
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile synthesis of pathogen specific bacterial cell wall building blocks†
Lukas Martin Wingen‡
a,
Christina Brauna,
Marvin Rauschbc,
Harald Gross d,
Tanja Schneiderb and
Dirk Menche*a
d,
Tanja Schneiderb and
Dirk Menche*a
aKekulé Institute of Organic Chemistry and Biochemistry, University of Bonn, D-53121 Bonn, Germany. E-mail: dirk.menche@uni-bonn.de
bInstitute for Pharmaceutical Microbiology, University Clinic Bonn, University of Bonn, D-53115 Bonn, Germany
cGerman Center for Infection Research (DZIF), Partner Site Bonn-Cologne, Germany
dPharmaceutical Institute, Dept. of Pharmaceutical Biology, University of Tübingen, D-72076 Tübingen, Germany
First published on 18th May 2022
Abstract
Full details on the design, strategies and tactics for development of a novel synthetic sequence to farnesyl lipid I and II analogs is reported. The modular route was based on a three coupling strategy involving an efficient solid phase synthesis of the elaborate peptide fragment, which proceeded with excellent yield and stereoselectivity and was efficiently applied for the convergent synthesis of 3-lipid I and II. Furthermore, the generality of this route was demonstrated by synthesis of 3-lipid I congeners that are characteristic for S. aureus and E. faecalis. All 3-lipid I and II building blocks were obtained in high purity revealing high spectroscopic resolution.
Introduction
Lipid I and biosynthetically derived lipid II represent the key precursors for bacterial cell wall biosynthesis (Fig. 1).1,2 On a molecular level, they deliver their glycosylated-pentapeptide “building blocks” into the growing peptidoglycan network and play functional roles in the coordination of this process.1 Furthermore, they regulate cell division and influence various other enzymatic processes.3 In addition, they present pivotal molecular targets for a broad range of antibiotics and antibacterial agents, adding to their importance for molecular studies.1,4 Consequently, functional studies with these key building blocks have attracted high interest from the perspective of antibacterial research and medicinal chemistry. However, such studies have been severely hindered by unfavourable physical properties of the original bactoprenol hydrocarbon side chain, leading to unwanted aggregation phenomena, precipitation as well as complex spectroscopic characteristics. Also, such analyses are further hampered by the low natural supply of these key functional compounds,2,5 which also could not be resolved by synthetic approaches due to the multiple steps required to access their elaborate architectures.5–7 Furthermore, existing synthetic strategies could not be adapted to pathogen specific modification, such as 5 or 6 (Fig. 1), where the authentic lysine moiety holds an additional peptidic sequence, despite their high structural relevance for cell wall crosslinks. | ||
| Fig. 1 Natural lipid I (1) and biosynthetically derived lipid II (2), key building blocks of bacterial cell wall biosynthesis: its farnesyl derivatives 3, 4 as improved agents for functional studies and interpeptidic analogs 5 and 6, bearing interpeptidic sequences characteristic for S. aureus as well as E. faecalis and S. pneumoniae. | ||
To resolve the disadvantageous physical properties, a new group of lipid I analogs has been introduced, where the unfavorable authentic undecaprenyl side chain has been shortened, leading to derivatives, such as farnesyl congeners 3 and 4.8 In recent years it has become more and more clear, that these truncated analogs represent functional surrogates for cell wall biosynthesis.9 Consequently, the development of efficient synthetic routes towards these simplified versions is of high interest for functional studies. While several synthetic procedures were published,10–12 these existing routes still leave ample room for further improvement, with respect to overall yield, synthetic efficiency, experimental documentation, robustness and purity of final compounds, as well as application to strain dependent peptide modifications.
Herein we report in full details our considerations, strategies and tactics for development of a novel synthetic sequence to truncated lipid I and II analogs.13 The modular route is based on a novel solid phase approach to the pentapeptide chain, which proceeds with full stereochemical control, an improved synthesis of the pyrophosphate fragment and a chemoenzymatic attachment of the second carbohydrate (GlcNAc). Application of these routes allowed for a concise synthesis of farnesyl analogs 3 and 4, which were obtained with unprecedented purity, revealing excellent spectroscopic resolution. Furthermore, this sequence could be adopted for synthesis of novel pentaglycine derivative 5 specific for Staphylococcus aureus (S. aureus), as well as new analog 6 that is characteristic for Enterococcus faecalis (E. faecalis) and Streptococcus pneumoniae (S. pneumoniae).
Results and discussion
Synthetic strategy
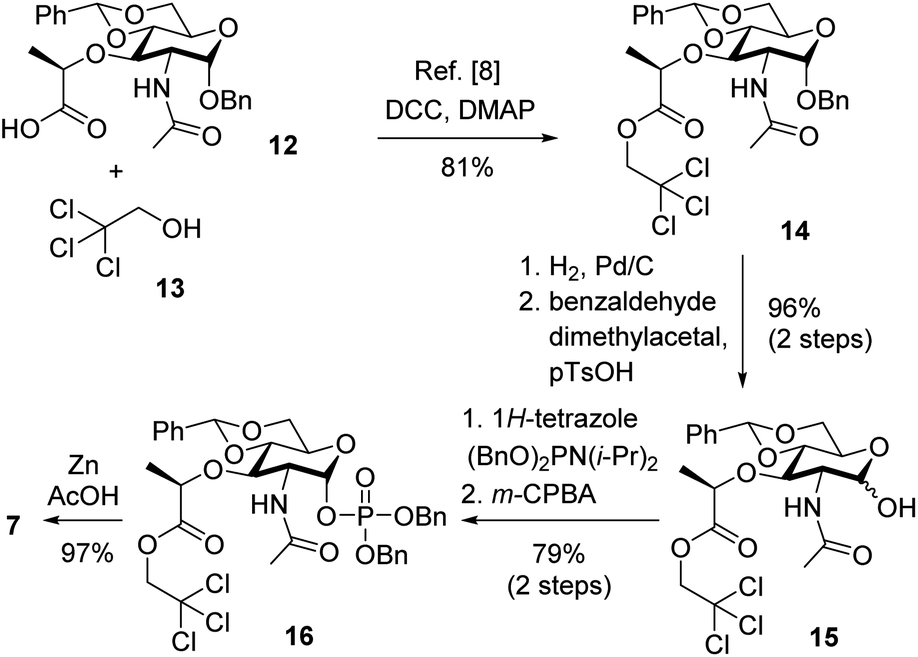
As shown in Fig. 1, the architecture of the targeted lipid analogs is characterized by three structurally distinctively different subunits, i.e. a pentapeptide, consisting of L-Ala–γ-D-Glu–L-Lys–D-Ala–D-Ala sequence, in combination with a muramic acid carbohydrate that is linked to a farnesyl chain via a pyrophosphate bridge. Consequently, our synthetic approach was based on initial formation of these three components in suitably protected form, revealing monosaccharide 7, farnesyl subunit 8 and peptide 11. In a similar fashion to a reported route,7 these should then be condensed by amide formation and pyrophosphate coupling. Final attachment of the second sugar (GlcNAc) may then be possible, bioenzymatically.14 Notably, such a strategy would be highly modular and be easily adopted to pathogen specific analogs, such as 5 or 6.While the monosaccharide 7 (Scheme 1) should be accessible by optimizing a previously reported procedure,15 synthesis of known pentapeptide 9 was reconsidered. In detail, it became apparent during this study that a more effective approach would involve a solid phase approach of only the tetrapeptide 11 and subsequent attachment of the final D-Ala building block 10 in solution phase, in contrast to a previous solid phase synthesis of full pentapeptide 9. Since the pentapeptide should include silyl protecting groups allowing for a final global deprotection step, the pentapeptide was initially synthesized via the solid phase approach, but it was found that the silyl protection of the D-Ala residue would lead to epimerization at C-4 (Scheme 4). Furthermore, a 2-chlorotrityl chloride (2CTC) resin was chosen to allow for facile peptide cleavage under mild conditions (hexafluoroisopropanol, HFIP), which would also be compatible with the silyl protecting groups selected for the two terminal carboxylates (D-Ala, L-Glu) and the side chain amine of lysine. This identical choice of protecting groups would also enable a joint removal at a late stage of the synthesis. Peptide coupling should then be realized using the respective synthesized silyl and Fmoc protected amino acids. Furthermore, synthesis of the peptide was planned in such a way that only slight modifications would allow access to stem-specific interpeptidic bridges. As in previous procedures and in view of the instability of an allylic pyrophosphate moiety, a late stage carbodiimidazole mediated coupling was envisaged for introduction of farnesyl phosphate 8.
 | ||
| Scheme 1 Modular three fragment retrosynthetic approach towards 3-lipid I analogs 3 and 4: novel solid phase based synthesis of pentapeptide. | ||
Synthesis of carbohydrate fragment 7
The final synthesis towards carbohydrate fragment 7 is shown in Scheme 2. While the overall route was adopted from previous described procedures,7 more technical modifications were implemented to improve the practicability and raise the overall yield. In detail, following a known procedure commercial carbohydrate carboxylate 12 was protected as trichloroethyl ester.15 Introduction of this ester proved to be more reliable as compared to introduction of a trimethylsilyl ester. Resulting carbohydrate 14 was then exposed to palladium in a hydrogen atmosphere to cleave the anomeric benzyl group, which also led to partial removal of the acetal group. While in principle a protocol for reattachment has been reported, which involves treatment with benzaldehyde dimethylacetal and catalytic amounts of p-toluenesulfonic acid in DMF,15 this method proved to be unreliable in our hands and modifications were evaluated. Finally, a solvent exchange to more easily removable MeCN and a slight increase of catalyst loading (0.1 to 0.3 equiv.) gave almost quantitative yields over two steps (96%) and a shortened reaction time (4 h). Additionally, the amount of benzaldehyde dimethylacetal may be dramatically reduced to 1.5 equiv. which further facilitates product isolation. Resulting alcohol 15 resided as a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β mixture,15 while these two anomers may be separated by HPLC giving pure α-anomer and partially enriched β-anomer, which was prone to reequilibration. Fully resolved NMR data for the α-anomer unequivocally confirmed the stereochemical assignment, as shown. The 3J-coupling constant for the respective proton was found to be 3.6 Hz, which points to a 3Jae coupling to the neighbouring axial proton. Furthermore the signal for the anomeric carbon atom was found to be at 92.3 ppm.
:1 α:β mixture,15 while these two anomers may be separated by HPLC giving pure α-anomer and partially enriched β-anomer, which was prone to reequilibration. Fully resolved NMR data for the α-anomer unequivocally confirmed the stereochemical assignment, as shown. The 3J-coupling constant for the respective proton was found to be 3.6 Hz, which points to a 3Jae coupling to the neighbouring axial proton. Furthermore the signal for the anomeric carbon atom was found to be at 92.3 ppm.
 | ||
| Scheme 2 Improved synthesis of protected saccharide building block 7.15 | ||
A nucleophilic introduction of the anomeric phosphate was then carried out with the anomeric mixture. Following a reported procedure,6,15,16 this involved treatment with dibenzyl N,N-diisopropylphosphoramidite and 1H-tetrazole, giving the corresponding phosphite (structure not shown). This presumably labile intermediate5 was then directly oxidized with m-CPBA to carbohydrate 16. In agreement with a previous observation,6,15,16 the α-anomer was obtained exclusively based on analysis of the fully resolved NMR data. In detail, assignment of the α-anomer was based on a 3J-coupling constant of 3.2 Hz and the shift of the respective carbon signal, located at 96.6 ppm. Presumably, the high selectivity arises from selective capture of the more reactive α-hydroxyl group.16 As an alternative to tetrazole, also more readily available 1,2,4-triazole was evaluated.5,16,17 However, this did not improve the process, presumably due to reduced acidity. Finally, completion of the synthesis involved treatment with zinc powder in acidic condition, rendering the desired monosaccharide scaffold 7 despite the harsh conditions in an excellent yield of 97%. This approach proved superior to likewise evaluated alternatives as it provides comparably good yields without involving an expensive catalyst like (Cp)2TiCl.18 Overall, carbohydrate fragment 7 was synthesized with a yield of 60% over six steps from commercially available compound 12, which compared favorably to the previous route.15 Furthermore, the two step-conversions of 14 to 15 and of 15 to 16 may be carried out in one-pot reactions, which further add to the efficiency of the process.
Synthesis of farnesyl fragment
Known farnesyl building block 8 was efficiently prepared by a method published by the group of Wessjohann,11 that is based on a one-pot conversion of the corresponding alcohol with tetrabutylammonium hydrogen phosphate (TBAP) and trichloroacetonitrile (TCA) giving the required phosphate in a reliable manner with useful yield (71%).Synthesis of amino acid building blocks
To synthesize the stem peptides of targeted lipid analogs, several amino acid building blocks were required. While some of them were commercially available, glutamic acid derivative 17, protected alanines 26 and 33 as well as lysine 31 and glycine 35 had to be synthesized in suitably protected form for the projected sequences.As shown in Scheme 3a, Fmoc–D-Glu(OH)–TMSE (17) was initially targeted by a reported procedure7 from 18 which in turn was accessible by TMSE protection from the corresponding commercial α-carboxylate.7 However, selective hydrogenolysis of the benzyl group proved more challenging than expected, leading to various degrees of concomitant removal of also the more stable Fmoc-group under conventional hydrogenation conditions (23% yield of 17). While this outcome could only be partially remedied (up to 41%) by careful reaction monitoring, it was found that addition of CaCl2 could increase the lifetime of the Fmoc protective group, presumably due to prevention of basic conditions.19 In detail, addition of CaCl2 to a final concentration of 0.25 M had a positive effect and desired product was isolated in 54% yield. Finally, expected stability of targeted 17 towards a slightly acidic environment and the fact that benzyl protecting groups are acid labile motivated addition of 100 μL (0.38 equiv.) of acetic acid and the yield was further increased to 84%. While cooling and solvent exchange were also evaluated, in the end it turned out that a higher yielding, preparative less laborious and more economical strategy could be implemented, which involved selective cleavage of the tBu-ester of 19. This ester was readily available by TMSE-protection from the corresponding commercial α-carboxylate by a Steglich esterification (96%, not shown, see ESI section†). As shown in the respective table, a variety of reagents were evaluated to effectuate specific removal of this ester without affecting the TMSE group. Various acidic conditions led to unfavorable or low selectivity (entries 1 and 2).20 Also, use of triethylsilane previously used as a carbocation scavenger in the deprotection of tert-butyl esters,21 did not circumvent this lack of selectivity (entry 3). A reported procedure using an excess of ZnBr2 furnished only compound 20 (entry 4) and using a previously described aqueous phosphoric acid procedure as much milder alternative showed no reaction at all (entry 5).22 Finally, an excess of TMSOTf and 2,6-lutidine was able to unmask the side chain carboxylic acid in a selective manner rendering glutamic acid building block 17 in 97% yield (entry 6).23 Notably, no column chromatography was necessary for this step and simple extractions were adequate to give the desired compound in high purity. This novel two-step procedure compared favorably to the reported method,7 with respect to yield, cost of starting material and reagent, and does not require a chromatography in the second step (Table 1).7
 | ||
| Scheme 3 Efficient synthesis of required amino acid building blocks 17 (part a), 26 (part b), 31, 33 and 35 (part c). | ||
 | ||
| Scheme 4 Solid phase synthesis of pentapeptide 39 and epimerization during TMSE protection of terminal D-Ala. Reagents and conditions: (a) HO–D-Ala–Fmoc (41), DIPEA (b) Ac2O, N-methylimidazole, DMF (c), 20% piperidine/DMF (d) HBTU, HOBt, amino acid, DIPEA, DMF (e) 20% HFIP/DCM. | ||
| Entry | Conditions | 17/20 | Yield | |
|---|---|---|---|---|
| 1 | TFA/DCM (1:1) |
1:4 |
n.d. | 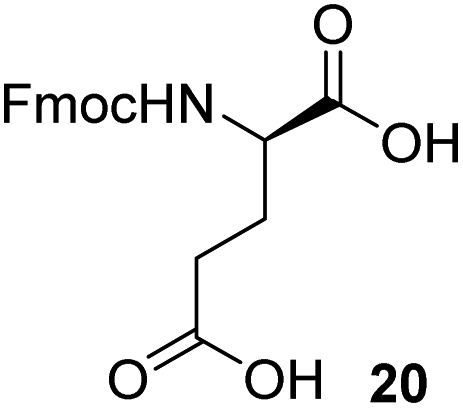 |
| 2 | TFA/DCM (1:9) |
∼1:1 |
n.d. | |
| 3 | Et3SiH, TFA/DCM (1:4) |
∼1:1 |
n.d. | |
| 4 | ZnBr2, DCM | <1:20 |
n.d. | |
| 5 | H3PO4/DCM (1:1) |
— | — | |
| 6 | TMSOTf, 2,6-lutidine | >20:1 |
97% |
The amino acid building block H2N–D-Ala–TMSE (26) in turn was initially synthesized in a two-step procedure from commercial 21 involving a TMSE-esterification and subsequent Fmoc cleavage (Scheme 3b). While the former conversion could be realized in high yield using the Steglich procedure (96%), conventional protocols using piperidine/DMF gave only moderate amounts of desired amine 26, presumably, due to loss of material during work up. Although various conditions like extractions, NEt3 addition during chromatography, different solvents or alternative chromatography materials (aluminum oxide, different pH, silica) were employed, the overall yield could not be improved beyond 52%. Also, a recently reported procedure using polymer-bound piperazine 24 (ref. 24) did not show any conversion. Finally, highest yield (87%) was obtained by a unconventional procedure reported by Gioia et al.25 involving NEt3 in ionic liquid 25 as solvent, which allowed for facile purification. However, elongated reaction times (2 d) required for full conversion was considered as a too disadvantageous leading to the design and implementation of an alternative approach. This involved Boc protected D-Ala 27, which was esterified as a TMSE ester in quantitative amounts towards 28,26 followed by deprotection of the Boc group using a 25% TFA/DCM mixture, which proceeded with excellent selectivity towards carbamate cleavage. After evaporation of all volatiles, amine 26 was synthesized in a quantitative yield over two steps without the need of column chromatography.
Finally, TMSO-carbonyl (=Teoc) protected lysine (31) and alanine (33), as required for synthesis of interpeptidic analogs (see below), were best prepared from the native amino acids using succinimide reagent 30 with NEt3 in a solvent mixture of 1,4-dioxane/water.7
Synthesis of pentapeptide 44
A reliable, scalable and high yielding synthesis of the required pentapeptides presents one of the most crucial aspects in the development of lipid I/II analogs and it was decided that a solid phase approach would best meet these requirements. Given apparent limitations of existing solid phase approaches,7 a de novo design was pursued within this project.For solid phase a 2-chlorotrityl-chloride (2CTC) 36 resin was chosen in order to enable a facile cleavage under mild conditions (hexafluoroisopropanol, HFIP),27 which was also expected to be compatible with the silyl protecting groups that had been chosen for the carboxylates of D-alanine 26, D-glutamic acid 17 as well as the side chain amine of L-lysine 31. After some experimentation, protocols for resin functionalization, deprotection, coupling, washing and cleavage were developed and then strictly followed. Optimal conditions involved loading with 0.5 equiv. of the first amino acid in presence of DIPEA in DMF (1 h, rt), capping of the unreacted CTC-functionalities with acetic anhydride and N-methylimidazole in DMF and stepwise elaboration of the peptide involving Fmoc cleavage of the solid bound material with 20% piperidine/DMF and HBTU/HOBt mediated attachment of the next amino acid and final cleavage of the fully elaborated peptide from the resin with 20% HFIP in DCM.
Following this sequence of repetitive deprotection and coupling with amino acid building blocks 41, 31, 17 and 42 gave resin bound pentapeptide 38. Final treatment with HFIP and precipitation in Et2O liberated pentapeptide 39 in high yield over these 11 steps (59%). The overall process may be carried out in less than 15 h, which adds to the efficiency of this solid phase approach.
