
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular polymer/peptide hybrid hydrogels with tunable stiffness mediated by interchain acid-amide hydrogen bonds†
Yu-Shen Liu ab,
Rajan Deepan Chakravarthyab,
Abdelreheem Abdelfatah Saddikabc,
Mohiuddin Mohammedab and
Hsin-Chieh Lin*ab
ab,
Rajan Deepan Chakravarthyab,
Abdelreheem Abdelfatah Saddikabc,
Mohiuddin Mohammedab and
Hsin-Chieh Lin*ab
aDepartment of Materials Science and Engineering, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan. E-mail: hclin45@nycu.edu.tw
bDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu 300, Taiwan
cMaterials Science and Engineering Laboratory, Department of Chemistry, Faculty of Science, Assiut University, Assiut 71516, Egypt
First published on 11th May 2022
Abstract
Hydrogels are a class of biomaterials used in the field of tissue engineering and drug delivery. Many tissue engineering applications depend on the material properties of hydrogel scaffolds, such as mechanical stiffness, pore size, and interconnectivity. In this work, we describe the synthesis of peptide/polymer hybrid double-network (DN) hydrogels composed of supramolecular and covalent polymers. The DN hydrogels were prepared by combining the self-assembled pentafluorobenzyl diphenylalanyl aspartic acid (PFB-FFD) tripeptide for the first network and the polymeric PNIPAM-PEGDA copolymer for the second network. During this process, self-assembled peptide nanostructures are cross-linked to the polyacrylamide group in the polymer network through non-covalent interactions. The PNIPAM-PEGDA:PFB-FFD hydrogel exhibited higher mechanical stiffness (G′ ∼2 kPa) than the PNIPAM-PEGDA copolymer. Moreover, PNIPAM-PEGDA:PFB-FFD hydrogel shows a decrease in pore size (∼1.2 μm) compared to the original copolymer (∼5.2 μm), with the structural framework of highly interconnected fibrous peptide network. The mechanical stiffness of hydrogels was systematically investigated by rheological analysis in response to various variables, including UV exposure time, concentration of peptides, and amino acid functionalization. Modulating the time of UV irradiation resulted in PNIPAM-PEGDA:PFB-FFD hydrogels with a four-fold increase in stiffness. The influence of amino acid side chains and terminal charge of peptides on the strength of DN hydrogels was also investigated using pentafluorobenzyl diphenylalanyl lysine (PFB-FFK). Interestingly, PFB-FFK, which has an amine group on the side chain, does not exhibit the DN structures. The mechanical properties and pore sizes of PNIPAM-PEGDA:PFB-FFK hydrogel were very similar to those of the PNIPAM-PEGDA copolymer due to poor cross-linking. The biocompatibility of the hydrogel materials was tested with the hMSC cell line using the MTT method, and the results indicate that the materials are non-toxic and potentially useful for biological applications.
Introduction
Hydrogel is a highly versatile, cross-linked, three-dimensional polymer network that can accommodate significant quantities of aqueous and biological fluids.1 Due to their high-water content, hydrogels are also flexible like biological tissue.2 They can serve as drug delivery systems,3–5 scaffolds for tissue engineering,6,7 sensors and actuators,8–10 soft machines,11–13 and wearable electronics.14–16 Most hydrogels have, however, been either too soft or mechanically brittle, which limits their potential applications.17 Additionally, the heterogeneity of the structures and the lack of efficient dissipation mechanisms severely restrict their applications.18PNIPAM-based hydrogels are considered smart materials because they can undergo temperature-dependent phase transitions in aqueous solutions at 32–34 °C, which is closer to physiological body temperature.19 In spite of the advantages, poly(NIPAM) hydrogels have three major shortcomings, namely poor mechanical properties, low biodegradability, and limited drug loading.20 PNIPAM has also been copolymerized with other polymer segments to achieve numerous functions in addition to its thermoresponsive property. A variety of applications have also been reported by grafting with macromolecules. Researchers have also prepared synthetic hydrogels containing PNIPAM along with natural components such as gelatin, chitosan, cellulose, dextran, polysaccharides, and polyamino acids to improve hydrogel performance.21 In many of them, PNIPAM chains are directly attached to natural polymers. In addition, peptides have also been combined with PNIPAM to fabricate the multicomponent hydrogels via covalent or non-covalent interactions.22,23 Although some progress has been made, the exploration of smart hydrogels will be further enhanced by incorporating ultrashort peptides with different functions.
The mechanical properties, scaffold pore size, and interconnectivity are undoubtedly critical for most bio applications. Crosslinking plays a key role in the mechanical properties of hydrogels.24 Polymeric hydrogels stemming from supramolecular crosslinks through specific, directional, and dynamic non-covalent interactions have led to the development of novel polymeric hydrogels. Several approaches to supramolecular crosslinking are currently available, ranging from ionic chemistry to molecular host–guest interactions. Hydrogen bonding is one of the most important non-covalent interactions that impart functions such as self-healing, shape memory, and dynamic energy dissipation to materials. Hydrogen bonding between amides and carboxylic acids is a molecular recognition process in which amide hosts selectively bind to carboxylic acid guest species.25 Such guest-recognition hydrogen bonding for supramolecular cross-links is relatively rare, but this approach might provide unique properties to functional hydrogels.
