
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of 2-bromopropionyl esterified cellulose nanofibrils as hydrophobic coating and film†
Mengzhe Guo and
You-Lo Hsieh*
and
You-Lo Hsieh*
Department of Biological and Agricultural Engineering, University of California, Davis, California 95616, USA. E-mail: ylhsieh@ucdavis.edu; Tel: +1 530 752 0843
First published on 17th May 2022
Abstract
Hydrophobic 2-bromopropionyl esterified cellulose nanofibrils (Br-CNFs) have been facilely produced via one-pot esterification of cellulose with 2-bromopropionyl bromide (BPB) then in situ disintegrated by ultrasonication in the same reaction media. Br-CNFs optimally produced by this robust esterification-ultrasonication approach, i.e., 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 BPB to anhydroglucose (AGU) molar ratio, 23 °C, 6 h and ultrasonication (50% amplitude, 30 min), were 4.6 nm thick, 29.3 nm wide, and 1 μm long in 71% yield and 48% crystallinity. Successful 2-bromopropionyl esterification of cellulose was confirmed by FTIR and 1H NMR. The degree of substitution (DS) of surface hydroxyl to 2-bromopropanoate was determined to be between 0.53 (DSs) based on XRD and Br-CNF dimensions and 0.56 (DSNMR) from solution-state 1H NMR. Br-CNF dispersions in DMF exhibited Newtonian behaviors at concentrations below and shear thinning behaviors above 0.5%, enabling homogeneous deposition at dilute concentrations up to 0.01% into a few nm ultra-thin layers as well as blade coating of gel into ca. 100 μm thick film, all similarly hydrophobic with surface water contact angles (WCAs) in the range of 70–75°. The ultra-high modulus and strength film from gel coating further showed the potential for dual high-strength and hydrophobic applications of Br-CNFs.
:1 BPB to anhydroglucose (AGU) molar ratio, 23 °C, 6 h and ultrasonication (50% amplitude, 30 min), were 4.6 nm thick, 29.3 nm wide, and 1 μm long in 71% yield and 48% crystallinity. Successful 2-bromopropionyl esterification of cellulose was confirmed by FTIR and 1H NMR. The degree of substitution (DS) of surface hydroxyl to 2-bromopropanoate was determined to be between 0.53 (DSs) based on XRD and Br-CNF dimensions and 0.56 (DSNMR) from solution-state 1H NMR. Br-CNF dispersions in DMF exhibited Newtonian behaviors at concentrations below and shear thinning behaviors above 0.5%, enabling homogeneous deposition at dilute concentrations up to 0.01% into a few nm ultra-thin layers as well as blade coating of gel into ca. 100 μm thick film, all similarly hydrophobic with surface water contact angles (WCAs) in the range of 70–75°. The ultra-high modulus and strength film from gel coating further showed the potential for dual high-strength and hydrophobic applications of Br-CNFs.
Introduction
Cellulose is not only the most abundant natural polymer on earth with renewable annual production of 75 to 100 billion tons,1 but also the most chemically homogeneous and intrinsically semi-crystalline. The crystalline domains can be isolated as a few to tens nm wide and hundreds nm long rod-like cellulose nanocrystals (CNCs) or thinner and longer cellulose nanofibrils (CNFs). These so called nanocelluloses have gained increasing attention due to their ultra-high elastic modulus (150 GPa for CNCs, 28 GPa for CNFs),2,3 low axial thermal expansion coefficient (10−7 K−1 for CNCs, 5 × 10−6 K−1 for CNFs),4,5 and biocompatibility.6,7 CNCs and CNFs have been most commonly produced from cellulose by removing the non-crystalline regions via acid hydrolysis,8–12 modifying them by oxidation,12–18 disintegration via mechanical forces,12–17,19–22 or a combination of the latter two.13–17 These nanocelluloses8–22 are all hydrophilic, some with anionic charges, making them easily dispersible in aqueous media, but incompatible with less polar and non-polar organic liquids and most synthetic polymers.To render these hydrophilic nanocelluloses compatible with organic media and polymers for broader applications, various physical or chemical means have been explored23–33 and reviewed.34 CNCs were freeze-dried then ultrasonicated23–25 and CNFs were acetone exchanged then homogenized26 to be dispersible in DMF. Chemical reactions, such as esterification,27–29 acetylation,30,31 silanation,32 and amidation33 have been applied to convert the hydrophilic hydroxyls27–32 and carboxyls33 of CNCs27–29 and CNFs30–33 to the more hydrophobic long alkyl chains27–29,32,33 or acetyl groups.30,31 Alkyl bromide has also been introduced to sulfuric acid hydrolyzed CNC35 and TEMPO-oxidized and homogenized CNF36 using 2-bromoisobutyryl bromide (BIB) aided by 4-dimethylaminopyridine (DMAP) catalyst to improve their respective dispersity in DMF35 and anisole.36 To date, efforts to generated hydrophobic nanocelluloses have been limited, mainly from modification of already fabricated nanocelluloses.27–36 On product of solvent cast CNC37 and vacuum filtrated CNF,38 nanopaper was esterified37 or acetylated38 to be more hydrophobic.
Producing organically dispersible or hydrophobic nanocelluloses from direct modification of cellulose is scarce. Esterification of cellulose with acetic anhydride followed by multiple processes of refining, cryo-crushing and homogenization has shown to generate ethanol and acetone suspensible cellulose and 10–50 nm wide CNFs, but the extent of CNFs was not reported.39 To diversify chemistry, we have successfully established the one-pot solventless telomerization of 1,3-butadiene on cellulose to convert the hydroxyls to 2,7-octadienyl ether (ODE), an 8-carbon diene, then mechanical blending of aqueous ODE-cellulose suspensions to generate hydrophobic ODE-nanocellulose in the precipitates that were 27 to 41% dispersible in DMF, DMSO, and chloroform.40 To advance this sequential functionalization-disintegration of cellulose approach to produce hydrophobic nanocelluloses, one-pot synthesis of hydrophobic cellulose followed by direct disintegration in the same organic media into hydrophobic and organic dispersible nanocelluloses would simplify the process further.
Herein, a one-pot synthesis of hydrophobic cellulose coupled with in situ disintegration in the same organic liquid was developed and streamlined. First and foremost, rationally designed 2-bromopropionyl esterification was applied to convert the accessible cellulose C2, C3 and C6 hydroxyls into organic compatible 2-bromopropanoates. While both 2-bromopropionyl bromide (BPB)41 and 2-bromoisobutyryl bromide (BIB)42 were effective in acylating ionic liquid dissolved wood pulp cellulose to become DMF soluble, the more chemically stable BPB with secondary carbon as relatively poor nucleophile was selected to be the bromine provider for the heterogeneous esterification of cellulose solids. The extent of esterification of cellulose necessary to allow disintegration in organic media was studied by sequentially varying bromine provider BPB quantity to anhydroglucose unit (AGU) molar ratios (1:1 to 10:1), reaction times (1 to 12 h), then temperatures (23 to 90 °C). DMF, a common solvent for cellulose ester,43,44 was used as the media for reaction as well as dispersing and disintegrating 2-bromopropionyl esterified cellulose (Br-Cell) into nanocelluloses (Br-NCs) by ultrasonication. Ultrasonication that has shown to be effective to disintegrate TEMPO-oxidized wood cellulose (0.01 w/v%) in aqueous media into 3.6 (±0.3) nm wide CNF with ca. 100 length-to-width ratio45 was carried out at a range of power levels by varying amplitudes and lengths of time. The optimal esterification reaction and ultrasonication conditions were determined by evaluating the quantities and qualities of DMF-dispersible Br-NCs imaged by atom force microscopy (AFM) and transmission electron microscopy (TEM). The structures of Br-CNFs were further characterized by Fourier-transform infrared (FTIR) and liquid phase proton nuclear magnetic resonance (1H NMR) spectroscopy. Thermal properties and crystallinity of Br-CNFs were characterized by thermogravimetric analysis (TGA) and X-ray diffraction (XRD), respectively. Moreover, the viscosity and wetting behaviors of thin layer hydrophobic Br-NCs on HOPG or blade coated film on glass were evaluated by water contact angle (WCA) measurement for potential surface modification and coating applications.