At this stage, the terminal carboxylate had to be protected as a TMSE ester towards 40 to allow for selective attachment of the sugar fragment at a later stage of the synthesis (see below). However, in contrast to previous observations with amino acid building blocks, Steglich esterification of 39 with 2-(trimethylsilyl)ethanol (22), suffered from long reaction times, incomplete conversions and epimerization (see Table 2, entry 1). Possibly, this may arise from stabilization of the generated O-acylisourea by formation of various hydrogen bonds with parts of the peptide, which was supported by mass spectrometric analysis. Alternatively, also an N-acylurea byproduct may be involved. Also, other esterification methods were considered.28,29 However, they likewise resulted in epimerization (entries 2 and 3) or did not lead to any conversion at all (entry 4).28,29
| Entry | Conditions | 40/(4epi)-40 |
|---|---|---|
| 1 | 22 (1.3 equiv.), DCC, [DMAP] | ∼1:1 |
| 2 | 22 (2.0 equiv.), PyBOP/HOBt, DIPEA | ∼1:1 (42%) |
| 3 | 22 (10.0 equiv.), TFFH, DIPEA | ∼1:1 |
| 4 | 22 (2.0 equiv.), MNBA, [DMAP], DIPEA | — |
Separate analysis of the two products, which may be separated by HPLC (see chromatogram), revealed identical molecular ion MS data, but small NMR differences, especially in the region of the terminal stereogenic center. Also, optical rotation values were different (see Experimental section for details), which supports an epimerization. Presumably, this may result from decreased acidity of the respective activated ester intermediate, leading to deprotonation and subsequent scrambling of the α-center. Alternatively, also an oxazole intermediate may be possible.30
At this stage it was rationalized that this unfavourable issue may be resolved by stronger nucleophilicity of amines as compared to alcohols and therefore coupling of free amino acid building block H2N–D-Ala–TMSE (26) in solution was evaluated. Consequently, instead of the full pentapeptide 39 only tetrapeptide 43 was targeted. As shown in Scheme 5, it was prepared on solid phase in an analogous fashion as before. However, in contrast to the previous sequence, the terminal D-alanine was attached after liberation from solid support by coupling in solution with already TMSE-protected D-alanine 26. In detail, after cleavage from the solid phase, tetrapeptide 43 was obtained in excellent 82% yield over nine steps starting from resin 36. Subsequent attachment of final amino acid D-Ala 26, already incorporating the desired silyl protection, was carried out with PyBOP/HOBt. Gratifyingly, this coupling proceeded not only in high yield (95%) but also without loss of stereochemical purity, as unambiguously proven by HPLC analysis. Finally, Fmoc removal gave desired pentapeptide 40 in 76% yield. In total, this novel route giving key stem peptide 40 in high purity and yield (59%) over eleven steps, proved reliable, scalable, fast and compares favorably to previous lengthy solution phase sequences10,16 or solid phase procedures giving low yield (15%) and limited purity.7
 | ||
| Scheme 5 Stereoselective preparation of protected pentapeptide 40 by solid phase synthesis of tetrapeptide 43 and attachment of the final D-Ala residue in solution. Reagents and conditions: (a) HO–D-Ala–Fmoc (41), DIPEA (b) Ac2O, N-methylimidazole, DMF (c), 20% piperidine/DMF (d) HBTU, HOBt, amino acid, DIPEA, DMF (e) 20% HFIP/DCM. | ||
Synthesis of decapeptide 45
At this stage our efforts were directed to evaluate the generality and adaptability of this solid phase peptide approach also to pathogen specific analogs. As a first target we chose decapeptide 45 which is characteristic for S. aureus. Synthesis of 45 which required access to the characteristic pentaglycine side chain in suitably protected form (48). As shown in Scheme 6, our first approach was to introduce a Teoc protecting group to commercially available pentaglycine 46, as this silyl protecting group would allow for a late stage attachment of the interpeptidic side chain (vide infra). | ||
| Scheme 6 Unsuccessful direct protection of pentaglycine 46. | ||
However, desired product 48 could not be generated under a variety of reaction conditions, presumably due to the very poor solubility of 46 in a variety of solvents. In contrast, Teoc protection of glycine proceeded uneventfully (Scheme 3). Given the reliability and robustness of our novel solid phase approach, we therefore planned to attach the pentaglycine moiety in a stepwise fashion on the resin. Gratifyingly, this could be realized. Following the sequence shown in Scheme 7 this involved again a final attachment of the terminal D-Ala in solution phase. In detail, the successful solid-phase route relied on initial attachment of the penultimate D-Ala amino acid to the chlorotrityl resin and stepwise elaboration using Fmoc protected amino acids. However, in contrast to the previous sequence towards pentapeptide 40, a modified lysine building block 49 was utilized, now bearing the Fmoc group at the ε-amine and an orthogonal allyloxycarbonyl (alloc) moiety for the α-amine. After coupling of this lysine derivative, the Fmoc group in the side chain was then removed first. Four glycine residues (50) were then attached in an iterative fashion using standard coupling and deprotection protocols, before the final glycine was introduced as its silyl protected building block 35. At this stage, the success of this procedure was verified, by a test cleavage revealing heptapeptide 52, as expected. After this confirmation of complete incorporation of the full pentaglycine residue, solid bound peptide 51 was further elaborated. This involved cleavage of the alloc protecting group with Pd(PPh)4 and phenylsilane (PhSiH3), and attachment of amino acids 17 and 42. Finally, the resulting nonapeptide was cleaved from the resin giving peptide 54 in excellent yield (83%) over these nineteen steps on the solid phase support. In the end, remaining (TMSE) protected D-alanine (26) was coupled in solution, before the Fmoc group was cleaved using piperidine. In total, desired decapeptide 45 was obtained in high overall yield (40%) over 21 steps starting from commercial resin 36.
 | ||
| Scheme 7 Solid phase synthesis of decapeptide 45 characteristic for S. aureus. Reagents and conditions: (a) HO–D-Ala–Fmoc (41), DIPEA (b) Ac2O, N-methylimidazole, DMF (c), 20% piperidine/DMF (d) HBTU, HOBt, amino acid, DIPEA, DMF (e) 20% HFIP/DCM. | ||
Synthesis of heptapeptide 55
In order to further evaluate the generality of this solid phase sequence, we then opted for synthesis of heptapeptide 55, incorporating an L-Ala–L-Ala side chain that is characteristic for E. faecalis and S. pneumoniae. Accordingly, our general solid phase approach was modified as shown in Scheme 8. In detail, this involved again modified lysine building block 49, bearing a terminal Fmoc group and an orthogonal alloc moiety at the α-amine. After selective Fmoc cleavage, the two L-Ala fragments could be successfully introduced, before the α-amine group was liberated in an orthogonal fashion [Pd(PPh)4; (PhSiH3)]. Amino acids 17 and 42 were then attached to the peptidic backbone. After removal from the solid phase, desired hexapeptide 56 was obtained in high overall yield (75%), demonstrating the generality of this solid phase sequence. Finally, completion of the synthesis of 55 involved attachment of the terminal D-Ala motif and Fmoc removal giving desired branched heptapeptide in high yield (51%) over 15 steps.8 | ||
| Scheme 8 Solid phase synthesis of heptapeptide 55 characteristic for E. faecalis and S. pneumoniae. Reagents and conditions: (b) 20% piperidine/DMF, (c) HBTU, HOBt, amino acid, DIPEA, DMF, (d) 20% HFIP/DCM. | ||
Fragment coupling and completion of the syntheses of 3-lipid analogs
For fragment union, our strategy relied on first coupling the peptide fragments with the monosaccharide building block and subsequently attaching the farnesyl phosphate subunit. As shown in Scheme 9, we first targeted original lipid I analog 3.6,14,31,32 Accordingly, respective peptide 44 was connected to carbohydrate 7 with PyBOP/HOBt giving glycopeptide precursor 60 in high yield (81%). In contrast to a previous report,15 a joint removal of the benzyl groups and the acetal moiety using hydrogenolysis could not be realized. Even after increase of the reaction time from reported 30 min to 5 h, only the free phosphate was observed, without cleavage of the acetal group. Apparently, such a joint removal appears challenging, as also Arimoto et al. reported a two-step procedure for their depsi-lipid I analog.10 In an analogous fashion for 60, the benzyl group was first removed by hydrogenolysis, before the acetal was cleaved using acidic conditions giving desired deprotected 61. Pyrophosphate linkage of farnesyl phosphate 8 and phosphate 61 was then achieved using carbonyldiimidazole coupling.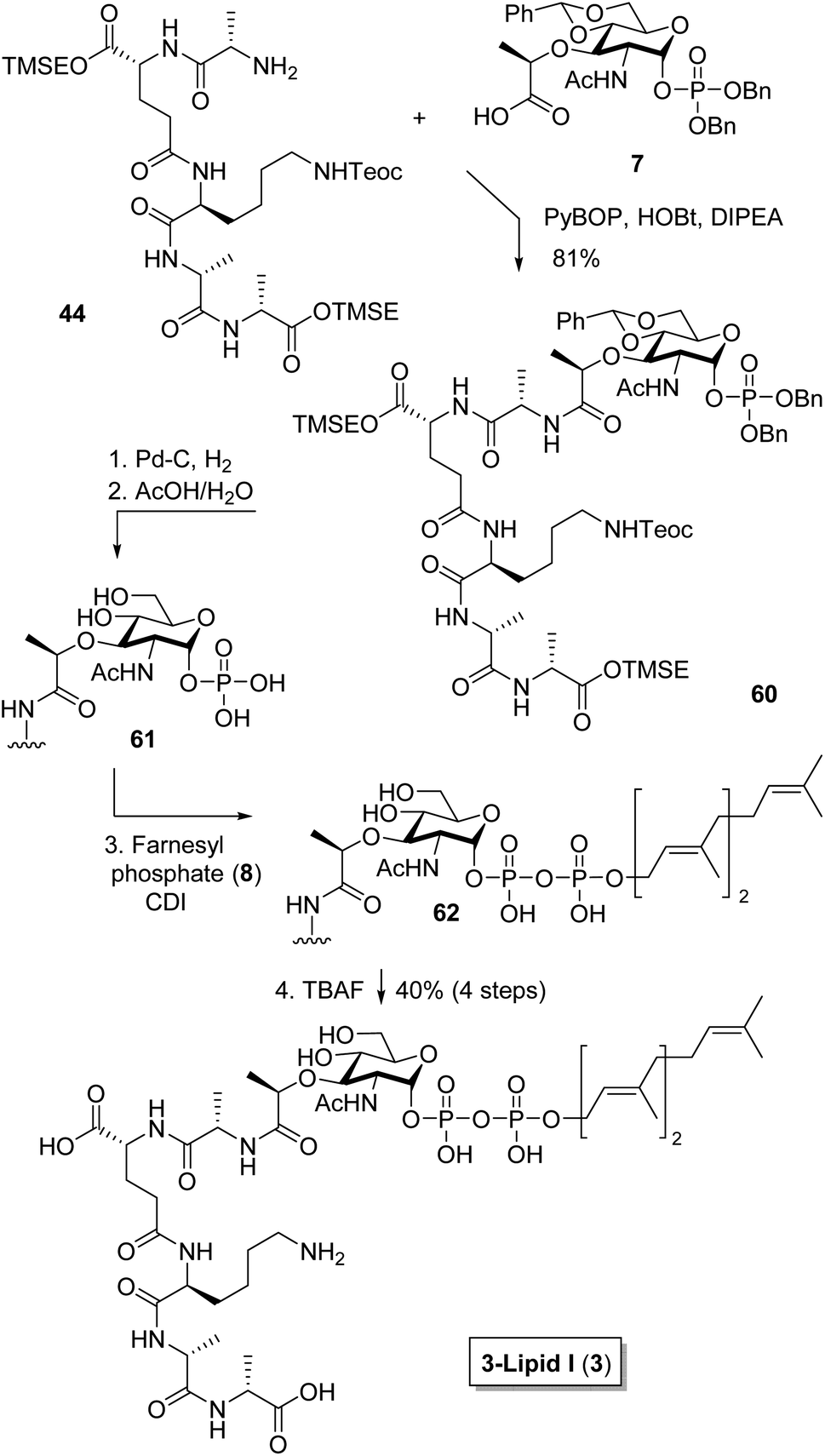 | ||
| Scheme 9 Completion of the total synthesis of 3-lipid I. | ||
Finally, all silyl protecting groups could be cleaved in a joint fashion using tetrabutylammonium fluoride, proving the success of our synthetic design. For full purification of farnesyl lipid I analog 3, a three step method was developed. First, gel filtration with Sephadex® LH-20 was applied, followed by ion-exchange chromatography (Dowex® 50WX8) to substitute residual tetrabutylammonium counterions with ammonium, which otherwise proved difficult to remove by HPLC. Final HPLC separation yielded highly pure farnesyl lipid I analog 3 in 40% yield over four steps starting from glycopeptide precursor 60. In total, farnesyl lipid I 3 was obtained in pure form in 16 steps in the longest linear sequence from commercially available HO–D-Ala–Fmoc (41) with an excellent overall yield of 19%, which compared favorably to the previous synthetic route starting from Sasrin resin–D-Ala–Fmoc 63.7,15,31 The sequence proved well scalable and highly pure 3-lipid I (3) was obtained.
Encouraged by the successful final steps towards farnesyl lipid I analog 3, an analogous three component sequence was applied to complete the total synthesis of S. aureus lipid I analog 5. As shown in Scheme 10, characteristic decapeptide 45 was coupled to carbohydrate component 7 using a previously established PyBOP/HOBt procedure to give glycopeptide precursor 64 in 86% yield, which was even slightly higher than the yield after coupling with the stem pentapeptide 44 (81%). Caused by the longer peptide chain, solubility in solvents like EtOAc and Et2O decreased, which can cause problems in handling and reactions but turned out to be advantageous here, since precipitation led to more facile purification. Removal of the phosphate benzyl groups by hydrogenolysis, acidic cleavage of the acetal and CDI mediated pyrophosphate coupling with farnesyl phosphate 8 gave silyl protected 65, which was semi-purified using Sephadex® LH-20. Again, final global deprotection using TBAF freed the peptide from its silyl groups and farnesyl lipid I analog 5, containing the S. aureus pentaglycine, was gained in pure form after utilization of the three step purification developed above, consisting out of gel filtration with Sephadex® LH-20, ion-exchange chromatography (Dowex® 50WX8) and a final HPLC purification. Overall, lipid I analog 5 was obtained in an overall yield of 8% in its longest linear sequence over 26 steps starting from HO–D-Ala–Fmoc (41).
 | ||
| Scheme 10 Completion of the total synthesis of 3-lipid I (S. aureus). | ||
The generality of this final coupling strategy was then further confirmed by synthesis of lipid I analog 6 containing the interpeptidic sequence characteristic for pathogen E. faecalis and S. pneumoniae. As shown in Scheme 11, the PyBOP/HOBt mediated amide coupling between the respective peptidic fragment and the sugar moiety proceeded again with high efficiency. In detail, glycopeptide precursor 66 was obtained in essentially quantitative yield, following a slightly improved procedure. This involved cold diethyl ether as solvent, which does dissolve the reactants but may not solubilize the product. Therefore, simple filtration gave glycopeptide precursor 66 in high purity after washing. Finally, farnesyl lipid I analog 6 was obtained in a straightforward fashion using the four-step sequence established above, demonstrating the general utility of this approach.8 In total, this novel lipid analog was obtained in 20 steps and likewise very high overall yield (25%) and high purity.
 | ||
| Scheme 11 Completion of the total synthesis of 3-lipid I (E. faecalis, S. pneumoniae). | ||
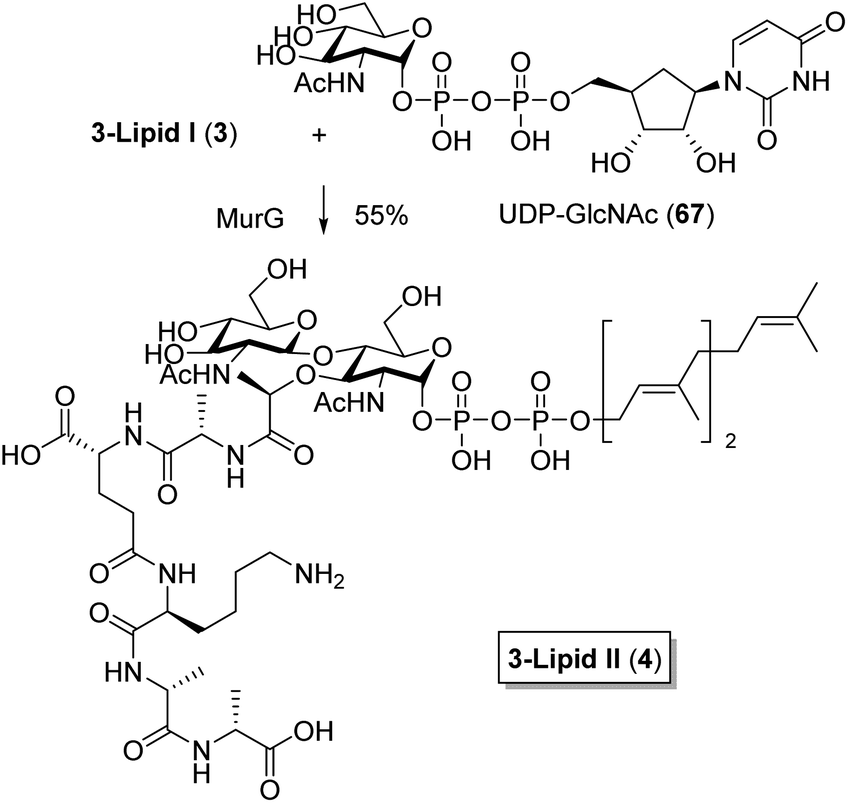
Finally we confirmed the biochemical compatibility of the shortened farnesyl group by conversion of 3-lipid I (3) to 3-lipid II (4). As shown in Scheme 12, this previously reported conversion,31 involves MurG catalyzed chemoenzymatic attachment of β-1-4-linked N-acetylglucosamine. It was carried out with 1 mg of 3 and lipid II analog 4 was obtained in pure form with a useful yield of 55%, which represented a sufficient amount for characterization including the assignment of NMR signals. The generality of this approach was further confirmed by also converting an analytic sample of 5 to the corresponding lipid II analog (not shown).
 | ||
| Scheme 12 Chemoenzymatic conversion of 3-lipid I (3) to 3-lipid II (4) using UDP–GlcNAc (66). | ||
Initial NMR studies
Finally, the NMR resolution of all new analogs 3–6 and usefulness for solution NMR measurements were analysed. Gratifyingly, optimum 1H signal dispersion was realised in D2O for all shortened lipid compounds at the highest available field strengths (500 and 700 MHz). As shown for the NMR spectra in the ESI section† excellent resolution was obtained for all lipid-analogs, allowing for complete assignment of all 1H and 13C resonances based on two dimensional techniques (see ESI section: Table S1†). Furthermore, farnesyl lipid I analog 3 was analyzed in more detail, including temperature dependent NMR measurements, which may give insights into effects like hydrogen bonding, stability of the compound and conformational changes.33 As shown in Fig. S1 (see ESI section),† there are detectable differences in the chemical shifts at various temperatures. With increasing temperature, a downfield shift was observed. Additionally, broadening of the signals was seen especially for the temperature shift from 278 K to 282 K. If the temperature was increased further, the signals got sharp again. 31P NMR spectra were recorded at the same temperatures as well (Fig. S2†). These measurements showed an even more interesting effect. While the signals for the pyrophosphate were not visible at 278 K, sharp signals appeared at 282 K, which broadened again and vanished completely if the temperature was increased further. Surprisingly, this observation is the opposite of the 1H spectra in which the signals at 282 K were the most broadened. The changes in the signals can be caused by various effects like conformational changes, exchange processes or temperature dependent hydrogen bonding. Possibly, the phosphate counter ions also have an effect of causing the disappearing signals, especially at lower temperatures. During these measurements it was also noted that synthesized farnesyl lipid I analog 3 was stable at elevated temperatures, as the 1H spectra at 363 K did not showed significant degradation. These initial NMR studies make it now even more promising to apply these analogs as chemical tools for structural as well as functional studies.Conclusion
In summary, an optimized, efficient, scalable and reproducible route to farnesyl lipid I analogs 3, 5 and 6 was developed. For this purpose, syntheses of the three components being carbohydrate 7, peptide 44 and farnesyl phosphate 8 were closely examined. Regarding carbohydrate 7, various technical modifications and optimizations, for example by exchange of the reaction solvent or by adjusting the reagent equivalents, led to a considerable increase of the overall yield from literature reported 41% to 60% over six steps (Scheme 2).15 Application of a recently reported one-pot procedure for the conversion of alcohols to their phosphates11 allowed to further simplify the route towards the precious cell wall precursors, giving required farnesyl phosphate 8 from farnesol in 71% yield using only one step.Considerable efforts were then invested before a novel, efficient and scalable solid phase synthesis of the characteristic stem pentapeptide was developed, which compares favorably to previous lengthy solution phase sequences as well as low reported yield (15%) for a solid phase approach.6,7,10,15,16,36 This optimized route relied on preparation of the tetrapeptide HO–D-Ala–L-Lys(N-Teoc)–D-γ-Glu(O-TMSE)–L-Ala–Fmoc (43) using 2-chlorotrityl chloride (2CTC) solid phase support and subsequent attachment of the remaining silyl protected D-Ala amino acid (26) in solution phase, which turned out to be crucial to avoid an unfavorable epimerization. Following this novel sequence synthesis of pentapeptide 44, was obtained in an excellent yield of 59% over 11 steps. This route was then further elaborated and applied for synthesis of pathogen specific interpeptidic analogs, i.e. the pentaglycine variation found in S. aureus and the L-Ala–L-Ala modification observed in E. faecalis and S. pneumoniae, were targeted, which are considered most important among the interpeptidic variations described for specific pathogens.37 In detail, a novel solid phase sequence was designed to allow for the introduction of interpeptidic modifications, which involved an alternative lysine building bearing an ε-Fmoc and an α-alloc group to allowing for an orthogonal strategy. Overall this route enabled the synthesis of required decapeptide 45 in 40% yield over 21 steps and necessary heptapeptide 55 in 51% yield over 15 steps, demonstrating the modularity and true applicability of this sequence to various pathogen specific analogs.
