 For such a purpose, well-tempered metadynamics simulations are the most suitable since one can explore the physically interesting regions of the free energy surface.48
Results and discussion
Profiles which prove the convergence of MD simulations can be observed in Fig. S4 and S5 of the ESI† and final dimensions of the simulation boxes and concentrations of all compounds can be observed in Tables S1–S3.†Characterization of intermolecular interactions
Radial distribution functions (RDFs) between molecular centers of mass were computed between peptides in order to see if there are any indications for them to aggregate (see Fig. 2). In general, a value of RDF (g(r)) higher than 1 at a short distance (below 1 nm) can indicate an association between molecules, particularly if such a value is among the highest values. The function is computed according to the following eqn (1):51
 | (1) |
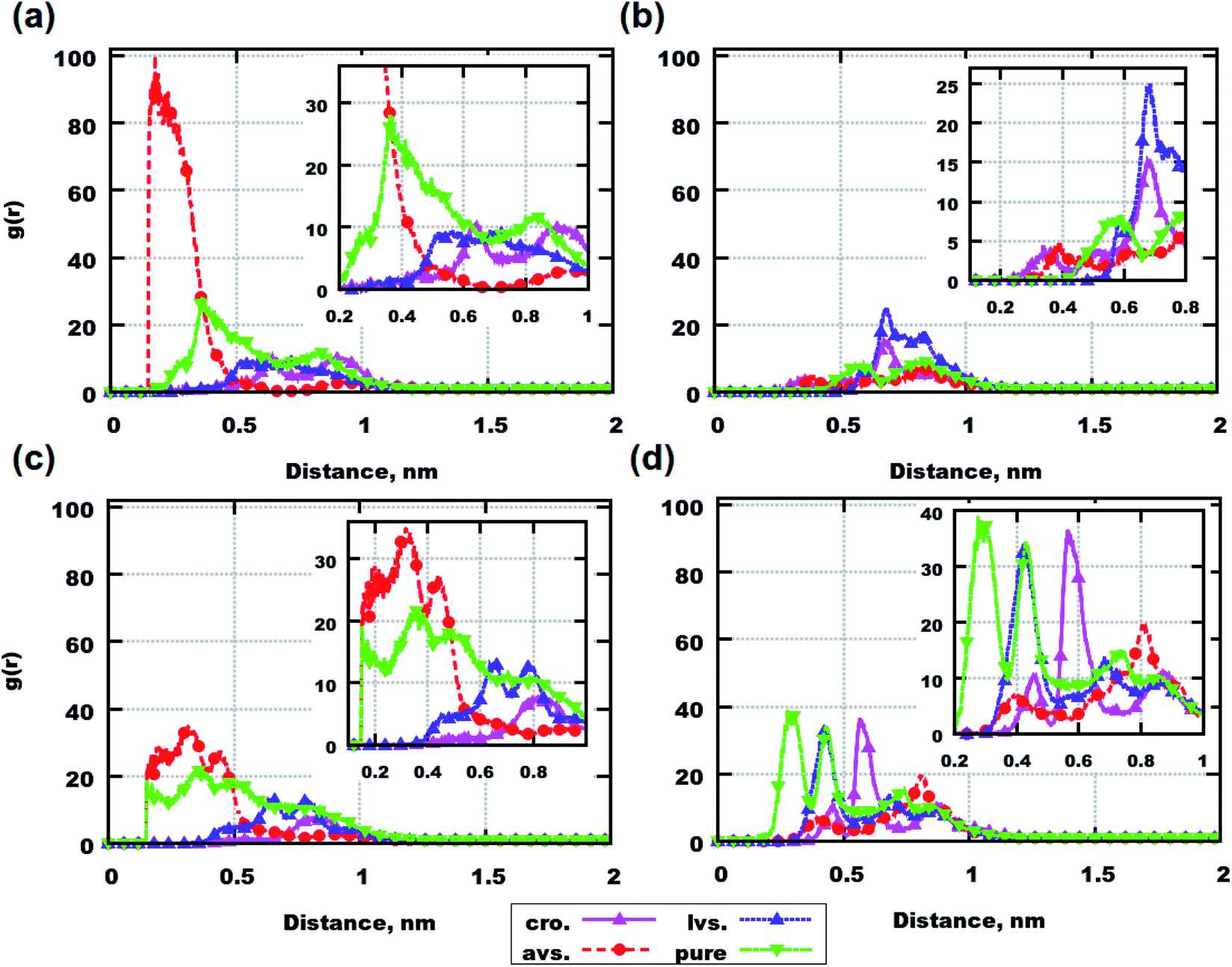 | ||
| Fig. 2 Radial distribution functions between the molecular centers of mass of peptides. (a) Systems containing 6 Aβ(25–35) with and without drugs. (b) Systems containing 6 Aβ(31–35) with and without drugs. (c) Systems containing 8 Aβ(25–35) with and without drugs. (d) Systems containing 8 Aβ(31–35) with and without drugs. Abbreviations mean the following: “cro.” – systems containing cromolyn, “avs.” – systems containing atorvastatin, “lvs.” – systems containing lovastatin, “pure” – systems with only peptides. | ||
The plotted data demonstrates that atorvastatin can promote the aggregation of Aβ(25–35) (see Fig. 2(a) and (c)) as well as the aggregation of Aβ(31–35) in the case of the system with 6 molecules (see Fig. 2(b)). On the other hand, for the system with 8 molecules of Aβ(31–35), atorvastatin can be seen as an inhibitor of peptide accumulation according to Fig. 2(d). Cromolyn and lovastatin reduce the peptide aggregation in all systems, according to the RDF profiles. Additionally, in all simulations, a parameter such as the radius of gyration does not seem to play a role in the aggregation according to Tables S5 and S6 in the ESI,† i.e. average values for all drugs and peptides are similar (those average values were computed during the last 250 ns of each simulation over all conformations for a particular molecule).
Considering the behavior of drug molecules in mixtures with and without peptides, cromolyn demonstrates the highest tendency to build clusters according to the RDFs in Fig. S6 of the ESI.† Comparing systems containing 6 peptides, it is clear that in the presence of Aβ(31–35), the drug aggregates more than in simulations with Aβ(25–35). In systems with 8 molecules, the presence of peptides reduces the aggregation of all investigated drug molecules. Therefore, out of the presented data, we can conclude that the aggregations of drugs and peptides are interconnected.
Nevertheless, Aβ(25–35), Aβ(31–35) and medicines are large molecules. The coordinates of their centers of mass can vary depending on the molecular conformations. Consequently, the data from RDF calculations gives only a preliminary idea about the clustering of molecules.