Experimental section
Materials
Cellulose was isolated from rice straw (Calrose variety) by a previously reported three-step 2:1 v/v toluene/ethanol extraction, acidified NaClO2 (1.4%, pH 3–4, 70 °C, 5 h) delignification, alkaline hemicellulose dissolution (5% KOH, 90 °C, 2 h) process and lyophilized (Labconco lyophilizer).46 2-Bromopropionyl bromide (BPB, 97%, Alfa Aesar), 4-dimethylaminopyridine (DMAP, 99%, Acros Organics), potassium bromide (KBr, spectroscopic grade 99.9+%, Fisher Scientific), acetone (histological grade, Fisher Scientific), N,N-dimethylformamide (DMF, certified grade, Fisher Scientific) and trifluoroacetic acid (99%, Sigma Aldrich) were used as received without further purification. All water used was purified by Milli-Q Advantage water purification system (Millipore Corporate, Billerica, MA). For AFM imaging, mica (highest-grade V1 mica discs, 10 mm, Ted Pella, Inc. Redding, CA) and highly oriented pyrolytic graphite (HOPG, grade ZYB) were used. For TEM, carbon grids (300-mesh copper, formvar-carbon, Ted Pella Inc.) were used. For UV-vis spectrophotometry, 1 mm UV-vis standard cell quartz cuvettes (Fisher Scientific) were used.
Synthesis of 2-bromopropionyl esterified cellulose (Br-Cell)
Esterification of cellulose was performed using 2-bromopropionyl bromide (BPB) at varying BPB to cellulose anhydroglucose unit (AGU) ratios (1:1 to 10:1 BPB:AGU), reaction times (1 to 12 h), and temperatures (23 to 90 °C) (Scheme 1). Based on 162 g mol−1 for AGU, there is 6.2 mmol AGU per gram of cellulose40 or 0.2 mmol amorphous AGU per gram of cellulose using an estimated 0.67 crystallinity for rice straw cellulose from the average 0.618 (ref. 46) and 0.722 (ref. 12) by XRD. Freeze-dried cellulose (0.50 g, 3.1 mmol AGU) was added to DMF (30 mL) and stirred until homogeneously dispersed. At 5:1 BPB:AGU, BPB (3.33 g, 15.4 mmol) and DMAP (0.05 g, 0.4 mmol) catalyst were dissolved in DMF (10 mL) in an ice bath under constant vortexing, then added to the cellulose dispersion to start the reaction and stopped by vacuum filtration. The reacted cellulose solids were rinsed with acetone three times to remove residual BPB, DMAP and DMF, then dried at 55 °C overnight to obtain Br-Cell.
 | ||
| Scheme 1 2-Bromopropionyl esterification of cellulose and ultrasonication of Br-Cell to generate hydrophobic Br-CNFs in the supernatant and Br-Cell microfibrils in the precipitate. | ||
The extent of reaction in Br-Cell was determined by mass gain and expressed as Br add-on (σ, mol g−1):
 | (1) |
Generation of 2-bromopropionyl esterified nanocelluloses (Br-NCs) by ultrasonication
Br-Cell (0.1 g) was resuspended in 100 mL DMF at 0.1 w/v% and ultrasonicated (Qsonica Q700, 50/60 Hz) at varied amplitudes (25–100%) and times (10–120 min) in an ice bath and 10 minute time intervals to disintegrate the microfibers. All ultrasonicated dispersions were centrifuged (Eppendorf 5804R, 5k rpm, 10 min) to collect the clear Br-NC containing supernatants and Br-Cell precipitates for further characterization. Mass of air-dried Br-Cell precipitates were determined gravimetrically and subtracted from initial Br-Cell mass to derive the Br-NC quantities in supernatants. The Br-NC as percentage of the initial Br-Cell was reported.Characterizations
The morphologies of dried Br-Cells were imaged by optical microscopy (Leica DM2500). Br-Cell was redispersed at 0.1 w/v% in DMF and 10 μL droplets were deposited on glass slides to measure the width and length (n > 100) of microfibers. Their averages and standard deviations were reported. Br-NCs in DMF dispersions were imaged by AFM and TEM on different substrates. Br-NCs (10 μL, 0.0005 w/v%) were deposited on freshly cleaved hydrophilic mica or relatively hydrophobic highly oriented pyrophoric graphite (HOPG), then air-dried in fume hood for 6 h and profiled by AFM in the tapping mode with scan size and rate set to 5 μm × 5 μm and 512 Hz. Br-NCs (10 μL, 0.0005 w/v%) were deposited onto both glow and non-glow discharged carbon-coated TEM grids, and excess liquid was removed after 5 min by blotting with a filter paper. The specimens were negatively stained with aqueous uranyl acetate (2 w/v%) and blotted to remove excess solution with filter paper, repeated five times then dried under the ambient condition for 15 min. The samples were observed using a Philip CM12 transmission electron microscope at a 100 kV accelerating voltage. The lengths and widths of CNFs were measured using ImageJ Analyzer (ImageJ, NIH, USA) and their means and standard deviations reported.For 1H NMR, 40 mL acetone was added into Br-CNF3 in DMF dispersion (10 mL, 0.50 w/v%) and centrifuged (5k rpm, 10 min) to decant the supernatant. This process repeated three times to prepare Br-CNF3 acetone gel. Br-CNF3 acetone gel (ca. 5 mg) was added into 1 mL DMSO-d6, then sonicated (10 min, Branson 2510), and vacuum evaporated at 50 °C or 80 °C for 1 h. This sonication-evaporation process was repeated three times to remove residual acetone. After centrifugation (5k rpm, 10 min), Br-CNF3 in DMSO-d6 supernatant was collected for 1H NMR (Bruker AVIII 800 MHz 1H NMR spectrometer) characterization. Around 1 mL supernatant was placed in one NMR tube with 50 μL trifluoroacetic acid added to shift all OHs peak downfield to above 4.5 ppm.
Transparent FTIR pellets were prepared by mixing 3 mg of oven dried Br-Cell, Br-CNF3 and Br-Cell3 precipitates with 300 mg KBr after 1 min pressurization under 800 MPa barrel chamber, then scanned by Thermo Nicolet 6700 spectrometer under ambient conditions from an accumulation of 64 scans at a 4 cm−1 resolution from 4000 to 400 cm−1. TGA were performed on a TGA-50 thermogravimetric analyzer (Shimadzu, Japan) by heating 5 mg dry sample at 10 °C min−1 from 25 to 500 °C under purging N2 (50 mL min−1).
The crystalline structures were determined by XRD using a PANalytical X'pert Pro powder diffractometer with a Ni-filtered Cu Kα radiation (λ = 1.5406 Å) at 45 kV anode voltage and 40 mA current. Br-Cell3 powder was rinsed three times with acetone and oven-dried (55 °C) overnight. Br-CNF3 film was generated from 0.5 w/v% DMF dispersions by evaporating DMF in fume hood for 7 d. The samples were fixed on stage by double-sided tape, then diffractograms were recorded from 5 to 40° at a scan rate of 2° min−1. Crystallinity index (CrI) was calculated using the intensity of the 200 peak (I200, 2θ = 22.5°) and the intensity minimum between the peaks at 200 and 110 (Iam, 2θ = 19.0°) as follows47
 | (2) |
The crystallite dimension (Dhkl) was calculated using the Scherrer equation48
 | (3) |
Br-CNF3 at concentrations of 0.0005 to 0.01 w/v% were deposited on freshly exfoliated graphite and allowed to air-dry for 6 h. 0.5 w/v% Br-CNF3 DMF dispersion was concentrated to 2.5 w/v% Br-CNF3 organogel by ambient drying in fume hood for 4 d, then 5 mL gel (1 mm thickness) was coated on glass using a Doctor-Blade film coater (INTSUPERMAI Adjustable Film Applicator Coater KTQ-II) as one hundred μm thick film after ambient drying overnight. Water contact angles (WCAs) of sessile drops (5 μL) on fresh mica, exfoliated graphite, carbon, glow-discharged carbon and film coated glass, as well as single and double deposited Br-CNF3 on graphite were measured using the ImageJ Analyzer and the average values were calculated from both sides of a sessile drop reported in total of 5 images for each (n = 5). The root mean square (RMS) of Br-CNF deposited graphite surfaces were measured from microscopic peaks and valleys of AFM images.