After coupling of the fragments and deprotection, farnesyl lipid I analog 3 was purified with a newly developed three step purification including gel filtration (Sephadex® LH-20), ion exchange chromatography (Dowex® 50WX8) and HPLC separation giving the desired product 3 in high purity over 16 steps (longest linear sequence, 19% yield). This sequence proved well scalable and a batch of 11 mg lipid I analog 3 was readily obtained in high purity. Following analogous three component coupling sequences then enabled completion of the first total syntheses of S. aureus interpeptidic analog 5 (8% over 26 steps longest linear sequence) and of E. faecalis and S. pneumoniae analog 6 (25% over 20 steps longest linear sequence).
Furthermore, shortened lipid I analogs were accepted as substrates by the natural glycosyltransferase MurG, which catalyses attachment of the β-1-4-linked N-acetylglucosamine, demonstrating the biochemical compatibility of the shortened side chain, which also allowed preparative synthesis of 3-lipid II analog 4.
Finally, detailed NMR analyses of all lipid I and lipid II analogs revealed high spectroscopic resolution in D2O and useful stability at various temperatures, demonstrating that they present valuable tool compounds for structural studies and may now be used to address biochemical questions and to gain new structural insights into bacterial cell wall biosynthesis. Present studies along these lines will be reported in due course.
Experimental
General synthetic procedures
All reactions were performed under an atmosphere of argon in flame-dried glassware which had been cooled under argon unless stated otherwise. All flasks were equipped with rubber septa and reactants were handled using standard Schlenk techniques. Temperatures above rt (23 °C) refer to oil bath temperatures which were controlled by a temperature modulator. For cooling, the following baths were used: acetone/dry ice (−78 °C), ACN/dry ice (−40 °C), water/ice (0 °C). All reagents were purchased from commercial suppliers (Sigma-Aldrich, Alfa Aesar, TCI, Iris Biotech, Acros) in the highest purity grade available and used without further purification unless otherwise stated. Anhydrous solvents (THF, Et2O, DCM, MeCN and toluene) were freshly obtained from a solvent drying system MB SPS-800 (MBraun) and stored over molecular sieves (3 or 4 Å). Reactions were monitored via TLC on silica gel 60 F254 precoated plates (0.2 mm SiO2, Macherey-Nagel) and visualized using UV light and/or staining with a solution of CAM (1 g Ce(SO4)2, 2.5 g (NH4)6Mo7O24, 8 mL conc. H2SO4 in 100 mL H2O) and subsequent heating. For column chromatography, silica gel (pore size 60 Å, 40–63 μm) obtained from Merck or Aldrich was used. Compounds were eluated using the stated mixtures under positive pressure of nitrogen or air. Solvents for column chromatography were distilled prior to use. For ion exchange chromatography Dowex® 50WX8, 100–200 mesh by Acros was used. Gel permeation chromatography was performed using Sephadex® LH-20 purchased from GE Healthcare.Optical rotations were measured with a PerkinElmer 341 or a Anton Paar MCP 150 polarimeter in a 10 mm cuvette and are uncorrected. All NMR spectra were recorded on Bruker spectrometers at the University of Bonn under supervision of Dr Senada Nozinovic with operating frequencies of 125 (13C), 150 (13C), 175 (13C), 400 (1H), 500 (1H) and 700 MHz (1H) in deuterated solvents obtained from Deutero, Carl Roth, VWR and Sigma-Aldrich. Spectra were measured at room temperature unless otherwise stated and chemical shifts are reported in ppm relative to (Me)4Si (δ = 0 ppm) and were calibrated to the residual signal of undeuterated solvents. Data for 1H NMR spectra are reported as follows: chemical shift (multiplicity, coupling constants in hertz, number of hydrogens, assignments; atom numbering for 1H and 13C signals can be found in the respective drawings in the ESI section†). Abbreviations are as follows: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), m (multiplet), br (broad), pt (pseudo triplet). Mass spectra (MS) and high resolution-mass spectra (HR-MS) were recorded on the documented systems in Table S1† at the University of Bonn under supervision of Dr Marianne Engeser. Ionization processes and mol peaks were given (Tables 3 and 4).
| Name | Manufacturer | Ionization type |
|---|---|---|
| MAT 95 XL | Thermo Finnigan (Bremen) | EI |
| MAT 90 | Thermo Finnigan (Bremen) | EI, LIFDI, CI, FAB |
| MALDI autoflex II TOF/TOF | Bruker Daltonik (Bremen) | MALDI |
| micrOTOF-Q | Bruker Daltonik (Bremen) | ESI, APCI, nano-ESI, MS/MS, LC-MS, DC-MS |
| Orbitrap XL | Thermo Fisher Scientific (Bremen) | ESI, APCI, APPI, nano-ESI |
| Apex IV FT-ICR | Bruker Daltonik (Bremen) | ESI, nano-ESI, MALDI, EI, CI |
| System A (analytical) | System A (preparative) | |
|---|---|---|
| Series | PLATINblue | Smartline |
| Pumps | Binary, HPG P1 system, 5 mL | Binary, S-1800, 100 mL |
| Pressure | 1000 bar | 400 bar |
| Autosampler | AS1 with 10 μL injection loop | Assistant 6000 with a feed pump S-100 |
| Mixing chamber | Static, SmartMix 100 | Static, SmartMix 350 |
| Column heater | T1 | |
| Detection type | PDA UV/Vis detection PDA1, D2/Hg halogen lamps, 190–1000 nm | UV-detector S-2550, 190–600 nm |
| Degasser | Analytical 2-channel-online-degasser | Preparative 2-channel-online-degasser |
Semi-preparative and analytical HPLC analyses were performed on Knauer Wissenschaftliche Geräte GmbH systems by Andreas Schneider. The solvents for HPLC were purchased in HPLC grade. The chromatograms were recorded by UV-detection at 240, 215, 210 and 205 nm.
Synthesis of compound 14
To a solution of acid 12 (1.00 g, 2.12 mmol) and 4-(dimethylamino)-pyridine (DMAP) (25.9 mg, 212 μmol) in THF (16 mL) was added 2,2,2-trichloroethanol (13) (0.48 mL, 4.24 mmol) followed by N,N′-dicyclohexylcarbodiimide (656 mg, 3.18 mmol). After stirring for 5 h at rt the mixture was filtered through a cotton plug and the precipitate was washed with EtOAc (2 × 20 mL). The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (15% EtOAc/CH2Cl2) to yield 1.03 g (1.71 mmol, 81%) of a white, crystalline solid.Rf 0.44 (15% EtOAc/CH2Cl2). [α]25D = +93° (c = 0.90 in CH2Cl2). 1H NMR (500 MHz, CDCl3) δ = 7.46–7.28 (m, 10H, Harom.), 7.07 (d, J = 5.5 Hz, 1H, NH), 5.58 (s, 1H, H-7), 5.34 (d, J = 3.4 Hz, 1H, H-1), 4.97 (d, J = 11.9 Hz, 1H, H-11), 4.68 (d, J = 11.8 Hz, 1H, H-15), 4.66 (q, J = 7.1 Hz, 1H, H-8), 4.60 (d, J = 11.9 Hz, 1H, H-11′), 4.52 (d, J = 11.9 Hz, 1H, H-15′), 4.21 (dd, J = 10.1, 4.6 Hz, 1H, H-6), 4.01–3.98 (m, 2H, H-3, H-5), 3.76 (dd, J = 10.3 Hz, 10.3 Hz, 1H, H-6′), 3.73 (dd, J = 9.2 Hz, 9.2 Hz, 1H, H-4), 2.03 (s, 3H, CH3-14), 1.50 (d, J = 7.0 Hz, 3H, CH3-9). 13C NMR (126 MHz, CDCl3) δ = 173.7 (C-10), 170.9 (C-13), 137.5 (Carom.), 137.3 (Carom.), 129.2 (Carom.), 128.6 (Carom.), 128.5 (Carom.), 128.1 (Carom.), 128.0 (Carom.), 126.0 (Carom.), 101.6 (C-7), 97.5 (C-1), 94.6 (C-12), 83.4 (C-4), 75.2 (C-8), 75.1 (C-3), 74.3 (C-11), 70.5 (C-15), 69.1 (C-6), 63.1 (C-5), 54.2 (C-2), 23.3 (C-14), 18.8 (C-9). HRMS (ESI) m/z: calcd for C27H30Cl3NO8Na [M + Na]+: 624.0929, found: 624.0932. The spectroscopic data were in agreement with those previously reported.13,15
Synthesis of compound 15
To a solution of carbohydrate 14 (500 mg, 829 μmol) in EtOAc (40 mL) was added Pd–C (650 mg, 10% Pd). The reaction vessel was filled with hydrogen. After stirring for 20 min at rt the suspension was filtered through Celite and the precipitate was washed with methanol (2 × 20 mL). The solvent was removed under reduced pressure and acetonitrile (30 mL) was added followed by benzaldehyde dimethyl acetal (187 μL, 1.24 mmol) and a solution of p-TsOH in acetonitrile (1 mL of a 232 mM solution, 232 μmol) dried over 3 Å MS. After stirring for 4 h at rt the reaction was neutralized with Net3 and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (90% EtOAc/cyclohexane) to yield 409 mg (798 μmol, 96%) of a white, crystalline solid as a mixture of α and β anomers (ratio α:β = 4:1). The mixture was used for the next reactions without separation. For analytical purpose, separation of the α and β anomer was achieved with HPLC (50% ACN/water, retention time 11.5 min (β anomer), 16.1 min (α anomer), using a KNAUER Eurospher II 100-5 C18; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 210 nm).
Rf (α anomer) 0.33, Rf (β anomer) 0.26 (90% EtOAc/cyclohexane). α anomer: [α]25D = +59° (c = 0.73 in CH2Cl2). 1H NMR (700 MHz, CD2Cl2) δ = 7.47–7.45 (m, 2H, Harom.), 7.40–7.36 (m, 3H, Harom.), 7.19 (d, J = 4.5 Hz, 1H, NH), 5.60 (s, 1H, H-7), 5.58 (dd, J = 3.9 Hz, 3.6 Hz, 1H, H-1), 5.01 (d, J = 12.0 Hz, 1H, H-11), 4.68 (q, J = 7.1 Hz, 1H, H-8), 4.65 (d, J = 12.0 Hz, 1H, H-11′), 4.24 (dd, J = 10.3, 5.0 Hz, 1H, H-6), 4.03 (td, J = 10.3 Hz, 5.0 Hz, 1H, H-5), 3.94–3.86 (m, 1H, H-3), 3.87–3.83 (m, 1H, H-2), 3.75 (dd, J = 10.3 Hz, 10.3 Hz, 1H, H-6′), 3.73 (dd, J = 10.3 Hz, 10.3 Hz, 1H, H-4), 3.68 (dd, J = 3.9 Hz, 1.0 Hz, 1H, OH), 2.01 (s, 3H, CH3-14), 1.51 (d, J = 7.1 Hz, 3H, CH3-9). 13C NMR (176 MHz, CD2Cl2) δ = 174.3 (C-10), 171.5 (C-13), 138.1 (Carom.), 129.5 (Carom.), 128.8 (Carom.), 126.5 (Carom.), 101.9 (C-7), 95.1 (C-12), 92.3 (C-1), 83.8 (C-4), 75.7 (C-8), 75.2 (C-3), 74.7 (C-11), 69.6 (C-6), 63.3 (C-5), 55.2 (C-2), 23.5 (C-14), 19.1 (C-9). HRMS (ESI) m/z: calcd for C20H24Cl3NO8Na [M + Na]+: 534.0460, found: 534.0463. The spectroscopic data were in agreement with those previously reported.15
Synthesis of compound 16
Alcohol 15 (100 mg, 195 μmol) was dissolved in dry CH2Cl2 (5 mL) and a 0.45 M solution of 1H-tetrazole in acetonitrile (1.63 mL, 731 μmol) was added. The reaction was cooled to −40 °C and dibenzyl(N,N-diisopropyl)phosphoramidite (164 μL, 488 μmol) was added. After 1 h warming to rt the reaction was stirred for another hour. Then, mCPBA (101 mg, 585 μmol) was added at −60 °C and the reaction was stirred for 30 min at 0 °C followed by 30 min stirring at rt. The mixture was diluted with 5 mL CH2Cl2 and washed two times with aq. Na2SO3 (10 mL, 10%), two times with aq. sat. NaHCO3 (10 mL) and two times with water (10 mL). After drying over MgSO4 the mixture was filtered, concentrated and purified by flash chromatography (65% EtOAc/cyclohexane) to yield 101 mg of a white, crystalline solid (79%, 585 μmol).Rf 0.22 (70% EtOAc/cyclohexane). [α]25D = +67° (c = 0.66 in CH2Cl2). 1H NMR (700 MHz, CD2Cl2) δ = 7.45–7.44 (m, 2H, Harom.), 7.42–7.33 (m, 13H, Harom.), 7.15 (d, J = 4.9 Hz, 1H, NH), 6.04 (dd, J = 6.1, 3.2 Hz, 1H, H-1), 5.57 (s, 1H, H-7), 5.06 (d, J = 8.3 Hz, 4H, 2× CH2–Ph), 5.01 (d, J = 12.0 Hz, 1H, H-11), 4.65 (q, J = 7.0 Hz, 1H, H-8), 4.62 (d, J = 12.0 Hz, 1H, H-11′), 4.08 (dd, J = 10.3, 4.9 Hz, 1H, H-6), 4.00–3.96 (m, 1H, H-2), 3.92 (ddd, J = 10.3, 10.3, 4.9 Hz, 1H, H-5), 3.82 (dd, J = 9.9, 9.9 Hz, 1H, H-3), 3.77 (dd, J = 10.3 Hz, 9.9 Hz, 1H, H-4), 3.72 (dd, J = 10.3, 10.3 Hz, 1H, H-6′), 1.87 (s, 3H, H-14), 1.51 (d, J = 7.1 Hz, 3H, H-9). 13C NMR (176 MHz, CD2Cl2) δ = 174.2 (C-10), 171.3 (C-13), 137.8 (Carom.,quart.–Ph), 136.4 (d, J = 7.2 Hz, Carom.,quart.–Bn), 136.4 (d, J = 7.2 Hz, Carom.,quart.–Bn), 129.6 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 129.1 (Carom.), 128.8 (Carom.), 128.4 (Carom.), 128.4 (Carom.), 126.5 (Carom.), 102.1 (C-7), 96.6 (d, J = 6.7 HZ, C-1) 95.0 (C-12), 82.9 (C-4), 75.8 (C-8), 74.8 (C-11), 74.7 (C-3), 70.0 (d, J = 4.1, CH2–Ph), 70.0 (d, J = 4.1, CH2–Ph), 69.0 (C-6), 65.1 (C-5), 54.5 (C-2), 23.2 (C-14), 19.0 (C-9). 31P NMR (284 MHz, CD2Cl2) δ = −2.4. HRMS (ESI) m/z: calcd for C34H37Cl3NO11PH [M + H]+: 772.1243, found: 772.1236. The spectroscopic data were in agreement with those previously reported.15
Synthesis of compound 7
Carbohydrate 16 (160 mg, 207 μmol) was dissolved in a mixture of 90% AcOH/water (15 mL) and zinc powder (120 mg, 1.84 mmol) was added. The suspension was stirred vigorously for 3 h at rt. After filtering and washing with MeOH the solvent was removed under reduced pressure and the crude product was purified by flash chromatography (0.1% AcOH/10% MeOH/CH2Cl2) to yield 129 mg of a white, crystalline solid (97%, 201 μmol).Rf 0.29 (0.1% AcOH/10% MeOH/CH2Cl2). [α]25D = +61° (c = 0.46 in MeOH). 1H NMR (700 MHz, CD3OD) δ = 7.48–7.45 (m, 2H, Harom.), 7.41–7.35 (m, 13H, Harom.), 6.06 (dd, J = 6.0, 3.3 Hz, 1H, H-1), 5.62 (s, 1H, H-7), 5.11–5.08 (m, 4H, 2× CH2–Ph), 4.42 (q, J = 7.1 Hz, 1H, H-8), 4.03–4.01 (m, 1H, H-6), 3.87–3.84 (m, 1H, H-2), 3.79–3.71 (m, 4H, H-3, H-4, H-5, H-6′), 1.87 (s, 3H, H-12), 1.37 (d, J = 7.1 Hz, 3H, H-9). 13C NMR (176 MHz, CD3OD) δ = 178.4 (C-10, not resolved but HMBC correlation), 173.9 (C-11), 138.9 (Carom.,quart.–Ph), 137.0 (d, J = 2.4 Hz, Carom.,quart.–Bn), 137.0 (d, 2.4 Hz, Carom.,quart.–Bn), 130.0 (Carom.), 129.9 (Carom.), 129.9 (Carom.), 129.8 (Carom.), 129.8 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 128.4 (Carom.), 127.2 (Carom.), 102.7 (C-7), 97.3 (d, J = 6.7, C-1), 83.0 (C-4), 77.0 (C-8), 75.1 (C-3), 71.2 (d, J = 2.2 Hz, CH2–Ph), 71.1 (d, J = 2.3 Hz, CH2–Ph), 69.1 (C-6), 66.1 (C-5), 55.8 (C-2), 22.6 (C-12), 19.2 (C-9). 31P NMR (284 MHz, CD2Cl2) δ = −3.0. HRMS (ESI) m/z: calcd for C32H36NO11Pna [M + Na]+: 664.1918, found: 664.1914. The spectroscopic data were in agreement with those previously reported.15
Synthesis of compound 19
Fmoc–D-Glu(O-tBu)–OH (1.50 g, 3.53 mmol) and DMAP (86.1 mg, 705 μmol) were dissolved in 20 mL EtOAc and cooled to 0 °C. 2-(Trimethylsilyl)ethanol (758 μL, 5.29 mmol) and DCC (1 M in CH2Cl2, 4.23 mL, 4.23 mmol) were added. After stirring for 2 h at rt, the reaction was filtered over Celite and the residue was washed with EtOAc (2 × 20 mL). The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (15% EtOAc/cyclohexane) to yield 1.77 g (3.37 mmol, 96%) of a white, amorphous solid.Rf 0.32 (15% EtOAc/cyclohexane). [α]25D = +14° (c = 1.98 in MeOH). 1H NMR (700 MHz, CD3OD) δ = 7.79 (d, J = 7.5 Hz, 2H, Harom.), 7.66 (t, J = 8.3 Hz, 2H, Harom.), 7.38 (t, J = 7.2 Hz, 2H, Harom.), 7.30 (t, J = 8.0 Hz, 2H, Harom.), 4.38–4.32 (m, 2H, H-10), 4.23–4.19 (m, 4H, H-2, H-4, H-11), 2.33 (t, J = 7.0 Hz, 2H, H-6), 2.15–2.07 (m, 1H, H-5), 1.92–1.83 (m, 1H, H-5), 1.44 (s, 9H, 3× CH3–Boc), 1.04–0.96 (m, 2H, H-1), 0.04 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, CD3OD) δ = 173.8 (C-7), 173.7 (C-3), 158.6 (C-9), 145.3 (Carom.), 145.2 (Carom.), 142.6 (Carom.), 128.8 (Carom.), 128.2 (Carom.), 128.1 (Carom.), 126.3 (Carom.), 126.2 (Carom.), 120.9 (Carom.), 81.8 (C-8), 68.0 (C-10), 64.6 (C-2), 54.8 (C-4), 48.4 (C-11), 32.6 (C-6), 28.4 (3× CH3–Boc), 27.8 (C-5), 18.2 (C-1), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C29H40NO6Si [M + H]+: 526.2619, found: 526.2598. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 17
Fmoc–D-Glu(O-tBu)–OTMSE (19) (1.00 g, 1.90 mmol) was dissolved in CH2Cl2 (30 mL). 2,6-Lutidine (4.42 mL, 38.0 mmol) and trimethylsilyl trifluoromethanesulfonate (TMSOTf, 3.44 mL, 19.0 mmol) were added at 0 °C and stirred for 30 min. The reaction was stirred for further 30 min at rt followed by slowly addition of aq. sat. NH4Cl (30 mL). The aqueous phase was extracted with EtOAc (3 × 40 mL). The organic phase was washed with 30% AcOH/water, dried over MgSO4 and the solvent was removed under reduced pressure to yield 866 mg (1.84 mmol, 97%) of a white, amorphous solid.Rf 0.44 (10% EtOAc/CH2Cl2). [α]25D = +19° (c = 1.04 in MeOH). 1H NMR (500 MHz, CD3OD) δ = 7.79 (d, J = 7.5 Hz, 2H, Harom.), 7.67 (t, J = 6.3 Hz, 2H, Harom.), 7.39 (t, J = 7.5 Hz, 2H, Harom.), 7.31 (td, J = 7.5, 1.1 Hz, 2H, Harom.), 4.39–4.32 (m, 2H, H-9), 4.24–4.19 (m, 4H, H-2, H-4, H-10), 2.40 (t, J = 7.4 Hz, 2H, H-6), 2.19–2.12 (m, 1H, H-5), 1.95–1.88 (m, 1H, H-5), 1.02–0.99 (m, 2H, H-1), 0.04 (s, 9H, Si(CH3)3). 13C NMR (126 MHz, CD3OD) δ = 176.3 (C-7), 173.8 (C-3), 158.6 (C-8), 145.3 (Carom.), 145.2 (Carom.), 142.6 (Carom.), 128.2 (Carom.), 128.1 (Carom.), 126.3 (Carom.), 126.2 (Carom.), 120.9 (Carom.), 68.0 (C-9), 64.7 (C-2), 55.0 (C-4), 48.4 (C-10), 31.1 (C-6), 27.7 (C-5), 18.2 (C-1), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C25H31NO6SiNa [M + Na]+: 492.1813, found: 492.1817. The spectroscopic data were in agreement with those previously reported.
