
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of potent and selective HDAC6 inhibitors led to unexpected opening of a quinazoline ring†
Davide Moi‡
ab,
Andrea Citarella‡ab,
Davide Bonannia,
Luca Pinzia,
Daniele Passarella b,
Alessandra Silvanib,
Clelia Gianninib and
Giulio Rastelli*a
b,
Alessandra Silvanib,
Clelia Gianninib and
Giulio Rastelli*a
aDepartment of Life Sciences, University of Modena and Reggio Emilia, Via Campi 103, Modena 41125, Italy. E-mail: giulio.rastelli@unimore.it
bDepartment of Chemistry, University of Milan, Via Golgi 19, Milano 20133, Italy
First published on 13th April 2022
Abstract
Histone deacetylase (HDAC) inhibitors are highly involved in the regulation of many pharmacological responses, which results in anti-inflammatory and anti-cancer effects. In the present work, chemoinformatic analyses were performed to obtain two potent and selective aminotriazoloquinazoline-based HDAC6 inhibitors. We unexpectedly obtained an aminotriazole from a water-driven ring opening of the triazoloquinazoline scaffold. Both compounds were evaluated as HDAC6 inhibitors, resulting in subnanomolar inhibitory activity and high selectivity with respect to class I HDAC1 and HDAC8. Importantly, the compounds were about 3- and 15-fold more potent compared to the reference compound trichostatin A. Additionally, the predicted binding modes were investigated with docking. Considering that the aminotriazole scaffold has never been embedded into the chemical structure of HDAC6 inhibitors, the present study suggests that both the aminotriazoloquinazoline and aminotriazole classes of compounds could be excellent starting points for further optimization of potential anticancer compounds, introducing such novel groups into a relevant and new area of investigation.
Introduction
Epigenetic regulation is generally defined as the change in gene function deriving from DNA modification operated through chromatin remodeling, RNA regulation and histone modification. Several enzymes are involved in epigenetic regulation.1 Histone deacetylases (HDACs) play a crucial role in this process, catalyzing the removal of acetyl groups from lysine residues in histone and several non-histone proteins.2–4 Currently, eighteen HDACs have been identified and grouped into four different classes, according to their cellular localization and enzymatic activity. Class I HDACs includes four isoforms (i.e., HDAC1, 2, 3, 8), which are mainly located in the nucleus and can act on histones and transcription factors. Class II HDACs consists of two subclasses, i.e., IIa (HDAC4, 5, 7, 9) and IIb (HDAC 6 and 10). Of note, several members of class II HDACs can shuttle between the nucleus and the cell cytoplasm, where they can modulate the activity of various non-histone proteins. Class III histone deacetylases includes seven isoforms named sirtuins (SIRT1-7), which present several differences with respect to the other HDACs. Moreover, the activity of sirtuins is mainly involved in the regulation of metabolic processes as insulin secretion, ammonia detoxification and metabolic inflammation.5 Class IV includes only HDAC11, which is involved in the regulation of many biological processes in cells.6 Classes I, II and IV require Zn2+ for catalysis, while sirtuins employ NAD+ (nicotinamide adenine dinucleotide) as a cofactor.3 Recent studies have shown that Zn-dependent HDACs, and especially class I and class IIb isoforms, are overexpressed in many types of tumors, such as breast and liver cancer, multiple myeloma and neuroblastoma.7 HDAC6 plays a crucial role in protein degradation, cell shape, migration and regulation of immunomodulatory factors8 and is a well-established target for the development of anticancer compounds. Interestingly, the HDAC6 inhibitors reported so far share recurring features: (i) a zinc binding group (ZBG), which forms a stable coordination complex with the catalytic Zn2+ ion; (ii) a hydrophobic linker that fits into the catalytic tunnel of the active site, and; (iii) a CAP moiety interacting with the residues that line the entrance of the pocket.9,10 This latter region has been extensively probed for the design of HDAC6 selective inhibitors. Indeed, HDAC6 presents a CAP region significantly different in terms of shape and volume with respect to that of the other Zn-dependent histone deacetylases. As for the ZBG, the most relevant and explored one is the hydroxamic acid (HA), which is a powerful chelator of Zn2+. The formation of HA–Zn2+ complexes generally occur via a bidentate mechanism, resulting in significant enzyme inhibition.10 Several structurally different CAP groups have been explored for the design of HDAC6 inhibitors, the quinazoline motif demonstrating to be a valuable scaffold to obtain potent inhibitors. A few key examples of HDAC6 inhibitors bearing the quinazoline scaffold are reported in Fig. 1.11–14 For example, compounds 1, 2 and 3 consist of a quinazoline core decorated at position 4 with a substituted aniline, which in turn is connected to an aliphatic chain linked to the ZBG through ethereal (1) or amidic (2 and 3) bonds. Changing the position of the ZBG led to 4, a potent cinnamyl derivative. The most significant structural modification was obtained by switching the ZBG to position 2, affording 5, which, to the best of our knowledge, is the most active quinazoline inhibitor reported so far (IC50 = 12 nM).13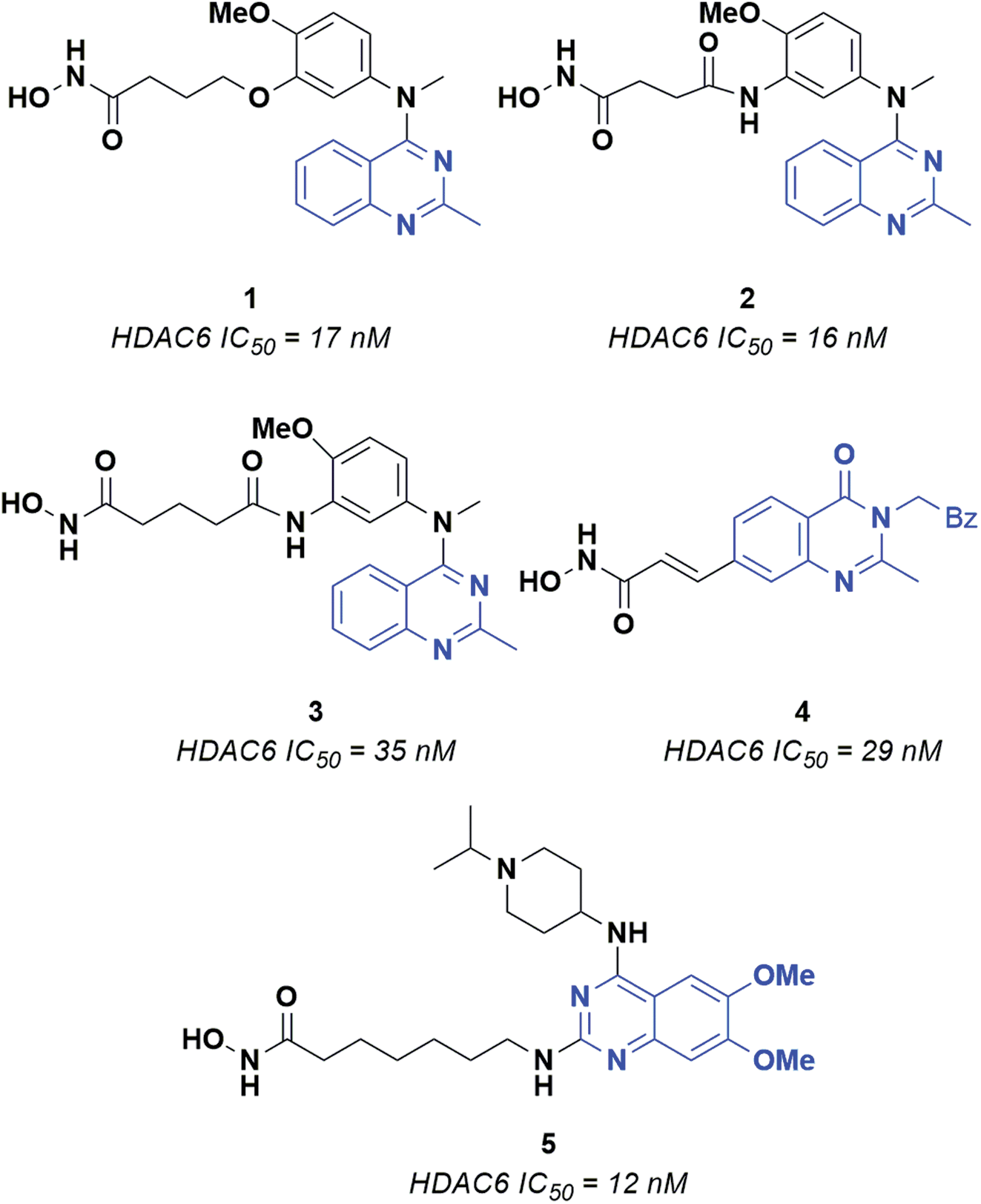 | ||
| Fig. 1 Known quinazoline-capped inhibitors of HDAC6. | ||
Based on these premises and relying on our previous work on HDAC6 inhibitors,2,15–19 in this study we report the computational design and the synthesis of two structurally novel derivatives containing an aminotriazoloquinazoline (11a) and aminotriazole scaffold (18). The two synthesized compounds were tested in vitro to evaluate their inhibitory activity against HDAC6. Docking investigations were performed to provide structural explanations of the observed inhibitory activity. Of note, this is the first study that explores the aminotriazoloquinazoline and aminotriazole scaffolds as CAP groups of HDAC6 inhibitors. Moreover, for the first time we observed the opening of the triazoloquinazoline ring in water, which was previously investigated only in the presence of carbon nucleophiles. The results are particularly appealing in light of the potent sub-nanomolar HDAC6 inhibitory activity displayed by the two compounds.
Results and discussion
Design of aminotriazoloquinazoline compounds based on chemoinformatics analyses
Compound 11a (Fig. 2) was assembled from a set of chemical scaffolds selected among those more frequently occurring in potent HDAC6 inhibitors, by adopting an approach similar to that of our previous studies.20 To this aim, HDAC6 inhibitors collected from ChEMBL were firstly fragmented as detailed in the ESI,† identifying a set of substructures frequently present in the active and inactive compounds. Then, the identified chemical fragments were ranked according to their frequency of occurrence in active over inactive compounds. This allowed us to identify the benzohydroxamate, the 1,3-benzodioxole and the quinazoline moieties as valuable building blocks for the design and assembly of novel HDAC6 inhibitors (see Table S1† and Fig. 2). Interestingly, the benzohydroxamate moiety is frequently present in HDAC6 inhibitors. The hydroxamate coordinates the Zn2+,10 while the phenyl ring establishes favorable interactions with two phenylalanine residues of HDAC6 flanking the catalytic tunnel. Moreover, the 1,3-benzodioxole moiety has been explored for the optimization of structurally diverse classes of HDAC6 inhibitors, with promising results.21–23 The three chemical moieties emerging from these analyses were assembled into compound 11a, a substituted aminotriazoloquinazoline derivative. The connectivity between the three fragments was inspired by information available from HDAC6 crystal structures in complex with ligands and then verified by means of extensive similarity estimations (see ESI†) and docking (see below). Interestingly, this is the first time that the aminotriazoloquinazoline group is explored for the design of HDAC6 inhibitors. Of note, a visual inspection of the identified molecular fragments suggested that the benzohydroxamate moiety could be integrated in either position 8 (11a, Fig. 2) or 9 (11b, Fig. 2) of the aminotriazoloquinazoline group, without potentially altering the HDAC6 inhibitory activity.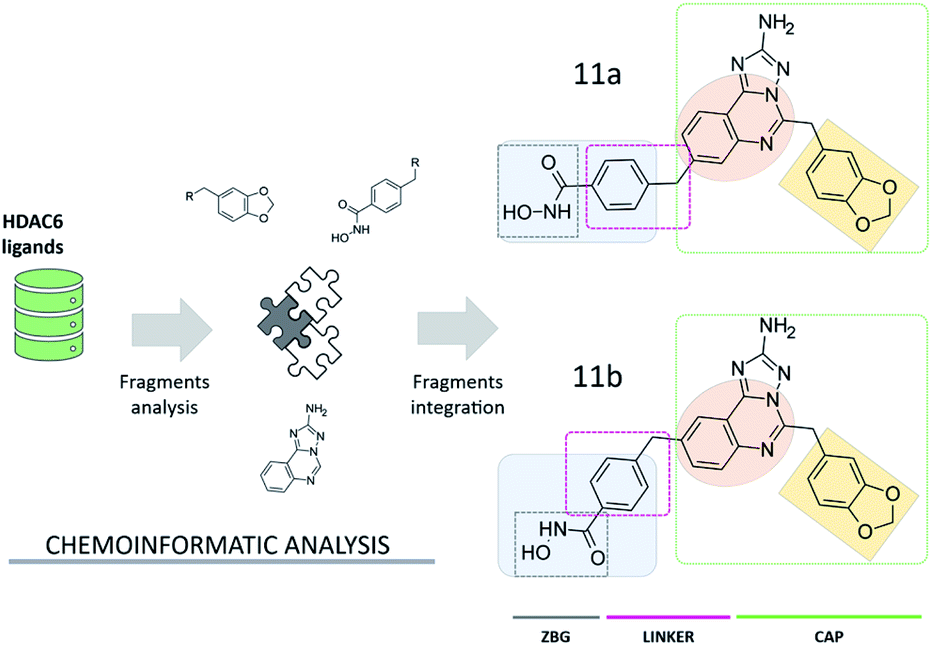 | ||
| Fig. 2 Design of aminotriazoloquinazoline-based HDAC6 inhibitors. The 1,3-benzodioxole (light orange) and aminotriazoloquinazoline (dark orange) moieties represent the CAP group of the ligands. | ||
Chemistry
The synthetic route to compound 11a was designed following a linear approach based on the construction of the aminotriazoloquinazoline core, a subsequent Suzuki–Miyaura cross-coupling for insertion of the benzylic linker and a final functional group interconversion to introduce the hydroxamic acid moiety (Scheme 1).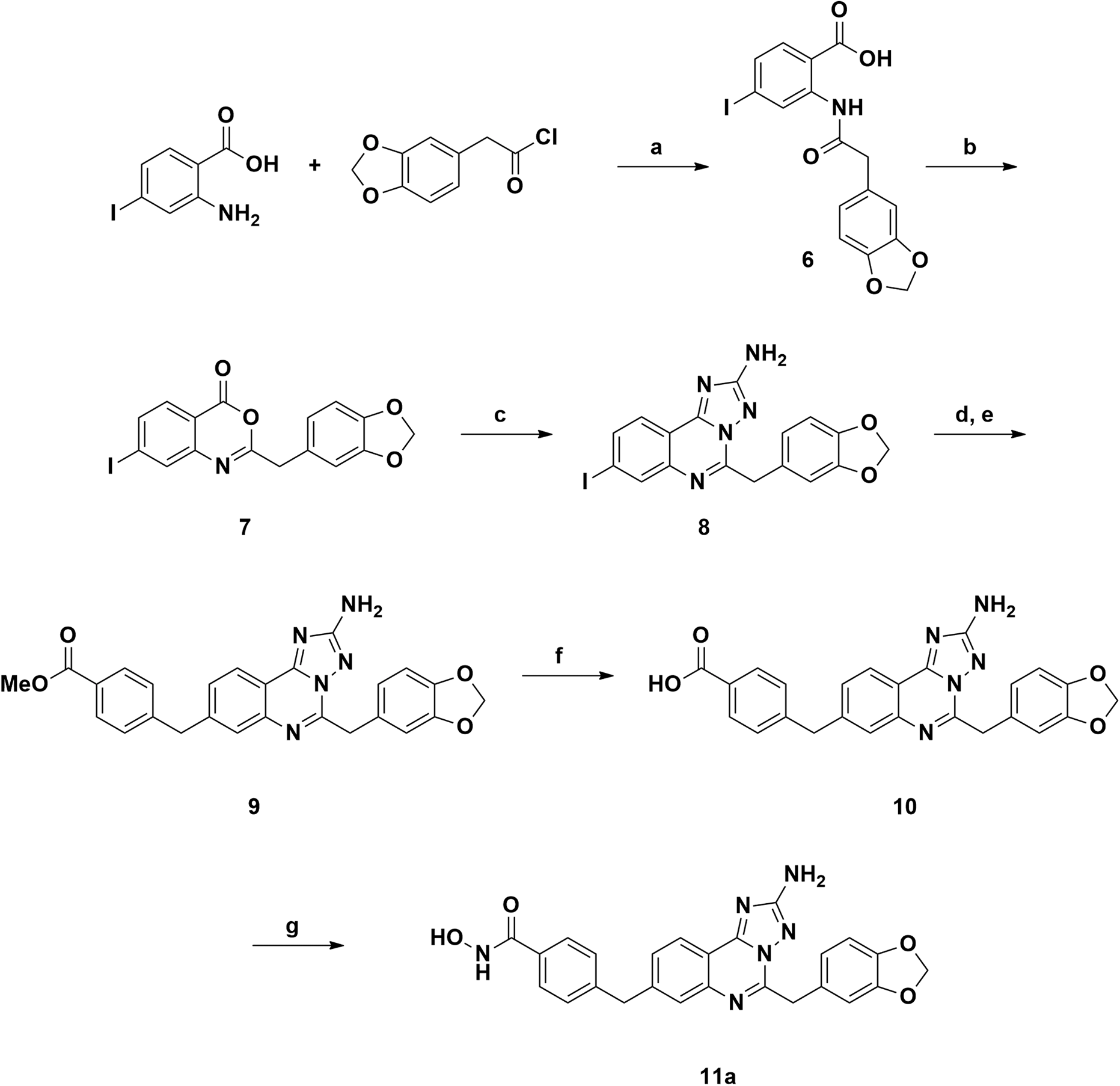 | ||
Scheme 1 Reagents and conditions: (a) NEt3, DCM, rt, overnight, yield 98%; (b) Ac2O, 150 °C, 10 min, yield 74%; (c) aminoguanidine bicarbonate, pyridine, 180 °C, microwave, 20 min, yield 93%; (d) bis(pinacolato)diboron, Pd(dppf)Cl2, AcOK, DMF, 120 °C, 4 h; (e) methyl 4-(bromomethyl)benzoate, Pd(PPh3)4, K2CO3, toluene/EtOH 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), 120 °C, 4 h, yield 70%; (f) LiOH, 1,4-dioxane/water 1:1 (v/v), rt, overnight, yield 95%; (g) NH2OTMS, HOBt, EDCI, DIPEA, DMF, rt, overnight, 51%. :1 (v/v), 120 °C, 4 h, yield 70%; (f) LiOH, 1,4-dioxane/water 1:1 (v/v), rt, overnight, yield 95%; (g) NH2OTMS, HOBt, EDCI, DIPEA, DMF, rt, overnight, 51%. | ||
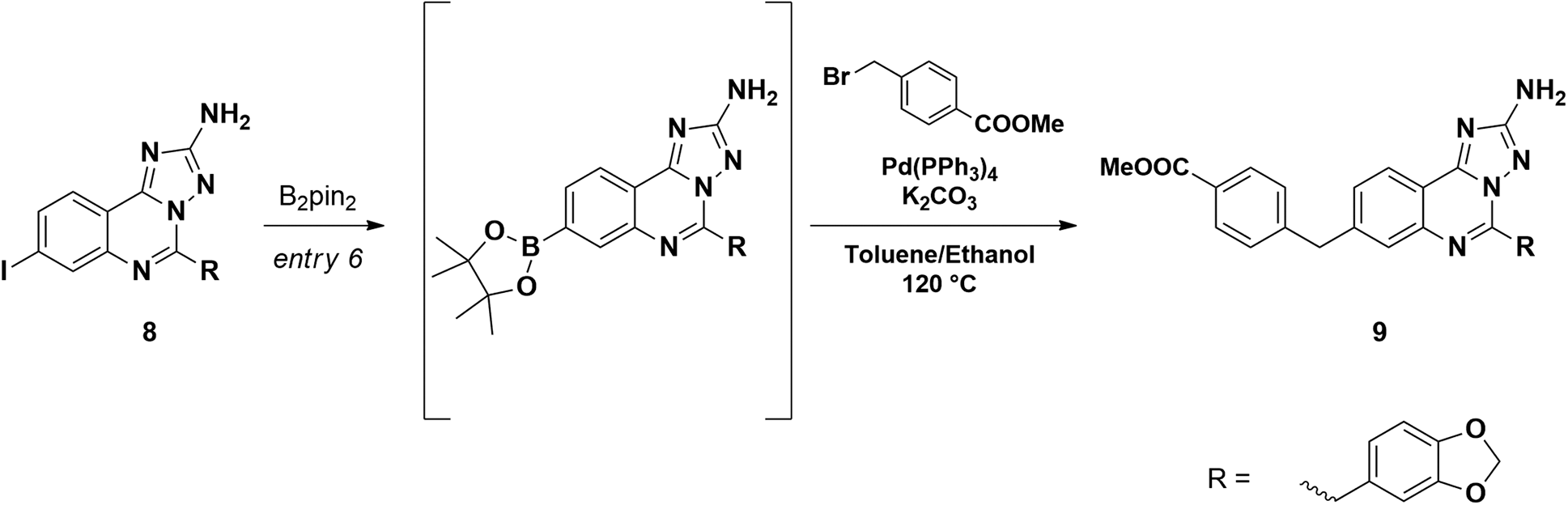
Starting from the commercially available 4-iodoanthranilic acid and 2-(benzo[d][1,3]dioxol-5-yl)acetyl chloride, the amide 6 was achieved in excellent yield, after precipitation with HCl. Treatment of 6 with acetic anhydride at reflux for 10 min gave quantitatively benzoxazinone intermediate 7, which was afterward reacted with aminoguanidine bicarbonate under microwave irradiation to provide the aminotriazoloquinazoline 8 in high yield.24 Several conditions for the conversion of the iodo derivative 8 into the corresponding pinacolate boronic ester were evaluated, as reported in Table 1. The use of Pd(PPh3)4 and the combination of Pd(OAc)2-PPh3 in dry toluene led to starting material recovery (entries 1–2), as well as the use of Pd(PPh3)4 in 1,4-dioxane/water 1:1 (v/v) or PdCl2(PPh3)2 in dry 1,4-dioxane (entries 3–4). The formation of the desired product was observed using Pd(dppf)Cl2 in 1,4-dioxane (Entry 5), while switching the solvent to dry DMF improved the conversion up to 63%, as calculated by 1H NMR (Entry 6). The crude mixture was analysed by LC-MS, which highlighted the predominance of the desired pinacolate boronic ester, together with a low percentage of the free boronic acid. The mixture was filtered off, the solvent was evaporated under reduced pressure and the crude was used in the following step without further purification. The Suzuki–Miyaura cross-coupling with methyl 4-bromomethyl benzoate, using Pd(PPh3)4 and K2CO3 in toluene/EtOH 3:1 (v/v), afforded the desired product 9 in good yield, after column chromatography purification. Methyl ester 9 was hydrolysed under basic conditions by aqueous LiOH and coupled with O-trimethylsilylhydroxylamine. To our advantage, coupling and –OH deprotection occurred in a single step operation, providing the final product 11a, after reverse phase column chromatography purification, with good yield.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Cat./ligand | Temp. | Time | Yield (9) |
| 1 | Toluene | Pd(OAc)2-PPh3 | 110 °C | 6 h | — |
| 2 | Toluene | Pd(PPh3)4 | 120 °C | 6 h | — |
| 3 | 1,4-Dioxane/water | Pd(PPh3)4 | 120 °C | 6 h | — |
| 4 | 1,4-Dioxane | PdCl2(PPh3)2 | 110 °C | 7 h | — |
| 5 | 1,4-Dioxane | Pd(dppf)Cl2 | 110 °C | 6 h | — |
| 6 | DMF | Pd(dppf)Cl2 | 120 °C | 4 h | 70 |
A synthetic strategy similar to that described above was adopted in the attempt to prepare the 9-substituted derivative (Scheme 2). However, the same hydrolysis conditions (step f) did not lead to the desired product 16. LC-MS analysis revealed the exclusive presence of a molecular structure (17) showing m/z [M + 18]+ with respect to the product of interest, suggesting the addition of water and the concomitant ring opening of the quinazoline ring. Interestingly this result has never been reported in the literature, although several examples of triazoloquinazoline ring opening have been observed only in the presence of carbon nucleophiles.25–27 Any attempt to discourage this side reaction, e.g., by carefully monitoring the reaction course and also reducing temperature to 0 °C, proved to be ineffective. Reasoning that the obtained acid derivative 17 could also be interesting for biological evaluation, we proceeded to convert it into the final hydroxamic acid 18, whose chemical structure was confirmed by two-dimensional NMR analysis.
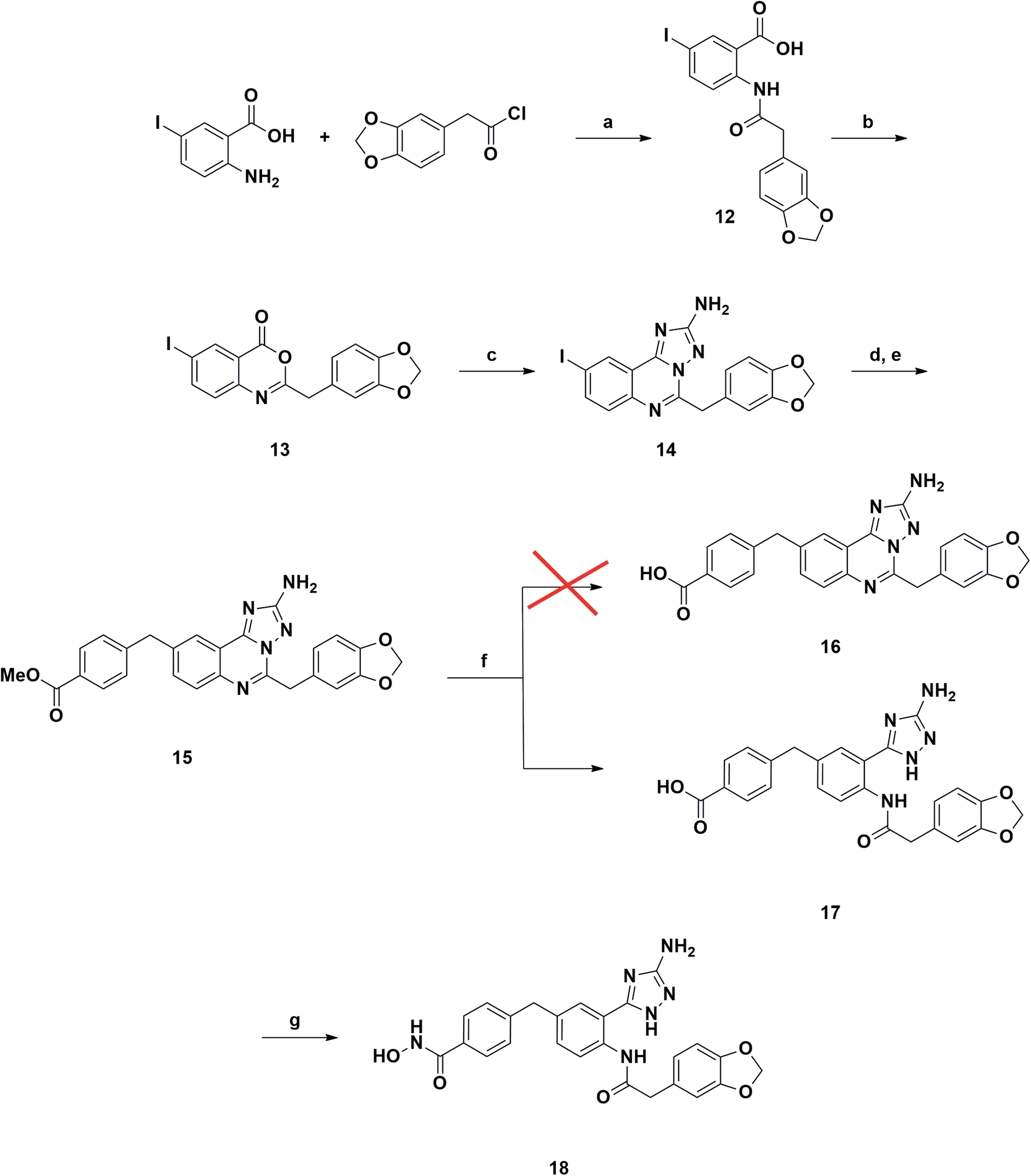 | ||
| Scheme 2 Reagents and conditions: (a) NEt3, DCM, rt, overnight, yield 93%; (b) Ac2O, 150 °C, 10 min, yield 84%; (c) aminoguanidine bicarbonate, pyridine, 180 °C, microwave, 20 min, yield 93%; (d) Bis(pinacolato)diboron, Pd(dppf)Cl2, AcOK, DMF, 120 °C, 4 h; (e) methyl 4-(bromomethyl)benzoate, Pd(PPh3)4, K2CO3, toluene/EtOH 3:1 (v/v), 120 °C, 4 h, yield 68%; (f) LiOH, 1,4-dioxane/water 1:1 (v/v), rt, overnight, yield 86%; (g) NH2OTMS, HOBt, EDCI, DIPEA, DMF, rt, overnight, yield 43%. | ||
In vitro inhibitory activity
The synthetized compounds 11a and 18 were tested in vitro to assess their inhibitory activity on purified recombinant HDAC6 enzyme. The results are reported in Table 2.| Compound | IC50 (nM) | ||
|---|---|---|---|
| HDAC6 | HDAC1 | HDAC8 | |
| 11a | 0.5 | 6080 | 653 |
| 18 | 0.1 | 1820 | 1560 |
| Trichostatin A | 1.9 | 3.25 | 648 |
Both 11a and 18 displayed potent, subnanomolar inhibitory activity towards HDAC6, resulting about 3- and 15-fold more potent than the reference compound trichostatin A (Table 2). Intriguingly, the unplanned compound 18 showed potent HDAC6 inhibitory activity. This result is particularly appealing, considering that aminotriazole compounds have never been reported as HDAC6 inhibitors. Furthermore, 11a and 18 were tested in vitro against purified recombinant HDAC1 and HDAC8 to evaluate their selectivity profile. Compound 11a resulted more than 12000-fold and 1000-fold selective for HDAC6 with respect to HDAC1 and HDAC8, respectively, while 18 was even more selective, being than 18000-fold and 15000-fold more active on HDAC6 than HDAC1 and HDAC8, respectively (Table 2). Consequently, our study suggests that both the aminotriazoloquinazoline and aminotriazole classes of compounds are novel and excellent starting points for further optimization for the development of highly potent and selective HDAC6 inhibitors.
Molecular docking in the HDAC6 active site
The newly synthesized compounds were docked into a representative crystal structure of HDAC6 as detailed in the ESI,† to evaluate whether they provide favorable docking scores and a binding mode consistent with those reported in crystal structure complexes. The complementarity of 11a and 18 with the HDAC6 binding site was thus evaluated through docking calculations performed on the 5EDU crystal structure of the human HDAC6 enzyme.28 For both ligands, we found that the ionized hydroxamate group coordinates the Zn2+ in bidentate mode,28 and it accepts two hydrogen bonds from Y782 and H610 through the carbonyl and the deprotonated hydroxyl, respectively (Fig. 3). Moreover, the phenyl ring of 11a and 18 establishes favourable π–π stacking interactions with the side chains of F620 and F680. Interestingly, the CAP groups of 11a and 18 are oriented differently (Fig. 3). The aminotriazoloquinazoline moiety of compound 11a stretches over F620 to donate a hydrogen bond to the side chain of D497, and the 1,3-benzodioxole accommodates in proximity of S564 and S568. On the contrary, the CAP group of 18 is directed towards F680, the amide carbonyl hydrogen bonds with the backbone nitrogen of F680, the 1,3-benzodioxole moiety forms a T-shaped stacking with the phenyl ring of F679, and the NH2 of the triazole hydrogen bonds with the side chain of D567. Compounds 11a and 18 achieved favourable docking scores of −10.4 and −11.0 kcal mol−1, respectively, compared to the self-docking result of −8.6 kcal mol−1.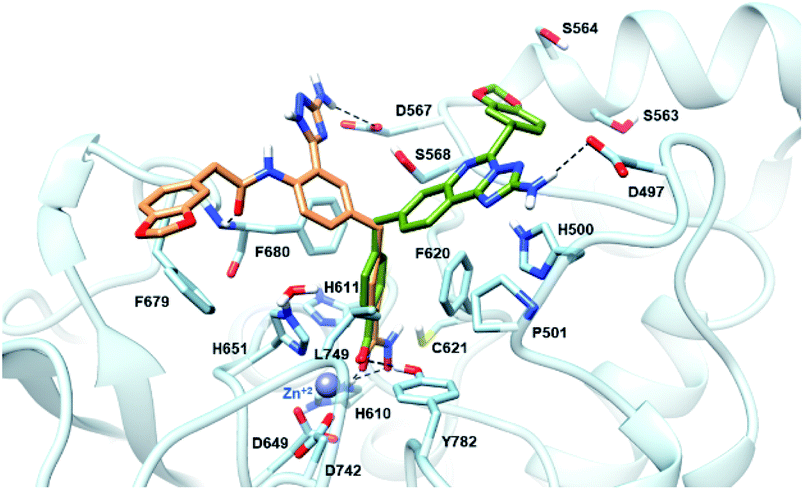 | ||
| Fig. 3 Binding mode predicted for compound 11a (sticks, coloured in green) and 18 (sticks, coloured in sienna) into the HDAC6 binding site (PDB ID 5EDU). The residues lining the enzyme binding site are shown in light blue sticks. The catalytic Zn2+ ion is depicted as a grey sphere, while the hydrogen bonds are shown as black dashed lines. | ||
Conclusions
In conclusion, a series of chemoinformatic analyses performed on reported histone deacetylase 6 inhibitors allowed the identification of the benzohydroxamate, 1,3-benzodioxole and quinazoline scaffolds as key moieties which were combined for the design of novel aminotriazoloquinazoline-based HDAC6 inhibitors. Two compounds were synthesized and tested in vitro to assess their inhibitory activity on purified recombinant HDAC6 enzyme. Aminotriazoloquinazoline 11a inhibited the enzyme (IC50 = 0.5 nM) 3 times more potently than the reference compound, while the unexpected ring-opened derivative 18 was even more active (IC50 = 0.1 nM), being about 15 times more active compared to thricostatin A. In addition, both 11a and 18 displayed high selectivity towards HDAC6 compared with the class I isoforms HDAC1 and HDAC8. The results indicate that the aminotriazoloquinazoline and aminotriazole scaffolds stand out as new starting points for the development of HDAC6 inhibitors, ranking in a promising area for future investigations.Experimental
Chemoinformatic analyses
Histone deacetylase 6 (HDAC6) inhibitors with activity data reported as IC50, Ki, Kd, EC50 were firstly downloaded from the ChEMBL database (https://www.ebi.ac.uk/chembl/, accessed on: September 28th, 2021).29 Then, duplicate records deriving from multiple assays on the same target were removed, retaining those with the best activity value. This allowed to obtain 3582 unique ligands, 2304 and 1278 compounds of which have reported activity data below 1 μM (herein labeled as “actives”) and higher than 1 μM (classified as “inactives”), respectively. Afterwards, an analysis of the molecular fragments composition was performed for the collected HDAC6 inhibitors. To this aim, the ligands were fragmented according to the BRICS,30 Bemis–Murcko31 and Recap32 algorithms implemented in the RDKit33 and OpenEye python toolkits,34 and by using Chomp (version 3.1.1.2, OpenEye)35 with default settings. Fragments whose substructure was not included in at least three active molecules and duplicates derived by the different fragmentation algorithms were discarded. Moreover, chemical moieties with a number of atoms lower than 4 or higher than 12 were also removed, obtaining a total of 544 unique fragments. The generated fragments were then used as queries for the identification of the substructures that are more frequently present in already reported HDAC6 compounds. The most interesting substructures emerging from the analysis and their related fragments (Table S2, in ESI†) were used as building blocks for the design of novel candidate compounds. The similarity degree between compound 11a, 11b and HDAC6 inhibitors extracted from ChEMBL was then evaluated by means of 2D fingerprints-based estimations. Similarity estimations were performed with MACCS and ECFP4 fingerprints (fp) implemented in RDKit python toolkits (https://www.rdkit.org),33 with settings consistent to those employed in our previous studies.36,37 Similarity records with Tanimoto coefficients below 0.8 and 0.3 for MACCSfp and ECFP4fp, respectively, were discarded. A visual inspection of the best-ranking ligand-based records (Table S1, in ESI†) was finally performed to evaluate whether compound 11a and 11b present key structural features and connectivity characterizing potent HDAC6 inhibitors, while showing a reasonable degree of chemical novelty.Docking
All the analyses were conducted on the 5EDU crystal structure of the human HDAC6 protein.28 The crystal structure of the protein underwent an optimization process using the Protein Preparation Wizard tool, implemented in Maestro of the Schrödinger Suite (release 2021-1).38 Missing hydrogen atoms were added, and bond orders were assigned. The prediction of protonation states for the protein residues was accomplished by using PROPKA, with the pH set to 7.4. Docking studies were performed by using XP protocol of the Glide program,39 keeping the ligands flexible. Default settings were used for the analyses and metal coordination constraint was applied during docking procedure to allow the ligands to coordinate to the catalytic Zn2+ ion.In vitro assay
In vitro tests on HDAC6, HDAC1 and HDAC8 were performed at Reaction Biology Corp. with modalities described below. The enzyme reactions were carried out in a solution of 50 mM Tris–HCl (pH 8.0), 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mg ml−1 BSA with the fluorogenic peptide from p53 residues 379–382 (RHKK-Ac-AMC) substrate for HDAC6 and HDAC1, and the fluorogenic peptide from p53 residues 379–382 (RHK-Ac-K-Ac-AMC) for HDAC8. The compounds were dissolved in DMSO and tested with 3-fold serial dilution starting from 100 μM. The enzymatic assays were performed by adding HDAC6 and the ligand solutions in the reaction buffer described above. After reaction termination, the fluorescence signal (Ex 360 nm/Em 460 nm) was recorded (in around 30 minutes) by adding a volume of 2 μM trichostatin A, 16 mg mL−1 trypsin in 50 mM Tris–HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2. IC50 values were calculated by using the GraphPad Prism 4 program based on a sigmoidal dose–response equation. The blank (DMSO) value concentration was entered as 1.00 × 10−12 M for curve fitting. Trichostatin A was used as a reference compound.General experimental procedures
Unless otherwise stated, reagents and solvents were purchased from Merck (Milan, Italy), Fluorochem (Hadfield, United Kingdom) or TCI (Zwijndrecht, Belgium) and used without further purification. All reactions were carried out in oven-dried glassware and dry solvents, under nitrogen atmosphere and were monitored by TLC on silica gel (Merck precoated 60 F254 plates), with detection by UV light (254 nm) or by permanganate, or by HPLC. HPLC was performed on Agilent 1100 Series System. Products were purified by flash column chromatography, using silica gel Merck 60 (230–400 mesh) as stationary phase. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance Spectrometer (400 MHz), using commercially available deuterated (chloroform-d, DMSO-d6) solvent at room temperature. Chemical shifts are reported in parts per million (δ ppm), compared to TMS as an internal standard. Multiplicities in 1H NMR are reported as follow: s – singlet, d – doublet, t – triplet, m – multiplet, br – broad. Data for 13C NMR are reported in chemical shift (δ ppm). High resolution mass spectra (HRMS) were recorded using the Q-ToF Synapt G2-Si HDMS Acquity UPLC I-Class Photodiode Detector Array (PDA) (Waters).:1 (v/v). The formed precipitate was filtered, washed with a solution of water/methanol 3:1 (v/v) and dried under vacuum to afford the desired 8 as a white non-crystalline solid (1.0 g, 93% yield). 1H NMR (400 MHz, DMSO-d6): 8.26 (s, 1H, Ar H), 7.95 (s, 2H, Ar H), 6.98 (s, 1H, Ar H), 6.83 (s, 2H, Ar H), 6.56 (s, 2H, NH2), 5.97 (s, 2H, O-CH2), 4.41 (s, 2H, Ar-CH2-Ar). 13C NMR (100 MHz, DMSO-d6): 165.8, 159.1, 148.7, 147.2, 146.9, 143.2, 136.0, 130.0, 129.0, 127.8, 122.4, 114.7, 109.7, 108.2, 100.9, 98.3, 37.9. MS (ESI), m/z [M + H]+: 446.16. HRMS (ESI), m/z [M + H]+: calculated for C17H13IN5O2+ 446.0109; found 446.0110. HPLC rt: 17.9 min.:1 v/v, 20 mL) and the reaction was heated to 120 °C. After 4 h, the suspension was filtered, and the filtrate was concentrated under reduced pressure. The crude was taken up in ethyl acetate (20 mL) and washed with brine (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4, filtered and evaporated to give the crude, which was purified by column chromatography using n-hexane/ethyl acetate (1:1, v/v) as eluent to afford 9 as a white solid (719 mg, 70%); mp. = 227–232 °C. 1H NMR (400 MHz, DMSO-d6): 8.12 (d, 3JH,H = 8.2 Hz, 1H, Ar H), 7.90 (d, 3JH,H = 8.0 Hz, 2H, Ar H), 7.74 (s, 1H, Ar H), 7.54 (d, 3JH,H = 8.2 Hz, 1H, Ar H), 7.45 (d, 3JH,H = 8.0 Hz, 2H, Ar H), 6.98 (s, 1H, Ar H), 6.83 (s, 2H, Ar H), 6.46 (s, 2H, NH2), 5.96 (s, 2H, O-CH2), 4.41 (s, 2H, Ar-CH2-Ar), 4.23 (s, 2H, Ar-CH2-Ar), 3.82 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6): 163.7, 150.4, 148.0, 147.9, 146.0, 143.6, 142.0, 140.1, 133.8, 131.2, 129.1, 129.0, 128.7, 128.6, 123.0, 122.7, 115.3, 110.0, 107.4, 101.1, 52.2, 42.0, 38.1. MS (ESI), m/z [M + H]+: 468.29. HRMS (ESI), m/z [M + H]+: calculated for C26H22N5O4+ 468.1667; found 468.1664. HPLC rt: 19.6.:1, v/v, 12 mL) was added LiOH (287 mg, 12 mmol, 8 equiv.) and the mixture was left stirring at room temperature overnight. The reaction was concentrated under reduced pressure, the residue was taken up in water and adjusted to pH 4 with 2 N HCl. The formed precipitate was filtered, washed with water and dried under vacuum to afford 10 as a white non-crystalline solid (650 mg, 95%). 1H NMR (400 MHz, DMSO-d6): 8.12 (d, 3JH,H = 8.2 Hz, 1H, Ar H), 7.89 (m, 3JH,H = 8.0 Hz, 2H, Ar H), 7.75 (s, 1H, Ar H), 7.55 (dd, 3JH,H = 8.6 Hz, 4JH,H = 1.5 Hz, 1H, Ar H), 7.45 (m, 3JH,H = 8.0 Hz, 2H, Ar H), 6.98 (s, 1H, Ar H), 6.83 (s, 2H, Ar H), 6.47 (s, 2H, NH2), 5.97 (s, 2H, O-CH2), 4.39 (s, 2H, Ar-CH2-Ar), 4.23 (s, 2H, Ar-CH2-Ar). 13C NMR (100 MHz, DMSO-d6): 169.0, 157.5, 156.3, 147.2, 146.1, 144.6, 135.1, 134.9, 130.6, 129.3, 129.1, 129.0, 128.6, 127.2, 127.1, 122.4, 119.4, 109.6, 108.1, 100.8, 44.5, 40.1. MS (ESI), m/z [M + H]+: 454.19. HRMS (ESI), m/z [M + H]+: calculated for C25H20N5O4+ 454.1510; found 454.1506. HPLC rt: 13.79 min.:1 (v/v). The formed precipitate was filtered, washed with a solution of water/methanol 3:1 (v/v) and dried under vacuum to afford the desired 14 as a white non-crystalline solid (1.0 g, 93% yield); mp. = 218–222 °C. 1H NMR (400 MHz, DMSO-d6): 8.46 (s, 1H, Ar H), 8.05 (d, 3JH,H = 9.0 Hz, 1H, Ar H), 7.65 (d, 3JH,H = 9.0 Hz, 1H, Ar H), 6.98 (s, 1H, Ar H), 6.83 (s, 2H, Ar H), 6.54 (s, 2H, NH2), 5.96 (s, 2H, O-CH2), 4.40 (s, 2H, Ar-CH2-Ar). 13C NMR (100 MHz, DMSO-d6): 166.8, 148.9, 148.4, 147.2, 145.7, 141.5, 139.8, 131.0, 129.8, 129.0, 122.2, 117.3, 109.7, 108.2, 100.8, 92.7, 37.9. MS (ESI), m/z [M + H]+: 446.15. HRMS (ESI), m/z [M + H]+: calculated for C17H13IN5O2+ 446.0109; found 446.0104. HPLC rt: 18.6 min.:1 v/v, 20 mL) and the reaction was heated to 120 °C. After 4 h, the suspension was filtered, and the filtrate was concentrated under reduced pressure. The crude was taken up in ethyl acetate (20 mL) and washed with brine (3 × 15 mL). The organic layer was dried with anhydrous Na2SO4, filtered and evaporated to give the crude, which was purified by column chromatography using n-hexane/ethyl acetate (1:1, v/v) as eluent to afford 15 as a white solid (702 mg, 68%); mp. = 200–203 °C. 1H NMR (400 MHz, CDCl3): 8.12 (m, 1H, Ar H), 7.96 (d, 3JH,H = 8.6 Hz, 2H, Ar H), 7.88 (s, 1H, Ar H), 7.56 (dd, 3JH,H = 8.7 Hz, 4JH,H = 2.3 Hz, 1H, Ar H), 7.28 (d, 3JH,H = 8.6 Hz, 2H, Ar H), 7.01 (d, 4JH,H = 1.6 Hz, 1H, Ar H), 6.95 (dd, 3JH,H = 8.0 Hz, 4JH,H = 1.6 Hz, 1H, Ar H), 6.73 (dd, 3JH,H = 8.0 Hz, 1H, Ar H), 5.90 (s, 2H, O-CH2), 4.62 (bs, 2H, NH2), 4.46 (s, 2H, Ar-CH2-Ar), 4.20 (s, 2H, Ar-CH2-Ar), 3.89 (s, 3H, OCH3). 13C NMR (100 MHz, CDCl3): 167.0, 164.8, 151.4, 148.0, 147.8, 146.8, 145.6, 142.0, 140.1, 133.0, 130.2, 129.1, 129.0, 128.7, 128.6, 122.9, 122.7, 116.4, 110.0, 108.4, 101.1, 52.2, 42.0, 38.8. MS (ESI), m/z [M + H]+: 468.35. HRMS (ESI), m/z [M + H]+: calculated for C26H22N5O4+ 468.1667; found 468.1664. HPLC rt: 19.7 min.:1, v/v, 10 mL) was added LiOH (287 mg, 12 mmol, 8 equiv.) and the mixture was left stirring at room temperature overnight. The reaction was concentrated under reduced pressure, the residue was taken up in water and adjusted to pH 4 with 2 N HCl. The formed precipitate was filtered, washed with water and dried under vacuum to afford 17 as a white solid (590 mg, 86%); mp. = <260 °C with decomposition. 1H NMR (400 MHz, DMSO-d6): 7.97 (s, 1H, Ar H), 7.89 (d, 3JH,H = 8.2 Hz, 2H, Ar H), 7.82 (d, 3JH,H = 8.0 Hz, 1H, Ar H), 7.69 (d, 3JH,H = 8.0 Hz, 1H, Ar H), 7.42 (d, 3JH,H = 8.2 Hz, 2H, Ar H), 6.97 (s, 1H, Ar H), 6.82 (s, 2H, Ar H), 5.96 (s, 2H, O-CH2), 4.39 (s, 2H, Ar-CH2-Ar), 4.26 (s, 2H, Ar-CH2-Ar). 13C NMR (100 MHz, DMSO-d6): 167.8, 166.3, 150.8, 148.1, 147.8, 146.9, 146.4, 141.6, 140.9, 133.3, 129.8, 129.5, 128.6, 122.8, 122.7, 116.1, 110.3, 108.8, 101.5, 41.1, 38.5. MS (ESI), m/z [M + H]+: 472.26. HRMS (ESI), m/z [M + H]+: calculated for C25H22N5O5+ 472.1615; found 472.1620. HPLC rt: 14.22 min.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro [AIRC IG 23635].References
- R. Jaenisch and A. Bird, Nat. Genet., 2003, 33, 245–254 CrossRef CAS PubMed.
- D. Bonanni, A. Citarella, D. Moi, L. Pinzi, E. Bergamini and G. Rastelli, Curr. Med. Chem., 2022, 29, 1474–1502 CrossRef PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- A. C. West and R. W. Johnstone, J. Clin. Invest., 2014, 124, 30–39 CrossRef CAS PubMed.
- X. Ye, M. Li, T. Hou, T. Gao, W. G. Zhu and Y. Yang, OncoTargets Ther., 2017, 8, 1845–1859 CrossRef PubMed.
- S.-S. Liu, F. Wu, Y.-M. Jin, W.-Q. Chang and T.-M. Xu, Biomed. Pharmacother., 2020, 131, 110607 CrossRef CAS PubMed.
- C. Zhao, H. Dong, Q. Xu and Y. Zhang, Expert Opin. Ther. Pat., 2020, 30, 263–274 CrossRef CAS PubMed.
- C. Seidel, M. Schnekenburger, M. Dicato and M. Diederich, Epigenomics, 2015, 7, 103–118 CrossRef CAS PubMed.
- M. T. Tavares, A. P. Kozikowski and S. Shen, Eur. J. Med. Chem., 2021, 209, 112887 CrossRef CAS PubMed.
- A. Citarella, D. Moi, L. Pinzi, D. Bonanni and G. Rastelli, ACS Omega, 2021, 6, 21843–21849 CrossRef CAS PubMed.
- J. Chen, Z. Sang, Y. Jiang, C. Yang and L. He, Chem. Biol. Drug Des., 2019, 93, 232–241 CrossRef CAS PubMed.
- V. N. Osipov, D. S. Khachatryan and A. N. Balaev, Med. Chem. Res., 2020, 29, 831–845 CrossRef CAS.
- H. Zheng, Q. Dai, Z. Yuan, T. Fan, C. Zhang, Z. Liu, B. Chu, Q. Sun, Y. Chen and Y. Jiang, Bioorg. Med. Chem., 2022, 53, 116524 CrossRef CAS PubMed.
- Z. Yang, T. Wang, F. Wang, T. Niu, Z. Liu, X. Chen, C. Long, M. Tang, D. Cao, X. Wang, W. Xiang, Y. Yi, L. Ma, J. You and L. Chen, J. Med. Chem., 2016, 59, 1455–1470 CrossRef CAS PubMed.
- D. Passarella, D. Comi, A. Vanossi, G. Paganini, F. Colombo, L. Ferrante, V. Zuco, B. Danieli and F. Zunino, Bioorg. Med. Chem. Lett., 2009, 19, 6358–6363 CrossRef CAS PubMed.
- E. Riva, S. Gagliardi, C. Mazzoni, D. Passarella, A. Rencurosi, D. Vigo and M. Martinelli, J. Org. Chem., 2009, 74, 3540–3543 CrossRef CAS PubMed.
- M. Géraldy, M. Morgen, P. Sehr, R. R. Steimbach, D. Moi, J. Ridinger, I. Oehme, O. Witt, M. Malz, M. S. Nogueira, O. Koch, N. Gunkel and A. K. Miller, J. Med. Chem., 2019, 62, 4426–4443 CrossRef PubMed.
- C. Zagni, A. Citarella, M. Oussama, A. Rescifina, A. Maugeri, M. Navarra, A. Scala, A. Piperno and N. Micale, Int. J. Mol. Sci., 2019, 20, 1–15 Search PubMed.
- P. Linciano, L. Pinzi, S. Belluti, U. Chianese, R. Benedetti, D. Moi, L. Altucci, S. Franchini, C. Imbriano, C. Sorbi and G. Rastelli, J. Enzyme Inhib. Med. Chem., 2021, 36, 2080–2086 CrossRef CAS PubMed.
- L. Pinzi, F. Foschi, M. S. Christodoulou, D. Passarella and G. Rastelli, ChemistryOpen, 2021, 10, 1177–1185 CrossRef CAS PubMed.
- J. Yang, G. Cheng, Q. Xu, S. Luan, S. Wang, D. Liu and L. Zhao, Bioorg. Med. Chem. Lett., 2018, 26, 1418–1425 CrossRef CAS PubMed.
- Z. Yuan, Q. Sun, D. Li, S. Miao, S. Chen, L. Song, C. Gao, Y. Chen, C. Tan and Y. Jiang, Eur. J. Med. Chem., 2017, 134, 281–292 CrossRef CAS PubMed.
- V. Zwick, C. A. Simões-Pires, A. Nurisso, C. Petit, C. Dos Santos Passos, G. M. Randazzo, N. Martinet, P. Bertrand and M. Cuendet, Bioorg. Med. Chem. Lett., 2016, 26, 4955–4959 CrossRef CAS PubMed.
- E. Casale, N. Amboldi, M. G. Brasca, D. Caronni, N. Colombo, C. Dalvit, E. R. Felder, G. Fogliatto, A. Galvani, A. Isacchi, P. Polucci, L. Riceputi, F. Sola, C. Visco, F. Zuccotto and F. Casuscelli, Bioorg. Med. Chem. Lett., 2014, 22, 4135–4150 CrossRef CAS PubMed.
- A. Petrič, M. Tišler and B. Stanovnik, Monatsh. Chem., 1985, 116, 1309–1319 CrossRef.
- L. Antipenko, A. Karpenko, S. Kovalenko, A. Katsev, E. Komarovska-Porokhnyavets, V. Novikov and A. Chekotilo, Chem. Pharm. Bull., 2009, 57, 580–585 CrossRef CAS PubMed.
- P. Molina, A. Arques, I. Cartagena and M. Valcarcel, J. Heterocycl. Chem., 1986, 23, 43–48 CrossRef CAS.
- Y. Hai and D. W. Christianson, Nat. Chem. Biol., 2016, 12, 741–747 CrossRef CAS PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrián-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magariños, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45(D1), D945–D954 CrossRef CAS PubMed.
- J. Degen, C. Wegscheid-Gerlach, A. Zaliani and M. Rarey, ChemMedChem, 2008, 3, 1503–1507 CrossRef CAS PubMed.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39(10), 2887–2893 CrossRef CAS PubMed.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, J. Chem. Inf. Comput. Sci., 1998, 38(3), 511–522 CrossRef CAS PubMed.
- RDKit: Open-Source Cheminformatics, available online: https://www.rdkit.org Search PubMed.
- OpenEye Toolkits 2020.2.2 OpenEye Scientific Software, Santa Fe, NM, available online: https://www.eyesopen.com Search PubMed.
- CHOMP 3.1.1.2: OpenEye Scientific Software, Santa Fe, NM, available online: https://www.eyesopen.com Search PubMed.
- F. Caporuscio, A. Tinivella, V. Restelli, M. S. Semrau, L. Pinzi, P. Storici, M. Broggini and G. Rastelli, Future Med. Chem., 2018, 10(13), 1545–1553 CrossRef CAS PubMed.
- L. Pinzi, A. Tinivella, L. Gagliardelli, D. Beneventano and G. Rastelli, Nucleic Acids Res., 2021, 49(W1), W326–W335 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01753a |
| ‡ Equally contributing authors. |
| This journal is © The Royal Society of Chemistry 2022 |