Br-CNF3 in DMF dispersions were serial diluted from 0.5 w/v% to 0.25, 0.13 and 0.06 w/v% then scanned by UV-vis spectroscopy (Thermo Scientific, Evolution 600) from 325 to 800 cm−1 at 4 cm−1 s−1. Viscosities of Br-CNF3 DMF dispersions were measured at 25 °C with shear rates from 1 to 220 s−1 using a Brookfield DV3T rheometer.
Results and discussion
2-Bromopropionyl esterification of cellulose
Cellulose was isolated from rice straw at 35.0 ± 3.3% (n = 10) yield, comparable to previous reported value,42 and freeze-dried to a white fluffy mass. 2-Bromopropionyl esterification of cellulose was conducted under varying BPB:AGU molar ratios (1:1 to 10:1), reaction times (1 to 12 h), and temperatures (23 to 90 °C) to evaluate their effects on the extent of hydroxyl to ester conversion or Br add-on (σ, mmol g−1) (Scheme 1). At 23 °C for 12 h, a 5 time increase of BPB:AGU ratio from 1:1 to 5:1 led to a 12 time increase in the Br add-on (σ) from 0.6–6.0 mmol g−1 while further doubling the ratio only increase σ by another 20% to 7.2 mmol g−1 (Fig. 1a). The optimal 5:1 BPB:AGU ratio was selected to vary the length of reaction at 23 °C. Br add-on (σ) increased from 3.4 to 5.7 mmol g−1, showing close to linear relationship with reaction time from 1 to 6 h, then only slightly to 6.0 mmol g−1 at 12 h, essentially unaffected by further doubling of reaction time (Fig. 1b). Under the optimal 5:1 BPB:AGU ratio and 6 h time, increasing temperatures from 23 to 70 C° improved Br add-on (σ) from 5.7 to 8.7 mmol g−1, but further increase to 90 °C lowered the σ to 3.4 mmol g−1 (Fig. 1c). The significantly reduced σ from esterification at 90 °C suggested possible dissolution of the excessively esterified cellulose. Overall, Br-Cell with 0.6 to 8.7 mmol g−1 σ has been facilely produced and easily controlled by the esterification conditions, i.e., 1:1 to 5:1 BPB:AGU ratios, 1 to 6 h, and 23 to 70 °C. The highest 8.7 mmol g−1 Br add-on was achieved by esterification conducted at 5:1 BPB:AGU ratio and 70 °C for 6 h.
 | ||
| Fig. 1 Br add-on (σ) in Br-Cell as affect by reaction conditions: (a) BPB:AGU molar ratio (23 °C, 12 h); (b) reaction time (5:1 BPB:AGU, 23 °C); and (c) temperature (5:1 BPB:AGU, 6 h); (d) 1% DMF dispersion immediate and 10 min after vortexing; (e) microfiber dimensions. | ||
The DMF dispersibility of 1% Br-Cells with four ester levels, i.e., Br-Cell1 (0.6 mmol g−1), Br-Cell2 (3.4 mmol g−1), Br-Cell3 (5.7 mmol g−1), and Br-Cell4 (8.7 mmol g−1), was then observed. The least esterified Br-Cell1 did not disperse and remained settled even at a lower 0.1% whereas those more esterified Br-Cell2, 3 and 4 were more dispersible in DMF but to different degrees (Fig. 1d). The Br-Cell2 and Br-Cell3 DMF dispersions appeared homogeneous and translucent, but Br-Cell4 phase separated immediately (t = 0). After 10 min, Br-Cell3 also settled similarly as Br-Cell4 while Br-Cell2 remained somewhat dispersed. Optical microscopic observation showed all four Br-Cells to be microfibers in similar 4–5 μm widths while their lengths reduced by nearly 30% from 221 to ca. 158 μm compared to the original cellulose for all except for the significantly shorter 36 μm long Br-Cell4. While no change in microfiber width, the 84% reduction in their length of Br-Cell4 (Fig. S1†) with the highest σ (8.7 mmol g−1) suggested this more extensive esterification condition may be close to the onset of chain scission with potential cellulose dissolution. One control reaction, i.e., without BPB, was performed at 23 °C for 6 h to produce only 1.1% mass loss, insignificant under the reaction conditions and from the filtration and evaporation process. Thus, the Br add-on (σ, mmol g−1) based on mass gain as in eqn (1) is appropriate to estimate Br add-on of esterified cellulose.
Br-NCs by ultrasonication
Four Br-Cells with σ from 0.6 to 8.7 mmol g−1 were ultrasonicated in DMF (0.1 w/v%) at 25 to 100% amplitudes for 10 to 120 min to collect the Br-NC containing supernatants. Upon ultrasonication at 50% amplitude for 30 min, the least esterified Br-Cell1 (σ = 0.6 mmol g−1) produced only 10.6% Br-NCs, essentially the same as the 10.4% NCs from unmodified cellulose under the same ultrasonication condition (Fig. S2†), indicating esterification at 0.6 mmol g−1 to be insufficient to facilitate disintegration and/or dispersion. The NCs from unmodified cellulose appeared as few larger and thicker (5–20 nm) nanoparticles (NPs) on mica but numerous much smaller and thinner (3 nm) NPs on graphite, indicative of their more hydrophobic surfaces. Among the three more esterified Br-Cells, increasing Br-NCs were produced with increasing ultrasonication amplitudes from 25 to 100% at 60 min, with the highest 97.3% yield for Br-Cell3 (5.7 mmol g−1) compared to the modest 56.9 and 73.1% from either the respective less esterified Br-Cell2 (3.4 mmol g−1) and the more esterified Br-Cell4 (8.7 mmol g−1) (Fig. S3†).Since a very close second highest yield of 93.8% was produced from Br-Cell3 at half of the amplitude, 50% amplitude was used to conserve energy and observe the effect of ultrasonication time (10 to 120 min) on the morphology of Br-NCs generated from Br-Cell2–4 to conserve energy (Fig. 2). Br-NC yields increased with longer ultrasonication (50% amplitude) for all three Br-Cells; Br-NC yields from Br-Cell3 (5.7 mmol g−1) was highest and most time-dependent, ranging from 38.2% to 97.3%, followed by slight time-dependent and modest 29.3 to 49.3% Br-NC yields for Br-Cell4 (8.7 mmol g−1) and the least time-dependent and lowest 19.0 to 25.8% Br-NC yields for Br-Cell2 (3.4 mmol g−1). However, not all Br-NCs were fibrillar. For the most DMF dispersible Br-Cell2 (Fig. 1d), nearly all Br-NCs were fibrillar, from entangled to more individualized in reducing thickness of 6.5 to 1.2 nm, but in low yields. The most esterified Br-Cell4 was disintegrated into mostly 2-bromopropionyl esterified cellulose nanoparticulates (Br-NPs), also in decreasing sizes, with only few fibrils from longer ultrasonication. Ultrasonication of Br-Cell3 produced 38.2% Br-NCs in the forms of both Br-CNFs and Br-NPs at 10 min, 70.9% Br-CNFs at 30 min, and 93.8% at 60 min all as Br-NPs. As expected, ultrasonication transfers sound energy to disintegrate Br-Cell microfibers into Br-NCs with increasing power by either higher amplitudes or longer time. The forms and sizes of NCs, however, were found highly dependent on the extent of esterified cellulose. The less esterified (3.4 mmol g−1) produced all Br-CNFs but at low yields (19.0 to 25.8%) whereas the most esterified (8.7 mmol g−1) produced majority of Br-NPs at modest yields (29.3 to 49.3%). Therefore, Br-Cell3 was deemed optimally esterified (5.7 mmol g−1) with sufficient 2-bromoesters to be disintegrated by ultrasonication (50%, 30 min) into mostly Br-CNF in 70.9% yield, and 4.6 nm average thickness. Noteworthy, longer ultrasonication of the optimally esterified Br-Cell3 produced 93.8% and 97.0% Br-NPs at respective 1 h and 2 h. Those Br-NPs may be attributed to shortened Br-CNF due to chain scission and further disintegration of less esterified cellulose from extensive sonication. Therefore, this esterification-ultrasonication approach to functionalize cellulose and disintegrate in functionalize nanocelluloses are highly effective to produce Br-CNFs and potentially Br-NPs efficiently.