Another way to complement the investigation of intermolecular interactions is to compute the number of hydrogen bonds and contacts between molecules in the systems. Table 1 demonstrates the average number of hydrogen bonds for systems containing 6 molecules of peptides and drugs. 6 molecules of lovastatin do not show any hydrogen bonding between themselves, whereas cromolyn and atorvastatin have 3 and 4 intermolecular hydrogen bonds, respectively. Lovastatin molecules bind to fewer water molecules (34 hydrogen bonds) than cromolyn and atorvastatin (47 and 44 hydrogen bonds, respectively). Considering the hydrogen bonding between peptides, it can be concluded from the table that for Aβ(25–35), the number of such bonds does not vary much depending on the presence of a drug, i.e. the differences are rather insignificant. For Aβ(31–35), lovastatin is seen to be a promoter of the hydrogen bonding between peptides, while cromolyn and atorvastatin do not have any significant effect. However, this fact is not seen as a negative effect of lovastatin, because hydrogen bonds are not the only indicators of aggregation and, additionally, Aβ(31–35) is known to be toxic without demonstrating any aggregative properties according to experimental studies by Pike et al.31 and Misiti et al.35 The number of hydrogen bonds between peptides and drugs was the highest in the systems with cromolyn for both peptides, while lovastatin bound the least to both peptides. Moreover, the presence of the drugs affects the hydration of the peptides. In systems with atorvastatin and cromolyn, the number of hydrogen bonds between water and Aβ(25–35) is lower than in the system with pure Aβ(25–35) and the system with lovastatin. Aβ(31–35) has the smallest number of hydrogen bonds with water in the mixture with lovastatin, while in the systems with cromolyn and atorvastatin, that number was rather similar.
| System | Peptide–peptide | Peptide–drug | Peptide–water | Drug–drug | Drug–water | Total number of contacts |
|---|---|---|---|---|---|---|
| cro. | — | — | — | 3 ± 3 | 47 ± 4 | — |
| avs. | — | — | — | 4 ± 3 | 44 ± 4 | — |
| lvs. | — | — | — | 0 ± 3 | 34 ± 4 | — |
| Aβ(25–35) & cro. | 24 ± 6 | 10 ± 3 | 150 ± 8 | 4 ± 3 | 35 ± 4 | 792 ± 30 |
| Aβ(25–35) & avs. | 26 ± 6 | 8 ± 3 | 149 ± 8 | 2 ± 3 | 35 ± 4 | 651 ± 30 |
| Aβ(25–35) & lvs. | 23 ± 6 | 5 ± 3 | 157 ± 8 | 0 ± 3 | 27 ± 4 | 729 ± 30 |
| Aβ(25–35) | 26 ± 6 | — | 162 ± 8 | — | — | 905 ± 23 |
| Aβ(31–35) & cro. | 5 ± 6 | 6 ± 3 | 79 ± 8 | 4 ± 3 | 39 ± 4 | 500 ± 3 |
| Aβ(31–35) & avs. | 7 ± 6 | 3 ± 3 | 81 ± 8 | 4 ± 3 | 35 ± 4 | 503 ± 3 |
| Aβ(31–35) & lvs. | 14 ± 6 | 2 ± 3 | 70 ± 8 | 0 ± 3 | 30 ± 4 | 504 ± 3 |
| Aβ(31–35) | 8 ± 6 | — | 86 ± 8 | — | — | 504 ± 1 |
The total number of contacts between peptides was calculated not only for ones caused by hydrogen bonding, but also for hydrophobic parts. From Table 1, it can be concluded that atorvastatin decreases the number of inter- and intra-peptide contacts most for Aβ(25–35). Lovastatin comes out as a second good candidate for peptide separation, while cromolyn shows the lowest ability to separate Aβ(25–35) peptides. In the case of Aβ(31–35), the presence of the drug has no significant effect on the total number of contacts.
For systems containing 8 molecules of drugs and peptides, the trends in the number of hydrogen bonds and total number of contacts are different (see Table 2). In systems with Aβ(25–35), atorvastatin and cromolyn inhibit the hydrogen bonding between peptides, while lovastatin promotes it. Peptide–drug hydrogen bonding is similar as in systems with 6 molecules, where cromolyn binds the most to peptides. Water binds to peptides most in the system with atorvastatin and least in the simulation with lovastatin. The total number of contacts between peptides is smallest in the system with lovastatin.
| System | Peptide–peptide | Peptide–drug | Peptide–water | Drug–drug | Drug–water | Total number of contacts |
|---|---|---|---|---|---|---|
| cro. | — | — | — | 5 ± 3 | 63 ± 4 | — |
| avs. | — | — | — | 6 ± 3 | 55 ± 4 | — |
| lvs. | — | — | — | 0 ± 3 | 44 ± 4 | — |
| Aβ(25–35) & cro. | 29 ± 6 | 17 ± 3 | 202 ± 8 | 6 ± 3 | 39 ± 4 | 837 ± 36 |
| Aβ(25–35) & avs. | 29 ± 6 | 9 ± 3 | 210 ± 8 | 5 ± 3 | 46 ± 4 | 799 ± 36 |
| Aβ(25–35) & lvs. | 35 ± 6 | 6 ± 3 | 196 ± 8 | 0 ± 3 | 36 ± 4 | 705 ± 36 |
| Aβ(25–35) | 33 ± 6 | — | 215 ± 8 | — | — | 1092 ± 38 |
| Aβ(31–35) & cro. | 17 ± 6 | 8 ± 3 | 83 ± 8 | 5 ± 3 | 52 ± 4 | 634 ± 8 |
| Aβ(31–35) & avs. | 17 ± 6 | 5 ± 3 | 86 ± 8 | 4 ± 3 | 51 ± 4 | 627 ± 8 |
| Aβ(31–35) & lvs. | 14 ± 6 | 4 ± 3 | 97 ± 8 | 0 ± 3 | 39 ± 4 | 671 ± 8 |
| Aβ(31–35) | 11 ± 6 | — | 108 ± 8 | — | — | 672 ± 1 |
In simulations with Aβ(31–35), the number of hydrogen bonds between peptides is higher in the presence of drugs than in the pure peptide system. In the presence of cromolyn, the number of peptide–water hydrogen bonds is lowest and cromolyn itself binds most to the Aβ(31–35) peptide. Atorvastatin and cromolyn decrease the total number of peptide–peptide contacts, while lovastatin does not influence the peptide separation.
The data presented above provides statistical information about the different types of interactions in the systems, but does not provide structural insights about the interactions. To obtain such structural information, it is valuable to investigate structural snapshots of the systems. Fig. 3 shows simulated systems containing 6 molecules of peptides and/or medicines. This figure demonstrates that all the drug molecules have a tendency to build clusters in water, but only the peptide Aβ(25–35) forms clusters without any medicine, in agreement with experimental findings.31,35 Moreover, both Aβ(25–35) and Aβ(31–35) have a tendency to aggregate around those clusters of drugs. When peptides are located on the surface of the same drug cluster, their RDF can show that they accumulate there, since their centers of mass are located close to each other. The ability to “collect” cytotoxic Aβ peptides can be considered to be a good property of a medicine against neurodegenerative diseases, since those peptide aggregates are inhibited from growing further by the surrounding drug molecules.