Synthesis of compound 28
Boc–D-Ala–OH (27) (1.50 g, 7.93 mmol) and DMAP (194 mg, 1.59 mmol) were dissolved in EtOAc (40 mL) and cooled to 0 °C. 2-(Trimethylsilyl)ethanol (22) (1.70 mL, 11.9 mmol) and DCC (1 M in CH2Cl2, 9.51 mL, 9.51 mmol) were added. After stirring for 2 h at rt, the reaction was filtered through Celite and the residue was washed with EtOAc (2 × 20 mL). The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (10% EtOAc/cyclohexane) to yield 2.27 g (7.84 mmol, 99%) of a white, amorphous solid.Rf 0.17 (10% EtOAc/cyclohexane). [α]25D = +31° (c = 2.00 in MeOH). 1H NMR (700 MHz, CD3OD) δ = 4.25–4.18 (m, 2H, H-2), 4.09 (q, J = 7.2 Hz, 1H, H-4), 1.44 (s, 9H, 3× CH3–Boc), 1.33 (d, J = 7.2 Hz, 3H, H-5), 1.04–1.00 (m, 2H, H-1), 0.06 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, CD3OD) δ = 175.2 (C-3), 157.9 (C-6), 80.5 (C-7), 64.4 (C-2), 50.8 (C-4), 28.7 (3× CH3–Boc), 18.2 (C-1), 17.7 (C-5), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C13H27NO4SiNa [M + Na]+: 312.1597, found: 312.1602. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 26
Boc–D-Ala–TMSE 28 (220 mg, 760 μmol) was dissolved in 20% TFA/CH2Cl2 (4.5 mL). After stirring for 1 h at rt, toluene (15 mL) was added and the solvent was removed under reduced pressure to yield 144 mg (760 μmol, quant.) of a white, amorphous solid.Rf 0.16 (10% MeOH/CH2Cl2). [α]25D = +1.2° (c = 0.82 in MeOH). 1H NMR (500 MHz, CD3OD) δ = 4.35–4.31 (m, 2H, H-2), 4.00 (q, J = 7.2 Hz, 1H, H-4), 1.51 (d, J = 7.2 Hz, 3H, H-5), 1.09–1.06 (m, 2H, H-1), 0.07 (s, 9H, Si(CH3)3). 13C NMR (126 MHz, CD3OD) δ = 171.7 (C-3), 65.8 (C-2), 50.0 (C-4), 18.2 (C-1), 16.6 (C-5), −1.6 (3× CH3). HRMS (ESI) m/z: calcd for C8H20NO2Si [M + H]+: 190.1258, found: 190.1263. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 31
Fmoc–L-Lys(N-Boc)–OH (29) (1.50 g, 3.20 mmol) was dissolved in CH2Cl2 (20 mL) and TFA (20 mL) was added. The mixture was stirred for 20 min at rt. After removing the solvent under reduced pressure DMF (20 mL) and diisopropylethylamine (2.72 mL, 16.0 mmol) were added. 2-(Trimethylsilyl)ethyl p-nitrophenyl carbonate (47) (1.09 g, 3.84 mmol) was dissolved in DMF (6 mL) and transferred to the reaction solution. After stirring for 2 h at rt the solvent was removed under reduced pressure and the crude product was purified by flash chromatography (eluting first with EtOAc, then with 10% MeOH/CH2Cl2/0.1% AcOH) to yield 1.79 g (3.81 mmol, 99%) of a colorless, amorphous solid.Rf 0.42 (10% MeOH/CH2Cl2). [α]25D = −1.1° (c = 2.42 in MeOH). 1H NMR (500 MHz, CD3OD) δ = 7.80 (d, J = 7.5 Hz, 2H, Harom.), 7.67 (t, J = 7.4 Hz, 2H, Harom.), 7.39 (t, J = 7.3 Hz, 2H, Harom.), 7.31 (td, J = 7.5, 1.1 Hz, 2H, Harom.), 4.37–4.34 (m, 2H, H-8), 4.23 (t, J = 6.9 Hz, 1H, H-9), 4.18–4.08 (m, 3H, H-11, H-2), 3.09 (t, J = 6.9 Hz, 2H, H-6), 1.89–1.82 (m, 1H, H-3), 1.73–1.65 (m, 1H, H-3), 1.55–1.48 (m, 2H, H-5), 1.45–1.39 (m, 2H, H-4), 0.95 (t, J = 8.6 Hz, 2H, H-12), 0.02 (s, 9H, TMS). 13C NMR (126 MHz, CD3OD) δ = 159.3 (C-10), 158.7 (C-7), 145.4 (Carom.), 142.6 (Carom.), 128.8 (Carom.), 128.2 (Carom.), 126.3 (Carom.), 120.9 (Carom.), 67.9 (C-8), 63.7 (C-11), 55.9 (C-2), 48.5 (C-9), 41.4 (C-6), 32.7 (C-3), 30.6 (C-5), 24.1 (C-4), 18.7 (C-12), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C27H36N2O6SiNa [M + Na]+: 535.2235, found: 535.2231. The spectroscopic data were in agreement with those previously reported.15
Synthesis of compound 43
with: E = absorption of the sample solution at 301 nm, Vstock = volume of the stock solution [mL], D = dilution factor, ε = molar absorption coefficient at 301 nm = 6054 [L mol−1 cm−1], m = sample weight of the resin [mg], l = optical path length of the cell = 1 [cm].

Rf 0.19 (10% MeOH/CH2Cl2). [α]25D = −9.0° (c = 0.44 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 12.48 (br, s, 1H, COOH), 8.25 (d, J = 7.7 Hz, 1H, NH(Glu)), 8.17–8.13 (m, 1H, NH(Ala)), 7.90–7.88 (m, 3H, 2× Harom., NH(Lys)), 7.75–7.69 (m, 2H, Harom.), 7.47 (d, J = 7.9 Hz, 1H, NH(Ala)), 7.41 (t, J = 7.4 Hz, 2H, Harom.), 7.32 (t, J = 7.4 Hz, 2H, Harom.), 6.95–6.93 (m, 1H, NH(Teoc)), 4.28–4.24 (m, 3H, H-4, H-12), 4.22–4.16 (m, 4H, H-8, H-13, H-10, H-2), 4.14–4.10 (m, 2H, H-23), 4.01–3.98 (m, 2H, H-20), 2.93–2.89 (m, 2H, H-18), 2.22–2.13 (m, 2H, H-6), 1.99–1.92 (m, 1H, H-7), 1.81–1.73 (m, 1H, H-7), 1.61–1.55 (m, 1H, H-15), 1.47–1.43 (m, 1H, H-15), 1.36–1.32 (m, 2H, H-17), 1.24 (d, J = 7.2 Hz, 3H, H-14), 1.24 (d, J = 7.2 Hz, 3H, H-25), 1.22–1.17 (m, 2H, H-16), 0.95–0.93 (m, 2H, H-24), 0.91–0.88 (m, 2H, H-21), 0.01 (s, 9H, Si(CH3)3), −0.00 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 174.0 (C-1), 172.4 (C-9), 171.5 (C-22), 171.1 (C-5), 170.8 (C-3), 156.0 (C-19), 155.3 (C-11), 149.3 (Carom.), 140.4 (Carom.), 127.3 (Carom.H), 126.8 (Carom.H), 125.0 (Carom.H), 119.8 (Carom.H), 65.4 (C-12), 62.3 (C-23), 61.0 (C-20), 51.9 (C-4), 51.5 (C-8), 49.6 (C-10), 47.2 (C-2), 46.4 (C-13), 39.8 (C-18), 31.8 (C-15), 31.0 (C-6), 28.9 (C-17), 26.9 (C-7), 22.3 (C-16), 18.4 (C-14), 17.1 (C-25), 16.9 (C-21), 16.5 (C-24), −1.4 ((Lys)Si(CH3)3), −1.5 ((Glu)Si(CH3)3). HRMS (ESI) m/z: calcd for C43H66N5O11Si2 [M + H]+: 884.4279, found: 884.4279. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 40
To a solution of Fmoc–L-Ala–γ-D-Glu(O-TMSE)–L-Lys(N-Teoc)–D-Ala–COOH (43) (25.0 mg, 28.7 μmol), PyBOB (22.1 mg, 42.4 μmol) and HOBt (5.73 mg, 42.4 μmol) in DMF (0.5 mL) was added dropwise a solution of H2N–D-Ala-O–TMSE 26 (7.49 mg, 39.6 μmol), DIPEA (24.0 μL, 141 μmol) in DMF (0.5 mL). After stirring for 25 min at rt the solvent was removed under reduced pressure and the crude product was purified by HPLC (85% can/water, retention time 14.6 min, using a KNAUER Eurospher II 100-5 C18; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 265 nm) to yield 28.3 mg (26.8 μmol, 95%) of a white, amorphous solid.Rf 0.34 (10% MeOH/CH2Cl2). [α]25D = +10° (c = 0.60 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.24 (d, J = 7.7 Hz, 1H, NH(Glu)), 8.16 (d, J = 7.5 Hz, 1H, NH(D-Ala)), 8.14 (d, J = 7.5 Hz, 1H, NH(D-Ala)), 8.00 (d, J = 7.3 Hz, 1H, NH(Lys)), 7.89 (d, J = 7.4 Hz, 2H, Harom.), 7.75–7.69 (m, 2H, Harom.), 7.47 (d, J = 7.5 Hz, 1H, NH(L-Ala)), 7.41 (t, J = 7.4 Hz, 2H, Harom.), 7.32 (t, 2H, J = 7.4 Hz, Harom.), 6.94 (t, J = 5.4 Hz, 1H, NH(Teoc)), 4.30 (q, J = 7.5 Hz, 1H, H-6), 4.24 (d, J = 7.0 Hz, 2H, H-16), 4.21–4.07 (m, 12H, H-12, H-8, H-4, H-14, H-2, H-17, H-28), 4.01–3.99 (m, 2H, H-25), 2.92–2.90 (m, 2H, H-23), 2.21–2.12 (m, 2H, H-10), 1.95–1.90 (m, 1H, H-11), 1.80–1.75 (m, 1H, H-11), 1.59–1.54 (m, 1H, H-20), 1.47–1.44 (m, 1H, H-20), 1.35 (p, J = 6.8 Hz, 2H, H-22), 1.28 (d, J = 7.3 Hz, 3H, H-18), 1.23 (d, J = 7.5 Hz, 3H, H-30), 1.19 (d, J = 7.0 Hz, 3H, H-19), 1.26–1.15 (m, 2H, H-21), 0.94–0.88 (m, 6H, H-1, H-26, H-28), 0.01 (s, 9H, Si(CH3)3), 0.01 (s, 9H, Si(CH3)3), 0.00 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 172.2 (C-14), 171.9 (C-3), 171.3 (C-5), 171.0 (C-27), 171.0 (C-7), 171.0 (C-9), 156.2 (C-24), 155.4 (C-15), 143.6 (Carom.), 140.6 (Carom.), 127.5 (Carom.H), 126.9 (Carom.H), 125.2 (Carom.H), 120.0 (Carom.H), 65.5 (C-16), 62.4 (C-28), 62.3 (C-2), 61.2 (C-25), 52.7 (C-8), 51.6 (C-12), 49.7 (C-14), 47.6 (C-4), 47.4 (C-6), 46.5 (C-17), 39.7 (C-23), 31.3 (C-20), 31.1 (C-11), 29.1 (C-22), 26.9 (C-11), 22.5 (C-21), 18.5 (C-30), 17.9 (C-19), 17.3 (C-26), 16.7 (C-29), 16.6 (C-1), 16.6 (C-18), −1.6 (Si(CH3)3), −1.6 (Si(CH3)3), −1.7 (Si(CH3)3). HRMS (ESI) m/z: calcd for C51H81N6O12Si3Na [M + Na]+: 1077.5191, found: 1077.5195. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 44
Fmoc–L-Ala–γ-D-Glu(O-TMSE)–L-Lys(N-Teoc)–D-Ala–D-Ala–O-TMSE 40 (100 mg, 94.7 μmol) was dissolved in 20% piperidine/DMF (1 mL). After stirring for 30 min at rt, the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (10% MeOH/CH2Cl2) to yield 60.3 mg (72.4 μmol, 76%) of a white, amorphous solid.Rf 0.23 (10% MeOH/CH2Cl2). [α]25D = +23° (c = 0.44 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.15 (d, J = 7.9 Hz, 1H, NH(D-Ala)), 8.14 (d, J = 7.9 Hz, 1H, NH(D-Ala)) 8.08 (d, J = 6.8 Hz, 1H, NH(Glu)), 8.00 (d, J = 7.3 Hz, 1H, NH(Lys)), 6.94 (t, J = 5.5 Hz, 1H, NH(Teoc)), 4.31–4.27 (m, 1H, H-6), 4.21–4.17 (m, 2H, H-4, H-12), 4.17–4.08 (m, 5H, H-8, H-2, H-25), 4.02–3.97 (m, 2H, H-22), 3.29 (q, J = 6.9 Hz, 1H, H-14) 2.91 (dt, J = 6.8 Hz, 2H, H-20), 2.52 (m, 2H, NH2), 2.21–2.13 (m, 2H, H-10), 1.97–1.92 (m, 1H, H-11), 1.81–1.76 (m, 1H, H-11), 1.59–1.54 (m, 1H, H-17), 1.49–1.44 (m, 1H, H-17), 1.35 (p, J = 7.5 Hz, 2H, H-19), 1.28 (d, J = 7.3 Hz, 3H, H-15), 1.26–1.22 (m, 2H, H-18), 1.19 (d, J = 7.1 Hz, 3H, H-16), 1.11 (d, J = 6.9 Hz, 3H, H-27), 0.96–0.88 (m, 6H, H-1, H-23, H-26), 0.02 (s, 18H, 2× Si(CH3)3), 0.01 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 175.7 (C-13), 172.1 (C-3), 171.7 (C-5), 171.6 (C-24), 171.2 (C-9), 171.1 (C-7), 156.0 (C-21) 62.3 (C-25), 62.2 (C-2), 61.0 (C-22), 52.6 (C-8), 51.1 (C-12), 49.8 (C-14), 47.4 (C-4), 47.2 (C-6), 39.7 (C-20), 31.2 (C-17), 31.0 (C-10), 28.9 (C-19), 26.9 (C-11), 22.3 (C-18), 21.3 (C-27), 17.7 (C-16), 17.1 (C-23), 16.5 (C-26), 16.5 (C-1), 16.4 (C-15), −1.7 (2× Si(CH3)3), −1.8 (Si(CH3)3), −1.8 (Si(CH3)3). HRMS (ESI) m/z: calcd for C36H73N6O10Si3 [M + H]+: 833.4690, found: 833.4690. The spectroscopic data were in agreement with those previously reported.34
Synthesis of compound 35
Glycine (34) (250 mg, 3.33 mmol) was dissolved in 50% dioxane/water (8 mL) and NEt3 (1.38 mL, 9.99 mmol) and N-(2-(trimethylsilyl)ethoxycarbonyloxy)succinimide (950 mg, 3.66 mmol) were added. The solution was stirred at rt and diluted with water (20 mL). Citric acid 0.5 M (aq.) was added until the pH turned 4. The aqueous phase was extracted with diethylether (3 × 40 mL) and the combined ether layer was washed twice with water (15 mL). The organic phase was dried over MgSO4 and the solvent was removed under reduced pressure to yield 714 mg (3.26 mmol, 98%) of a white, amorphous solid.Rf 0.41 (10% MeOH/0.1% AcOH/CH2Cl2). 1H NMR (500 MHz, CD3OD) δ = 4.18–4.14 (m, 2H, H-4), 3.80 (s, 2H, H-2), 1.02–0.99 (m, 2H, H-5), 0.05 (s, 9H, Si(CH3)3). 13C NMR (126 MHz, CD3OD) δ = 173.7 (C-1), 159.4 (C-3), 64.2 (C-4), 43.0 (C-2), 18.6 (C-5), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C8H16NO4Si [M − H]−: 218.0854, found: 218.0851. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 54
with: E = absorption of the sample solution at 301 nm, Vstock = volume of the stock solution [mL], D = dilution factor, ε = molar absorption coefficient at 301 nm = 6054 [L mol−1 cm−1], m = sample weight of the resin [mg], l = optical path length of the cell = 1 [cm].