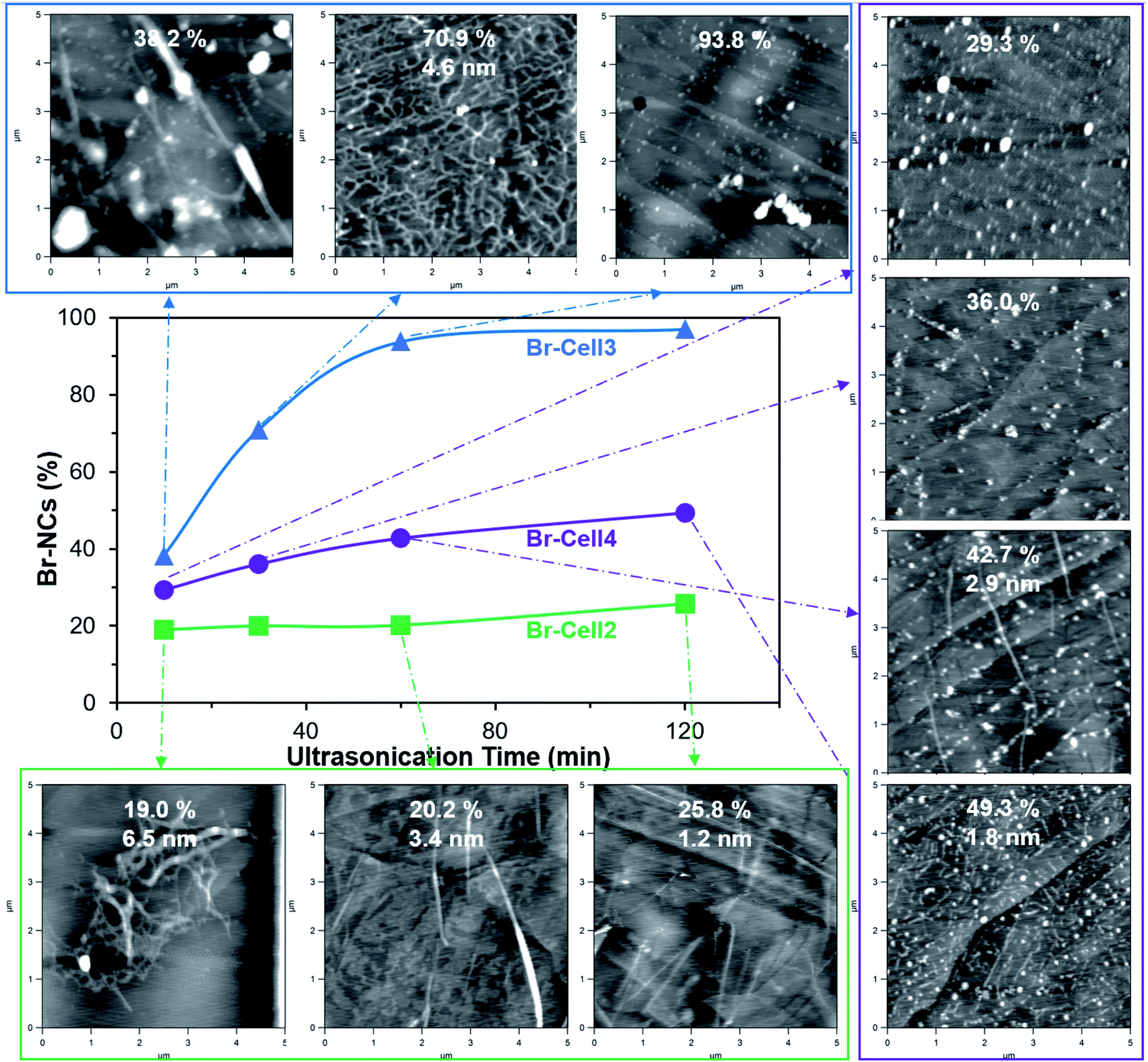 | ||
| Fig. 2 Yields and morphology of Br-NCs (5k rpm, 10 min) from ultrasonication (50% amplitude, 10–120 min) of Br-Cell2 (3.4 mmol g−1), Br-Cell3 (5.7 mmol g−1), and Br-Cell4 (8.7 mmol g−1). AFM were imaged on highly oriented pyrophoric graphite (HOPG) with corresponding yield and Br-CNF thickness. | ||
Br-CNF3 morphology characterized by AFM and TEM
Br-CNF3 generated from optimal ultrasonication (50% amplitude, 30 min) of Br-Cell3 were further imaged by AFM on freshly exfoliated graphite and by TEM on glow discharged carbon grid to display interconnecting nanofibrils with 4.6 nm average thickness (Fig. 3a), 29.2 nm average width, and varying lengths in the order around 1 μm (Fig. 3b). The irregular widths under TEM may be due to the reassociation of the more esterified surface chains along Br-CNF surfaces. Also, under both AFM and TEM, the inter-connecting fibrillar network left few isolated fibrils, making differentiating fibril ends difficult and causing inaccurate estimation of Br-CNF3 length. Similar inter-connecting fibrillar structures were also observed at much more diluted 0.0001 and 0.00005 w/v% concentrations (Fig. S3a–c†), confirming the inter-connecting fibrillar structures to be independent of concentrations and seemingly mono-layer. The inter-connecting fibrillar network dried from the most concentrated Br-CNF (0.0005 w/v%, on graphite) appeared thinner and more fragmented after exposing to air for 24 h (Fig. S4d†), possibly due to reactions between 2-bromoesters and moisture in the air, degrading Br-CNF. | ||
| Fig. 3 Morphology of Br-CNF3 (0.0005 w/v%, 10 μL) supernatant (5k rpm, 10 min) from ultrasonication (50% amplitude, 30 min): (a) AFM image on graphite, height profile, and height distribution; (b) TEM images on glow-discharged carbon grid with width distribution. | ||
To further elucidate the interaction among Br-CNFs, a second Br-CNF3 droplet was placed on top of the completed dry first (10 μL, 0.0005 w/v%) (Fig. S5†). More heterogenous, condensed and inter-connecting CNFs were observed at the center than near the edge of the first dried droplet (Fig. S5b–d†). The significant association among Br-CNFs from second deposition (Fig. S5b†) as compared to isolated fibrils from the initial single droplet gave evidence to preferential and stronger association among Br-CNFs over affinity of Br-CNF to graphite surface. Association among Br-CNFs may include dipole–dipole interactions between surface 2-bromoesters, hydrogen bonding among unsubstituted surface hydroxyls, and potential chemical reaction between 2-bromoesters and remaining hydroxyls in preference to adhesion to the graphite surface from the sequential deposition.