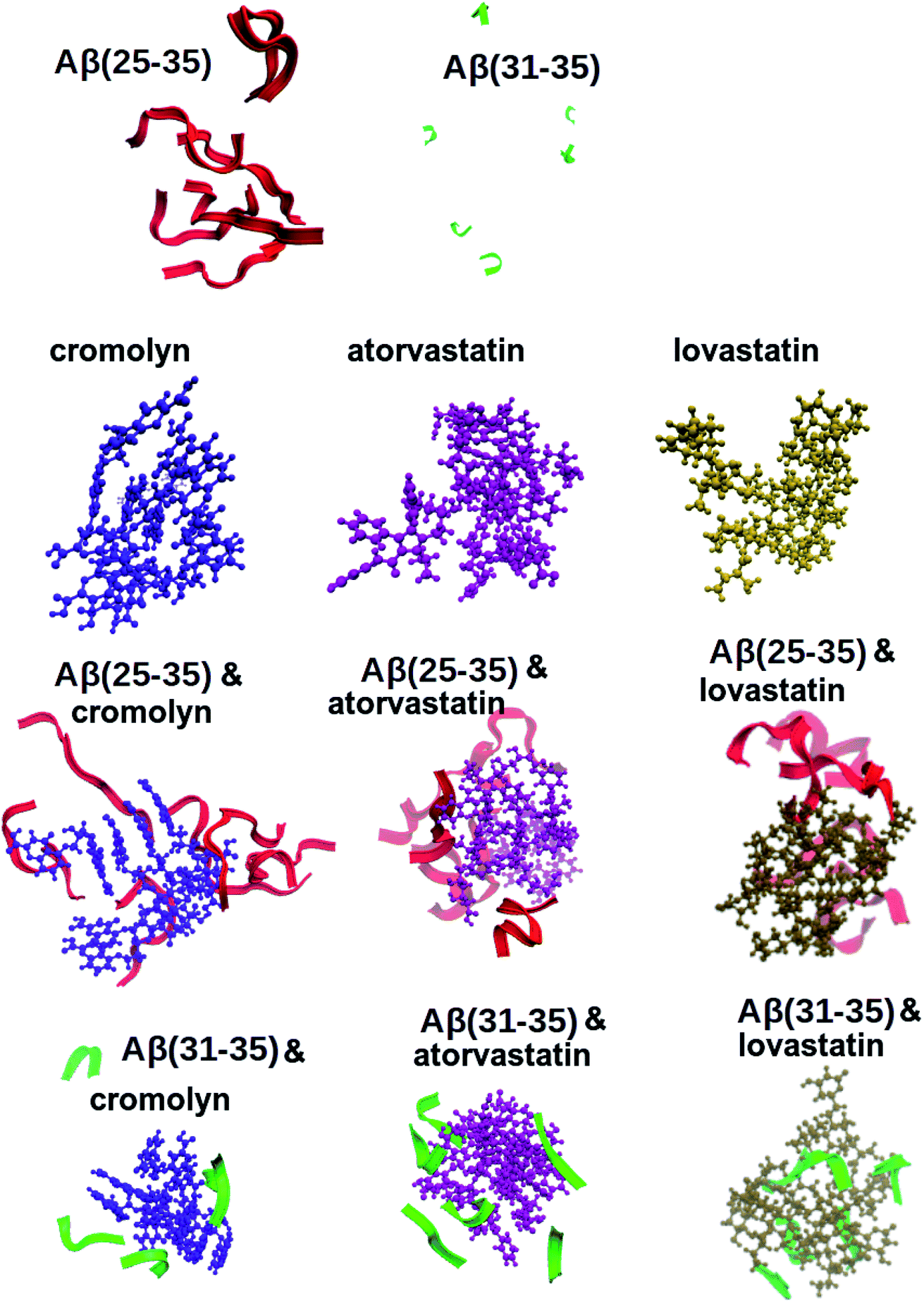 | ||
| Fig. 3 Snapshots of simulated systems containing 6 molecules of each drug and 6 molecules of Aβ(25–35)/Aβ(31–35). Systems with 8 molecules had similar snapshots. All snapshots were done after 250 ns using VMD software.52 | ||
However, the cytotoxicity is not only related to peptide aggregation in general. From both computational and experimental studies, it is known that Aβ(31–35) is a peptide which does not build clusters.31,42 The cytotoxicity of Aβ(25–35) is related to other features of the peptide as well, from the order of the sequence to the presence of MET35 in the C-terminal. In fact, the presence and the position of MET35 are seen as the reason for the toxicity of both Aβ(31–35) and Aβ(25–35).37,38,53 This fact makes it interesting to investigate how the number of contacts between certain amino-acid residues is affected by the simulated drug molecules.
Fig. 4 demonstrates how the total number of contacts between amino-acid residues varies depending on the presence of drug molecules. In simulations with 6 molecules of peptides and drugs, it can be clearly seen that atorvastatin (Fig. 4(a)) is the most promising medicine for the separation of Aβ(25–35) at the simulated concentration. Moreover, since the presence of MET35 in the C-terminal is strongly related to the toxicity of Aβ(25–35), it is important to mention that in the mixture with atorvastatin, the total number of contacts for MET35 is dramatically smaller than in the systems with the two other drugs. For the same peptide, the second best candidate is lovastatin, whereas cromolyn exhibits the smallest positive effect. For systems with 6 Aβ(31–35), none of the drugs is effective in decreasing the total number of contacts between amino-acid residues (Fig. 4(b)).
 | ||
| Fig. 4 Number of contacts with other peptides for amino-acid residues during the last 250 ns of simulation. (a) Systems containing 6 Aβ(25–35) with and without drugs. (b) Systems containing 6 Aβ(31–35) with and without drugs. (c) Systems containing 8 Aβ(25–35) with and without drugs. (d) Systems containing 8 Aβ(31–35) with and without drugs. For error estimates, see Tables 1 and 2 Abbreviations mean the following: “cro.” – systems containing cromolyn, “avs.” – systems containing atorvastatin, “lvs.” – systems containing lovastatin, “pure” – systems with only peptides. Snapshots of contact maps can be observed in Fig. S17 and S18 in the ESI.† | ||
In simulations with 8 molecules, lovastatin is shown to be more effective for the detachment of Aβ(25–35) (Fig. 4(c)), while atorvastatin and cromolyn can be seen as alternative drugs. This can also be concluded from the standard deviations from Table 2: atorvastatin and cromolyn demonstrate similar performances. For the separation of Aβ(31–35), atorvastatin and cromolyn are the best medicines, while lovastatin demonstrates no effect at all at the simulated ratio of compounds (Fig. 4(d)). Additionally, the total number of contacts for MET35 is lower for atorvastatin than for the two other drugs.
From the decreasing number of contacts per amino-acid residue arises a question regarding the binding of those residues to the simulated drugs. Particularly, MET35 is of interest.54 Fig. 5 presents the RDFs between the centers of mass of MET35 and the drug molecules. For systems with 6 Aβ(25–35) (Fig. 5(a)), cromolyn appears to associate strongest, while lovastatin and particularly atorvastatin are interacting less with MET35. In the simulations with 6 Aβ(31–35) (Fig. 5(b)), atorvastatin binds slightly more strongly to MET35 than cromolyn, while lovastatin binds less.
 | ||
| Fig. 5 Radial distribution functions between the molecular centers of mass of methionine (MET35) and drugs. (a) Systems containing 6 Aβ(25–35). (b) Systems containing 6 Aβ(31–35). (c) Systems containing 8 Aβ(25–35). (d) Systems containing 8 Aβ(31–35). Abbreviations mean the following: “cro.” – systems containing cromolyn, “avs.” – systems containing atorvastatin, “lvs.” – systems containing lovastatin. | ||
In the case of 8 Aβ(25–35), atorvastatin binds considerably more strongly to MET35 than lovastatin and cromolyn (Fig. 5(c)). In fact, cromolyn does not show any binding at all to MET35. For systems with 8 Aβ(31–35), cromolyn appears to bind the strongest to MET35, while atorvastatin and lovastatin associate less with the amino-acid residue. Such different binding to MET35 in comparison with systems containing 6 molecules can be explained from the RDFs of the drug molecules in Fig. S6 of the ESI.† Cromolyn aggregates more than lovastatin and atorvastatin in all simulations. Moreover, in the system with 8 Aβ(25–35), it shows binding to most of the amino-acid residues (Fig. S7 and S16 in ESI†). Additional molecules of cromolyn and Aβ(25–35) could lead to an increase of the size of the drug cluster as well as conformational changes of the peptides themselves, which could be a reason for why binding to MET35 was not observed in simulations with 8 Aβ(25–35).