Peptide-based hydrogels are important in tissue engineering and regenerative medicine due to their superior biocompatibility and low toxicity.26,27 Short peptides derived from naturally occurring amino acids have also been presented in the extensive literature as viable candidates for molecular self-assembly to fabricate the supramolecular structures such as fibrils, belts, tubes, and vesicles. In addition, short peptides are more stable and non-immunogenic than their long counterparts, and hydrogels of short self-assembling peptides show promise for drug delivery and regenerative medicine.28 Ultrashort peptides containing two to three amino acids are inexpensive, easy to prepare, and can be produced on a large scale. This study aims to synthesize peptide-polymer hybrid hydrogels combining tripeptide and PNIPAM-PEGDA with emphasis on supramolecular cross-links. We also systematically investigate the effects of the amino acid side chains and the terminal charge of the peptides on the self-assembly and mechanical properties of the hydrogel materials. Although hydrogels of peptide/PNIPAM have been described in a few reports, the hybrid system with tripeptide has not been reported to our knowledge. The peptide-polymer hybrid hydrogels described here can be tailored to change their mechanical properties over a wide range.
Results and discussion
Design principle of peptide/polymer hybrid double network hydrogels
To develop a hydrogel with high mechanical stiffness and cross-linking density with an interconnecting network, we created a polymer-supramolecular double network (DN) hydrogel with dynamic non-covalent crosslinking. Based on the concept shown in Fig. 1, we design and synthesize a pair of PFB tripeptides to prepare a supramolecular peptide/polymer hybrid hydrogels. The hydrogel design used for this study consists of two segments: (i) PNIPAM-PEGDA copolymer, with large pore sizes on the order of few micrometers; (ii) tripeptide segment that can self-assemble into nanofibrous and nano porous morphology. We also investigate the effect of aspartic acid and lysine groups for creating supramolecular crosslinking with PNIPAM-PEGDA copolymer. We selected two known short peptide sequences PFB-FFD and PFB-FFK to establish the intermolecular interactions between the polymer and the peptide network (Fig. 2). These materials can be prepared by solid-phase peptide synthesis (SPPS).29 The gel was obtained by one-pot polymerization of NIPAM, PEGDA (NIPAM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEGDA wt% 10:1) and PFB-FFD or PFB-FFK (peptide 24 mg mL−1) in dimethyl sulfoxide (DMSO) solution and the results are summarized in Table 1. PEGDA serves as a crosslinker for PNIPAM. Then, ammonium persulfate was added as a photoinitiator and polymerization was carried out under UV light for 20 h. After the polymerization process was completed, the pre-gel was treated with water for 3 h to initiate the self-assembly of peptide molecules. The resulting material was then dialyzed in deionized water (DI water) to remove unpolymerized species and DMSO solvent. The PNIPAM-PEGDA copolymer gel was weak and brittle. We found a similar result when mixing with PFB-FFK tripeptide which could be due to weak hydrogen bonding. Surprisingly, the addition of PFB-FFD peptide material can significantly improve the stability and toughness of the gel due to the supramolecular interaction between the amide and acid groups of NIPAM and PFB-FFD.
:PEGDA wt% 10:1) and PFB-FFD or PFB-FFK (peptide 24 mg mL−1) in dimethyl sulfoxide (DMSO) solution and the results are summarized in Table 1. PEGDA serves as a crosslinker for PNIPAM. Then, ammonium persulfate was added as a photoinitiator and polymerization was carried out under UV light for 20 h. After the polymerization process was completed, the pre-gel was treated with water for 3 h to initiate the self-assembly of peptide molecules. The resulting material was then dialyzed in deionized water (DI water) to remove unpolymerized species and DMSO solvent. The PNIPAM-PEGDA copolymer gel was weak and brittle. We found a similar result when mixing with PFB-FFK tripeptide which could be due to weak hydrogen bonding. Surprisingly, the addition of PFB-FFD peptide material can significantly improve the stability and toughness of the gel due to the supramolecular interaction between the amide and acid groups of NIPAM and PFB-FFD.