FTIR spectroscopy and thermal analysis of Br-Cell, Br-CNF3 and Br-Cell3 precipitates
The presence of the new 2-bromopropionyl carbonyl peak at 1740 cm−1 in the FTIR spectra of all four Br-Cells confirmed the successful conversion of cellulose hydroxyls to 2-bromopropionyl group (Fig. 4a). The carbonyl peak (1740 cm−1) intensities increased whereas the cellulose C–H stretching peak (2900 cm−1) reduced in intensities with the increasing Br add-on (σ) from 0.6–8.7 mmol g−1. The ester sp3 C–C stretching at 2780 cm−1 appeared for Br-Cell2 and increased with increasing σ, while the most esterified Br-Cell4 also showed intense ester sp2 C–C stretching at 2970 cm−1. The carbonyl peak (1740 cm−1) on Br-CNF3 remained unchanged from Br-Cell3, indicating no significant side reaction or degradation on the alkyl bromines by ultrasonication. The stronger carbonyl peak (1740 cm−1) in Br-CNF3 compared to Br-Cell3 precipitates indicated that Br-CNF3 in supernatant represented the more esterified fraction (70.9%) whereas the less esterified could be disintegrated by ultrasonication and remained in the precipitate. The persistent cellulose crystalline peak (1430 cm−1) in both Br-Cell and Br-CNFs suggested that the bulk of cellulose crystalline domains was not affected by esterification and ultrasonication. The lowered intensity of the absorbed moisture peak (1632 cm−1) for the highly esterified Br-Cell4 was also expected. Furthermore, weaker hydrogen bonding O–H stretching peak (3100–3800 cm−1) in all Br-Cells than underivatized cellulose supported the successful cellulose hydroxyl conversion to esters. The reduced hydrogen bonding interactions between cellulose chains could also aid the opening of (110) or (1−10) planes in Br-Cell via ultrasonication to generate more Br-NCs. | ||
| Fig. 4 Characterizations of cellulose, Br-Cell1-4, Br-CNF3 and Br-Cell3 precipitate: (a) FT-IR spectra; (b) TGA; and (c) DTGA curves. Moisture (%) was the mass loss at 140 °C. | ||
With increasing esterification levels, moisture absorption of Br-Cell reduced from 6.08% to 0.02% (Fig. 4b) which was consistent with effect of converting hydrophilic hydroxyls to esters (Fig. 4a). The underivatized cellulose was stable at up to 260 °C and rapidly lost significant mass to give 2.6% char at 500 °C. With the increase of Br add-on from 0 to 8.7 mmol g−1, both the onset and max degradation temperatures lowered for Br-Cell1, 2 and 3, then slightly increased for Br-Cell4 (Fig. 4c). The lowered onset and max degradation temperatures may be due to the insertion of less thermal stable esters; while the opposite increasing onset and max degradation temperatures of Br-Cell4 may be explained by the highly substituted 2-bromoesters behaved as vapor-phase flame-retardant moieties to suppress decomposition of cellulose, a potential worthy of further study in the future. The significantly higher moisture contents of Br-CNF3 (8.5%) and Br-Cell3 precipitate (4.5%) than that of precursor Br-Cell3 (0.65%) gave evidence to generation of new hydrophilic surfaces due to the opening of cellulose (110) and (1−10) planes from ultrasonication. The lower onset and max degradation temperatures of Br-CNF3 (205 and 234 °C) than its precursor Br-Cell3 (219 and 241 °C) could be due to the three order of magnitude smaller fiber dimensions (4.6 nm thickness) and much higher specific surfaces.
Degree of substitution of surface OH by solution-state 1H-NMR and model simulation
For solution-state 1H NMR of Br-CNF3, solvent was first changed from DMF to acetone then to DMSO-d6. The intermediate acetone exchange was repeated in three times by evaporation at either 50 or 80 °C to show cellulosic protons with characteristic methyl proton (Ha) or methylene proton (Hb) peaks of the alkyl bromide groups (Fig. 5a). For spectra of both Br-CNF (Fig. 5b,c), the furthest downfield peak at δ 4.20–4.52 and δ 4.53 are assigned to the cellulosic anomeric proton (H1) similar to the chemical shift at δ 4.5 reported in dissolved cellulose.49 The H6 and H6′ peaks for 50 °C treated Br-CNF3 appeared at δ 3.71–4.06 whereas two clear peaks appeared at δ 3.97 and δ 3.83 for 80 °C treated Br-CNF3. Both ranges were relatively downfield due to esters' de-shielding effect, but comparable to those in δ 3.65–3.88 range for dissolved MCC in NaOD/D2O.50 Multiple overlapping peaks between δ 3.29–3.70 and δ 3.42–3.57 were assigned to H3, H4 and H5, matching those at δ 3.34–3.66 of TEMPO-CNF in D2O.51 The furthest upfield cellulosic peak at δ 3.16 and δ 3.04 coincided with the chemical shift of H2 at δ 3.29 for cellulose/NaOD/D2O45 and δ 3.05 for cellulose dissolved in DMA-d9/LiCl.52 Doublet methyl proton peaks (Ha) at δ 2.12 and δ 1.29 were both comparable to δ 1.6 (ref. 37) of soluble esterified cellulose. Most significantly, both doublet methyl (Ha) and quartet methylene proton (Hb) peaks at δ 4.53–4.87 and δ 4.11 indicated the successful insertion of 2-bromoesters. | ||
| Fig. 5 (a) Structure and proton assignment of Br-CNF3; Br-CNF3 1H NMR spectra in DMSO-d6 via solvent exchange from acetone evaporated at (b) 50 °C and (c) 80 °C vacuum chamber; (d) integral values of H1-6′, Ha and Hb with corresponding DSHa, DSHb and DSNMR. DSHa and DSHb were calculated using eqn (4) and (5) whereas DSNMR was their average. | ||
The degree of substitution of surface hydroxyls to 2-bromoesters (DSNMR) were quantified based on the assumption that all anomeric protons and all Ha and Hb protons of amorphous and crystalline surface AGUs of Br-CNF3 are detectable by 1H NMR. The cellulose anomeric proton was the sum of the integrated areas for all anomeric H1 to H6′ proton peaks averaged by 7 then normalized by reference methylene proton Hb. Esters could be estimated by integration of the areas of methyl Ha or methylene Hb divided by their respective 3 and 1 protons. The ratio of esterified C2, C3 and C6 OHs per surface AGU could be determined mathematically by the area ratio of ester calculated from Ha or Hb over normalized anomic proton. Since each AGU has 3 OHs, DSHa and DSHb, representing the fraction of OH substituted by ester determined by proton Ha or Hb, could be calculated by dividing ratio of esterified OHs per surface AGU by 3 according to eqn (4) or (5).
 | (4) |
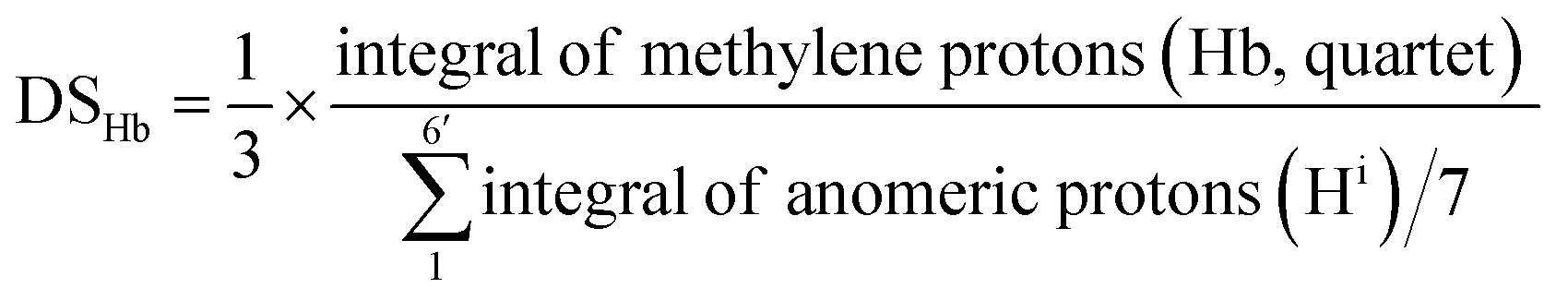 | (5) |
For Br-CNF3 prepared at 80 °C, majority of Br-CNF3 were in the precipitates of DMSO-d6 suspension after centrifugation and decanted, causing much lower proton signal compared to Br-CNF3 prepared at 50 °C (Fig. 5b and c). At 80 °C, Br-CNFs may associate with each by potential endothermic53 interfibrillar N-substitution between alkyl bromide and OHs to precipitate, leaving only small portion of dissolved cellulose in DMSO-d6. Thus, 80 °C treated spectra showed more distinguishable anomic proton peaks with inconsistent 0.75 DSHa and 0.57 DSHb or possibly less reliable 0.66 DSNMR (Fig. 5d). Br-CNF prepared at 80 °C showed a relative upfield Ha proton peak at δ 1.29 compared to δ 2.12 at 50 °C to indicate the decreased de-shielding effect of ester caused by releasing HBr at 80 °C. In addition, the cellulose dissolution could also be observed from the significant lower integral values (Fig. 5b) of H1(0.04), H2 (0.06), H6 (0.13) and H6′(0.08) compared to H3, 4, 5 (3.81). In comparison, 0.53 DSHa and 0.58 DSHb calculated from 50 °C treated Br-CNF3 spectra within the 2σ range of total 7.4% benchtop NMR uncertainty54 were averaged to be 0.56 DSNMR. In preparing Br-CNF3 in DMSO-d6 for 1H NMR, Br-CNF3 was first solvent exchanged from DMF to acetone where the more hydrophilic or less esterified may be left in DMF, thus not included for 1H NMR. Therefore, the 0.56 DSNMR derived may represent the more hydrophobic or more highly esterified CNF, thus higher than the DS of overall Br-CNF3 population.