Characterization data for compound 52 (test cleavage)
Rf 0.30 (20% MeOH/CH2Cl2). [α]25D = −3.5° (c = 0.28 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.22–8.16 (br, m, 2H, 2× NH(Gly)), 8.10 (t, J = 5.2 Hz, 2H, 2× NH(Gly)), 8.04 (s, 1H, NH(Ala)), 7.73 (t, J = 5.6 Hz, 1H, NH(Lys)), 7.27–7.23 (m, 2H, NH(Lys), NH(Teoc)), 5.93–5.87 (m, 1H, H-24), 5.29 (dd, J = 17.2 Hz (trans coupling), 1.5 Hz, 1H, H-25), 5.17 (d, J = 10.5 Hz (cis coupling), 1.2 Hz, 1H, H-25), 4.46 (d, J = 5.2 Hz, 2H, H-23), 4.17–4.12 (m, 1H, H-2), 4.05–4.02 (m, 2H, H-20), 3.98–3.96 (m, 1H, H-4), 3.75 (d, J = 5.7 Hz, 2H, H-14), 3.75 (d, J = 5.7 Hz, 2H, H-16), 3.73 (d, J = 5.8 Hz, 2H, H-12), 3.65 (d, J = 5.9 Hz, 2H, H-10), 3.62 (d, J = 6.0 Hz, 2H, H-18), 3.02 (q, J = 6.7 Hz, 2H, H-8), 1.63–1.54 (m, 1H, H-5), 1.52–1.47 (m, 1H, H-5), 1.40–1.34 (m, 2H, H-7), 1.31–1.26 (m, 2H, H-6), 1.24 (d, J = 7.2 Hz, 3H, H-26), 0.93–0.91 (m, 2H, H-21), 0.02 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 174.0 (C-1), 171.5 (C-3), 169.7 (C-17), 169.3 (C-15), 169.3 (C-13), 169.0 (C-11), 168.4 (C-9), 156.7 (C-19), 155.7 (C-22), 133.6 (C-24), 116.9 (C-25), 64.4 (23), 61.9 (C-20), 54.4 (C-4), 47.7 (C-2), 43.5 (C-18), 42.1 (C-16), 42.0 (C-14), 42.0 (C-12), 42.0 (C-10), 38.3 (C-8), 31.8 (C-5), 28.6 (C-7), 22.7 (C-6), 17.6 (C-21), 17.3 (C-26), −1.4 (Si(CH3)3). HRMS (ESI) m/z: calcd for C29H49N8O12Si [M + H]+: 729.3245, found: 729.3241.Characterization data for compound 54
Rf 0.21 (15% MeOH/CH2Cl2). [α]25D = −2.8° (c = 0.70 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.26 (d, J = 7.5 Hz, 1H, NH(Glu)), 8.19 (br, s, 2H, 2× NH(Gly)), 8.10–8.07 (m, 3H, 2× NH(Gly), NH–D-Ala), 7.91 (d, J = 8.2 Hz, 1H, NH(Lys)), 7.89 (d, J = 7.5 Hz, 2H, Harom.), 7.74–7.71 (m, 3H, NH(Lysside chain), 2× Harom.), 7.48 (d, J = 7.0 Hz, 1H, NH–L-Ala), 7.41 (t, J = 7.8 Hz, 2H, Harom.), 7.32 (t, J = 7.4 Hz, 2H, Harom.), 7.23 (t, J = 5.9 Hz, 1H, NH(Teoc)), 4.25–4.10 (m, 9H, H-33, H-12, H-13, H-10, H-2, H-4, H-8), 4.04–4.02 (m, 2H, H-30), 3.75 (d, J = 5.7 Hz, 2H, H-24), 3.75 (d, J = 5.7 Hz, 2H, H-26), 3.73 (d, J = 5.8 Hz, 2H, H-22), 3.65 (d, J = 5.8 Hz, 2H, H-20), 3.62 (d, J = 5.9 Hz, 2H, H-28), 3.01 (q, J = 6.4 Hz, 2H, H-18), 2.22–2.14 (m, 2H, H-6), 1.98–1.93 (m, 1H, H-7), 1.81–1.75 (m, 1H, H-7), 1.62–1.57 (m, 1H, H-15), 1.47–1.46 (m, 1H, H-15), 1.36 (p, J = 7.1 Hz, 2H, H-17), 1.24 (d, J = 7.0 Hz, 3H, H-35), 1.23 (d, J = 7.3 Hz, 3H, H-14), 1.28–1.19 (m, 2H, H-16), 0.95–0.90 (m, 4H, H-31, H-34), 0.02 (s, 9H, Si(CH3)3), 0.00 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 174.0 (C-1), 172.7 (C-9), 171.7(C-3), 171.3 (C-32), 171.1 (C-5), 169.7 (C-27), 169.3 (C-25), 169.3 (C-23), 169.0 (C-21), 168.4 (C-19), 156.7 (C-29), 155.6 (C-11), 143.8 (Carom.), 140.7 (Carom.), 127.6 (Carom.H), 127.1 (Carom.H), 125.3 (Carom.H), 120.1 (Carom.H), 65.7 (C-12), 62.6 (C-33), 61.9 (C-30), 52.3 (C-4), 51.7 (C-8), 49.8 (C-10), 47.6 (C-2) 46.6 (C-13), 43.5 (C-28), 42.1 (C-24), 42.1 (C-22), 42.0 (C-26), 42.0 (C-20), 38.4 (C-18), 31.9 (C-15), 31.3 (C-6) 28.7 (C-17), 27.1 (C-7), 22.6 (C-16), 18.6 (C-14), 17.3 (C-35), 16.8 (C-31), 16.8 (C-34), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3). HRMS (ESI) m/z: calcd for C53H79N10O16Si2 [M + H]+: 1167.5220, found: 1167.5216. The spectroscopic data were in agreement with those previously reported.8Synthesis of alanine derivative of 54 (compound 68)
Fmoc–L-Ala–γ-D-Glu(O-TMSE)–L-Lys((Gly)5–Teoc)–D-Ala–COOH (54) (60.0 mg, 51.3 μmol), PyBOB (40.1 mg, 76.9 μmol), HOBt (10.4 mg, 76.9 μmol) and H2N–D-Ala–O-TMSE 26 (13.6 mg, 71.8 μmol) were dissolved in DMF (2 mL) and DIPEA (34.9 μL, 205 μmol) was added immediately. After stirring for 40 min at rt the solvent was removed under reduced pressure and the crude product was dissolved in a minimum DMF and added to ice cold diethyl ether. The formed precipitate was filtered and washed with cold ether and dichloromethane, redissolved in MeOH and the solvent was removed under reduced pressure to yield to yield 38.5 mg (28.7 μmol, 56%) of a white, amorphous solid.Rf 0.27 (10% MeOH/CH2Cl2). [α]25D = +7.4° (c = 0.68 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.24 (d, J = 7.6 Hz, 1H, NH(Glu)), 8.17–8.12 (m, 4H, 2× NH–D-Ala, 2× NH(Gly)), 8.08–8.04 (m, 2H, 2× NH(Gly)), 8.01 (d, J = 7.4 Hz, 1H, NH(Lys)), 7.89 (d, J = 7.6 Hz, 2H, Harom.), 7.73–7.70 (m, 3H, NH(Lysside chain), 2× Harom.), 7.47 (d, J = 7.0 Hz, 1H, NH–L-Ala), 7.41 (t, J = 7.4 Hz, 2H, Harom.), 7.32 (t, J = 7.3 Hz, 2H, Harom.), 7.22 (t, J = 6.0 Hz, 1H, NH(Teoc)), 4.31 (q, J = 7.6 Hz, 1H, H-6), 4.25–4.07 (m, 11H, H-2, H-38, H-16, H-17, H-14, H-12, H-8, H-4), 4.04–4.02 (m, 2H, H-35), 3.75 (d, J = 5.6 Hz, 2H, H-31), 3.75 (d, J = 5.6 Hz, 2H, H-29), 3.73 (d, J = 5.8 Hz, 2H, H-27), 3.65 (d, J = 5.8 Hz, 2H, H-25), 3.62 (d, J = 6.0 Hz, 2H, H-33), 3.02–2.99 (m, 2H, H-23), 2.22–2.12 (m, 2H, H-10), 1.95–1.90 (m, 1H, H-11), 1.80–1.76 (m, 1H, H-11), 1.60–1.56 (m, 1H, H-20), 1.50–1.45 (m, 1H, H-20), 1.39–1.35 (m, 2H, H-22), 1.28 (d, J = 7.3 Hz, 3H, H-18), 1.27–1.20 (m, 2H, H-21), 1.23 (d, J = 7.1 Hz, 3H, H-40), 1.19 (d, J = 7.1 Hz, 3H, H-19), 0.94–0.90 (m, 6H, H-1, H-36, H-39), 0.02 (s, 9H, Si(CH3)3), 0.01 (s, 9H, Si(CH3)3), 0.00 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 172.7 (C-13), 172.4 (C-3), 172.0 (C-5), 171.7 (C-7), 171.5 (C-37), 171.4 (C-9), 169.7 (C-32), 169.3 (C-30), 169.3 (C-28), 169.0 (C-26), 168.4 (C-24), 156.7 (C-34), 155.6 (C-15), 143.8 (Carom.), 140.7 (Carom.), 127.6 (Carom.), 127.1 (Carom.), 125.3 (Carom.), 120.1 (Carom.), 65.7 (C-16), 62.6 (C-38), 62.5 (C-2), 61.9 (C-35), 52.8 (C-8), 51.7 (C-12), 49.8 (C-14), 47.7 (C-4), 47.5 (C-6), 46.6 (C-17), 43.5 (C-33), 42.1 (C-29), 42.1 (C-27), 42.0 (C-31), 42.0 (C-25), 38.4 (C-23), 31.4 (C-20), 31.2 (C-10), 28.8 (C-22), 27.0 (C-11), 22.7 (C-21), 18.7 (C-40), 18.1 (C-19), 17.3 (C-18), 16.8 (C-36), 16.7 (C-1), 16.7 (C-39), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3), −1.5 (Si(CH3)3). HRMS (ESI) m/z: calcd for C61H97N11O17Si3Na [M + Na]+: 1362.6264, found: 1362.6238. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 45
Fmoc–L-Ala–γ-D-Glu(O-TMSE)–L-Lys((Gly)5–Teoc)–D-Ala–D-Ala–O-TMSE (68) (35.0 mg, 26.1 μmol) was dissolved in 20% piperidine/DMF (2 mL). After stirring for 30 min at rt the solvent was removed under reduced pressure. Cold diethyl ether was added and the resulting precipitate was filtered, washed with cold diethyl ether, dissolved in MeOH and the solvent was removed under reduced pressure to yield 25.5 mg (22.8 μmol, 87%) of a white, amorphous solid.Rf 0.53 (22% MeOH/CH2Cl2). [α]25D = +10° (c = 0.99 in MeOH). 1H NMR (700 MHz, (CD3)2SO) δ = 8.22–8.11 (m, 5H, NH(Glu), 2× NH–D-Ala, 2× NH(Gly)), 8.07 (dt, J = 11.3, 5.7 Hz, 2H, 2× NH(Gly)), 8.01 (d, J = 7.4 Hz, 1H, NH(Lys)), 7.73 (t, J = 5.5 Hz, 1H, NH(Lysside chain)), 7.22 (t, J = 5.9 Hz, 1H, NH(Teoc)), 4.29 (p, J = 7.1 Hz, 1H, H-6), 4.20–4.08 (m, 7H, H-2, H-4, H-8, H-12, H-35), 4.06–3.99 (m, 2H, H-32), 3.75 (d, J = 5.5 Hz, 2H, H-28), 3.75 (d, J = 5.5 Hz, 2H, H-26) 3.73 (d, J = 5.8 Hz, 2H, H-24), 3.65 (d, J = 5.8 Hz, 2H, H-22), 3.62 (d, J = 6.0 Hz, 2H, H-30), 3.29 (q, J = 6.9 Hz, 1H, H-14), 3.01 (q, J = 6.9 Hz, 2H, H-20), 2.52 (m, 2H, NH2), 2.20–2.14 (m, 2H, H-10), 1.97–1.95 (m, 1H, H-11), 1.80–1.78 (m, 1H, H-11), 1.61–1.56 (m, 1H, H-17), 1.49–1.46 (m, 1H, H-17), 1.39–1.34 (m, 2H, H-19), 1.28 (d, J = 7.3 Hz, 3H, H-15), 1.28–1.19 (m, 2H, H-18), 1.19 (d, J = 7.1 Hz, 3H, H-16), 1.16 (d, J = 6.9 Hz, 3H, H-37), 0.96–0.91 (m, 6H, H-1, H-26, H-33), 0.02 (s, 9H, Si(CH3)3), 0.02 (s, 9H, Si(CH3)3), 0.02 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, (CD3)2SO) δ = 172.4 (C-3), 172.0 (C-5), 171.8 (C-7), 171.5 (C-34), 171.4 (C-9), 169.7 (C-29), 169.3 (C-27), 169.3 (C-25), 169.0 (C-23), 168.4 (C-21), 156.7 (C-31), 62.7 (C-35), 62.5 (C-2), 61.9 (C-32), 52.8 (C-8), 51.5 (C-12), 49.8 (C-14), 47.7 (C-4), 47.5 (C-6), 43.5 (C-30), 42.1 (C-26), 42.1 (C-24), 42.0 (C-28), 42.0 (C-22), 38.4 (C-20), 31.4 (C-17), 31.2 (C-10), 28.8 (C-19), 27.1 (C-11), 22.7 (C-18), 20.9 (C-37) 18.0 (C-16), 17.3 (C-15), 16.8 (C-33), 16.8 (C-1), 16.7 (C-39), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3), −1.5 (Si(CH3)3). HRMS (ESI) m/z: calcd for C46H88N11O15Si3 [M + H]+: 1118.5764, found: 1118.5750. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 33
L-Ala (250.0 mg, 2.81 mmol, 1.00 equiv.) was dissolved in 50% dioxane/water (6.7 mL) and triethylamine (1.2 mL, 8.43 mmol, 3.00 equiv.) and Teoc–OSu (802 mg, 3.1 mmol, 1.10 equiv.) were added. The reaction mixture was stirred overnight at room temperature and then diluted with water (17 mL). Afterwards a 0.5 M solution of citric acid in water was added until the pH turned 4. The aqueous phase was extracted with diethyl ether (3 × 35 mL) and the organic phase was washed with water (2 × 13 mL). The combined organic phases were dried over magnesium sulfate and the solvent was removed under reduced pressure to yield 656 mg (2.81 mmol, quant.) of a colorless, amorphous solid. The product was used without further purification.Rf = 0.55 (10% MeOH/0.1% AcOH/CH2Cl2). [α]25D = +3.4° (c = 1.20 in MeOH). 1H-NMR (500 MHz, CD3OD) δ = 15.93 (br, 1H, COOH), 7.05 (d, J = 7.2 Hz, 1H, NH), 4.17 (t, J = 6.8 Hz, 1H, H-2), 4.14 (t, J = 7.1 Hz, 2H, H-4), 1.37 (d, J = 7.3 Hz, 3H, H-6), 1.02–0.98 (m, 2H, H-5), 0.05 (s, 9H, TMS). 13C-NMR (126 MHz, CD3OD) δ = 176.6 (C-1), 158.7 (C-3), 64.0 (C-4), 50.7 (C-2), 18.6 (C-5), 17.9 (C-6), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C9H20NO4Si [M + H]+: 323.1633, found: 323.1639.
Synthesis of compound 56
Rf = 0.24 (10% MeOH/CH2Cl2). [α]25D = −5.8° (c = 0.88 in MeOH). 1H-NMR (700 MHz, (CD3)2SO) δ = 12.53 (br, 1H, COOH), 8.26 (d, J = 7.6 Hz, 1H, NH–Glu), 8.09 (m, J = 7.6 Hz, 1H, NH–D-Ala), 7.91 (d, J = 8.4 Hz, 1H, NH–Lys), 7.89 (d, J = 7.5 Hz, 2H, Harom.), 7.83 (m, 1H, NH–Ala), 7.77 (m, 1H, NH(Lysside chain)), 7.73 (dd, J = 7.8 Hz, 1.8 Hz, 2H, Harom.), 7.47 (d, J = 7.8 Hz, 1H, NH–Ala), 7.41 (td, J = 7.4 Hz, 1.1 Hz, 2H, Harom.), 7.32 (td, J = 7.4 Hz, 1.2 Hz, 2H, Harom.), 7.20 (d, J = 7.5 Hz, 1H, NH(Teoc)), 4.25 (s, 1H, H-4), 4.24 (s, 2H, H-12), 4.22–4.15 (m, 4H, H-2, H-8, H-10, H-13), 4.15–4.09 (m, 2H, H-29), 4.05–3.97 (m, 2H, H24), 3.06–3.00 (m, 1H, H-18), 2.98–2.92 (m, 1H, H-18), 2.23–2.13 (m, 2H, H-6), 1.99–1.92 (m, 1H, H-7), 1.83–1.75 (m, 1H, H-7), 1.63–1.56 (m, 1H, H-15), 1.49–1.42 (m, 1H, H-15), 1.39–1.31 (m, 2H, H-17), 1.24 (d, J = 5.1 Hz, 3H, H-31), 1.23 (d, J = 5.1 Hz, 3H, H-14), 1.25–1.22 (dd, J = 7.2 Hz, 5.1 Hz, 2H, H-20), 1.17 (d, J = 7.2 Hz, 8H, H-16, H-26, H-27), 0.95–0-90 (dt, J = 8.7 Hz, 16.4 Hz, 4H, H-25, H-30), 0.01 (s, 9H, TMS), −0.00 (s, 9H, TMS). 13C-NMR (176 MHz, (CD3)2SO) δ = 173.9 (C-1), 172.7 (C-9), 171.8 (C-3), 171.4 (C-28), 171.1 (C-5), 169.0 (C-21), 168.4 (C-19), 156.8 (C-23), 155.9 (C-11), 143.8 (Carom.), 140.7 (Carom.), 127.6 (Carom.–H), 127.1 (Carom.–H), 125.3 (Carom.–H), 120.1 (Carom.–H), 65.7 (C-12), 62.6 (C-29), 61.8 (C-24), 52.2 (C-4), 51.7 (C-8), 50.5 (C-20), 50.3 (C-22), 49.8 (C-10), 48.1 (C-2), 46.6 (C-13), 38.3 (C-18), 31.9 (C-15), 31.3 (C-6), 28.6 (C-17), 27.1 (C-7), 22.6 (C-16), 18.5 (C-14), 18.5 (C-26), 17.8 (C-27), 17.3 (C-31), 16.8 (C-25), 16.8 (C-30), −1.5 (3× CH3), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C49H76N7O13Si2 [M + H]+: 1026.5034, found: 1026.5034.