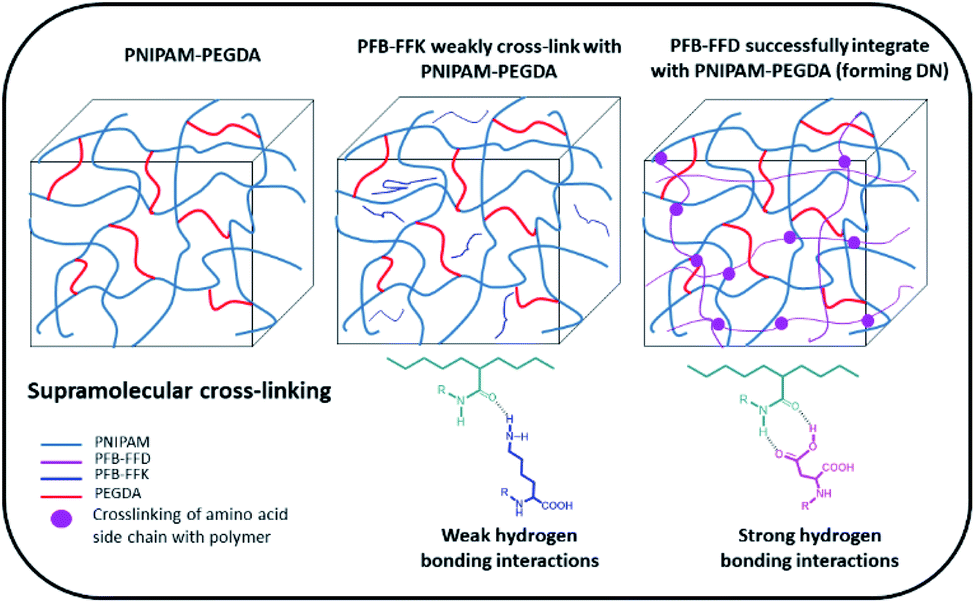 | ||
| Fig. 1 Schematics of hydrogels and the possible cross-linking mechanism between polymer/peptide units via intermolecular hydrogen bonding between amine-amide or acid-amide moieties. | ||
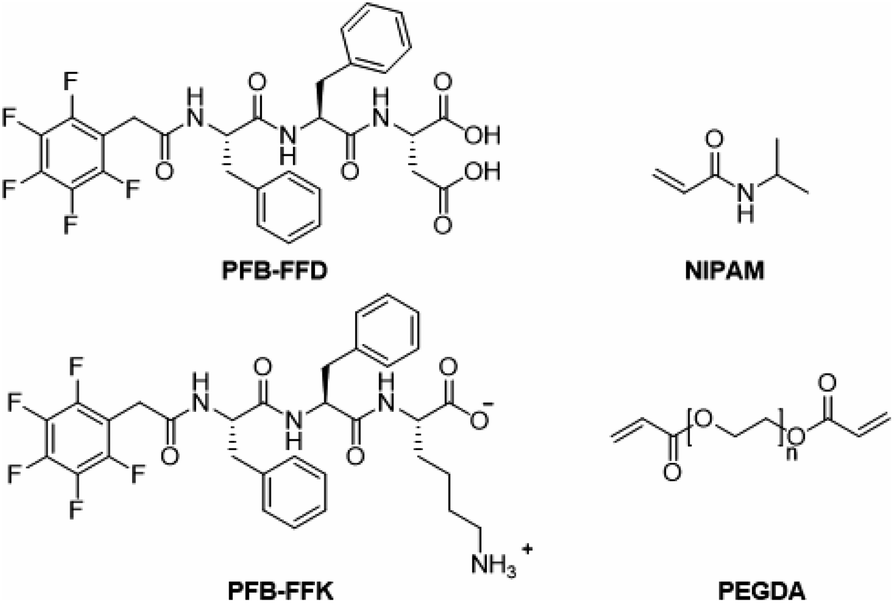 | ||
| Fig. 2 Chemical structures of pentafluorobenzyl diphenylalanyl aspartic acid PFB-FFD; pentafluorobenzyl diphenylalanyl lysine PFB-FFK; N-isopropylacrylamide (NIPAM); Poly (ethylene glycol) diacrylate (PEGDA). | ||
| Entry | Appearancea | Pore sizeb (μm) | Modulic G′, G′′ (Pa) | Critical straind γc (%) |
|---|---|---|---|---|
| a OG: opaque gel.b SEM data.c The values at 10 rad s−1.d The crossover transition point from primary elastic to primary viscous is shown in parenthesis. | ||||
| PNIPAM-PEGDA | OG | 5.2 ± 0.3 | 3.5 × 102, 0.26 × 102 | 54.3 (242) |
| PNIPAM-PEGDA:PFB-FFD | OG | 1.2 ± 0.3 | 2.1 × 103, 1.9 × 102 | 46.6 (791) |
| PNIPAM-PEGDA:PFB-FFK | OG | 5.1 ± 0.2 | 5.0 × 102, 0.23 × 102 | 73.8 (482) |
Morphology and rheological properties of the hydrogels
To determine the morphological structures of the hydrogels and to understand the relative differences in microstructures upon the addition of peptides, scanning electron microscopes (SEM) and transmission electron microscopes (TEM) were used, and the results are shown in Fig. 3. As shown by the SEM image, all hydrogels exhibited porous structures in the micrometre range, (Fig. 3a–c). The data showed that PNIPAM-PEGDA consisted of a porous structure with a diameter of 5.2 ± 0.3 μm. Similar results were obtained for the PFB-FFK peptide/polymer hybrid system with a pore size of 5.1 ± 0.2 μm. However, the pore size of the PFB-FFD-based DN hydrogel system decreased dramatically to 1.2 ± 0.3 μm, which could be due to strong crosslinking between peptide and polymer network. TEM analysis proved that the PFB-FFD peptide/polymer hydrogel network consisted of fibrous networks that were highly cross-linked (Fig. 3d and S1†). The self-assembled PFB-FFD had successfully integrated into the hydrogel network and did not diffuse into the water during the dialysis process. These results indicate that the addition of peptides with an acid side chain can induce strong hydrogen bonds leading to a DN hydrogel system with the polymer.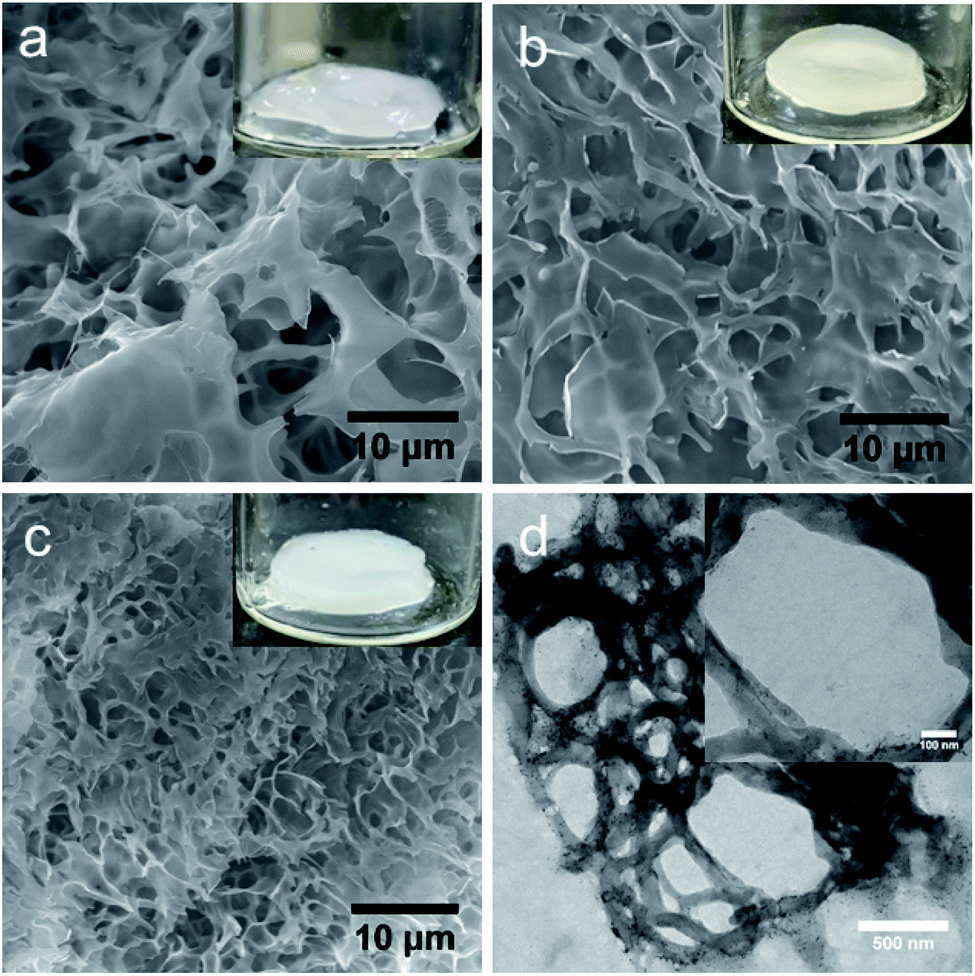 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of the hydrogel (inset shows optical image of the gel); (a) PNIPAM-PEGDA, (b) PNIPAM-PEGDA: PFB-FFK, (c) PNIPAM-PEGDA:PFB-FFD. TEM image of (d) PNIPAM-PEGDA:PFB-FFD hydrogel (inset shows nanostructures of the self-assembled peptides). | ||