Both Br-Cell3 and Br-CNF3 displayed 2θ peaks at 14.6, 16.5, and 22.5° corresponding to the respective (1−10), (110), and (200) monoclinic Iβ lattice planes of cellulose (Fig. 6a and b). The lowered crystallinity of Br-Cell3 (CrI = 0.50) compared to original cellulose (CrI = 0.69) without reduction of crystallite dimension (Fig. S1a and c†) gave evidence to 2-bromopropionyl esterification of exposed cellulose chains on crystalline surfaces (Fig. 6c). The 0.48 CrI of Br-CNF3 was only very slightly lower than the 0.50 CrI of Br-Cell3. The 1.45 nm crystallite size of Br-CNF3, calculated via Scherrer eqn (3), was only one third of Br-Cell (4.77 nm), a clear evidence of disintegration of crystalline regions in Br-Cell into smaller domains by ultrasonication without affecting overall crystallinity. The much higher absorbed moisture (8.5%) in Br-CNF3 than that in Br-Cell3 microfibers (0.65%) (Fig. 4b) also supports the notion that ultrasonication of Br-Cell3 has created additional hydrophilic surfaces on Br-CNF3 by breaking the original crystalline structure along the (1−10) and/or (110) lattice planes.
 | ||
| Fig. 6 Crystalline structure of cellulose, Br-Cell and Br-CNF3: (a) XRD spectra; (b) Br-CNF cross-section with cellulose chains represented by green rectangles, esterified (1−10) and (110) plane surfaces with thickness (T), width (W), and length (L) indicated; inset shows cellulose Iβ monoclinic unit cell; (c) crystallinity (CrI) and crystal parameters calculated by the Scherrer eqn (3); (d) degree of substitution (DSσ) calculated from Br-CNF dimensions based on the cross-section shown in c. | ||
A model representing the lateral cross-section of individual Br-CNF with hydrophilic (110) and (1−10) planes as surfaces was thus used (Fig. 6b), displaying thickness (T), width (W), and length (L) and the cellulose Iβ monoclinic unit cell dimensions along the (100) and (010) planes as a and b, respectively. The number of total cellulose chains in the crystalline cross-section and the number of surface cellulose chains Ns are expressed respectively as
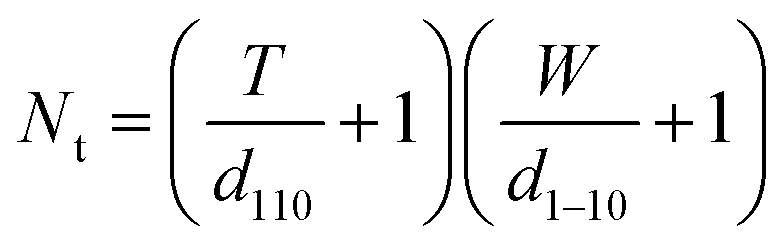 | (6) |
 | (7) |
 | (8) |
Both amorphous and crystalline surfaces OHs should be counted as part of CNF surface OHs. Since all OHs in amorphous regions are exposed, the amorphous OHs per AGU (Rm) is 3 OHs/AGU. The ratio of total available OHs per AGU (ROH) is the weight average of those on the crystalline surfaces (Rc) and on amorphous region (Rm) expressed as
| ROH = Rc × CrI + Rm × (1 − CrI) | (9) |
 | (10) |
Interfacial and surface properties of Br-CNF3
The interfacial interactions among Br-CNF3 and four substrates with varied hydrophilicity/hydrophobicity in a range of concentrations (0.0005 to 0.01 w/v%) were observed. The substrates using in AFM and TEM imaging and their water contact angles (WCAs) were mica (16.8°), freshly exfoliated graphite (71.8°), glow discharged carbon grid (68.2°), and carbon grid (115.3°) (Fig. 7a). On hydrophilic mica, most Br-CNF3 appeared as either loosely or extensively agglomerated short fibrils in 1.2 nm average thickness (Fig. 7b). On the moderately hydrophobic graphite and glow discharged carbon grid, inter-connecting fibrillar network was prevalent (Fig. 7c and d) as noted earlier (Fig. 3a–c) whereas only large NPs were observed on the hydrophobic carbon grid (Fig. 7e). These observations indicated Br-CNF3 in the supernatant were mostly moderately hydrophobic with better compatibility to graphite and glow discharged carbon, appearing as 4.6 nm thick and 29.3 nm wide fibrils. Meanwhile some Br-CNF3 were sufficiently hydrophilic to be partially dispersed as thin fibrils on mica while none was as hydrophobic as carbon. Both higher moisture absorption of Br-CNF3 than Br-Cell3 (Fig. 4b) and thinner hydrophilic CNFs (Fig. 7b) are consistent with the opening of hydrophilic (110) planes as new surfaces from ultrasonication of Br-Cell into Br-CNF. These observations also further supported the hydrophilic model for Br-CNF (Fig. 6c). Moreover, the presence of Br-CNF as large NPs on carbon grid may be due to their aggregation via hydrogen bonding of surface OHs and dipole–diploe interactions of newly surface functionalized 2-bromoesters. | ||
| Fig. 7 Br-CNF3 on substrates: (a) individual substrates with WCAs indicated; AFM (b, c) and TEM (d, e) images of Br-CNF3 (10 μL, 0.0005 w/v%) on corresponding substrates above; (f–i) AFM images and WCAs of Br-CNF3 air-dried on graphite at: (f) 0.0005 w/v%, (g) 0.001 w/v %, (h) 0.005 w/v%, and (i) 0.01 w/v% concentrations. | ||
With increasing Br-CNF3 concentrations from 0.0005 to 0.01 w/v%, WCAs on Br-CNF3 deposited graphite increased from 55.6° to 73.0° (Fig. 7f–i). Br-CNF3 appeared as inter-connecting fibrils at the lower 0.0005 and 0.001 w/v% and as entangled fibrillar networks with few particulates at higher 0.005 and 0.01 w/v%. The initial deposition of 0.0005 w/v% Br-CNF3 dispersion partially covered the graphite to increase its hydrophilicity, lowering WCA from 71.8° (Fig. 7b) to 55.6° (Fig. 7f). The hydrophilicity decreased slightly with increasingly coverage at 0.001 w/v% (60.8° WCA) and further when nearly full coverage at 0.005 w/v% (75.4° WCA) (Fig. 7k), with slightly increased surface roughness. Further increased Br-CNF3 to 0.01 w/v% did not alter WCA, but slightly reduced the surface roughness as expected with fuller coverage. Therefore, diluted Br-CNF3 deposited at concentrations from 0.0005 to 0.01 w/v% is capable of monolayer to few layers to alter surface wettability and may be used in potentially surface coating and super-thin film applications.