Considering the associations of drugs with other amino-acid residues, it can be pointed out that cromolyn demonstrates binding to most of them (Fig. S7–S16 in ESI†), which is a positive feature, since the presence of ASN27 (Fig. S9 in ESI†) is seen as one of the reasons for the toxicity of Aβ(25–35).55 Both statins demonstrate weaker associations with amino-acid residues from the sequence (31–35) and with ALA30 (Fig. S12 in ESI†).
Nevertheless, the binding to certain amino-acid residues, preventing the aggregation and decreasing the total number of contacts between peptides, is not the only way of acting against the cytotoxicity of Aβ peptides. Often, their toxicity and behaviors are also related to their secondary structures.
Secondary structures of the peptides
The secondary structures of proteins and peptides strongly affect their functions in biological cells. Molecular conformations also play roles in such features as aggregation, binding to certain molecules etc.Determining secondary structures is a complex process for short peptides since the number of amino acid residues (a peptide’s length) is a crucial parameter here. Random coils and α- and 310-helices can be defined when there are at least 4 residues in a peptide sequence, while a π-helix contains 5.56–58 The so-called hydrogen bonded turn can be built of 3–5 amino acid residues and an extended conformation has 2. From this point of view, both peptides are suitable for secondary structure calculations using the STRIDE algorithm.59,60
Comparing Aβ(25–35) with Aβ(31–35) peptides, it should be noted that the first one is known to aggregate, while the latter one has no such tendency. In simulations with 6 molecules (Fig. 6(a) and (b)), it is clear that there is a larger variety of secondary structures for Aβ(25–35) than for Aβ(31–35). Such a variety is also related to the difference in the lengths of sequences of the peptides and their chemistry: Aβ(25–35) contains both hydrophobic and hydrophilic parts, while Aβ(31–35) is mainly hydrophobic.
 | ||
| Fig. 6 Amounts of secondary structures during the last 250 ns of simulation computed using VMD software.52 (a) Systems containing 6 Aβ(25–35) with and without drugs. (b) Systems containing 6 Aβ(31–35) with and without drugs. (c) Systems containing 8 Aβ(25–35) with and without drugs. (d) Systems containing 8 Aβ(31–35) with and without drugs. Abbreviations mean the following: “cro.” – systems containing cromolyn, “avs.” – systems containing atorvastatin, “lvs.” – systems containing lovastatin, “pure” – systems with only peptides. On the x-axis, codes for secondary structures are: T – turn, E – extended conformation, B – isolated β-bridge, H – α-helix, G – 310-helix, I – π-helix, C – coil. The full set of secondary structures can be observed in Fig. S19–S30 of the ESI.† | ||
In the case of Aβ(25–35), significant numbers of extended conformations, isolated β-bridges, 310-helices and even α-helices are observed. In particular, combinations of extended conformations and isolated β-bridges are related to peptide aggregation.31,42,61,62 A larger amount of such secondary structures is observed in Fig. 6(a) and (c) for Aβ(25–35), which can be correlated with its aggregation (see the RDFs in Fig. 2(a) and (c)).31,42,61,62 For Aβ(31–35) in Fig. 6(b) and (d), clear differences can be seen in the distribution of secondary structures: extended conformations and isolated β-bridges are observed for 8 molecules in (d), which can also be interconnected with the aggregation of Aβ(31–35) (see the RDFs in Fig. 2(b) and (d)).
These results regarding the peptides’ secondary structures and their aggregation can be related to earlier findings by Pike et al.,31,61,62 where the presence of extended conformations and β-bridges was associated with a more pronounced aggregation of Aβ(25–35). In the case of our simulations, the appearance of such conformations is also associated with the aggregation of Aβ(31–35). Moreover, in the previous work on cannabidiol,42 the presence of such secondary structures was also interconnected with peptide aggregations.
Nevertheless, despite the fact that alterations in the secondary structures of the peptides can explain their aggregation behavior, it is difficult to draw conclusions from the presented data regarding the best drug candidate against neurotoxicity. We can only conclude that every studied drug can strongly affect the peptides’ conformational distributions.
Potential of mean force profiles and free energies
Free energy calculations give thermodynamic insights into the process of the peptides’ aggregations. The lower the value of free energy, the higher the probability that two molecules bind to each other. Moreover, such a binding probability can also be seen from the potential of mean force (PMF) profiles: the lower the depth of the curve below a zero value at a short distance, the higher the probability that binding occurs between the studied compounds.Fig. 7 presents the PMF profiles for systems containing peptides42 and peptides with drugs. From Fig. 7(a), it can be concluded that cromolyn is the best promoter of binding between Aβ(25–35), while lovastatin (with not a significant difference compared to atorvastatin) appears to be an inhibitor of peptide aggregation.
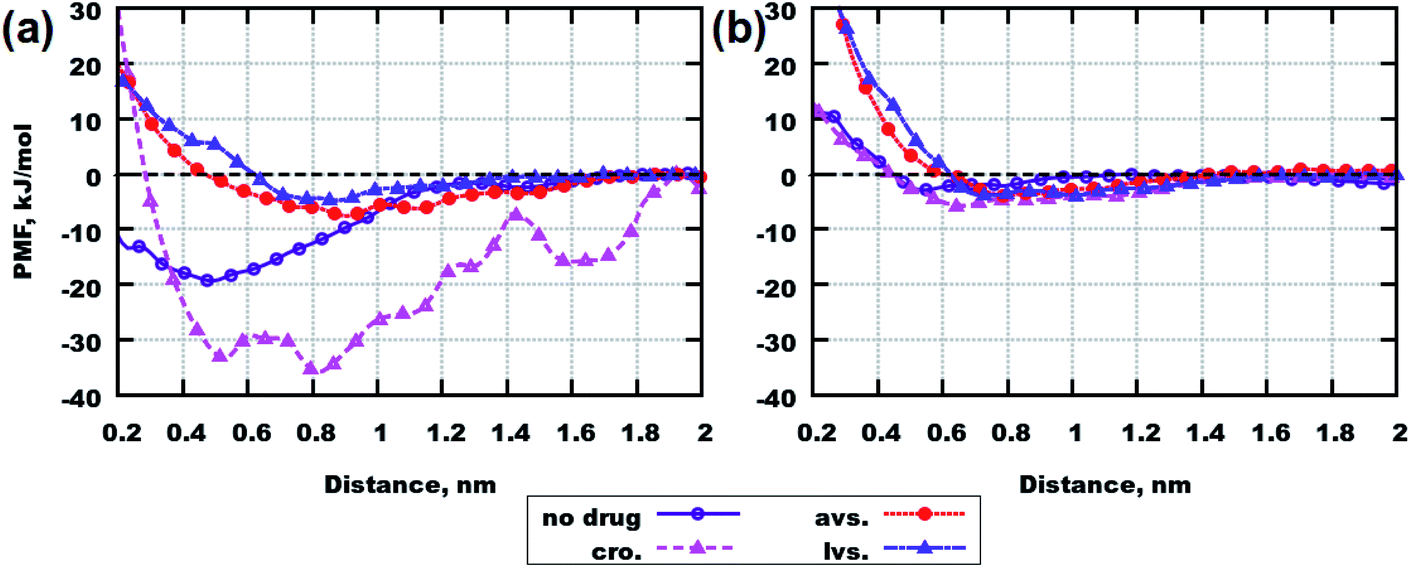 | ||
| Fig. 7 PMF profiles for the first collective variable (CV1). (a) Systems with Aβ(25–35). (b) Systems with Aβ(31–35). Abbreviations mean the following: “no drug” – systems containing only peptides (data is taken from Chrobak et al.42), “cro.” – systems containing cromolyn, “avs.” – systems containing atorvastatin, “lvs.” – systems containing lovastatin. | ||
Also for Aβ(31–35), cromolyn can be regarded as a promoter of peptide clustering, although its effect is much weaker in this case, with only a minor difference compared to the two other drugs (see Fig. 7(b)). These results are consistent with the results from classical MD simulations, despite the low number of hydrogen bonds between peptides in Table 1.