The mechanical properties of the hydrogels were measured using oscillatory rheology, and the results are summarized in Fig. 4 and S2–S6 (ESI).† Storage modulus (G′) and loss modulus (G′′) were determined as a function of frequency and strain. All hydrogels have elastic properties as their storage moduli (G′) are higher than their loss moduli (G′′). The frequency sweep rheology of PNIPAM-PEGDA exhibited low G′ values of 0.3 kPa. Moreover, the hydrogel retained its morphological structures up to a critical strain of about 54.3% (Fig. S3, ESI†). The addition of PFB-FFD peptide to the polymer system leads to a significant increase in G′ values to 2.1 kPa with the critical strain value of 46.6%. Furthermore, the hydrogel showed a crossover transition between gel-like and liquid-like states at a strain value of about 791%, indicating significant hydrogen bonding between the peptide and polymeric network. When the hydrogel is prepared with PFB-FFK, the G′ value drops to 0.5 kPa with a strain value of 73.8%. However, the crossover point occurs at a strain value of 482%, which may be attributed to weaker hydrogen bonds and network stability. These results indicated that the amino acid side chain had a significant impact on the formation of DN hybrid hydrogels. In addition, oscillatory rheological tests with temperature sweep were performed to investigate the thermoresponsive behavior of the PNIPAM-PEGDA:PFB-FFD hydrogel. The storage modulus (G′) value increased from 1.9 kPa at 25 °C to 5.9 kPa at 45 °C (Fig. 4b). This result is consistent with the established phase transition behavior of PNIPAM hydrogels. Further, the hydrogels showed a greater dependence on the peptide concentration. For example, hydrogels prepared with a PFB-FFD concentration of 0.6 wt% showed a lower critical strain of about 13.6%, indicating a higher brittleness. This might be due to a lower cross-link density of the peptide-polymer hydrogel. Under this condition, an average storage modulus value G′ of 0.87 kPa was obtained. Moreover, the presence of salt affects the viscoelastic behavior of the hydrogel due to the metal-ion crosslinking of peptide molecules. The calcium salt was chosen to study the influence of the metal ion on the hydrogelation, based on a previous report.30 The addition of 10 mM Ca2+ to the pre-gel mixture decreases the critical strain (γc) to 21.7%, indicating a weaker cross-linking network between the peptide and the polymer (Table S1, ESI†). However, further research is needed to gain a deeper understanding of this phenomenon.
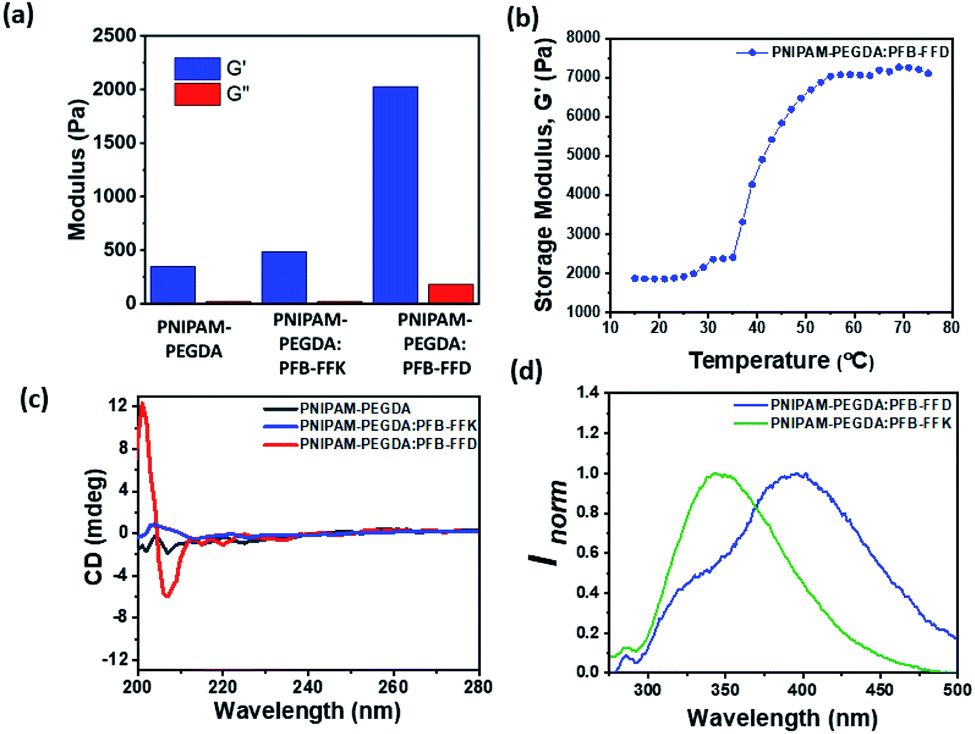 | ||