Dispersing behaviors and rheology of Br-CNF3
Dispersing behaviors of Br-CNF3 and their corresponding rheology at higher concentrations were investigated for additional potential formulation and application. First, disintegration of Br-Cell3 in DMF by ultrasonication (50%, 30 min) was scaled up from 0.1 to 1.0 w/v% or ten times. With increasing Br-Cell3 quantities, the resulting Br-CNF3 concentrations in the supernatants increased from 0.07 to 0.5 w/v%, while the Br-Cell3 to Br-CNF3 conversion or the yield reduced from 70.9 to 50.7% (Fig. 8a). All Br-CNF dispersions up to 0.5 w/v% appeared relatively transparent (Fig. 8b and c). The Br-CNF dispersions from 0.06 to 0.25 w/v% exhibited Newtonian behaviors, i.e., their viscosities were independent of shear rates (Fig. 8d). At 0.5 w/v%, Br-CNF3 dispersion exhibited a shear thinning region at low shear rates below 150 s−1 and a Newtonian region above. This implied that strong inter-fibrillar interaction of Br-CNF3 at 0.5 w/v% leads to higher viscosity in the low shear region below 150 s−1. With increasing shear rates, fibrillar Br-CNF3 became more oriented in the direction of shear flow until reached the Newtonian region. The highest concentration that Br-CNF3 can be directly and homogeneously dispersed into DMF was 0.5 w/v%, above which Br-CNF3 formed gels that contained crystalline micro-fibers (Fig. 8e). Br-CNF3 (0.5 w/v%) DMF dispersion was further concentrated to 2.5 w/v% gel by evaporation (ambient temperature, 4 d), then blade coated as 1 mm thick gel on glass and air-dried overnight to ca. 100 μm thick film. The WCA of the coated Br-CNF3 film was 69.8° (Fig. 8f), close to the WCAs of thin layers deposited from 0.005 w/v% (75.4°, Fig. 7h) and 0.01 w/v% (73.0°, Fig. 7i). Similar WCAs for thin layers and thick blade coated film implied the organization of Br-CNF3 on these surfaces to be independent of thickness from several nm to one hundred μm and may applied as hydrophobic coating in either manner. Furthermore, the coated thin film had an impressively high modulus of 198 MPa, as well as 6.7 MPa tensile stress and 10.7% strain-to-failure (Fig. S7†). Therefore, not only the Br-CNF3 dispersion's shear-thinning behavior above 0.5 w/v% demonstrate its potential as a rheology modifier in organic formulation, but the ultra-high modulus and strength of its thin film also present its potential as strength enhancement additive in coating and film applications. | ||
| Fig. 8 Scaled up generation of Br-CNF3 from ultrasonication (50% amplitude, 30 min) and their viscosity and gel characteristics: (a) Br-CNF yields and supernatant concentrations as related to initial Br-Cell3 concentrations in DMF; (b) UV-vis spectra; (c) images of Br-CNF supernatants and gel at above 0.5 w/v%; (d) viscosity at varied shear rates; (e) optical microscopy of 0.5 w/v% Br-CNF gel; (f) WCA of film by blade coating 1 mm Br-CNF3 (2.5 w/v%) gel on glass, and air dried. | ||
Conclusions
This study has proven the concept for facile one-pot synthesis of functionalized cellulose and in situ ultrasonication in the same organic liquid to disintegrate functionalized cellulose directly into hydrophobic nanocelluloses. Organic compatible 2-bromopropionyl esterified cellulose nanofibrils (Br-CNFs) have been successfully synthesized via rationally designed 2-bromopropionyl esterification of cellulose and in situ ultrasonication in DMF. This esterification-ultrasonication approach can be tuned to produce either Br-CNFs or Br-NPs efficiently. The optimally esterified Br-Cell3 (5:1 BPB:AGU ratio, 23 °C, 6 h) contained 5.7 mmol g−1 esters to be disintegrated by ultrasonication (50% amplitude, 30 min) to yield 70.9% Br-CNF3 in average 4.6 nm thickness, 29.3 nm width, up to 1 μm length, and 47.8% crystallinity. While esterification lowered the overall crystallinity (69% to 50%), ultrasonication reduced the crystalline size (from 4.77 nm to 1.45 nm) of Br-CNF, exposing new (110) and (1−10) hydrophilic planes as evident by the increased moisture absorption (0.65% to 8.5%). The successful conversion of surface OHs to esters was confirmed by the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1740 cm−1 in FTIR and chemical shifts for methyl proton (Ha) and methylene proton (Hb) at δ 2.12 and δ 4.53–4.87 in 1H NMR, respectively. The degree of substitution (DS) of Br-CNF was determined to be between the underestimated 0.53 DSσ based on CrI and cross-sectional dimension model and the overestimated 0.56 DSNMR from solution-state 1H NMR. Br-CNF3 dispersions exhibited Newtonian behaviors at concentrations below and shear thinning behaviors at above 0.5 w/v%. Dilute 0.005 and 0.01 w/v% Br-CNF3 dispersions could be homogeneously deposited as few nm ultra-thin layers to exhibit WCAs in the range of 73–75°. Moreover, blade coating of gel (2.5 w/v%) could also dried to 100 μm thick hydrophobic (70° WCA) film, showing comparable hydrophobicity irrespective of thickness. All were similarly hydrophobic as cellulose acetates and polyesters. The shear-thinning behavior of Br-CNF dispersions demonstrate their potential application as viscosity modifiers in variety of mechanical fluids. The ultra-high modulus and strength film from gel coating further showed the potential for dual high-strength and hydrophobic applications.
O at 1740 cm−1 in FTIR and chemical shifts for methyl proton (Ha) and methylene proton (Hb) at δ 2.12 and δ 4.53–4.87 in 1H NMR, respectively. The degree of substitution (DS) of Br-CNF was determined to be between the underestimated 0.53 DSσ based on CrI and cross-sectional dimension model and the overestimated 0.56 DSNMR from solution-state 1H NMR. Br-CNF3 dispersions exhibited Newtonian behaviors at concentrations below and shear thinning behaviors at above 0.5 w/v%. Dilute 0.005 and 0.01 w/v% Br-CNF3 dispersions could be homogeneously deposited as few nm ultra-thin layers to exhibit WCAs in the range of 73–75°. Moreover, blade coating of gel (2.5 w/v%) could also dried to 100 μm thick hydrophobic (70° WCA) film, showing comparable hydrophobicity irrespective of thickness. All were similarly hydrophobic as cellulose acetates and polyesters. The shear-thinning behavior of Br-CNF dispersions demonstrate their potential application as viscosity modifiers in variety of mechanical fluids. The ultra-high modulus and strength film from gel coating further showed the potential for dual high-strength and hydrophobic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the California Rice Research Board (RU-9) and USDA National Institute of Food and Agriculture (CA-D-6706) is greatly appreciated.References
- L. Chen, J. Zhu, C. Baez, P. Kitin and T. Elder, Green Chem., 2016, 18, 3835–3843, 10.1039/C6GC00687F.
- S. Iwamoto, W. Kai, A. Isogai and T. Iwata, Biomacromolecules, 2009, 10, 2571–2576, DOI:10.1021/bm900520n.
- H. Fukuzumi, T. Saito and A. Isogai, Carbohydr. Polym., 2013, 93, 172–177, DOI:10.1016/j.carbpol.2012.04.069.
- J. A. Diaz, X. Wu, A. Martini, J. P. Youngblood and R. J. Moon, Biomacromolecules, 2013, 14, 2900–2908, DOI:10.1021/bm400794e.
- B. Puangsin, Q. Yang, T. Saito and A. Isogai, Int. J. Biol. Macromol., 2013, 59, 208–213, DOI:10.1016/j.ijbiomac.2013.04.016.
- Y.-S. Ye, H.-X. Zeng, J. Wu, L.-Y. Dong, J.-T. Zhu, Z.-G. Xue, X.-P. Zhou, X.-L. Xie and Y.-W. Mai, Green Chem., 2016, 18, 1674–1683, 10.1039/C5GC01979F.
- B. Thomas, M. C. Raj, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625, DOI:10.1021/acs.chemrev.7b00627.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500, DOI:10.1021/cr900339w.
- D. Bondeson, A. Mathew and K. Oksman, Cellulose, 2006, 13, 171, DOI:10.1007/s10570-006-9061-4.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2007, 9, 57–65, DOI:10.1021/bm700769p.
- A. Dufresne, Mater. Today, 2013, 16, 220–227, DOI:10.1016/j.mattod.2013.06.004.
- F. Jiang and Y.-L. Hsieh, Carbohydr. Polym., 2013, 95, 32–40, DOI:10.1016/j.carbpol.2013.02.022.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691, DOI:10.1021/bm060154s.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85, 10.1039/C0NR00583E.
- M. S. Wang, F. Jiang, Y.-L. Hsieh and N. Nitin, J. Mater. Chem. B, 2014, 2, 6226–6235, 10.1039/C4TB00630E.
- F. Jiang and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2016, 4, 1041–1049, DOI:10.1021/acssuschemeng.5b01123.
- F. Jiang, S. Han and Y.-L. Hsieh, RSC Adv., 2013, 3, 12366–12375, 10.1039/c3ra41646a.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989, DOI:10.1021/bm0497769.
- J. Li, X. Wei, Q. Wang, J. Chen, G. Chang, L. Kong, J. Su and Y. Liu, Carbohydr. Polym., 2012, 90, 1609–1613, DOI:10.1016/j.carbpol.2012.07.038.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson and O. Ikkala, Biomacromolecules, 2007, 8, 1934–1941, DOI:10.1021/BM061215P.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795, DOI:10.1021/la702481v.
- C. Aulin, E. Johansson, L. Wågberg and T. Lindström, Biomacromolecules, 2010, 11, 872–882, DOI:10.1021/bm100075e.
- A. Pei, J.-M. Malho, J. Ruokolainen, Q. Zhou and L. A. Berglund, Macromolecules, 2011, 44, 4422–4427, DOI:10.1021/ma200318k.
- M. L. Auad, V. S. Contos, S. Nutt, M. I. Aranguren and N. E. Marcovich, Polym. Int., 2008, 57, 651–659, DOI:10.1002/pi.2394.
- D. Viet, S. Beck-Candanedo and D. G. Gray, Cellulose, 2007, 14, 109–113, DOI:10.1007/s10570-006-9093-9.
- S. Fujisawa, T. Ikeuchi, M. Takeuchi, T. Saito and A. Isogai, Biomacromolecules, 2012, 13, 2188–2194, DOI:10.1021/bm300609c.
- M. Fumagalli, F. Sanchez, S. Molina-Boisseau and L. Heux, Cellulose, 2015, 22, 1451–1457, DOI:10.1007/s10570-015-0585-3.
- Y. Wang, X. Wang, Y. Xie and K. Zhang, Cellulose, 2018, 25, 3703–3731, DOI:10.1007/s10570-018-1830-3.
- X. Kang, P. Sun, S. Kuga, C. Wang, Y. Zhao, M. Wu and Y. Huang, ACS Sustainable Chem. Eng., 2017, 5, 2529–2534, DOI:10.1021/acssuschemeng.6b02867.
- A. Ashori, M. Babaee, M. Jonoobi and Y. Hamzeh, Carbohydr. Polym., 2014, 102, 369–375, DOI:10.1016/j.carbpol.2013.11.067.
- A. Tripathi, M. Ago, S. A. Khan and O. J. Rojas, ACS Appl. Mater. Interfaces, 2018, 10, 44776–44786, DOI:10.1021/acsami.8b17790.
- F. Valdebenito, R. García, K. Cruces, G. Ciudad, G. Chinga-Carrasco and Y. Habibi, ACS Sustainable Chem. Eng., 2018, 6, 12603–12612, DOI:10.1021/acssuschemeng.8b00771.
- R. K. Johnson, A. Zink-Sharp and W. G. Glasser, Cellulose, 2011, 18, 1599–1609, DOI:10.1007/s10570-011-9579-y.
- L. Sun, X. Zhang, H. Liu, K. Liu, H. Du, A. Kumar, G. Sharma and C. Si, Curr. Org. Chem., 2021, 25, 417–436, DOI:10.2174/1385272824999201210191041.
- G. Morandi, L. Heath and W. J. L. Thielemans, Langmuir, 2009, 25, 8280–8286, DOI:10.1021/la900452a.
- C.-F. Huang, J.-K. Chen, T.-Y. Tsai, Y.-A. Hsieh and K.-Y. A. Lin, Polymer, 2015, 72, 395–405, DOI:10.1016/j.polymer.2015.02.056.
- A. Operamolla, S. Casalini, D. Console, L. Capodieci, F. Di Benedetto, G. V. Bianco and F. Babudri, Soft Matter, 2018, 14, 7390–7400, 10.1039/C8SM00433A.
- A. G. Cunha, Q. Zhou, P. T. Larsson and L. A. Berglund, Cellulose, 2014, 21, 2773–2787, DOI:10.1007/s10570-014-0334-z.
- M. Jonoobi, J. Harun, A. P. Mathew, M. Z. B. Hussein and K. Oksman, Cellulose, 2010, 17, 299–307, DOI:10.1007/s10570-009-9387-9.
- J. Fukuda and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2021, 9, 6489–6498, DOI:10.1021/acssuschemeng.1c02092.
- X. Sui, J. Yuan, M. Zhou, J. Zhang, H. Yang, W. Yuan, Y. Wei and C. Pan, Biomacromolecules, 2008, 9, 2615–2620, DOI:10.1021/bm800538d.
- P. Chmielarz, Express Polym. Lett., 2017, 11, 140, DOI:10.3144/expresspolymlett.2017.15.
- T. Yang, P. Xiao, J. Zhang, R. Jia, H. Nawaz, Z. Chen and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 11, 4302–4310, DOI:10.1021/acsami.8b15642.
- X. Cao, S. Sun, X. Peng, L. Zhong, R. Sun and D. Jiang, J. Agric. Food Chem., 2013, 61, 2489–2495, DOI:10.1021/jf3055104.
- T. Saito, R. Kuramae, J. Wohlert, L. A. Berglund and A. Isogai, Biomacromolecules, 2013, 14, 248–253, DOI:10.1021/bm301674e.
- P. Lu and Y.-L. Hsieh, Carbohydr. Polym., 2012, 87, 564–573, DOI:10.1016/j.carbpol.2011.08.022.
- L. Segal, J. J. Creely, A. Martin Jr and C. Conrad, Text. Res. J., 1959, 29, 786–794, DOI:10.1177/004051755902901003.
- P. Scherrer, Nachr. Ges. Wiss. Göttingen, 1918, 2, 96–100 Search PubMed.
- K. Kowsaka, K. Okajima and K. Kamide, Polym. J., 1988, 20, 1091–1099, DOI:10.1295/polymj.20.1091.
- A. Isogai, Cellulose, 1997, 4, 99–107, DOI:10.1023/A:1018471419692.
- F. Jiang, J. L. Dallas, B. K. Ahn and Y.-L. Hsieh, Carbohydr. Polym., 2014, 110, 360–366, DOI:10.1016/j.carbpol.2014.03.043.
- R. Nardin and M. Vincendon, Macromolecules, 1986, 19, 2452–2454, DOI:10.1021/ma00163a022.
- J. D. Roberts and M. C. Caserio, Basic principles of organic chemistry, WA Benjamin, Inc., 1977, https://authors.library.caltech.edu/25034/5/BPOCchapter4.pdf Search PubMed.
- Y. Lee, Y. Matviychuk and D. J. Holland, J. Magn. Reson., 2020, 320, 106826, DOI:10.1016/j.jmr.2020.106826.
Footnote |
| † Electronic supplementary information (ESI) available: Optical images of Br-Cells, ultrasonication of unmodified cellulose, Br-NC yields from varied ultrasonication of Br-Cells. AFMs of Br-CNF dried 6 h from 0.00005 to 0.0005% and for 24 h, AFM of double deposition of Br-CNF3, deconvolution of XRD of cellulose, engineering stress–strain curves for blade coating CNFs. See https://doi.org/10.1039/d2ra00722c |
| This journal is © The Royal Society of Chemistry 2022 |