Regarding the interactions between the drugs and the peptides, cromolyn seems to bind most to both Aβ(25–35) and Aβ(31–35) (see Fig. S31 in ESI†), while atorvastatin and lovastatin bind more weakly.
The PMF curves give us an approximate idea of the binding process. A better understanding can be obtained from the values of the binding free energies. Such values were computed according to eqn (2):
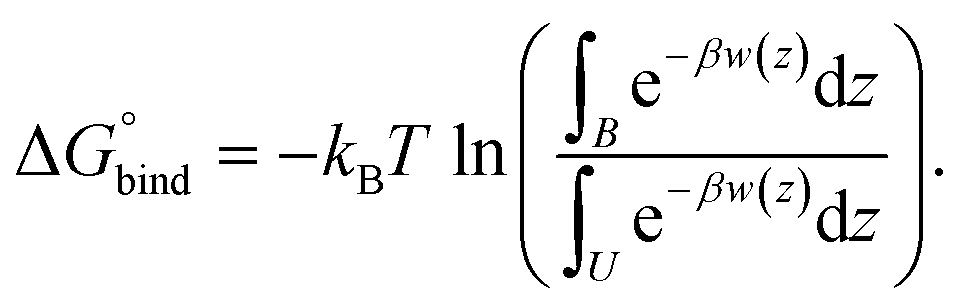 | (2) |
In this equation, kB is Boltzmann’s constant, β = 1/(kBT), w(z) is the PMF at a certain distance z, and T is the temperature. Letters B and U denote “bound” (at a distance closer than 1.5 nm) and “unbound” states (at a distance longer than 1.5 nm), which means that the molecular centers of mass are very close to each other or far enough away (to be considered unbound), respectively.
According to the data presented in Table 3, cromolyn binds the strongest to both peptides. With its presence, peptides aggregate more strongly, since the values of free energies are the lowest. Lovastatin acts best against the aggregation of peptides as well as it binds less to them, compared to atorvastatin. However, it is important to mention that the differences in the free energies and PMF profiles for statins are not significant if one compares them with each other. These findings are coherent with the results from classical MD simulations, where we observed that cromolyn is the drug which was binding the strongest to the amino-acid residues. Moreover, the data for the total number of contacts suggested as well that cromolyn has the smallest ability to separate the peptides.


Discussion
Indeed, the cytotoxicity of peptides is a complex phenomenon. Therefore, determining a potential drug candidate is not an easy procedure and cannot be based mainly on its anti-aggregative properties. This is evidenced by the fact that Aβ(31–35) is toxic, but does not form clusters.Furthermore, selecting the best medicine against peptide aggregation cannot be done only from RDF data, but hydrogen bonds and the total number of contacts must also be considered. If all these results are taken into account, it is clear that the most efficient drug for separating the peptides is atorvastatin, as it demonstrates a good efficacy in all simulations except the one with 6 Aβ(31–35).
Atorvastatin also demonstrates an important ability to bind to MET35 in all the simulated systems, containing it, since the RDFs have peaks with values higher than 1 at distances below 0.6 nm, while cromolyn does not bind to MET35 in the system with 8 Aβ(25–35). The latter fact puts cromolyn out of our considerations. Then, in the sense of associating with MET35, lovastatin can certainly be considered as the second potential medicine: despite the lower total score from the MET35 binding property, this drug could bind to this amino-acid residue in all simulated systems.
Considering the binding to ASN27, cromolyn appears to be the only drug fulfilling this requirement. Nevertheless, this requirement is not strongly considered in this study, since the number of experimental studies regarding the relation between the cytotoxicity of Aβ peptides and the presence of ASN27 in their sequences is not substantial.
From a thermodynamical point of view, lovastatin comes out as the most prominent drug against peptide aggregation, with atorvastatin as the second potential candidate. In the presence of cromolyn, Aβ(25–35) aggregates the strongest, while Aβ(31–35) demonstrates similar binding as in the presence of atorvastatin.
Another interesting property of all the simulated medicines is their ability to bind peptides on their own clusters. Such a property was also observed in simulations with cannabidiol.42 The positive outcome of this can be that all the peptides aggregate on the drugs instead of the surfaces of neuronal membranes, which could save neurons from their death. If this is an important ability of the drugs, then the well-tempered metadynamics simulations indicate that cromolyn would be the most efficient drug to form such clusters and keep the peptides away from the neural membranes. However, since this phenomenon is not investigated in this work, we can only speculate about the possible benefit.
To finalize this discussion, it would be valuable to suggest the best potential drug against the cytotoxicity of Aβ peptides from the computational data, which could be further considered for in vitro and in vivo studies. For this purpose, we created a grading system which gave scores from 1 (the weakest effect) to 3 (the strongest effect) to certain properties of drug molecules with respect to their actions against the cytotoxicity of the simulated peptides. The score is equal to zero if there is no effect at all. If the number of hydrogen bonds and contacts between peptides was the smallest for a certain drug, then that drug would get a score of 3 and the other two drugs would get scores of 2 and 1 with a weakening ability to prevent binding. However, if a particular property was similar for two or all three drugs, then the same score would be given to those. A similar process was used for grading the binding to amino-acid residues: the highest value of RDFs at the shortest distance would give the highest score to a medicine. The ability of a drug to separate peptides was judged from the highest value of the binding free energy, which would mean that the probability of spontaneous binding between two peptides would be smallest (exergonic and endergonic processes).
Table 4 demonstrates how this grading system was applied for the drug selection. The differences in the scores between the three drugs are not big, since all the drugs exhibit both good and bad properties, where none of them fulfill all the criteria for preventing cytotoxicity. However, atorvastatin appears to be the best drug (it has the highest total score), since it can fulfill such requirements as inhibiting peptide aggregation, binding to MET35, absorbing Aβ(25–35) and Aβ(31–35) on the surface of the drug cluster and acting towards changing the secondary structure of the peptides. The strongest disadvantage of atorvastatin is its poor permeation through the blood–brain barrier. As mentioned by Shepardson et al.,19 the question of whether this drug can pass through the barrier is not clear, since two different works63,64 have resulted in different answers to this question. If atorvastatin could definitely pass through the blood–brain barrier or if there was a way to deliver the drug through it, then it would be the suggested medicine for future investigations. However, if not, lovastatin, which can definitely go through the blood–brain barrier according to Shepardson et al.,19 seems to be the most promising medicine. Therefore, lovastatin is recommended for further investigations, while atorvastatin is suggested to be considered if its ability to penetrate the blood–brain barrier could be confirmed.
| Effect (simulation information) | Scores for drugs | ||
|---|---|---|---|
| Cromolyn | Atorvastatin | Lovastatin | |
| Aβ(25–35) separation (6 Aβ(25–35)) | 1 | 3 | 2 |
| Aβ(31–35) separation (6 Aβ(31–35)) | 1 | 1 | 1 |
| Aβ(25–35) separation (8 Aβ(25–35)) | 1 | 2 | 3 |
| Aβ(31–35) separation (8 Aβ(31–35)) | 2 | 3 | 1 |
| Aβ(25–35) binding MET35 (6 Aβ(25–35)) | 3 | 1 | 2 |
| Aβ(31–35) binding MET35 (6 Aβ(31–35)) | 2 | 3 | 1 |
| Aβ(25–35) binding MET35 (8 Aβ(25–35)) | 0 | 3 | 2 |
| Aβ(31–35) binding MET35 (8 Aβ(31–35)) | 3 | 2 | 1 |
| Number of AA residues to bind (6 Aβ(25–35)) | 3 | 2 | 2 |
| Number of AA residues to bind (6 Aβ(31–35)) | 3 | 3 | 3 |
| Number of AA residues to bind (8 Aβ(25–35)) | 3 | 2 | 2 |
| Number of AA residues to bind (8 Aβ(31–35)) | 3 | 3 | 3 |
| Aβ(25–35) separation (metadynamics) | 1 | 2 | 3 |
| Aβ(31–35) separation (metadynamics) | 1 | 2 | 3 |
| Total score | 27 | 32 | 29 |
Cromolyn got the lowest total score, but perhaps can still be considered as a medicine which can affect the cytotoxicity of Aβ peptides. However, it might be better to investigate its fluorinated version,65,66 since it has demonstrated better potential in the treatment of AD.65,67
Additionally, our classical MD simulations were well-equilibrated and performed at a constant number of water molecules, but with slightly different amounts of peptides and drugs (either 6 or 8 molecules of each compound). The fact that with such small alterations of concentration, different effects of drugs on peptide behavior are observed suggests that in further in vitro, in vivo or clinical studies, the effects of small changes in concentration should be taken into account.
Conclusions
In this work, the goal was to make a comparison between three drug candidates as potential medicines against the cytotoxicity of Aβ(31–35) and Aβ(25–35).All the investigated compounds exhibit mechanistic properties which could inhibit the toxicity of Aβ peptides in one way or another. Atorvastatin was shown to be the most promising drug against the cytotoxicity of Aβ(25–35) and Aβ(31–35). Nevertheless, due to its disputed ability to penetrate the blood–brain barrier, it might be not suggested for usage in current formulations. However, improving the delivery of atorvastatin would make it a very attractive drug against neurodegenerative diseases.
Lovastatin fulfilled all requirements to be considered in treatments of neurodegenerative diseases. It can pass through the blood–brain barrier and has similar properties to atorvastatin. Lovastatin is certainly recommended for further investigations as a potential drug against the cytotoxicity of Aβ peptides.
Cromolyn may also be regarded as a potential drug candidate against the cytotoxicity of Aβ(25–35) and Aβ(31–35). However, it did not appear to be the best agent against peptide aggregation, as indicated by both MD and well-tempered metadynamics simulations. Perhaps it would be worth studying its chlorinated version.
As a future perspective, it would be interesting to also consider other statins as medicines against neurodegenerative diseases.
Method
Models for drug molecules
Before running MD simulations, starting models need to be developed. The general amber force field (GAFF)68,69 is a suitable one for organic drug molecules. Nevertheless, for the medicines investigated in this work, parameters for all potentials are available, except for the electrostatic potential, which is described by eqn (3). In the equation, qi and qj are the partial atomic charges, ε is a dielectric constant, and Ri,j is the distance between two particles. The partial atomic charges have to be derived in order to start running actual simulations.
 | (3) |
For retrieving partial atomic charges, twenty different conformations were computed for each molecule by randomly changing the torsion angles. For each optimized conformation, ab initio calculations were performed using the Hartree–Fock method with the 6-31G(d) basis set and applying the restrained electrostatic potential (RESP)70 fitting method, as done in the original description of GAFF. The final partial atomic charges were retrieved through averaging across the twenty conformations. The computations were made using Gaussian 16 software.71 The final partial atomic charges can be observed in Fig. S1–S3 of the ESI.†
Classical molecular dynamics simulations
Firstly, the compositions of the simulated systems were set up; these are shown in Table 5. In order to build up the simulations with peptides and drugs, systems containing only peptides were used from our previous work by Chrobak et al.42 The FF for peptides was amber99sb-ildn72 and the water model was TIP3p.73 Thereafter, drug molecules were added using an artificial van der Waals radius of 0.5 nm in order to avoid molecules clustering or overlapping at the beginning of the simulations. Chlorine counter ions were added in systems with Aβ(25–35) in order to neutralize the charge of the peptide. Finally, water molecules were added randomly inside the box.| System | Number of counter ions | Number of water molecules |
|---|---|---|
| 6 drug mol | 0 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 8 drug mol | 0 | 10000 |
| 6 Aβ(25–35) & 6 drug mol | 6 | 10000 |
| 6 Aβ(31–35) & 6 drug mol | 0 | 10000 |
| 8 Aβ(25–35) & 8 drug mol | 8 | 10000 |
| 8 Aβ(31–35) & 8 drug mol | 0 | 10000 |
Simulation boxes containing only drugs were built similarly: the molecules were put at a minimum distance of 0.5 nm from each other in order to avoid overlaps and clustering. Thereafter, water was inserted in the resulting boxes.
All systems were equilibrated for 40 ns and then simulated for 250 ns with a time step of 2 fs in the NPT ensemble. The cut-off value was 0.9 nm with the so-called van der Waals modifier potential-shift.74 Long-range electrostatics was computed using particle mesh Ewald algorithm (PME)75 with the order equal to 4. The bonds were constrained during the simulations using the LINCS algorithm76,77 with 12 iterations. The simulation utilized isotropic pressure coupling and a velocity rescale thermostat,78 with a coupling constant of 0.5 ps, to keep the temperature regulated at 310 K. Furthermore, the Berendsen barostat79 was used, with a compressibility of 0.000045 bar−1 and a coupling constant of 10 ps, to keep the pressure at 1 atm. The integrator was leap-frog.80 All simulations were run by GROMACS-2019 software.81,82
Well-tempered metadynamics simulations
Free energy calculations were performed using well-tempered metadynamics48–50 simulations. In order to obtain a good sampling, 20 calculations were run in parallel where each one of them was 100 ns long. Every system contained 1 drug molecule and 2 peptides, and was created from equilibrated simulations with 6 molecules, where 5 drug molecules and 4 peptides were deleted. Moreover, for Aβ(25–35), only 2 counter ions were kept and the number of water molecules was 10000.