Synthesis of alanine derivative of 56 (compound 69)
The free acid 56 (210.3 mg, 204.6 μmol, 1.00 equiv.), PyBOP (160 mg, 307 μmol, 1.50 equiv.), HOBt (41.5 mg, 307 μmol, 1.50 equiv.) and 2-(trimethylsilyl)ethyl–D-alaninate (26) (55.7 mg, 287 μmol, 1.40 equiv.) were dissolved in DMF (7.0 mL). DIPEA (139 μL, 818 μmol, 4.00 equiv.) was added directly and the reaction mixture was stirred for 40 min at room temperature. The solvent was removed under reduced pressure and the residue dissolved in minimum DMF. Ice cold diethyl ether was added to the solution and the precipitate was filtered and washed with cold diethyl ether (two times) and CH2Cl2 (two times). The precipitate was dissolved in MeOH and the solvent removed under reduced pressure to yield 227 mg of a colorless, amorphous solid (190 μmol, 93%). The product was used for the next reaction without further purification.Rf = 0.34 (10% MeOH/CH2Cl2). [α]25D = +12° (c = 0.66 in MeOH). 1H-NMR (500 MHz, (CD3)2SO) δ = 8.24 (d, J = 7.3 Hz, 1H, NH–Glu), 8.16 (d, J = 6.9 Hz, 1H, NH–D-Ala), 8.14 (d, J = 7.9 Hz, 1H, NH–D-Ala), 7.97 (d, J = 8.3 Hz, 1H, NH–Lys), 7.88 (d, J = 7.6 Hz, 2H, Harom.), 7.80 (d, J = 7.3 Hz, 1H, NH–Ala), 7.78–7.75 (m, 1H, NH(Lysside chain)), 7.72 (t, J = 7.3 Hz, 2H Harom.), 7.46 (d, J = 7.9 Hz, 1H, NH–Ala), 7.41 (t, J = 7.6 Hz, 2H, Harom.), 7.32 (t, J = 6.9 Hz, 2H, Harom.), 7.19 (d, J = 7.8 Hz, 1H, NH(Teoc)), 4.30 (quint, J = 7.5 Hz, 1H, H-6), 4.26–4.23 (m, 2H, H-16), 4.23–4.15 (m, 4H, H-17, H-12, H-14, H-4), 4.13–4.06 (m, 5H, H-8, H-34, H-2), 4.06–3.96 (m, 2H, H-29), 3.63–3.58 (m, 2H, H-20), 3.17–3.10 (m, 2H, H-22), 3.03–3.00 (m, 2H, H-23), 2.21–2.12 (m, 2H, H-10), 1.97–1.89 (m, 1H, H-11), 1.83–1.76 (m, 1H, H-11), 1.28 (m, 3H, H-18), 1.25 (m, 2H, H-27, H-25), 1.22 (m, 3H, H-36), 1.20 (m, 3H, H-19), 1.17 (m, 8H, H-31, H-32, H-21), 0.95–0.89 (m, 6H, H-30, H-1, H-35), 0.01 (s, 9H, TMS), 0.01 (s 9H, TMS), −0.00 (s, 9H, TMS). 13C-NMR (126 MHz, (CD3)2SO) δ = 172.3 (C-3), 172.1 (C-13), 172.0 (C-5), 171.7 (C-7), 171.4 (C-9), 169.0 (C-26), 168.5 (C-24), 157.1 (C-28), 156.0 (C-15), 143.9 (Carom.), 140.7 (Carom.), 127.6 (Carom.–H), 127.0 (Carom.–H), 125.3 (Carom.–H), 120.1 (Carom.–H), 65.7 (C-16), 62.6 (C-34), 62.5 (C-2), 61.8 (C-29), 53.6 (C-8), 51.7 (C-12), 50.0 (C-14), 49.8 (C-6), 48.0 (C-25), 47.7 (C-4), 46.6 (C-17), 45.9 (C-27), 38.3 (C-23), 31.7 (C-20), 31.2 (C-10), 28.6 (C-22), 27.1 (C-11), 22.6 (C-21), 18.6 (C-31), 18.5 (C-19), 18.1 (C-32), 18.1 (C-36), 17.4 (C-18), 16.8 (C-30), 16.7 (C-1), 16.7 (C-35), −1.5 (3× CH3), −1.5 (3× CH3), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C57H92N8O14Si3Na [M + Na]+: 1219.5939, found: 1219.5934. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 55
Fmoc–L-Ala–γ-D-Glu(O-TMSE)–L-Lys((L-Ala)2–Teoc)–D-Ala–D-Ala–O-TMSE (69) (223 mg, 186 μmol, 1.00 equiv.) was dissolved in 20% piperidine/DMF (14.4 mL) and stirred for 2 h at room temperature. The solvent was removed under reduced pressure and cold diethyl ether was added. The formed precipitate was filtered and washed with diethyl ether. Afterwards it was dissolved in MeOH and the solvent was removed under reduced pressure to yield 136.1 mg of a colorless, amorphous solid (140 μmol, 75%), which was used in the next reaction without further purification.Rf = 0.28 (10% MeOH/CH2Cl2). [α]25D = +21° (c = 0.66 in MeOH). 1H-NMR (700 MHz, (CD3)2SO) δ = 8.18 (d, J = 6.9 Hz, 1H, NH–D-Ala), 8.14 (d, J = 7.9 Hz, 1H, NH–D-Ala), 8.01 (d, J = 7.5 Hz, 1H, NH–Glu), 7.80 (d, J = 7.7 Hz, 1H, NH–Lys), 7.76 (d, J = 5.7 Hz, 1H, NH(Lysside chain)), 7.50 (d, J = 8.2 Hz, 1H, NH–Ala), 7.19 (d, J = 7.4 Hz, 1H, NH(Teoc)), 4.30 (quint, J = 7.6 Hz, 1H, H-6), 4.23–4.16 (m, 2H, H-4, H-12), 4.16–4.11 (dd, J = 9.1 Hz, 7.8 Hz, 2H, H-8, H-14), 4.11–4.08 (td, J = 8.4 Hz, 1.3 Hz, 2H, H-31), 4.06–3.97 (m, 4H, H-2, H-26), 3.07–2.93 (m, 2H, H-20), 2.52 (t, J = 2.0 Hz, 2H, NH2), 2.23–2.13 (m, 2H, H-10), 1.99–1.93 (m, 1H, H-11), 1.83–1.77 (m, 1H, H-11), 1.77–1.73 (m, 1H, H-22), 1.65–1.61 (m, 1H, H-24), 1.61–1.52 (m, 1H, H-17), 1.50–1.43 (m, 1H, H-17), 1.41–1.33 (m, 2H, H-19), 1.28 (d, J = 7.3 Hz, 3H, H-15), 1.23 (d, J = 6.4 Hz, 3H, H-33), 1.18 (d, J = 7.2 Hz, 3H, H-16), 1.17 (dd, J = 7.1 Hz, 2.6 Hz, 8H, H-28, H-29, H-18), 0.96–0.91 (m, 6H, H-1, H-27, H-32), 0.03 (s, 9H, TMS), 0.02 (s, 9H, TMS), 0.02 (s, 9H, TMS). 13C-NMR (176 MHz, (CD3)2SO) δ = 172.4 (C-3), 172.1 (C-13), 172.0 (C-5), 171.8 (C-7), 171.6 (C-30), 171.3 (C-9), 169.1 (C-23), 168.5 (C-21), 156.8 (C-25), 62.7 (C-31), 62.5 (C-2), 61.8 (C-26), 53.0 (C-8), 51.6 (C-12), 50.0 (C-14), 48.1 (C-22), 47.7 (C- 4), 47.5 (C-6), 45.9 (C-24), 38.3 (C-20), 31.5 (C-17), 31.2 (C-10), 28.7 (C-19), 27.1 (C-11), 22.6 (C-18), 18.5 (C-28), 18.1 (C-16), 18.1 (C-33), 18.0 (C-29), 17.4 (C-15), 16.9 (C-27), 16.8 (C-1), 16.7 (C-32), −1.4 (3× CH3), −1.5 (3× CH3), −1.5 (3× CH3). HRMS (ESI) m/z: calcd for C42H83N8O12Si3 [M + H]+: 975.5433, found: 975.5436. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 8
Trichloroacetonitrile (TCA, 433 μL, 4.32 mmol) was added to farnesol (449 μL, 1.80 mmol). To this stirred solution was added tetrabutylammonium phosphate (TBAP, 0.4 M in acetonitrile, 8.99 mL, 3.60 mmol) over 1 h via syringe pump. The reaction was stirred for further 7 h at rt and the solvent was removed under reduced pressure. The crude product was first purified by flash chromatography (10% water/20% NH3 (conc.)/isopropanol) and then percolated through Dowex® 50WX8 with a 0.025 M NH4HCO3 solution. Dowex® 50WX8 was washed before usage with 3:1 NH3/water and 0.025 M NH4HCO3 solution until pH of the eluent turned 8. The solvent was removed by lyophilisation to yield 425 mg (1.27 mmol, 71%) of a white powder.
Rf 0.14 (10% water/20% NH3 (conc., aq.)/isopropanol). 1H NMR (500 MHz, D2O) δ = 5.40 (t, J = 6.4 Hz, 1H, H-2), 5.12 (t, J = 7.5 Hz, 1H, H-6), 5.08 (t, J = 6.8 Hz, 1H, H-10), 4.38 (s, 2H, H-1), 2.14–1.88 (m, 8H, H-4, H-5, H-8, H-9), 1.69 (s, 3H, H-13), 1.64 (s, 3H, H-15), 1.58 (s, 3H, H-14), 1.56 (s, 3H, H-12). 13C NMR (126 MHz, D2O) δ = 141.4 (C-3), 135.0 (C-7), 130.8 (C-11), 124.5 (C-10), 124.1 (C-6), 120.2 (d, J = 7.3 Hz, C-2), 61.9 (d, J = 4.8 Hz, C-1), 39.6 (C-4), 39.5 (C-5), 26.6 (C-8), 26.4 (C-9), 25.2 (C-15), 17.2 (C-12), 15.9 (C-13), 15.6 (C-14). 31P NMR (202 MHz, D2O) δ = 0.76. HRMS (ESI) m/z: calcd for C15H26O4P [M]2−: 301.1574, found: 301.1575. The spectroscopic data were in agreement with those previously reported.11
Synthesis of compound 60
H2N–L-Ala–γ-D-Glu(O-TMSE)–L-Lys(N-Teoc)–D-Ala–D-Ala–O-TMSE 44 (45.6 mg, 54.7 μmol), PyBOB (32.9 mg, 63.1 μmol), HOBt (8.53 mg, 63.1 μmol) and 7 (27.0 mg, 42.1 μmol) were dissolved in DMF (2 mL) and DIPEA (35.8 μL, 210 μmol) was added immediately. After stirring for 45 min at rt the solvent was removed under reduced pressure and the crude product was purified by flash chromatography (2.5% MeOH/CH2Cl2) to yield 49.8 mg (34.2 μmol, 81%) of a white, crystalline solid.Rf 0.35 (10% MeOH/CH2Cl2). [α]25D = +41° (c = 0.32 in MeOH). 1H NMR (700 MHz, CD3OD) δ = 7.49–7.48 (m, 2H, Harom.), 7.39–7.33 (m, 13H, Harom.), 5.85 (dd, J = 5.9, 3.5 Hz, 1H, H-19), 5.63 (s, 1H, H-22), 5.15–5.08 (m, 4H, 2× CH2–Ph), 4.38 (q, J = 7.1 Hz, 1H, H-16), 4.38 (q, J = 7.1 Hz, 1H, H-6), 4.36–4.31 (m, 3H, H-4, H-12, H-14), 4.20–4.11 (m, 8H, H-2, H-8, H-18, H-31, H-34), 4.04 (dd, J = 9.9, 4.1 Hz, 1H, H-23), 3.85–3.79 (m, 3H, H-17, H-20, H-21), 3.77 (dd, J = 9.9 Hz, J = 9.9 Hz, 1H, H-23), 3.11–3.07 (m, 2H, H-29), 2.26 (t, J = 7.2 Hz, 2H, H-10), 2.22–2.15 (m, 1H, H-11), 1.93–1.89 (m, 1H, H-11′), 1.85 (s, 3H, CH3–NHAc), 1.79–1.74 (m, 1H, H-26), 1.69–1.63 (m, 1H, H-26), 1.48 (p, J = 7.0 Hz, 2H, H-28), 1.39 (d, J = 7.3 Hz, 3H, CH3), 1.37 (d, J = 7.1 Hz, 3H, CH3), 1.35 (d, J = 7.2 Hz, 1H, CH3), 1.36 (d, J = 6.8 Hz 3H, CH3), 1.37–1.32 (m, 2H, H-27), 1.02–0.95 (m, 6H, H-1, H-32, H-35), 0.04 (s, 9H, Si(CH3)3), 0.04 (s, 9H, Si(CH3)3), 0.02 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, CD3OD) δ = 175.6 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 174.9 (CO), 174.7 (CO), 174.6 (CO), 174.4 (CO), 174.1 (CO), 173.8 (NHAc–CO), 172.9 (CO), 159.5 (not resolved in 13C-cpd but hmbc correlation, C-40), 138.9 (Carom.,quart.–Ph), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 130.0 (Carom.), 130.0 (Carom.), 129.9 (Carom.), 129.8 (Carom.), 129.8 (Carom.), 129.3 (Carom.), 129.2(Carom.), 129.2 (Carom.), 127.3 (Carom.), 102.8 (C-22), 97.9 (C-19), 82.3 (C-21), 78.3 (C-16), 76.4 (C-17), 71.2 (d, J = 6.0 Hz, CH2–Ph), 71.1 (d, J = 6.0 Hz, CH2–Ph), 69.1 (C-23), 66.0 (C-20), 64.7 (C-2), 64.5 (C-34), 63.7 (C-31), 55.4 (C-18), 55.1 (C-8) 53.1 (C-12), 50.4 (C-14/C-4/C-6), 50.1 (C-4/C-14/C-6), 49.7 (C-6/C-14/C-4), 41.3 (C-29) 32.4 (C-10), 32.2 (C-26), 30.6 (C-28), 28.2 (C-11), 24.1 (C-27), 22.9 (NHAc–CH3), 19.9 (CH3), 18.7 (C-32), 18.5 (CH3), 18.3 (C-35), 18.2 (C-1), 17.9 (CH3), 17.4 (CH3), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3), −1.5 (Si(CH3)3). 31P NMR (202 MHz, CD3OD) δ = −3.0. HRMS (ESI) m/z: calcd for C68H106N7O20Si3Pna [M + Na]+: 1478.6430, found: 1478.6431. The spectroscopic data were in agreement with those previously reported.15
O), 174.9 (CO), 174.7 (CO), 174.6 (CO), 174.4 (CO), 174.1 (CO), 173.8 (NHAc–CO), 172.9 (CO), 159.5 (not resolved in 13C-cpd but hmbc correlation, C-40), 138.9 (Carom.,quart.–Ph), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 130.0 (Carom.), 130.0 (Carom.), 129.9 (Carom.), 129.8 (Carom.), 129.8 (Carom.), 129.3 (Carom.), 129.2(Carom.), 129.2 (Carom.), 127.3 (Carom.), 102.8 (C-22), 97.9 (C-19), 82.3 (C-21), 78.3 (C-16), 76.4 (C-17), 71.2 (d, J = 6.0 Hz, CH2–Ph), 71.1 (d, J = 6.0 Hz, CH2–Ph), 69.1 (C-23), 66.0 (C-20), 64.7 (C-2), 64.5 (C-34), 63.7 (C-31), 55.4 (C-18), 55.1 (C-8) 53.1 (C-12), 50.4 (C-14/C-4/C-6), 50.1 (C-4/C-14/C-6), 49.7 (C-6/C-14/C-4), 41.3 (C-29) 32.4 (C-10), 32.2 (C-26), 30.6 (C-28), 28.2 (C-11), 24.1 (C-27), 22.9 (NHAc–CH3), 19.9 (CH3), 18.7 (C-32), 18.5 (CH3), 18.3 (C-35), 18.2 (C-1), 17.9 (CH3), 17.4 (CH3), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3), −1.5 (Si(CH3)3). 31P NMR (202 MHz, CD3OD) δ = −3.0. HRMS (ESI) m/z: calcd for C68H106N7O20Si3Pna [M + Na]+: 1478.6430, found: 1478.6431. The spectroscopic data were in agreement with those previously reported.15
Debenzylation of compound 60: synthesis of compound 70
To a solution of glycopeptide 60 (35.6 mg, 24.4 μmol) in MeOH (2.5 mL) was added Pd–C (40.0 mg, Pd-10%). The reaction vessel was filled with hydrogen. After stirring for 30 min at rt full conversion was detected by mass spectrometry and the suspension was filtered over Celite. The precipitate was washed with methanol and the solvent was removed under reduced pressure to yield 29.0 mg (24.4 μmol, quant.) of a colorless, crystalline solid, which was used for the next reaction without further purification or analysis.HRMS (ESI) m/z: calcd for C54H93N7O20Si3P [M − H]−: 1274.5526, found: 1274.5628. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 61
Phosphate 70 (27.0 mg, 21.2 μmol) was dissolved in 80% AcOH/water (3 mL) and stirred for 2 d at rt. Mass spectrometry showed full conversion at this point and toluene (20 mL) was added and the solvent was removed under reduced pressure to yield 25.1 mg (21.1 μmol, quant.) of a white, amorphous solid, which was used for the next reaction without further purification or analysis.HRMS (ESI) m/z: calcd for C47H89N7O20Si3P [M − H]−: 1186.5213, found: 1186.5334. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 62
Farnesol phosphate 8 (60.0 mg, 200 μmol) was coevaporated two times from toluene (1 mL) under argon and dissolved in 50% DMF/THF (1.4 mL). Carbonyldiimidazole (CDI, 130 mg, 799 μmol) was dissolved in 50% DMF/THF (1 mL) and added to the farnesol phosphate solution. The reaction was stirred for 2 h at rt and MeOH (35 μL) was added before stirring for further 45 min. The solvents were removed under reduced pressure and the synthesized phosphoimidazole intermediate A was coevaporated twice from toluene (1 mL) under argon before the addition of DMF (1 mL). Mass spectrometry showed the successfully synthesized A.HRMS (ESI) m/z: calcd for C18H28N2O3P [M − H]−: 351.1843, found: 351.1835.
In the meantime 61 (28.0 mg, 23.6 μmol) was coevaporated first from pyridine (200 μL) and then, twice from toluene (1 mL) under argon. DMF (1 mL) and the freshly prepared phosphoimidazole solution were added and the mixture was stirred for 3 d at rt. The solvent was removed under reduced pressure and the crude product was semi-purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) to yield 26.8 mg (<18.2 μmol, <77%) of a colorless, amorphous solid, which was used for the next reaction without further purification or analysis.
Rf 0.80 (4 MeOH/2 CHCl3/0.5 water). HRMS (ESI) m/z: calcd for C62H114N7O23Si3P2 [M − H]−: 1470.6755, found: 1470.6750. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 3
Protected, semi-pure 62 (26.8 mg, <18.2 μmol) was coevaporated from toluene (1 mL) twice and dissolved in DMF (0.5 mL). The solution was cooled to 0 °C and a 1 M solution of tetrabutylammonium fluoride in THF (TBAF, 419 μL, 419 μmol) was added. The reaction mixture was allowed to warm to rt and was stirred for 2 d at rt before removing the solvent under reduced pressure. The crude product was purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) and the tetrabutylammonium counterions were exchanged to ammonium ions using percolation through Dowex® 50WX8 with a 0.02 M NH4HCO3 solution. Dowex® 50WX8 was washed before usage with 3:1 NH3/water and 0.02 M NH4HCO3 solution until pH of the eluent turned 8. The solvent was removed by lyophilisation to yield 20.3 mg (<18.0 μmol) of the semi-purified 3. Final purification was achieved by HPLC (25–50% NH4HCO3 (0.1% aq.)/methanol, retention time 8.9 min, using a KNAUER Eurospher II 100-5 C8; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 205 nm) yielding 10.7 mg (7.28 μmol, 40% over four steps) of a white, amorphous solid.