| Fig. 4 (a) The average storage modulus and loss modulus of PNIPAM-PEGDA, PNIPAM-PEGDA:PFB-FFK and PNIPAM-PEGDA:PFB-FFD. (b) Temperature-dependent rheological analysis of PNIPAM-PEGDA:PFB-FFD. (c) CD spectra of PNIPAM-PEGDA in presence and absence of peptides at the concentration of 2 wt%. (d) Normalized fluorescence emission spectra of peptide/polymer hybrid hydrogels. | ||
Molecular interactions between NIPAM and PFB-tripeptides
To understand the molecular interactions between the polymer and supramolecular peptides as well as the secondary structure of the hydrogels, circular dichroism spectra were obtained (Fig. 4C). The CD spectra of both the PNIPAM-PEGDA gel and the PFB-FFK peptide/polymer hybrid system showed no noticeable signals in the near UV range 200–230 nm. The PFB-FFD -based DN hydrogels exhibited negative signals at λ = 205–210 nm, indicating possible β-sheet structures. To further evaluate the aromatic interactions of the hydrogels, the fluorescent emission spectra were obtained. The emission spectra of the monomeric peptides generally show a distinct band at approximately 310 nm with an excitation wavelength of 260 nm.29,31 The peptide/polymer hybrid system showed a significant red shift. The PFB-FFK system showed emission maxima at 345 nm and the PFB-FFD system at 395 nm respectively. The change in emission profile is due to the formation of strong aromatic–aromatic interactions. All these results indicated that the hydrogen bonding and aromatic interactions are the major driving forces for the formation of DN hybrid hydrogels.To further investigate thermal stability and thermally induced structural transitions at the molecular level, thermogravimetric analysis (TGA) was performed and the results are shown in Fig. S14.† TGA of peptide-polymer hybrid hydrogels showed a two-stage decomposition mechanism. The first stage, between 25–100 °C, is due to water evaporation in the hydrogels, and the second stage, between 300–430 °C is due to the decomposition of the hydrogel network. At high temperatures, the PNIPAM-PEGDA:PFB-FFD hydrogel reaches a plateau, which could be due to the higher carbon content introduced by PFB-FFD. These experiments suggest that PFB-FFD has a strong affinity toward the polymeric network.32
Analysis of the molecular interactions between the amide–amide and acid-amide functional groups was performed to identify the origin of the crosslinking mechanism in DN hydrogels.33 The amide–amide groups are capable of forming an eight-membered ring structure with two H-bonds (Fig. 5), but such interactions are weaker because the N–H bond is less polarized than the O–H bond. Moreover, the formation of a strong hydrogen-bonded network of the PNIPAM moiety in water precludes the possibility of eight-membered ring structures. The thermoresponsive behavior of the hydrogel is caused by its hydrated and dehydrated forms during temperature changes (Fig. S15†).34 The formation of acid–acid interactions is relatively less stable because of the repulsive interactions between the carboxylate groups. In contrast, there are no repulsive interactions in the acid-amide system. This results in stronger acid-amide hydrogen bonding interactions than amide–amide hydrogen bonds, which leads to improved mechanical properties for the DN hydrogels. To confirm this, we performed FTIR measurements for the PFB-FFD and the peptide/polymer DN systems. The FTIR spectrum of PFB-FFD showed characteristic amide bands between 1650-1610 cm−1. For the PFB-FFD-based DN hydrogel, the polymer signals predominate and shows intense signal at 1648 cm−1, which can be attributed to amide group of polyacrylamide. However, the intense peak of PFB-FFD located at 1493 cm−1 was completely absent in the DN hybrid system, which could be due to different molecular structures in the hydrogel system.
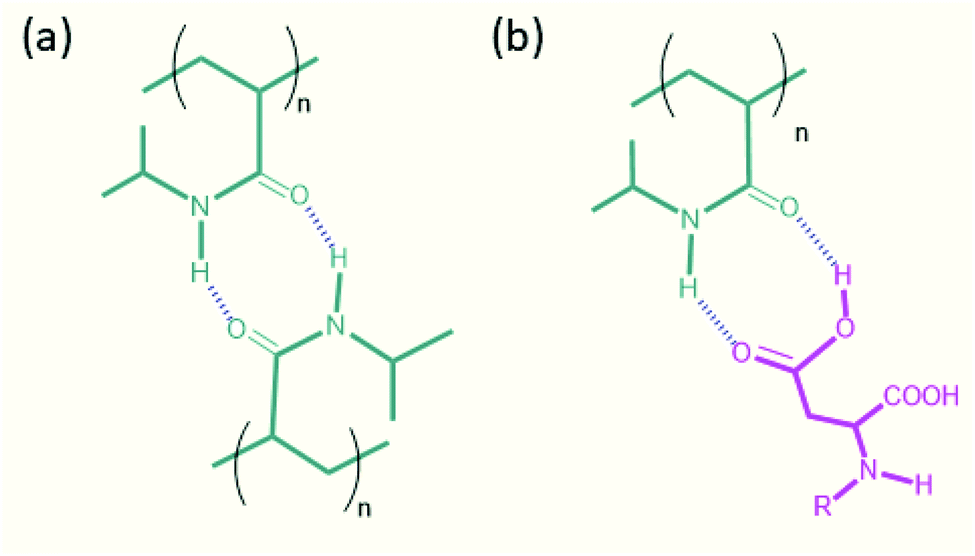 | ||
| Fig. 5 Proposed hydrogen bonding crosslinking mechanism for the formation of peptide/polymer DN hydrogel. | ||
Tunable mechanical properties and biocompatibility
Supramolecular hydrogels are effective scaffolds for tissue engineering. Hydrogels share some physical characteristics with the native extracellular matrix (ECM), widely used for cell-based therapies. Several studies have shown that the scaffold composition and mechanical properties affect the fate of the cells. We hypothesized that decreasing the polymerization time of PFB-FFD-polymer DN hydrogel could interfere with crosslinking, thereby reducing mechanical properties. We investigated this effect on PFB-FFD based DN hydrogel system. Four DN hydrogels were prepared (PNIPAM-PEGDA:PFB-FFD) by changing the UV-exposure time between 5 and 20 h, respectively. As shown in Fig. 6a, increasing the exposure time resulted in stiffer hydrogels. It was found that the hydrogels are elastic with G′ values significantly higher than G′′ over a broad frequency range, and the hydrogels with a tunable stiffness with a range of 0.6 to 2 kPa were achieved. Thus, by changing the UV exposure time, we can control the self-assembly of the hydrogel system thereby tuning the mechanical behavior.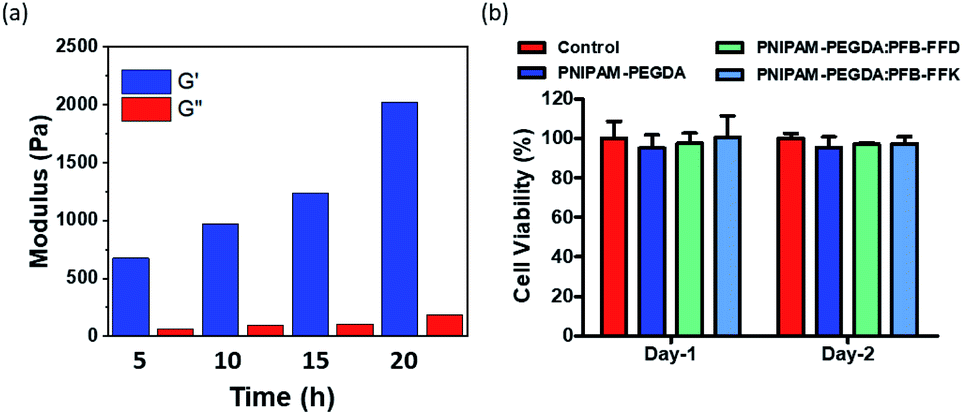 | ||
| Fig. 6 (a) The average storage modulus and loss modulus of PNIPAM-PEGDA: PFB-FFD DN hydrogels prepared at various time point. (b) Cell viability data of 3A6 cells incubated with PNIPAM-PEGDA hydrogels. | ||
The biocompatibility of the hydrogels was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay.35 The hydrogels leachable from the peptide-polymer hybrid hydrogels were exposed to human mesenchymal cells (hMSCs; 3A6 cells). As shown in Fig. 6b, the viability of the 3A6 cells treated with hydrogel leachable was more than 95% after 2 days of culture in all hydrogels compared with the control group, indicating that the hydrogels are not toxic to the 3A6 cells. Taken together, these results indicate that our gels are biocompatible and might serve as viable scaffolds for tissue engineering applications.
Conclusions
We have developed a PFB-based tripeptide system for promoting hydrogen bonding with polymer to form DN hydrogels. There appears to be a correlation between the mechanical properties of the hybrid hydrogels and supramolecular cross-linking of the functionalized peptide. We have illustrated that using the PFB-FFD tripeptide as an example. The terminal aspartic acid of PFB-FFD induces strong hydrogen bonding interactions between the polymer and peptide network, which could greatly improve the stiffness and toughness of the double network gel. Overall, this work highlights the importance of the polymer–supramolecular relationship on DN hydrogels and provides new insights into the design of hydrogels for biological applications.Experimental section
Materials and methods
:1 ratio was dissolved in DMSO at a concentration of 150 mg mL−1. The solution was irradiated with UV light for 20 h and the pre-gel was obtained. Polymerization was initiated by adding ammonium persulfate as a photoinitiator. Finally, the pre-gel obtained was dialyzed in DI water for 24 h to remove the unpolymerized monomers and DMSO.:1) were dissolved in DMSO at a concentration of 24 mg mL−1 and 150 mg mL−1 respectively. Polymerization was initiated by adding ammonium persulfate as a photoinitiator. The solution was exposed to UV light for 20 h to obtain the pre-gel. The pre-gel was then treated with water (1 mL) to initiate self-assembly of the peptide molecules for 3 h. Finally, the sample was dialyzed in DI water for 24 h to remove the unpolymerized monomers and DMSO.To investigate the relationship between gel strength and polymerization time, a time-dependent experiment was carried out using PFB-FFD, NIPAM, and PEGDA samples with polymerization times of 5, 10, 15, and 20 h, respectively.
000% at a frequency of 1 rad s−1. The temperature was maintained at room temperature during the measurement. The geometry gap was set to 100 μm.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by the Ministry of Science and Technology of the Republic of China, Taiwan (grant: MOST 110-2113-M-A49-015- and MOST 110-2124-M-A49-001-).Notes and references
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC.
- H. Cao, L. Duan, Y. Zhang, J. Cao and K. Zhang, Signal Transduction Targeted Ther., 2021, 6, 426 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- J. L. West and J. A. Hubbell, React. Polym., 1995, 25, 139–147 CrossRef CAS.
- G. W. Ashley, J. Henise, R. Reid and D. V. Santi, Proc. Natl. Acad. Sci., 2013, 110, 2318 CrossRef CAS PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- Y. Xing, E. Cheng, Y. Yang, P. Chen, T. Zhang, Y. Sun, Z. Yang and D. Liu, Adv. Mater., 2011, 23, 1117–1121 CrossRef CAS PubMed.
- G. Gerlach and K.-F. Arndt, Hydrogel Sensors and Actuators: Engineering and Technology, ed, G. Urban, Springer, Heidelberg, Germany, 2009 Search PubMed.
- A. Ehrenhofer, S. Binder, G. Gerlach and T. Wallmersperger, Adv. Eng. Mater., 2020, 22, 2000004 CrossRef CAS.
- A. Ehrenhofer, M. Elstner and T. Wallmersperger, Sens. Actuators, B, 2018, 255, 1343–1353 CrossRef CAS.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- D. Wirthl, R. Pichler, M. Drack, G. Kettlguber, R. Moser, R. Gerstmayr, F. Hartmann, E. Bradt, R. Kaltseis, C. M. Siket, S. E. Schausberger, S. Hild, S. Bauer and M. Kaltenbrunner, Sci. Adv., 2017, 3, e1700053 CrossRef PubMed.
- Z. Shen, X. Zhu, C. Majidi and G. Gu, Adv. Mater., 2021, 33, e2102069 CrossRef PubMed.
- J. Chen, Q. Peng, T. Thundat and H. Zeng, Chem. Mater., 2019, 31, 4553–4563 CrossRef CAS.
- Y. Zhang, M. Gong and P. Wan, Matter, 2021, 4, 2655–2658 CrossRef CAS.
- Q. Guan, G. Lin, Y. Gong, J. Wang, W. Tan, D. Bao, Y. Liu, Z. You, X. Sun, Z. Wen and Y. Pan, J. Mater. Chem. A, 2019, 7, 13948–13955 RSC.
- H. Fan and J. P. Gong, Macromolecules, 2020, 53, 2769–2782 CrossRef CAS.
- H. Li, H. Wang, D. Zhang, Z. Xu and W. Liu, Polymer, 2018, 153, 193–200 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef PubMed.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Angew. Chem., Int. Ed., 2015, 54, 7376–7380 CrossRef CAS PubMed.
- L. R. M. Lima, C. M. W. S. A. Cavalcante, M. J. M. Carneiro, J. F. S. Mendes, N. A. Sousa, R. S. Freire, V. P. T. Pinto, R. O. S. Fontenelle, J. P. A. Feitosa and R. C. M. de Paula, Carbohydr. Polym. Technol. Appl., 2021, 2, 100126 Search PubMed.
- J. Couet, J. D. J. S. Samuel, A. Kopyshev, S. Santer and M. Biesalski, Angew. Chem., Int. Ed., 2005, 44, 3297–3301 CrossRef CAS.
- K. Molawi and A. Studer, Chem. Commun., 2007, 5173–5175 RSC.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- P. L. Wash, E. Maverick, J. Chiefari and D. A. Lightner, J. Am. Chem. Soc., 1997, 119, 3802–3806 CrossRef CAS.
- W. Y. Seow and C. A. E. Hauser, Mater. Today, 2014, 17, 381–388 CrossRef CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- N. Yadav, M. K. Chauhan and V. S. Chauhan, Biomater. Sci., 2020, 8, 84–100 RSC.
- A. A. Saddik, R. D. Chakravarthy, M. Mohammed and H.-C. Lin, Soft Matter, 2020, 16, 10143–10150 RSC.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- S.-M. Hsu, Y.-C. Lin, J.-W. Chang, Y.-H. Liu and H.-C. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921–1927 CrossRef CAS.
- J. Mao, Q. J. Yu and S. Wang, Polym. Adv. Technol., 2021, 32, 1752–1762 CrossRef CAS.
- G. Beaudoin, A. Lasri, C. Zhao, B. Liberelle, G. De Crescenzo and X.-X. Zhu, Macromolecules, 2021, 54, 7963–7969 CrossRef CAS.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed.
- M. Mohammed, T.-S. Lai and H.-C. Lin, J. Mater. Chem. B, 2021, 9, 1676–1685 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01944b |
| This journal is © The Royal Society of Chemistry 2022 |