In order to run well-tempered metadynamics simulations, 2 collective variables (CVs) were chosen. One of them was the distance between the centers of mass of the peptides and the other one was the distance between the centers of mass of one of the peptides and a drug molecule (see Fig. 8). The distance between the center of mass of the second peptide and the drug molecule was ignored.
 | ||
| Fig. 8 Illustration of the chosen collective variables (CV) for calculations of the PMF. Here, CV1 is the distance between peptides and CV2 is the distance between one of the peptides and the drug molecule. | ||
Other settings in the calculations were as follows: the selected bias factor (γ) was 50, the temperature was 310 K, the width of the collective variable (σ) was 0.05 nm, and the height of the Gaussian function was 1.2 kJ mol−1. Gaussian functions were deposited every 500 steps (the PACE parameter). All simulations were performed in the NVT ensemble using the velocity rescale thermostat.78 The time step was 2 fs in every simulation and the leap-frog algorithm was utilized as an integrator for the Newtonian equations of motion with a cut-off value of 0.9 nm. The force fields used for the free energy calculations and the other settings were exactly the same as in the classical MD simulations (see the previous subsection). The MD engine was GROMACS-2019.4 (ref. 81–83) with plumed-2.5.4 for well-tempered metadynamics simulations.
Author contributions
W. C., D. W. P. and A. R. contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swedish National Infrastructure for Computing (SNIC) for computational resources in several centers. In the National Supercomputer Center (NSC), the Tetralith cluster was employed for calculations through projects SNIC2019/3-280, SNIC2019/3-553 and SNIC2019/7-36. In the High Performance Computing Center North (HPC2N), the Kebnekaise cluster was used for simulations with the project numbers SNIC2019/5-74 and SNIC2020/5-45 and the storage was given in terms of projects SNIC2020/10-22 and SNIC2020/6-53. In the Chalmers Centre for Computational Science and Engineering (C3SE), Hebbe and Vera clusters were utilized for calculations in projects SNIC/2018/3-490, SNIC2019/3-53 and C3SE/2020-1-15 with the storage given from the projects SNIC2020/6-12 and C3SE605/17-3. For the access to C3SE/2020-1-15 and C3SE605/17-3, we thank Professor Henrik Grönbeck from Chalmers University of Technology. The authors thank the Swedish Research Council for the financial support (grant numbers 2019-04020 and 2017-06716).References
- G. Thinakaran and E. H. Koo, J. Biol. Chem., 2008, 283, 29615–29619 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- C. Haass and D. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- The Alzheimer’s Association, Alzheimers Dement., 2021, 17, 19 Search PubMed.
- R. Yan and R. Vassar, Lancet Neurol., 2014, 13, 319–329 CrossRef CAS PubMed.
- Y. Kim, C. Kim, H. Y. Jang and I. Mook-Jung, J. Alzheimer’s Dis., 2016, 51, 1057–1068 CAS.
- J. T. Pedersen and E. M. Sigurdsson, Trends Mol. Med., 2015, 21, 394–402 CrossRef CAS PubMed.
- D. J. Conrado, S. Duvvuri, H. Geerts, J. Burton, C. Biesdorf, M. Ahamadi, S. Macha, G. Hather, J. Francisco Morales, J. Podichetty, T. Nicholas, D. Stephenson, M. Trame, K. Romero and B. Corrigan, Clin. Pharmacol. Ther., 2020, 107, 796–805 CrossRef PubMed.
- U. Rauch, J. Osende, J. H. Chesebro, V. Fuster, D. A. Vorchheimer, K. Harris, P. Harris, D. A. Sandler, J. T. Fallon, S. Jayaraman and J. J. Badimon, Atherosclerosis, 2000, 153, 181–189 CrossRef CAS PubMed.
- E. Stroes, Curr. Med. Res. Opin., 2005, 21, S9–S16 CrossRef CAS PubMed.
- S. Yusuf, J. Bosch, G. Dagenais, J. Zhu, D. Xavier, L. Liu, P. Pais, P. López-Jaramillo, L. A. Leiter, A. Dans, A. Avezum, L. S. Piegas, A. Parkhomenko, K. Keltai, M. Keltai, K. Sliwa, R. J. G. Peters, C. Held, I. Chazova, K. Yusoff, B. S. Lewis, P. Jansky, K. Khunti, W. D. Toff, C. M. Reid, J. Varigos, G. Sanchez-Vallejo, R. McKelvie, J. Pogue, P. Gao, R. Diaz and E. Lonn, N. Engl. J. Med., 2016, 374, 2021–2031 CrossRef CAS PubMed.
- T. C. Theoharides, W. Sieghart, P. Greengard and W. W. Douglas, Science, 1980, 207, 80–82 CrossRef CAS PubMed.
- U. Y. Ryo, B. Kang and R. G. Townley, JAMA, J. Am. Med. Assoc., 1976, 236, 927–931 CrossRef CAS.
- G. Watt and T. Karl, Front. Pharmacol., 2017, 8, 1–7 CAS.
- C. Zhang, A. Griciuc, E. Hudry, Y. Wan, L. Quinti, J. Ward, A. Forte, X. Shen, C. Ran, D. Elmaleh and R. Tanzi, Sci. Rep., 2018, 8, 1–9 Search PubMed.
- K. Fassbender, M. Simons, C. Bergmann, M. Stroick, D. Lütjohann, P. Keller, H. Runz, S. Kühl, T. Bertsch, K. Von Bergmann, M. Hennerici, K. Beyreuther and T. Hartmann, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5856–5861 CrossRef CAS PubMed.
- S. Shalaby and E. Louis, Neuroepidemiology, 2016, 47, 11–17 Search PubMed.
- N. E. Shepardson, G. M. Shankar and D. J. Selkoe, Arch. Neurol., 2011, 68, 1385–1392 CrossRef PubMed.
- P. Bar-On, L. Crews, A. O. Koob, H. Mizuno, A. Adame, B. Spencer and E. Masliah, J. Neurochem., 2008, 105, 1656–1667 CrossRef CAS PubMed.
- F. Arenas, C. Garcia-Ruiz and J. C. Fernandez-Checa, Front. Mol. Neurosci., 2017, 10, 382 CrossRef PubMed.
- I. I. Krivoi and A. M. Petrov, Int. J. Mol. Sci., 2019, 20, 1046 CrossRef CAS PubMed.
- C. D. Scala, J. Fantini, N. Yahi, F. J. Barrantes and H. Chahinian, Biomolecules, 2018, 8, 31 CrossRef PubMed.
- B. Schultz, D. Patten and D. Berlau, Transl. Neurodegener., 2018, 7, 1–11 CrossRef PubMed.
- C.-H. Lin, C.-H. Chang, C.-H. Tai, M.-F. Cheng, Y.-C. Chen, Y.-T. Chao, T.-L. Huang, R.-F. Yen and R.-M. Wu, Mov. Disord., 2020, 1229–1237 Search PubMed.
- N. Geifman, R. D. Brinton, R. E. Kennedy, L. S. Schneider and A. J. Butte, Alzheimer’s Res. Ther., 2017, 9, 10 CrossRef PubMed.
- F. Panza, D. Seripa, V. Solfrizzi, B. P. Imbimbo, M. Lozupone, A. Leo, R. Sardone, G. Gagliardi, L. Lofano, B. C. Creanza, P. Bisceglia, A. Daniele, A. Bellomo, A. Greco and G. Logroscino, Expert Opin. Emerging Drugs, 2016, 21, 377–391 CrossRef CAS PubMed.
- Y. Hori, S. Takeda, H. Cho, S. Wegmann, T. M. Shoup, K. Takahashi, D. Irimia, D. R. Elmaleh, B. T. Hyman and E. Hudry, J. Biol. Chem., 2015, 290, 1966–1978 CrossRef CAS PubMed.
- M. Lozupone, V. Solfrizzi, F. D’Urso, I. Di Gioia, R. Sardone, V. Dibello, R. Stallone, A. Liguori, C. Ciritella, A. Daniele, A. Bellomo, D. Seripa and F. Panza, Expert Opin. Emerging Drugs, 2020, 25, 319–335 CrossRef CAS PubMed.
- P. B. Eisenhauer, R. J. Johnson, J. M. Wells, T. A. Davies and R. E. Fine, J. Neurosci. Res., 2000, 60, 804–810 CrossRef CAS PubMed.
- C. J. Pike, A. J. Walencewicz-Wasserman, J. Kosmoski, D. H. Cribbs, C. G. Glabe and C. W. Cotman, J. Neurochem., 1995, 64, 253–265 CrossRef CAS PubMed.
- C. J. Pike, A. J. Walencewicz, C. G. Glabe and C. W. Cotman, Eur. J. Pharmacol., Mol. Pharmacol. Sect., 1991, 207, 367–368 CrossRef CAS.
- M. E. Clementi, S. Marini, M. Coletta, F. Orsini, B. Giardina and F. Misiti, FEBS Lett., 2005, 579, 2913–2918 CrossRef CAS PubMed.
- X.-Z. Yan, J.-T. Qiao, Y. Dou and Z.-D. Qiao, Neurosci., 1999, 92, 177–184 CrossRef CAS PubMed.
- F. Misiti, B. Sampaolese, M. Pezzotti, S. Marini, M. Coletta, L. Ceccarelli, B. Giardina and M. E. Clementi, Neurochem. Int., 2005, 46, 575–583 CrossRef CAS PubMed.
- S. Varadarajan, J. Kanski, M. Aksenova, C. Lauderback and D. A. Butterfield, J. Am. Chem. Soc., 2001, 123, 5625–5631 CrossRef CAS PubMed.
- F. Misiti, G. Martorana, G. Nocca, E. Di Stasio, B. Giardina and M. Clementi, Neurosci., 2004, 126, 297–303 CrossRef CAS PubMed.
- M. E. Clementi, G. E. Martorana, M. Pezzotti, B. Giardina and F. Misiti, Int. J. Biochem. Cell Biol., 2004, 36, 2066–2076 CrossRef CAS PubMed.
- Y. Yamaguchi and S. Kawashima, Eur. J. Pharmacol., 2001, 412, 265–272 CrossRef CAS PubMed.
- H.-H. G. Tsai, J.-B. Lee, S.-S. Tseng, X.-A. Pan and Y.-C. Shih, Proteins: Struct., Funct., Bioinf., 2010, 78, 1909–1925 CrossRef CAS PubMed.
- S.-W. Lee and Y.-M. Kim, Bull. Korean Chem. Soc., 2004, 25, 838–842 CrossRef CAS.
- W. Chrobak, D. W. Pacut, F. Blomgren, A. Rodin, J. Swenson and I. Ermilova, ACS Chem. Neurosci., 2021, 12, 660–674 CrossRef CAS PubMed.
- I. Ermilova and A. P. Lyubartsev, RSC Adv., 2020, 10, 3902–3915 RSC.
- H. Fatafta, M. Khaled, M. C. Owen, A. Sayyed-Ahmad and B. Strodel, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–10 CrossRef PubMed.
- N. Ntarakas, I. Ermilova and A. P. Lyubartsev, Eur. Biophys. J., 2019, 48, 813–824 CrossRef CAS PubMed.
- C. La Rosa, M. Condorelli, G. Compagnini, F. Lolicato, D. Milardi, T. N. Do, M. Karttunen, M. Pannuzzo, A. Ramamoorthy, F. Fraternali, F. Collu, H. Rezaei, B. Strodel and A. Raudino, Eur. Biophys. J., 2020, 49, 175–191 CrossRef CAS PubMed.
- R. Capelli, A. Bochicchio, G. Piccini, R. Casasnovas, P. Carloni and M. Parrinello, J. Chem. Theory Comput., 2019, 15, 3354–3361 CrossRef CAS PubMed.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Bonomi, A. Barducci and M. Parrinello, J. Comput. Chem., 2009, 30, 1615–1621 CrossRef CAS PubMed.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 1–13 Search PubMed.
- K. Reuter and J. Köfinger, Comput. Phys. Commun., 2019, 236, 274–284 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. E. Clementi and F. Misiti, Med. Sci. Monit., 2005, 11, BR381–BR385 CAS.
- D. A. Butterfield and R. Sultana, J. Amino Acids, 2011, 2011, 198430 Search PubMed.
- K. Sato, T. Maeda and M. Hoshi, Int. J. Pept. Res. Ther., 2012, 18, 341–345 CrossRef CAS.
- M. M. Gromiha, Protein bioinformatics: from sequence to function, Academic Press, 2010 Search PubMed.
- S. Ranganathan, K. Nakai and C. Schonbach, Encyclopedia of Bioinformatics and Computational Biology: ABC of Bioinformatics, Elsevier, 2018 Search PubMed.
- R. R. Sinden, DNA structure and function, Gulf Professional Publishing, 1994 Search PubMed.
- M. Heinig and D. Frishman, Nucleic Acids Res., 2004, 32, W500–W502 CrossRef CAS PubMed.
- M. Yahyavi, S. Falsafi-Zadeh, Z. Karimi, G. Kalatarian and H. Galehdari, Bioinformation, 2014, 10, 548 CrossRef PubMed.
- C. Hilbich, B. Kisters-Woike, J. Reed, C. L. Masters and K. Beyreuther, J. Mol. Biol., 1991, 218, 149–163 CrossRef CAS PubMed.
- L. W. Simpson, G. L. Szeto, H. Boukari, T. A. Good and J. B. Leach, Acta Biomater., 2020, 112, 164–173 CrossRef CAS PubMed.
- N. Kandiah and H. H. Feldman, J. Neurol. Sci., 2009, 283, 230–234 CrossRef CAS PubMed.
- R. H. Knopp and N. Eng, J. Med. Chem., 1999, 341, 498–511 CAS.
- T. M. Shoup, A. Griciuc, M. D. Normandin, L. Quinti, L. V. Walsh, M. Dhaynaut, S.-H. Moon, N. J. Guehl, P. Brugarolas, D. R. Elmaleh, G. E. Fakhri and R. E. Tanzi, J. Alzheimer’s Dis., 2021, 1–12 Search PubMed.
- Y.-J. Wang, A. Monteagudo, M. A. Downey, P. G. Ashton-Rickardt and D. R. Elmaleh, Sci. Rep., 2021, 11, 1 CrossRef PubMed.
- F. Belluti, A. Rampa, S. Gobbi and A. Bisi, Expert Opin. Ther. Pat., 2013, 23, 581–596 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- I. Ermilova, S. Stenberg and A. P. Lyubartsev, Phys. Chem. Chem. Phys., 2017, 19, 28263–28274 RSC.
- C. I. Bayly, P. Cieplak, W. Cornell and P. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, 2016 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins: Struct., Funct., Bioinf., 2010, 78, 1950–1958 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. D. Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- W. F. Van Gunsteren and H. J. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- C. Kutzner, S. Páll, M. Fechner, A. Esztermann, B. L. de Groot and H. Grubmüller, J. Comput. Chem., 2019, 40, 2418–2431 CrossRef CAS PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01963a |
| This journal is © The Royal Society of Chemistry 2022 |