Rf 0.46 (3 MeOH/3 CHCl3/1 water). [α]25D = +59° (c = 0.51 in MeOH). 1H NMR (500 MHz, D2O) δ = 5.50–5.47 (m, 1H, H-17), 5.46 (t, J = 7.1 Hz, 1H, H-31), 5.22 (t, 1H, J = 6.5 Hz, H-35), 5.20 (t, J = 7.8 Hz, 1H, H-39), 4.54–4.49 (m, 2H, H-30), 4.35 (q, J = 7.2 Hz, 1H, H-4), 4.29 (q, J = 7.2 Hz, 1H, H-12), 4.25–4.22 (m, 2H, H-6, H-14), 4.20–4.10 (m, 3H, H-2, H-10, H-16), 3.98–3.96 (m, 1H, H-18), 3.92–3.84 (m, 2H, H-20), 3.80 (dd, J = 9.6 Hz, J = 9.6 Hz, 1H, H-15), 3.66 (dd, J = 9.6 Hz, J = 9.6 Hz, 1H, H-19), 3.02 (t, J = 7.5 Hz, 2H, H-26), 2.32 (t, J = 7.8 Hz, 2H, H-8), 2.20–2.15 (m, 3H, H-9, H-34), 2.15–2.11 (m, 4H, H-33, H-38), 2.04 (t, J = 7.1 Hz, 2H, H-37), 2.02 (s, 3H, CH3–NHAc), 1.93–1.89 (m, 1H, H-9), 1.87–1.77 (m, 2H, H-23), 1.74 (s, 3H, H-43), 1.72–1.70 (m, 2H, H-25), 1.70 (s, 3H, H-42), 1.64 (s, 6H, H-41, H-44), 1.52–1.47 (m, 2H, H-24), 1.46 (d, J = 7.3 Hz, 3H, H-28), 1.43 (d, J = 6.8 Hz, 3H, H-29), 1.39 (d, J = 7.2 Hz, 3H, H-22), 1.35 (d, J = 7.2 Hz, 3H, H-21). 13C NMR (176 MHz, D2O) δ = 179.8 (C-1), 177.6 (C-27), 175.8 (C-11), 175.7 (C-7), 174.1 (C-5), 174.1 (NHAc–CO), 174.0 (C-3), 173.6 (C-13), 143.2 (C-32), 136.7 (C-36), 133.4 (C-40), 124.4 (C-39), 124.2 (C-35), 119.3 (C-31), 94.7 (C-17), 79.9 (C-15), 78.0 (C-4), 73.0 (C-18), 68.0 (C-19), 63.1 (C-30), 60.3 (C-20), 54.2 (C-10), 54.2 (C-6) 53.4 (C-16), 51.0 (C-2), 49.9 (C-12), 49.6 (C-14), 39.1 (C-26), 38.8 (C-33), 38.8 (C-37), 31.7 (C-8), 30.5 (C-23), 28.1 (C-9), 26.3 (C-25), 25.8 (C-38), 25.5 (C-34), 24.9 (C-42), 22.2 (NHAc–CH3), 22.0 (C-24), 18.6 (C-22), 17.4 (C-21), 17.0 (C-41), 16.8 (C-28), 16.5 (C-29), 15.7 (C-43), 15.3 (C-44). 31P NMR (284 MHz, D2O) δ = −10.9 (d, J = 14.0 Hz), −13.4 (d, J = 14.0 Hz). HRMS (ESI) m/z: calcd for C46H78N7O21P2 [M − H]−: 1126.4731, found: 1126.4715. The spectroscopic data were in agreement with those previously reported.31
Synthesis of compound 4
In vitro synthesis of compound 4 was performed in a total volume of 4.5 mL containing 1.50 mg (1.33 μmol) compound 3, 2 mM UDP–D-GlcNAc, 50 mM NaPi and 0.5 mM MgCl2, pH 6.5. The reaction was initiated by the addition of 300 μg of MurG–His6. After incubation for 4 h at 30 °C the reaction was quenched by the addition of 9 mL MeOH and evaporated to dryness. Dried samples were dissolved in distilled water for mass spectrometric analysis. The crude product was purified by HPLC (35–50% NH4HCO3 (0.1% aq.)/methanol, retention time 8.0 min, using a KNAUER Eurospher II 100-5 C8; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 205 nm) yielding 0.98 mg (0.73 μmol, 55%) of a white, amorphous solid. Recombinant MurG–His6 enzyme was overexpressed and purified as described for MurG35 and dialyzed against 10 mM NaPi buffer, pH 7.0.[α]25D = +11° (c = 0.18 in H2O). 1H NMR (700 MHz, D2O) δ = 5.50–5.47 (m, 1H, H-17), 5.46 (t, J = 6.9 Hz, 1H, H-31), 5.23 (t, J = 6.4 Hz, 1H, H-35), 5.20 (t, J = 6.4 Hz, 1H, H-39), 4.63 (d, J = 8.3 Hz, 1H, H-45), 4.51–4.49 (m, 2H, H-30), 4.35 (q, J = 7.1 Hz, 1H, H-4), 4.32–4.27 (m, 2H, H-12, H-14), 4.24–4.20 (m, 2H, H-6, H-10), 4.15–4.12 (m, 3H, H-2, H-16), 3.98–3.89 (m, 4H, H-18, H-19, H-20, H-50), 3.86–3.81 (m (pt), 1H, H-15), 3.79–3.74 (m, 3H, H-20′, H-49, H-50′), 3.58 (pt (dd), J = 8.6 Hz, J = 8.6 Hz, 1H, H-48), 3.46–3.91 (m, 2H, H-46, H-47), 2.97 (t, J = 7.0 Hz, 2H, H-26), 2.34–2.33 (m, 2H, H-8), 2.19–2.17 (m, 3H, H-9, H-34), 2.14–2.11 (m, 4H, H-33, H-38), 2.07 (s, 3H, CH3–NHAc), 2.05 (t, J = 7.7 Hz, 2H, H-37), 2.01 (s, 3H, CH3–NHAc), 1.93–1.89 (m, 1H, H-9′), 1.86–1.82 (m, 1H, H-23), 1.81–1.79 (m, 1H, H-23′), 1.74 (s, 3H, H43), 1.71 (s, 3H, H-42), 1.69–1.66 (m, 2H, H-25), 1.64 (s, 6H, H-41, H-44), 1.47 (d, J = 6.9 Hz, H-28), 1.46 (d, J = 6.8 Hz, 3H, H-29), 1.45–1.39 (m, 2H, H-24), 1.40 (d, J = 7.2 Hz, 3H, H-22), 1.35 (d, J = 7.2 Hz, 3H, H-21). 13C NMR (176 MHz, D2O) δ = 179.8 (C-1), 177.6 (C-27), 175.7 (C-11), 175.7 (C-7), 174.4 (NHAc–CO), 174.2 (NHAc–CO), 174.1 (C-5), 174.0 (C-3), 173.6 (C-13), 143.2 (C-32), 136.7 (C-36), 133.5 (C-40), 124.4 (C-39), 124.2 (C-35), 119.3 (C-31), 100.1 (C-45), 94.2 (C-17), 78.4 (C-14), 77.9 (C-15), 75.9 (C-46), 74.0 (C-48), 73.8 (C-18), 72.4 (C-19), 70.3 (C-47), 63.1 (C-30), 61.1 (C-50/C-20), 59.7 (C-50/C-20), 56.1 (C-49), 54.3 (C-6), 53.7 (C-10), 53.5 (C-16), 51.0 (C-2), 50.0 (C-12), 49.6 (C-4), 39.2 (C-26), 38.8 (C-33), 38.8 (C-37), 31.8 (C-8), 30.5 (C-23), 28.2 (C-9), 26.5 (C-25), 25.8 (C-38), 25.6 (C-34), 24.9 (C-42), 22.2 (C-24), 22.2 (NHAc–CH3), 22.1 (NHAc–CH3), 18.7 (C-22), 17.4 (C-21), 17.0 (C-41), 16.9 (C-28), 16.5 (C-29), 15.7 (C-43), 15.3 (C-44). 31P NMR (284 MHz, D2O) δ = −10.9 (d, J = 20.7 Hz), −13.4 (d, J = 19.2 Hz). HRMS (ESI) m/z: calcd for C54H90N8O26P2 [M − 2H]2−: 664.2726, found: 664.2719, calcd for C54H89N8O26P2 [M − 3H]3−: 442.5127, found: 442.5117. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 64
H2N–L-Ala–γ-D-Glu(O-TMSE)–L-Lys((Gly)5–Teoc)–D-Ala–D-Ala–O-TMSE 45 (21.1 mg, 18.9 μmol), PyBOB (13.4 mg, 25.7 μmol), HOBt (3.47 mg, 25.7 μmol) and acid 7 (11.0 mg, 17.4 μmol) were dissolved in DMF (1.5 mL) and DIPEA (11.7 μL, 68.6 μmol) was added immediately. After stirring for 45 min at rt the solvent was removed under reduced pressure. Cold EtOAc was added and the resulted precipitate was filtered off, washed with cold EtOAc, dissolved in MeOH and the solvent was removed under reduced pressure to yield 26.0 mg (14.9 μmol, 86%) of a colorless, amorphous solid.Rf 0.27 (10% MeOH/CH2Cl2). [α]25D = +25° (c = 0.36 in MeOH). 1H NMR (700 MHz, CD3OD) δ = 7.49–7.47 (m, 2H, Harom.), 7.43–7.34 (m, 13H, Harom.), 5.84 (dd, J = 5.8, 3.6 Hz, 1H, H-19), 5.63 (s, 1H, H-22), 5.15–5.08 (m, 4H, 2× CH2–Ph), 4.38 (q, J = 7.1 Hz, 1H, H-16), 4.38 (q, J = 7.1 Hz, 1H, H-6), 4.35–4.30 (m, 3H, H-4, H-12, H-14), 4.20–4.12 (m, 8H, H-2, H-8, H-18, H-41, H-44), 4.03 (dd, J = 9.8, 4.0 Hz, 1H, H-23), 3.92–3.77 (m, 15H, H-17, H-20, H-21, H-23, H-31, H-33, H-35, H-37, H-39), 3.21–3.19 (m, 2H, H-29), 2.28 (t, J = 7.2 Hz, 2H, H-10), 2.19–2.12 (m, 1H, H-11), 1.91–1.87 (m, 1H, H-11′), 1.85 (s, 3H, CH3–NHAc), 1.77–1.72 (m, 1H, H-26), 1.68–1.62 (m, 1H, H-26), 1.56–1.49 (m, 2H, H-28), 1.40 (d, J = 7.2 Hz, 3H, CH3), 1.37 (d, J = 7.2 Hz, 3H, CH3), 1.37–1.32 (m, 2H, H-27) 1.35 (d, J = 7.2 Hz, 3H, CH3), 1.34 (d, J = 6.8 Hz, 3H, CH3), 1.02–0.94 (m, 6H, H-1, H-42, H-45), 0.05 (s, 9H, Si(CH3)3), 0.04 (s, 9H, Si(CH3)3), 0.02 (s, 9H, Si(CH3)3). 13C NMR (176 MHz, CD3OD) δ = 175.6 (CO), 174.9 (CO), 174.7 (CO), 174.6 (CO), 174.5 (CO), 174.1 (CO), 173.8 (NHAc–CO), 173.4 (CO), 172.9 (CO), 172.7 (CO), 172.7 (CO), 172.1 (CO), 171.5 (CO), 159.4 (C-40), 138.9 (Carom.,quart.–Ph), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 137.0 (d, J = 6.5 Hz, Carom.,quart.–Bn), 130.1 (Carom.), 130.0 (Carom.), 129.9 (Carom.), 129.8 (Carom.), 129.8 (Carom.), 129.3 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 127.3 (Carom.), 102.8 (C-22), 97.9 (C-19), 82.3 (C-21), 78.4 (C-16), 76.5 (C-17), 71.2 (d, J = 6.0 Hz, CH2–Ph), 71.1 (d, J = 6.0 Hz, CH2–Ph), 69.1 (C-23), 66.0 (C-20), 64.7 (C-2), 64.5 (C-44), 64.5 (C-41), 55.5 (C-18), 55.0 (C-8), 53.2 (C-12), 50.3 (C-14/C-4/C-6), 50.1 (C-4/C-14/C-6), 49.7 (C-6/C-14/C-4), 45.0 (CH2–Gly), 44.0 (CH2–Gly), 43.9 (CH2–Gly), 43.8 (CH2–Gly), 43.7 (CH2–Gly), 40.0 (C-29), 32.4 (C-10), 32.2 (C-26), 29.8 (C-28), 28.2 (C-11), 24.1 (C-27), 22.9 (NHAc–CH3), 19.9 (CH3), 18.7 (C-42), 18.5 (CH3), 18.3 (C-45), 18.2 (C-1), 18.0 (CH3), 17.4 (CH3), −1.4 (Si(CH3)3), −1.4 (Si(CH3)3), −1.5 (Si(CH3)3). 31P NMR (284 MHz, CD3OD) δ = −2.6. HRMS (ESI) m/z: calcd for C78H121N12O25Si3pNa [M + Na]+: 1763.7503, found: 1763.7525. The spectroscopic data were in agreement with those previously reported.8
Debenzylation of compound 64: synthesis of compound 71
To a solution of glycopeptide 64 (24.2 mg, 13.9 μmol) in MeOH (2 mL) was added Pd–C (25.0 mg, Pd-10%). The reaction vessel was filled with hydrogen. After stirring for 30 min at rt full conversion was detected by mass spectrometry and the suspension was filtered over Celite. The catalyst was washed with methanol and the solvent was removed under reduced pressure to yield 19.5 mg (12.5 μmol, 90%) of a colorless, crystalline solid, which was used for the next reaction without further purification or analysis.HRMS (ESI) m/z: calcd for C64H108N12O25Si3P [M − H]−: 1559.6599, found: 1559.6516. The spectroscopic data were in agreement with those previously reported.8
Acetal cleavage of compound 71: synthesis of compound 72
Acetal 71 (16.7 mg, 10.7 μmol) was dissolved in 80% AcOH/water (3 mL) and stirred for 2 d at rt. Mass spectrometry showed full conversion at this point and toluene (30 mL) was added and the solvent was removed under reduced pressure to yield 15.7 mg (10.7 μmol, quant.) of a white, crystalline solid, which was used for the next reaction without further purification or analysis.HRMS (ESI) m/z: calcd for C57H104N12O25Si3P [M − H]−: 1471.6286, found: 1471.6201. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 65
Farnesol phosphate 8 (27.7 mg, 92.1 μmol) was coevaporated from toluene (1 mL) two times and dissolved in 50% DMF/THF (1.4 mL). Carbonyldiimidazole (CDI, 59.7 mg, 368 μmol) was dissolved in 50% DMF/THF (1 mL) and added to the farnesol phosphate solution. The reaction was stirred for 2 h at rt and MeOH (16 μL) was added before stirring further 45 min. The solvents were removed under reduced pressure and the synthesized phosphoimidazole intermediate was coevaporated from toluene (1 mL) twice before the addition of DMF (1 mL). Mass spectrometry showed the successful synthesis.HRMS (ESI) m/z: calcd for C18H28N2O3P [M − H]−: 351.1843, found: 351.1835.
In the meantime phosphate 72 (15.7 mg, 10.7 μmol) was coevaporated first from 95 μL pyridine and then twice from 1 mL toluene under argon. DMF (0.5 mL) and the freshly prepared phosphoimidazole solution were added and the reaction was stirred for 3 d at rt. The solvent was removed under reduced pressure and the crude product was semi-purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) to yield 10.9 mg (<6.20 μmol, <58%) of a colorless, amorphous solid, which was used for the next reaction without further purification or analysis.
Rf 0.62 (4 MeOH/2 CHCl3/0.5 water). HRMS (ESI) m/z: calcd for C72H129N12O28Si3P2 [M − H]−: 1755.7828, found: 1755.7826, calcd for C72H128N12O28Si3P2 [M − 2H]2−: 877.3877, found: 877.3879. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 5
Protected, semi-pure 65 (10.9 mg, <6.20 μmol) was coevaporated twice from 1 mL toluene under argon and dissolved in DMF (0.7 mL). The solution was cooled to 0 °C and a 1 M solution of tetrabutylammonium fluoride in THF (TBAF, 116 μL, 116 μmol) was added. The reaction mixture was allowed to warm to rt and was stirred for 2 d at rt before removing the solvent under reduced pressure. The crude product was purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) and the tetrabutylammonium counterions were exchanged to ammonium using percolation through Dowex® 50WX8 with a 0.02 M NH4HCO3 solution. Dowex® 50WX8 was washed before usage with 3:1 NH3/water and 0.02 M NH4HCO3 solution until pH of the eluent turned 8. The solvent was removed by lyophilisation to yield 8.20 mg (<5.80 μmol) of the semi-purified 5. Final purification was achieved using HPLC (25–50% NH4HCO3 (0.1% aq.)/methanol, retention time 8.1 min, using a KNAUER Eurospher II 100-5 C8; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 205 nm) yielding 4.4 mg (3.04 μmol, 22% over four steps) of a white, amorphous solid.8
Rf 0.38 (3 MeOH/3 CHCl3/1 water). [α]25D = +21° (c = 0.44 in MeOH). 1H NMR (700 MHz, D2O) δ = 5.50–5.48 (m, 1H, H-17), 5.46 (t, J = 6.7 Hz, 1H, H-31), 5.22 (t, J = 6.7 Hz, 1H, H-35), 5.20 (t, J = 6.7 Hz, 1H, H-39), 4.54–4.49 (m, 2H, H-30), 4.37 (q, J = 7.2 Hz, 1H, H-14), 4.30 (q, J = 7.2 Hz, 1H, H-4), 4.26–4.17 (m, 3H, H-6, H-10, H-12), 4.16–4.10 (m, 2H, H-2, H-16), 4.08 (s, 2H, H-52), 4.03 (H-48/H-50), 4.01 (H-48/H-50), 3.98–3.96 (m, 1H, H-18), 3.91 (s, 2H, H-46), 3.90–3.83 (m, 2H, H-20), 3.82 (s, 2H, H-45), 3.80 (dd, J = 9.6 Hz, J = 9.6 Hz, 1H, H-15), 3.66 (dd, J = 9.6 Hz, J = 9.6 Hz, 1H, H-19), 3.23 (t, J = 6.7 Hz, 2H, H-26), 2.37–2.28 (m, 2H, H-8), 2.20–2.16 (m, 3H, H-9, H-34), 2.15–2.11 (m, 4H, H-33, H-38), 2.04 (t, J = 7.1 Hz, 2H, H-37), 2.02 (s, 3H, CH3–NHAc), 1.93–1.88 (m, 1H, H-9), 1.83–1.73 (m, 2H, H-23), 1.74 (s, 3H, H-43), 1.72–1.70 (m, 2H, H-25), 1.71 (s, 3H, H-42), 1.64 (s, 6H, H-41, H-44), 1.56–1.51 (m, 2H, H-24), 1.46 (d, J = 7.2 Hz, 3H, H-22), 1.43 (d, J = 6.8 Hz, 3H, H-28), 1.38 (d, J = 7.2 Hz, 3H, H-29), 1.35 (d, J = 7.2 Hz, 3H, H-21). 13C NMR (176 MHz, D2O) δ = 179.9 (C-1), 177.7 (C-27), 175.7 (C-11), 175.6 (C-7), 174.2 (C-5), 174.1 (NHAc–CO), 174.0 (C-3), 173.6 (C-13), 172.2 (C-47/C-49/C-51), 172.1 (C-47/C-49/C-51), 172.0 (C-47/C-49/C-51), 171.0 (C-45), 169.9 (C-53) 143.2 (C-32), 136.7 (C-36), 133.4 (C-40), 124.4 (C-39), 124.2 (C-35), 119.3 (C-31), 94.7 (C-17), 79.9 (C-15), 78.0 (C-12), 73.0 (C-18), 68.0 (C-19), 63.2 (C-30), 60.3 (C-20), 54.3 (C-10), 54.3 (C-6), 53.4 (C-16), 51.0 (C-2), 49.8 (C-4), 49.5 (C-14), 42.6 (C-48/C-50), 42.5 (C-48/C-50), 42.5 (C-46), 42.4 (C-52), 41.2 (C-54), 39.0 (C-26), 38.8 (C-33), 38.8 (C-37), 31.8 (C-8), 30.7 (C-23), 28.1 (C-9), 27.8 (C-25), 25.8 (C-38), 25.5 (C-34), 24.9 (C-42), 22.4 (NHAc–CH3), 22.2 (C-24), 18.7 (C-28), 17.4 (C-21), 17.0 (C-41), 16.9 (C-22), 16.6 (C-29), 15.7 (C-43), 15.3 (C-44). 31P NMR (162 MHz, D2O) δ = −10.8 (d, J = 15.4 Hz), −13.3 (d, J = 15.4 Hz). HRMS (ESI) m/z: calcd for C56H93N12O26P2 [M − H]−: 1411.5805, found: 1411.5810, calcd for C56H92N12O26P2 [M − 2H]2−: 705.2866, found: 705.2877. The spectroscopic data were in agreement with those previously reported.8
Synthesis of compound 66
H2N–L-Ala–γ-D-Glu(O-TMSE)–L-Lys((L-Ala)2–Teoc)–D-Ala–D-Ala–OTMSE (55) (16.0 mg, 16.4 μmol, 1.10 equiv.), PyBOP (12.2 mg, 23.4 μmol, 1.50 equiv.) and the free acid 7 (9.6 mg, 14.9 μmol, 1.00 equiv.) were dissolved in DMF (1.34 mL) and DIPEA (10.5 μL, 61.8 μmol, 4.00 equiv.) was added directly. The reaction was stirred for 45 min at room temperature and then the solvent was removed under reduced pressure. The residue was dissolved in cold ethyl acetate and the formed precipitate filtered and washed with cold ethyl acetate. Afterwards it was dissolved in MeOH and the solvent was removed under reduced pressure to yield 6.5 mg of a colorless, crystalline solid (4.07 μmol, 27%).Rf = 0.31 (10% MeOH/CH2Cl2). [α]25D = +39° (c = 0.28 in MeOH). 1H-NMR (700 MHz, CD3OD) δ = 7.50–7.44 (m, 2H, Harom.), 7.40–7.33 (m, 13H, Harom.), 5.85 (dd, J = 3.7 Hz, 2.4 Hz, 1H, H-19), 5.63 (m, 1H, H-22), 5.14–5.08 (m, 4H, 2× CH2–Ph), 4.39 (d, J = 7.1 Hz, 1H, H-6), 4.38 (d, J = 7.1 Hz, 1H, H-16), 4.35–4.30 (m, 3H, H-4, H-12, H-14), 4.21–4.14 (m 8H, H-2, H-8, H-18, H-35, H-40), 4.03 (dd, J = 5.9 Hz, 3.9 Hz, 1H, H-23), 3.86–3.78 (m, 3H, H-17, H-20, H21), 3.78–3.72 (m, 1H, H-23), 3.17–3.14 (m, 2H, H-29), 2.38–2.29 (dt, J = 7.2 Hz, 6.7 Hz, 2H, H-10), 2.25–2.17 (m, 1H, H-11), 1.99–1.93 (m, 2H, H-33, H-31), 1.93–1.87 (m, 1H, H-11), 1.85 (s, 3H, CH3–NHAc), 1.82–1.71 (m, 1H, H-26), 1.69–1.61 (m, 1H, H-26), 1.55–1.48 (m, 2H, H-28), 1.43–1.40 (td, J = 7.5 Hz, 4.5 Hz, 3H, CH3), 1.40–1.38 (dd, J = 7.5 Hz, 3.0 Hz, 3H, CH3), 1.38–1.36 (m, 6H, 2× CH3), 1.34 (s, 3H, CH3), 1.33–1.32 (m, 2H, H-27), 1.33 (s, 3H, CH3), 1.02–0.98 (m, 6H, H-1, H-36, H-41), 0.04 (s, 9H, TMS), 0.04 (s, 9H, TMS), 0.02 (s, 9H, TMS). 13C-NMR (176 MHz, CD3OD) δ = 175.6 (CO), 174.9 (CO), 174.6 (CO), 174.6 (CO), 174.4 (CO), 174.1 (CO), 174.1 (CO), 173.8 (CO), 173.4 (CO), 172.9 (CO), 159.3 (CO), 158.9 (C-34), 138.9 (Carom.,quart.–Ph), 137.0 (Carom.,quart.–Bn), 137.0 (Carom.,quart.–Bn), 130.0 (Carom.), 130.0 (Carom.), 129.9 (Carom.), 129.8 (Carom.), 129.8 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 129.2 (Carom.), 127.3 (Carom.), 102.8 (C-22), 97.9 (C-19), 82.3 (C-21), 78.3 (C-16), 76.4 (C-17), 71.1 (CH2–Ph), 71.1 (CH2–Ph), 69.1 (C-23), 66.0 (C-20), 64.7 (C-2), 64.5 (C-35), 64.5 (C-40), 55.5 (C-18), 55.0 (C-8), 52.2 (C-12), 50.6 (C-14/C-4/C-6), 50.1 (C-4/C-14/C-6), 49.5 (C-6/C-14/C-4), 47.4 (C-31), 47.4 (C-33), 40.0 (C-29), 32.5 (C-10), 32.1 (C-26), 29.8 (C-28), 27.4 (C-11), 24.0 (C-27), 22.9 (NHAc–CH3), 19.9 (CH3), 18.7 (C-36), 18.5 (CH3), 18.3 (C-41), 18.2 (C-1), 18.1 (CH3), 18.0 (CH3), 18.0 (CH3), 17.4 (CH3), −1.4 ((CH3)3Si), −1.5 ((CH3)3Si), −1.5 ((CH3)3Si). HRMS (ESI) m/z: calcd for C74H116N9O22PSi3Na [M + Na]+: 1620.7178, found: 1620.7180.
Debenzylation of compound 66: synthesis of compound 73
Compound 66 (61.6 mg, 38.5 μmol, 1.00 equiv.) was dissolved in MeOH (3.85 mL) and Pd/C (62.8 mg, Pd-10%) was added. The flask was flooded with H2 and was stirred at rt o.n. until full conversion to 73 was detected by mass spectrometry. The reaction mixture was filtered over Celite and the residue was washed with methanol. The solvent was removed under reduced pressure to yield 44.4 mg (31.3 μmol, 81%) of a colorless, crystalline solid which was used without further purification or analysis.HRMS (ESI) m/z: calcd for C60H103N9O22PSi3 [M − H]−: 1416.6269, found: 1416.6289.
Acetal cleavage of compound 73: synthesis of compound 74
Acetal 73 (44.4 mg, 31.3 μmol) was dissolved in 80% AcOH/water (3.13 mL) and the reaction was stirred 2 d at rt. Mass spectrometry showed full conversion at this point and toluene (21 mL) was added and the solvent was removed under reduced pressure to yield 37.1 mg of a colorless, crystalline solid (27.9 μmol, 89%) which was used without further purification or analysis.HRMS (ESI) m/z: calcd for C53H98N9O22Psi3 [M − 2H]2−: 663.2965, found: 663.2962.
Synthesis of silyl-protected compound 75
Farnesol phosphate 8 (78.6 mg, 266.1 μmol) was coevaporated from toluene (2 mL) two times and dissolved in 50% DMF/THF (3.82 mL). Carbonyldiimidazole (CDI, 170.2 mg, 1.06 mmol) was dissolved in 50% DMF/THF (3.82 mL) and added to the farnesol phosphate solution. The reaction was stirred for 2 h at rt and MeOH (76.3 μL) was added before stirring further 45 min at rt. The solvents were removed under reduced pressure and the synthesized phosphoimidazole intermediate was coevaporated from toluene (2 mL) twice before the addition of DMF (3.1 mL). Mass spectrometry showed the successful synthesis.HRMS (ESI) m/z: calcd for C18H28N2O3P [M − H]−: 351.1843, found: 351.1835.
In the meantime phosphate 74 (37.1 mg, 27.9 μmol) was coevaporated first from 0.4 mL pyridine and then twice from 2 mL toluene under argon. DMF (3.1 mL) and the freshly prepared phosphoimidazole solution were added and the reaction was stirred for 3 d at rt. The solvent was removed under reduced pressure and the crude product was semi-purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) to yield 53.9 mg (33.4 μmol, 90%) of a colorless, amorphous solid (75), which was used for the next reaction without further purification or analysis.
Rf 0.78 (4 MeOH/2 CHCl3/0.5 water). HRMS (ESI) m/z: calcd for C68H123N9O24P2Si3 [M − 2H]2−: 791.8737, found: 791.8740.
Synthesis of compound 6
Protected, semi-pure 75 (53.9 mg, 33.4 μmol) was coevaporated twice from 1 mL toluene under argon and dissolved in DMF (0.5 mL). The solution was cooled to 0 °C and a 1 M solution of tetrabutylammonium fluoride in THF (TBAF, 0.70 mL, 701 μmol) was added. The reaction mixture was allowed to warm to rt and was stirred for 2 d at rt before removing the solvent under reduced pressure. The crude product was purified by gel permeation chromatography (Sephadex® LH-20, GE Healthcare, 260 × 20 mm, methanol) and the tetrabutylammonium counterions were exchanged to ammonium using percolation through Dowex® 50WX8 with a 0.02 M NH4HCO3 solution. Dowex® 50WX8 was washed before usage with 3:1 NH3/water and 0.02 M NH4HCO3 solution until pH of the eluent turned 8. The solvent was removed by lyophilisation to yield 21.9 mg (16.7 μmol, 50% over four steps) of 6 as a colorless, amorphous solid, which may further purified by HPLC (25–50% NH4HCO3 (0.1% aq.)/methanol, retention time 8.1 min, using a KNAUER Eurospher II 100-5 C8; 5 μm; 250 × 16 mm + precolumn 30 × 16 mm, 205 nm).
Rf = 0.87 (3 MeOH/3 CH2Cl2/1 water). [α]25D = +33° (c = 0.34 in MeOH). 1H (500 MHz, D2O) δ = 5.50–5.46 (m, 1H, H-17), 5.46 (t, J = 6.8 Hz, 1H, H-37), 5.23 (t, J = 6.8 Hz, 1H, H-41), 5.21–5.16 (m, 1H, H-45), 4.52 4.47 (m, 2H, H-36), 4.36 (q, J = 7.2 Hz, 1H, H-14), 4.31–4.29 (m, 1H, H-4), 4.29–4.24 (m, 3H, H-6, H-10, H-12), 4.23–4.21 (m, 11H, H-28), 4.21–4.19 (m, 1H, H-30), 4.19–4.14 (m, 2H, H-2, H-16), 3.99–3.94 (m, 1H, H-18), 3.91–3.85 (m, 2H, H-20), 3.80 (dd, J = 9.7 Hz, J = 9.7 Hz, 1H, H-15), 3.64 (pt, J = 9.7 Hz, 1H, H-19), 3.26–3.23 (m, 2H, H-26), 2.33–2.28 (m, 2H, H-8), 2.20–2.14 (m, 3H, H-9, H-40), 2.15–2.11 (m, 4H, H-39, H-44), 2.02 (t, J = 7.5 Hz, 2H, H-43), 2.00 (s, 3H, CH3–NHAc), 1.93–1.88 (m, 1H, H-9), 1.83–1.76 (m, 2H, H-23), 1.73 (s, 3H, H-50), 1.72–1.70 (m, 2H, H-25), 1.70 (s, 3H, H-48), 1.64 (s, 3H, H-47), 1.63 (s, 3H, H-49), 1.55 (d, J = 7.1 Hz, 3H, H-32), 1.53–1.50 (m, 2H, H-24), 1.45 (d, J = 7.2 Hz, 3H, H-22), 1.42 (d, J = 6.9 Hz, 3H, H-34), 1.38 (d, J = 7.1 Hz, 3H, H-35), 1.37 (d, J = 7.2 Hz, 3H, H-31), 1.34 (d, J = 7.1 Hz, 3H, H-21). 13C (176 MHz, D2O) δ = 181.4 (C-1), 177.9 (C-33), 175.9 (C-11), 175.8 (C-7), 174.8 (C-5), 174.1 (NHAc–CO), 174.1 (C-3), 173.7 (C-13), 172.1 (C-27), 172.0 (C-29), 143.2 (C-38), 136.8 (C-42), 133.4 (C-46), 124.5 (C-45), 124.4 (C-41), 119.3 (C-37), 94.8 (C-17), 79.9 (C-15), 78.2 (C-12), 73.1 (C-18), 68.2 (C-19), 63.2 (C-36), 58.2 (C-20), 54.3 (C-10), 54.3 (C-6), 53.3 (C-16), 52.2 (C-28), 51.1 (C-2), 50.2 (C-4), 49.9 (C-30), 49.5 (C-14), 38.9 (C-26), 38.9 (C-39), 38.9 (C-43), 31.9 (C-8), 30.7 (C-23), 28.2 (C-9), 27.7 (C-25), 25.9 (C-44), 25.5 (C-40), 25.0 (C-48), 22.5 (NHAc–CH3), 22.2 (C-24), 19.2 (C-32), 18.7 (C-34), 17.5 (C-21), 17.0 (C-47), 17.0 (C-22), 16.7 (C-35), 16.6 (C-31), 15.7 (C-50), 15.3 (C-49). 31P (284 MHz, D2O) δ = −10.8 (d, J = 14.8 Hz), −13.4 (d, J = 14.8 Hz).
HRMS (ESI) m/z: calcd for C53H87N9O25P2 [M − 2H]2−: 655.7650, found: 655.7651, calcd for C53H88N9O25P2 [M − H]−: 1312.5372, found: 1312.5381, calcd for C53H87DN9O25P2 [M − 2H + D]: 1313.5435, found: 1313.5421.
Synthesis of 3-lipid II (E. faecalis, S. pneumoniae)
In vitro synthesis of 3-lipid II (E. faecalis, S. pneumoniae) was performed in a total volume of 30 μL containing 10 μg compound 5, 2 mM UDP–D-GlcNAc, 50 mM NaPi and 0.5 mM MgCl2, pH 6.5. The reaction was initiated by the addition of 2 μg of MurG–His6. Afterincubation for 4 h at 30 °C the reaction was quenched by the addition of 60 μL MeOH and evaporated to dryness. Dried samples were dissolved in distilled water for mass spectrometric analysis. Recombinant MurG–His6 enzyme was overexpressed and purified as described for MurG35 and dialized against 10 mM NaPi buffer, pH 7.0.HRMS (ESI) m/z: calcd for C64H105N13O31P2 [M − 2H]2−: 806.8263, found: 806.8255; calcd for C64H104N13O31P2Na [M − 3H + Na]2−: 817.8173, found: 817.8166; calcd for C64H104N13O31P2 [M − 3H]3−: 537.5484, found: 537.5475. The spectroscopic data were in agreement with those previously reported.8
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the German Research Foundation (DFG), TRR261, project ID 398967434. Furthermore, we thank Andreas Schneider for excellent HPLC support and Sarah Jurytko for technical support.Notes and references
- A. Müller, A. Klöckner and T. Schneider, Nat. Prod. Rep., 2017, 34, 909–932 RSC , https://pubs.rsc.org/en/content/articlehtml/2017/np/c7np00012j.
- B. de Kruijff, V. van Dam and E. Breukink, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2008, 79, 117–121 CrossRef CAS PubMed.
- P. Hardt, I. Engels, M. Rausch, M. Gajdiss, H. Ulm, P. Sass, K. Ohlsen, H.-G. Sahl, G. Bierbaum, T. Schneider and F. Grein, Int. J. Med. Microbiol., 2017, 307, 1–10 CrossRef CAS PubMed.
- A. Bouhss, A. E. Trunkfield, T. D. H. Bugg and D. Mengin-Lecreulx, FEMS Microbiol. Rev., 2008, 32, 208–233 CrossRef CAS PubMed.
- S. A. Hitchcock, C. N. Eid, J. A. Aikins, M. Zia-Ebrahimi and L. C. Blaszczak, J. Am. Chem. Soc., 1998, 120, 1916–1917 CrossRef CAS.
- B. Schwartz, J. A. Markwalder and Y. Wang, J. Am. Chem. Soc., 2001, 123, 11638–11643 CrossRef CAS PubMed.
- H. Men, P. Park, M. Ge and S. Walker, J. Am. Chem. Soc., 1998, 120, 2484–2485 CrossRef CAS.
- L. M. Wingen, M. Rausch, T. Schneider and D. Menche, J. Org. Chem., 2020, 85, 10206–10215 CrossRef CAS PubMed.
- (a) C. A. Smith, J. Mol. Biol., 2006, 362, 640–655 CrossRef CAS PubMed; (b) H. Barreteau, A. Kovac, A. Boniface, M. Sova, S. Gobec and D. Blanot, FEMS Microbiol. Rev., 2008, 32, 168–207 CrossRef CAS.
- J. Nakamura, H. Yamashiro, H. Miya, K. Nishiguchi, H. Maki and H. Arimoto, Chem.–Eur. J., 2013, 19, 12104–12112 CrossRef CAS PubMed.
- L. M. Lira, D. Vasilev, R. A. Pilli and L. A. Wessjohann, Tetrahedron Lett., 2013, 54, 1690–1692 CrossRef CAS.
- (a) A. Katsuyama, K. Sato, F. Yakushiji, T. Matsumaru and S. Ichikawa, Chem. Pharm. Bull., 2018, 66, 84–95 CrossRef CAS PubMed , https://www.jstage.jst.go.jp/article/cpb/66/1/66_c17-00828/_article/-char/ja/; (b) S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef CAS PubMed.
- Part of these results have been previously communicated: (8).
- L. Chen, H. Men, S. Ha, X.-Y. Ye, L. Brunner, Y. Hu and S. Walker, Biochemistry, 2002, 41, 6824–6833 CrossRef CAS PubMed.
- S. Ha, E. Chang, M.-C. Lo, H. Men, P. Park, M. Ge and S. Walker, J. Am. Chem. Soc., 1999, 121, 8415–8426 CrossRef CAS.
- M. S. VanNieuwenhze, S. C. Mauldin, M. Zia-Ebrahimi, J. A. Aikins and L. C. Blaszczak, J. Am. Chem. Soc., 2001, 123, 6983–6988 CrossRef CAS PubMed.
- M. M. Sim, H. Kondo and C. H. Wong, J. Am. Chem. Soc., 1993, 115, 2260–2267 CrossRef CAS.
- (a) A. Gansäuer and T. Dahmen, Chimia, 2012, 66, 433–434 CrossRef PubMed; (b) A. J. Pearson and K. Lee, J. Org. Chem., 1994, 59, 2304–2313 CrossRef CAS.
- R. Pascal and R. Sola, Tetrahedron Lett., 1998, 39, 5031–5034 CrossRef CAS.
- (a) J. M. Benito and M. Meldal, QSAR Comb. Sci., 2004, 23, 117–129 CrossRef CAS; (b) L. Sambri, G. Bartoli, G. Bencivenni and R. Dalpozzo, Curr. Org. Synth., 2012, 9, 137–148 CrossRef CAS.
- A. Mehta, R. Jaouhari, T. J. Benson and K. T. Douglas, Tetrahedron Lett., 1992, 33, 5441–5444 CrossRef CAS.
- (a) R. Kaul, Y. Brouillette, Z. Sajjadi, K. A. Hansford and W. D. Lubell, J. Org. Chem., 2004, 69, 6131–6133 CrossRef CAS PubMed; (b) B. Li, M. Berliner, R. Buzon, C. K.-F. Chiu, S. T. Colgan, T. Kaneko, N. Keene, W. Kissel, T. Le, K. R. Leeman, B. Marquez, R. Morris, L. Newell, S. Wunderwald, M. Witt, J. Weaver, Z. Zhang and Z. Zhang, J. Org. Chem., 2006, 71, 9045–9050 CrossRef CAS PubMed.
- A. Trzeciak and W. Bannwarth, Synthesis, 1996, 1996, 1433–1434 CrossRef.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- M. L. Di Gioia, P. Costanzo, A. de Nino, L. Maiuolo, M. Nardi, F. Olivito and A. Procopio, RSC Adv., 2017, 7, 36482–36491 RSC.
- G. W. Liechti, E. Kuru, E. Hall, A. Kalinda, Y. V. Brun, M. VanNieuwenhze and A. T. Maurelli, Nature, 2014, 506, 507–510 CrossRef CAS PubMed.
- R. Bollhagen, M. Schmiedberger, K. Barlos and E. Grell, J. Chem. Soc., Chem. Commun., 1994, 2559 RSC.
- M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC.
- I. Shiina, M. Kubota and R. Ibuka, Tetrahedron Lett., 2002, 43, 7535–7539 CrossRef CAS.
- T. I. Al-Warhi, H. M. Al-Hazimi and A. El-Faham, J. Saudi Chem. Soc., 2012, 16, 97–116 CrossRef CAS.
- X. Y. Ye, M. C. Lo, L. Brunner, D. Walker, D. Kahne and S. Walker, J. Am. Chem. Soc., 2001, 123, 3155–3156 CrossRef CAS PubMed.
- C.-Y. Liu, C.-W. Guo, Y.-F. Chang, J.-T. Wang, H.-W. Shih, Y.-F. Hsu, C.-W. Chen, S.-K. Chen, Y.-C. Wang, T.-J. R. Cheng, C. Ma, C.-H. Wong, J.-M. Fang and W.-C. Cheng, Org. Lett., 2010, 12, 1608–1611 CAS.
- K. Trainor, J. A. Palumbo, D. W. S. MacKenzie and E. M. Meiering, Protein Sci., 2020, 29, 306–314 CrossRef CAS PubMed.
- Y. Zhang, E. J. Fechter, T.-S. A. Wang, D. Barrett, S. Walker and D. E. Kahne, J. Am. Chem. Soc., 2007, 129, 3080–3081 CrossRef CAS PubMed.
- T. Schneider, T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventós, S. Neve, B. Ravn, A. M. J. J. Bonvin, L. de Maria, A. S. Andersen, L. K. Gammelgaard, H.-G. Sahl and H.-H. Kristensen, Science, 2010, 328, 1168–1172 CrossRef CAS PubMed.
- M. S. VanNieuwenhze, S. C. Mauldin, M. Zia-Ebrahimi, B. E. Winger, W. J. Hornback, S. L. Saha, J. A. Aikins and L. C. Blaszczak, J. Am. Chem. Soc., 2002, 124, 3656–3660 CrossRef CAS PubMed.
- V. L. Healy, I. A. D. Lessard, D. I. Roper, J. R. Knox and C. T. Walsh, Chem. Biol., 2000, 7, R109–R119 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01915a |
| ‡ Present address: Boehringer Ingelheim, Ingelheim, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |