
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluvirucins B7–B10, new antifungal macrolactams from a marine-derived Nonomuraea sp. MYH522†
Hai Yu a,
Shuo Chena,
Hongji Lia,
Ruina Wanga,
Yuanying Jiangb,
Lan Yan*a and
Peng Sun*ab
a,
Shuo Chena,
Hongji Lia,
Ruina Wanga,
Yuanying Jiangb,
Lan Yan*a and
Peng Sun*ab
aSchool of Pharmacy, Naval Medical University, 325 Guo-He Road, Shanghai 200433, People's Republic of China. E-mail: sunpeng78@126.com; ylansmmu@sina.com
bTongji University School of Medicine, 1239 Siping Road, Shanghai, People's Republic of China
First published on 20th May 2022
Abstract
Marine rare actinomycetes are an important source of secondary metabolites. From a marine-derived actinomycete Nonomuraea sp. MYH522, four new macrolactams, fluvirucins B7–B10, together with known fluvirucin B6 were isolated. Their structures were determined based on comprehensive analysis of HRESIMS and NMR spectroscopic data as well as by comparing 13C NMR resonances and optical rotation values with those for related congeners. Fluvirucins are characterized by a 14-membered macrolactam attached by an aminosugar moiety. The discovery of fluvirucins B6–B10 enriched the N-acetylated derivatives of fluvirucins. The diverse alkyl substituents at C-2 and C-6 implied substrate promiscuity in fluvirucin polyketide biosynthesis. These compounds didn't exhibit any antibacterial or antifungal activities when used alone, which suggested the importance of the free amino group in the antimicrobial activity of fluvirucins. However, fluvirucins B6, B9, and B10 showed synergistic antifungal activity with fluconazole against fluconazole-resistant isolates of Candida albicans.
Introduction
Actinomycetes are characterized as one of the most important microorganisms for producing therapeutic agents in treating infections, pathogens, and cancer.1 Due to the decrease in discovering new chemical scaffolds, more attention has been drawn to rare actinomycetes or actinomycetes from previously underexplored environments. Marine actinomycetes are found to be a noteworthy source of new bioactive molecules.2–4 Moreover, rare actinomycetes, which are poorly assessed in contrast to the genus Streptomyces, are capable of producing a large number of new secondary metabolites.5–7Fluvirucins are a group of 14-membered macrolactams particularly produced by rare actinomycetes such as the genera of Actinomadura, Nonomuraea, and Nocardiopsis. It should be noted that numerous species of Actinomadura were reclassified as Nonomuraea.8 Since the first member Sch 38516 discovered in 1990, twenty fluvirucins have been reported.9–17 Fluvirucins are shown with various biological properties, i.e., antifungal,9,14,15 antibacterial,18–20 antiviral,10,20,21 and anthelmintic16,17 activities. The unique structure and the bioactivity of fluvirucins have also attracted attentions of chemists for total synthesis.22–24
During our ongoing investigation on new secondary metabolites from marine organisms,25–27 we encountered a rare actinomycete Nonomuraea sp. MYH522, which was isolated from a marine sponge in South China Sea. Chemical investigation of this strain led to the discovery of known fluvirucin B6 (1) together with four new analogs fluvirucins B7–B10 (2–5) (Fig. 1). Herein, we reported the isolation, structure elucidation, and biological evaluation of these compounds.
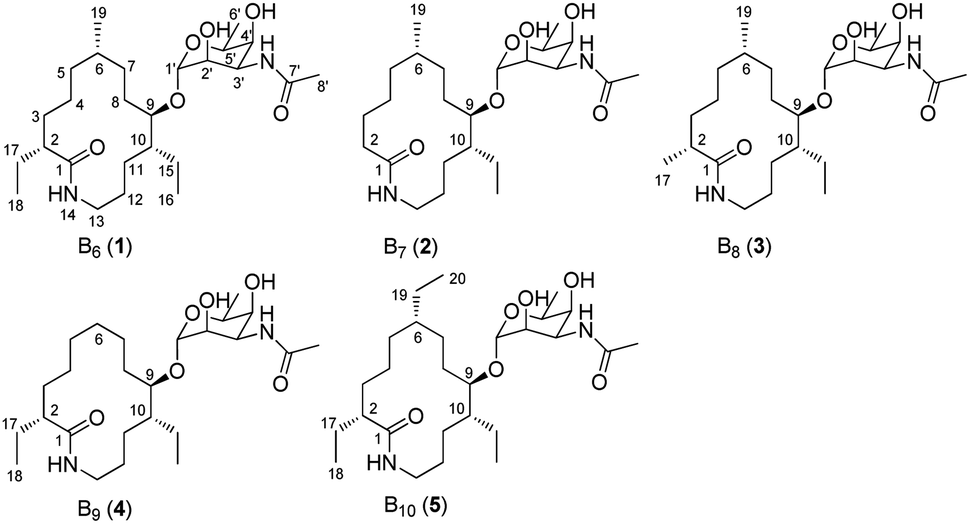 | ||
| Fig. 1 Structures of fluvirucins B6–B10 (1–5). | ||
Results
16S rDNA gene sequence and phylogenetic analysis
The 16S rDNA gene of strain MYH522 was amplified by PCR to obtain a 1450 bp sequence with NCBI GenBank accession number of NR117924.1. According to the results of nucleotide BLAST, MYH522 showed the highest sequence similarity to Nonomuraea jabiensis A4036 (99.24%). The related reference strains were used to multiple sequence alignment and phylogenetic analysis based on a neighbor-joining method via MEGA7. In the neighbor-joining tree, strain MYH522 fell into the cluster of the genus Nonomuraea (Fig. 2). Thus, the strain was notated as Nonomuraea sp. MYH522. | ||
| Fig. 2 Neighbor-joining phylogenetic tree of strain MYH522. | ||
Isolation and structural identification
The MYH522 strain was cultivated with 18 L fermentation broth and extracted with ethyl acetate (EtOAc). The extract was fractionated successively by column chromatography (CC) on silica gel, Sephadex LH-20, and RP-HPLC to produce a group of macrolactams. The major constituent was readily determined to be fluvirucin B6 (1) by comparison of 1H and 13C NMR data with those reported.18 Besides, four new analogs named fluvirucins B7–B10 (2–5) were co-isolated. The new compounds were subjected to structure elucidation by a set of spectroscopic experiments.Fluvirucin B7 (2) was obtained as an amorphous powder. The molecular formula was determined as C24H44N2O6 by the HRESIMS ion at m/z 457.3287 [M + H]+ (calcd 457.3278 for C24H45N2O6), which required four degrees of unsaturations. The IR spectrum of 2 displayed amide absorptions at 3302 cm−1 and 1643 cm−1. The structure of 2 was fully assigned by comparision with those of 1 and by further analyses of the 1D and 2D NMR data.18 The 1H spectrum displayed four methyl groups shown as one methyl singlet, one methyl triplet, and two methyl doublets, which were one methyl triplet less than those of 1. The 13C NMR and DEPT spectra indicated 24 signals that corresponded to 2 sp2 (2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 22 sp3 carbon atoms (4 CH3, 10 CH2, 3 CH, 4 OCH, and 1 OCO), accounting for two double bond equivalents (DBEs) (Tables 1 and 2). The remaining unsaturations were attributed to two cycles in the structure. The O-bearing methine protons resonating between δH 3.56 and δH 4.86 in conjunction with an acetal carbon (δC 99.1, C-1′, CH) suggested the presence of a sugar unit. Analysis of COSY spectrum delineated a long proton spin system of H2-2/H2-3/H2-4/H2-5/H-6/H2-7/H2-8/H-9/H-10/H2-11/H2-12/H2-13, H-6/H3-19, and H-10/H2-15/H3-16 (Fig. 3). The HMBC correlations from H2-2 (δH 2.18, 2.27) and H2-13 (δH 3.20, 3.35) to C-1 (δC 176.1, C) connected the long COSY fragment and permitted the establishment of a 14-membered macrolactam skeleton. The HMBC cross-peaks from a methyl doublet (δH 0.92, H3-19) to C-5 (δC 34.2, CH2), C-6 (δC 32.4, CH), and C-7 (δC 25.7, CH2) allowed the assignment of a methyl group at C-6. The methyl triplet (δH 0.90, H3-16) suggested the presence of an ethyl group, which was positioned at C-10 based on the HMBC correlations from H3-16 to C-15 (δC 22.8, CH2) and C-10 (δC 41.8, CH).
O) and 22 sp3 carbon atoms (4 CH3, 10 CH2, 3 CH, 4 OCH, and 1 OCO), accounting for two double bond equivalents (DBEs) (Tables 1 and 2). The remaining unsaturations were attributed to two cycles in the structure. The O-bearing methine protons resonating between δH 3.56 and δH 4.86 in conjunction with an acetal carbon (δC 99.1, C-1′, CH) suggested the presence of a sugar unit. Analysis of COSY spectrum delineated a long proton spin system of H2-2/H2-3/H2-4/H2-5/H-6/H2-7/H2-8/H-9/H-10/H2-11/H2-12/H2-13, H-6/H3-19, and H-10/H2-15/H3-16 (Fig. 3). The HMBC correlations from H2-2 (δH 2.18, 2.27) and H2-13 (δH 3.20, 3.35) to C-1 (δC 176.1, C) connected the long COSY fragment and permitted the establishment of a 14-membered macrolactam skeleton. The HMBC cross-peaks from a methyl doublet (δH 0.92, H3-19) to C-5 (δC 34.2, CH2), C-6 (δC 32.4, CH), and C-7 (δC 25.7, CH2) allowed the assignment of a methyl group at C-6. The methyl triplet (δH 0.90, H3-16) suggested the presence of an ethyl group, which was positioned at C-10 based on the HMBC correlations from H3-16 to C-15 (δC 22.8, CH2) and C-10 (δC 41.8, CH).
| No. | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1 | 176.1, C | 179.7, C | 178.9, C | 178.9, C |
| 2 | 36.2, CH2 | 43.0, CH | 50.9, CH | 51.2, CH |
| 3 | 26.5, CH2 | 36.2, CH2 | 34.1, CH2 | 34.6, CH2 |
| 4 | 26.3, CH2 | 26.1, CH2 | 26.5, CH2 | 26.3, CH2 |
| 5 | 34.2, CH2 | 35.2, CH2 | 27.9, CH2 | 33.4, CH2 |
| 6 | 32.4, CH | 32.4, CH | 27.8, CH2 | 39.8, CH |
| 7 | 25.7, CH2 | 26.1, CH2 | 27.2, CH2 | 23.2, CH2 |
| 8 | 21.9, CH2 | 22.8, CH2 | 20.0, CH2 | 22.6, CH2 |
| 9 | 78.5, CH | 78.7, CH | 78.4, CH | 78.5, CH |
| 10 | 41.8, CH | 42.3, CH | 42.3, CH | 42.0, CH |
| 11 | 26.9, CH2 | 26.3, CH2 | 26.2, CH2 | 26.3, CH2 |
| 12 | 25.8, CH2 | 28.5, CH2 | 28.6, CH2 | 28.7, CH2 |
| 13 | 39.7, CH2 | 39.8, CH2 | 39.7, CH2 | 39.7, CH2 |
| 15 | 22.8, CH2 | 22.3, CH2 | 22.2, CH2 | 22.0, CH2 |
| 16 | 10.3, CH3 | 9.4, CH3 | 9.3, CH3 | 9.2, CH3 |
| 17 | 18.9, CH3 | 27.7, CH2 | 27.5, CH2 | |
| 18 | 12.4, CH3 | 12.4, CH3 | ||
| 19 | 20.9, CH3 | 20.9, CH3 | 28.2, CH2 | |
| 20 | 12.8, CH3 | |||
| 1′ | 99.1, CH | 99.4, CH | 99.2, CH | 99.3, CH |
| 2′ | 71.1, CH | 71.2, CH | 71.1, CH | 71.2, CH |
| 3′ | 49.6, CH | 49.6, CH | 48.8, CH | 48.8, CH |
| 4′ | 72.2, CH | 72.3, CH | 72.2, CH | 72.2, CH |
| 5′ | 68.8, CH | 68.8, CH | 68.8, CH | 68.8, CH |
| 6′ | 17.0, CH3 | 17.0, CH3 | 17.1, CH3 | 17.0, CH3 |
| 7′ | 173.0, C | 173.0, C | 173.0, C | 173.0, C |
| 8′ | 22.6, CH3 | 22.7, CH3 | 22.7, CH3 | 22.7, CH3 |
| No. | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 2 | 2.27, m; 2.18, m | 2.28, ddt (12.9, 9.1, 4.4) | 2.10, m | 2.08, ddt (13.7, 8.9, 3.9) |
| 3 | 1.81, m; 1.53, m | 1.49, m; 1.42, m | 1.56, m; 1.46, m | 1.57, m; 1.48, m |
| 4 | 1.38, m; 1.24, m | 1.41, m; 1.16, m | 1.55, m; 1.18, m | 1.47, m; 1.14, m |
| 5 | 1.26, m; 1.24, m | 1.43, m; 1.06, m | 1.46, m; 1.21, m | 1.46, m; 1.02, m |
| 6 | 1.70, m | 1.68, m | 1.48, m; 1.48, m | 1.37, m |
| 7 | 1.53, m; 1.47, m | 1.42, m; 1.41, m | 1.36, m; 1.36, m | 1.47, m; 1.31, m |
| 8 | 1.68, m; 1.37, m | 1.56, m; 1.49, m | 1.36, m; 1.36, m | 1.55, m; 1.46, m |
| 9 | 3.63, m | 3.62, m | 3.62, m | 3.62, m |
| 10 | 1.54, m | 1.53, m | 1.55, m | 1.55, m |
| 11 | 1.47, m; 1.25, m | 1.40, m; 1.31, m | 1.56, m; 1.48, m | 1.47, m; 1.41, m |
| 12 | 1.47, m; 1.26, m | 1.62, m; 1.35, m | 1.62, m; 1.36, m | 1.64, m; 1.34, m |
| 13 | 3.35, m; 3.20, m | 3.57, m; 2.95, ddd (13.4, 5.5, 2.9) | 3.59, m; 2.98, ddd (13.4, 5.5, 2.8) | 3.60, m; 2.96, ddd (13.6, 5.4, 2.8) |
| 14 | 8.04, dd (5.9, 5.9) | 8.05, dd (8.0, 2.8) | ||
| 15 | 1.62, m; 1.47, m | 1.57, m; 1.53, m | 1.61, m; 1.39, m | 1.63, m; 1.33, m |
| 16 | 0.90, t (7.5) | 0.87, t (7.3) | 0.87, t (7.2) | 0.87, overlapped |
| 17 | 1.08, d (6.9) | 1.53, m; 1.39, m | 1.57, m; 1.38, m | |
| 18 | 0.87, t (7.2) | 0.87, overlapped | ||
| 19 | 0.92, d (7.0) | 0.91, d (6.7) | 1.26, m; 1.26, m | |
| 20 | 0.87, overlapped | |||
| 1′ | 4.86, d (1.5) | 4.87, d (1.6) | 4.87, s | 4.87, d (0.9) |
| 2′ | 3.56, m | 3.55, m | 3.55, m | 3.57, m |
| 3′ | 4.14, dd (3.0, 3.0) | 4.15, dd (3.0, 3.0) | 4.14, dd (3.0, 3.0) | 4.15, dd (3.0, 3.0) |
| 4′ | 3.56, m | 3.55, m | 3.55, m | 3.57, m |
| 5′ | 4.02, q (6.5) | 4.03, q (6.5) | 4.03, q (6.5) | 4.03, q (6.5) |
| 6′ | 1.21, d (6.5) | 1.21, d (6.5) | 1.21, d (6.5) | 1.21, d (6.5) |
| 8′ | 2.03, s | 2.03, s | 2.03, s | 2.03, s |
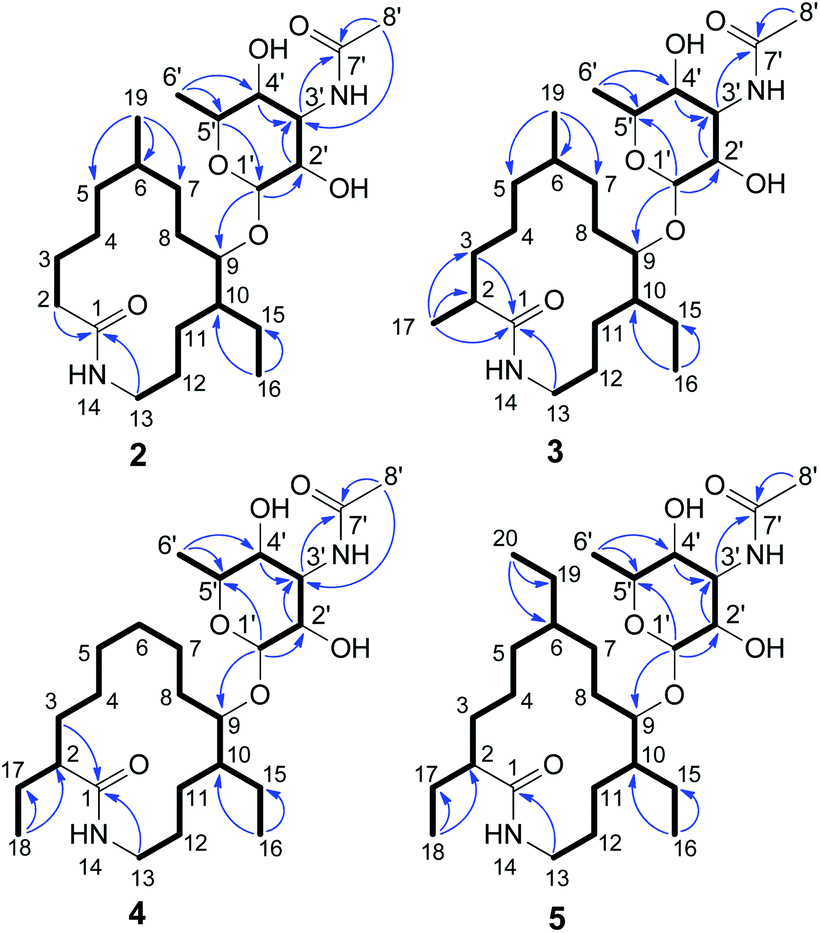 | ||
| Fig. 3 Key 1H–1H COSY correlations (bold) and HMBC correlations (blue arrows) of 2–5. | ||
The second fragment was interpreted starting from another proton sequence of H-1′/H-2′/H-3′/H-4′/H-5′/H3-6′ deduced by COSY spectrum. The HMBC correlations from H-1′ to C-5′, from H-2′ and H-4′ to C-3′ (δC 49.6, CH), and from H3-6′ to C-4′ and C-5′ suggested an amino sugar. An acetyl group (δH 2.03 H3-8′; δC 22.6 C-8′ CH3, 173.0 C-7′, C) was substituted at C-3′ NH based on the HMBC correlations from H-3′ (δH 4.14) and H3-8′ to C-7′ and the diagnostic long-range HMBC correlation from H3-8′ to C-3′. The relative configuration of amino sugar was determined by analysis of coupling constants and NOESY experiment. The obvious NOE correlation of H-3′/H-5′ indicated they were axial protons and in syn relationship (Fig. S1†). The anomeric proton (δH 4.86, d, J = 1.5) and carbon are resonated at deshielding region indicating an α-pyranoside configuration and an equatorial orientation of H-1′,10 which was suggested by the absence of NOE correlations of either H-1′/H-3′ or H-1′/H-5′. The small coupling constants of 3JH-2′,H-3′ (3.0) and 3JH-3′,H-4′ (3.0) indicated that both H-2′ and H-4′ were equatorially orientated. The amino sugar was then determined to be N-acetyl-4-epi-mycosamine. Finally, a HMBC correlation from H-1′ to C-9 (δC 78.5, CH) suggested the connectivity of two fragments, completing the structure assignment of 2.
Fluvirucin B8 (3) was isolated as an optically active powder. The HRESIMS gave a molecular formula of C25H46N2O6 (m/z 471.3444 [M + H]+, calcd for 471.3434), which are 14 atomic mass units more than that of 2. The 1H and 13C NMR spectroscopic data of 3 closely resembled those of 2. The obvious difference was observed for the presence of one additional methyl (δH 1.08, d, J = 6.9 Hz, H3-17; δC 18.9, C-17) in 3. The COSY cross-peak of H3-17/H-2 and HMBC correlations from H3-17 to C-1 (δC 179.7, C), C-2 (δC 43.0, CH), and C-3 (δC 36.2, CH2) allowed the assignment of the methyl group at C-2. Extensive analysis of 1D and 2D NMR spectra revealed that the rest part of 3 was the same as that of 2. Therefore, the structure of 3 was determined as depicted.
Fluvirucin B9 (4) was assigned a molecular formula as same as that of 3 on the basis of HRESIMS data. Comparison of 1H and 13C NMR spectra of 4 and 3 revealed a high similarity. The obvious difference was observed for the disappearance of one methyl and the presence of one more ethyl group in the 1H spectrum of 4. The methyl group of C-19 in 2 and 3 was replaced by a hydrogen, which led to a C-6 methylene. The ethyl group (δH 1.39, 1.53, H2-17 and 0.87, t, J = 7.2 Hz, H3-18; δC 27.7, CH2, C-17 and 12.4, CH3, C-18) was located at C-2 by interpretation of COSY cross-peaks of H3-18/H2-17/H-2 and HMBC correlations from H3-18 to C-17 and C-2 (δC 50.9, CH). The remaining part was determined to be as same as that of 3 on the basis of comprehensive analysis of 2D NMR data. The structure of 4 was then deduced as shown.
Fluvirucin B10 (5), an optically active powder, possessed a molecular formula of C27H50N2O6 on the base of HRESIMS data at m/z 499.3763 [M + H]+ (calcd for C27H51N2O6, 499.3747). The 1H and 13C NMR spectra of 5 was close to those of 4 with the exception of one more ethyl group (δH 1.26, H2-19 and 0.87, H3-20; δC 28.2, CH2, C-19 and 12.8, CH3, C-20). This is in consistent with the fact that molecular weight of 5 was 28 mass units more than that of 4. The ethyl group was positioned at C-6 by COSY cross-peaks of H3-20/H2-19/H-6 and by HMBC correlations from H3-20 to C-19 and C-6 (δC 39.8, CH). Cumulative analyses of the 1D and 2D NMR spectroscopic data allowed the structure assignment of 5.
The relative configurations at C-2, C-6, C-9, and C-10 of aglycons in fluvirucins B7–B10 could not be assigned independently on the basis of NMR spectroscopic data. Fluvirucins B6–B10 are featured with a N-acetylated amino sugar in contrast to other fluvirucins. Specifically, fluvirucins B6, B9, and B10 are N-acetylated derivatives of B1, B0, and B3/Sch 39185, respectively.10,14,16,18 The NMR spectroscopic data of B9 and B10 was compared with those of B0 and B3/Sch 39185. The 13C NMR resonances of B9 and B0 showed a high similarity in aglycone part (Table S1†). Moreover, the negative optical rotation (OR) value of fluvirucin B9 ([α]28.6D −29, c 1.0, MeOH) was close to that of B0 ([α]28.6D −38, c 0.5, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeOH/CHCl3).16 Meanwhile, the NMR and OR data of fluvirucin B10 ([α]30.9D −9.7, c 0.2, MeOH) were highly similar to those of fluvirucin B3/Sch 39185 ([α]26D −5.8, c 0.5, MeOH).10,14 All of fluvirucins have identical absolute configurations at C-2, C-6, C-9, and C-10, which were determined by X-ray crystallography for fluvirucins A1 and B1.9,28 Fluvirucins B7–B10 are likely to have the same configurations as known fluvirucins according to high similarity in NMR data of aglycon and OR values, and the assumed similar biogenetic pathway.
:1 MeOH/CHCl3).16 Meanwhile, the NMR and OR data of fluvirucin B10 ([α]30.9D −9.7, c 0.2, MeOH) were highly similar to those of fluvirucin B3/Sch 39185 ([α]26D −5.8, c 0.5, MeOH).10,14 All of fluvirucins have identical absolute configurations at C-2, C-6, C-9, and C-10, which were determined by X-ray crystallography for fluvirucins A1 and B1.9,28 Fluvirucins B7–B10 are likely to have the same configurations as known fluvirucins according to high similarity in NMR data of aglycon and OR values, and the assumed similar biogenetic pathway.
Antimicrobial activity
The isolated compounds were screened for antimicrobial activities against pathogenic bacteria including Staphylococcus aureus ATCC25923 and Escherichia coli ATCC25922, and Candida albicans (the standard clinical isolated strain SC5314, the fluconazole-resistant clinical isolates of 901 and 904).29 Compounds 1–5 did not exhibit any activities against S. aureus and E. coli up to 128 μg mL−1 (Table S2†). Moreover, 1–5 were not active against C. albicans SC5314, 901, or 904 at 64 μg mL−1 (Table 3). However, when combined with 8 μg mL−1 of fluconazole, compounds 1, 4, 5 showed synergistic antifungal activities against fluconazole-resistant isolates of C. albicans 901 and 904 during 24 hours.| Compounds | Used alone | With 8 μg mL−1 of fluconazole | |||
|---|---|---|---|---|---|
| SC5314 | 901 | 904 | 901 | 904 | |
| 1 | >64 | >64 | >64 | 0.125 | 0.125 |
| 2 | >64 | >64 | >64 | >64 | >64 |
| 3 | >64 | >64 | >64 | >64 | >64 |
| 4 | >64 | >64 | >64 | 0.125 | 0.125 |
| 5 | >64 | >64 | >64 | 0.125 | 0.125 |
| Fluconazole | 0.125 | >64 | >64 | >64 | >64 |
Discussion
Fluvirucins are characterized by 14-membered macrolactam attached with an aminosugar moiety. The structures of fluvirucins differ in the alkyl substituents and the sugar units. The alkyl substituents are usually located at C-2, C-6, and C-10, respectively, varying among methyl, ethyl, and hydroxyethyl groups (Fig. 4). The aminosugar, normally mycosamine or 4-epi-mycosamine, is appended to the macrocylic skeleton through a glycosidic linkage. According to the position of aminosugar attachment, fluvirucins are classified into “A” series having a sugar at C-3 (A1, A2) and “B” series with a sugar at C-9 (B0–B6).10,21 Rarely, the aminosugar is N-substituted by a 2-phenethylaminocarbonyl group (B4, B5) or an acetyl group (B6).18 The aminosugar monosaccharide may also be replaced by disaccharide (Sch 42729) or trisaccharide (Sch 42282).11,12 Besides, fluvirucins without any sugar unit (C1, C2) have been naturally isolated.19 The discovery of fluvirucins B7–B10 enriched the N-acetylated derivatives of fluvirucin. | ||
| Fig. 4 Structures of known fluvirucins. | ||
Biosynthetically, fluvirucins belong to an uncommon class of polyketides (PKS) using a specific β-alanine as starter unit. Fluvirucins B6–B10 share an ethyl group at C-10, but differ from each other in the alkyl substituents at C-2 and C-6. The alkyl substituents at both C-2 and C-6 vary between hydrogen atom, methyl, and ethyl groups which raises the question that the fluvirucin PKS may accept and process different substrates during chain extension. The biosynthetic gene clusters (BGC) for fluvirucins B1 (flu) and B2 (flv) have been completely identified and characterized.30,31 The flu and flv show a high similarity between each other containing three modular PKS genes (flu A–C and flv P1–P3) and β-amino acid forming genes. The Flu A–C as well as the homologous proteins of Flv P1–P3 are both divided into five modules (M1–M5). The substrate specificity of acyl transferase (AT) in each module is experimentally examined, showing M2_AT and M4_AT specific for malony-CoA, M3_AT for methylmalonyl-CoA, and M1_AT and M5_AT for ethylmalonyl-CoA. We re-analyzed the AT domains in Flu A–C and Flv P1–P3 using bioinformatic tools (Fig. 5).32,33 The M2_AT and M4_AT have the conserved “HAFH” motif (residues 194–197) that is specific to malonyl-CoA. Whereas, the M1_AT, M3_AT, and M5_AT have “VASH” or “YASH” motifs (residues 194–197), which are more specific for methylmalonyl-CoA (YASH motif) than ethylmalonyl-CoA (XAGH motif). The conserved motifs of AT domains could not explain the substrates promiscuity in fluvirucins. Probably, some residues dominating the canonical substrate specificity are not identified.
 | ||
| Fig. 5 Alignment of partial amino acid sequences of the AT domains of Flu PKSs (fluvirucin B1) and Flv PKSs (fluvirucin B2). Key catalytic residues are highlighted in bold, red for methylmalonyl-CoA or ethylmalonyl-CoA and blue for malonyl-CoA. | ||
Fluvirucins are known to have antimicrobial and anthelmintic activities.9,15,17,21 Compounds 1–5 displayed almost no antibacterial or antifungal activities when used alone at the tested concentrations. Decreased antimicrobial activity have been observed for other N-substituted fluvirucins or fluvirucins without aminosugar unit.11,18–20 The current results implied that the aminosugar especially the free amino group in fluvirucins was essential for antimicrobial activity. The synergistic antifungal activities of 1, 4, and 5 against the fluconazole-resistant C. albicans suggested that fluvirucins may help fluconazole availability in the C. albicans cells by damaging cell wall or increasing intracellular fluconazole concentration, or targeting other proteins on a parallel pathway that converges on an essential process. Probably, the ethyl group at C-2 plays an important role in the synergistic antifungal activity.
Conclusions
In summary, we have discovered fluvirucins B7–B10 as new N-acetylated derivatives from a marine rare actinomycete. The results not only enrich the family of fluvirucin but also imply the substrates promiscuity of fluvirucin PKS. The biological results indicate the importance of free amino group of fluvirucins in the antimicrobial activity. The synergistic antifungal activity of fluvirucins with fluconazole is worthy of further investigation.Experimental
General experimental procedures
Optical rotations were measured in MeOH on an Autopol-IV polarimeter at the sodium D line (589 nm). Infrared spectra were recorded in thin polymer film on a Nexus-470 FT-IR spectrophotometer (Nicolet, USA). The NMR spectra were recorded at 300 K on a Bruker AVANCE 600 NMR spectrometer. Chemical shifts are reported in parts per million (δ), with use of the residual CD3OD signals (δH = 3.31 ppm; δC = 49.0 ppm) as internal standard, and coupling constants (J) are in Hz; assignments are supported by COSY, HSQC, HMBC, and NOESY experiments. High-resolution mass spectral data were obtained using a Bruker APEXIII 7.0 T FT-MS spectrometer in m/z, resolution 5000; an isopropyl alcohol solution of sodium iodide (2 mg mL−1) was used as a reference. Semi-preparative HPLC was performed on an Agilent 1100 system with a UV detector using a YMC ODS-A column (5 μm, 250 × 10 mm). Silica gel (200–300 and 400–500 mesh; Yantai, China) was used for column chromatography. Precoated silica gel plates (HSGF254; Yantai, China) were used for TLC. Compounds were detected on TLC under UV light or by heating after spraying with anisaldehyde–sulfuric acid reagent. Antimicrobial activity assay was measured by a microplate reader SpectraMax M5.Strain isolation and identification
Strain MYH522 was isolated by ISP2 medium (yeast extract 4.0 g, malt extract 10.0 g, dextrose 4.0 g, agar 20.0 g, distilled water 1.0 L, pH 7.2) from a marine sponge, which was collected off Xisha Island in South China Sea.For phylogenetic study, strain MYH522 was grown in tryptic soy broth medium for 3 days at 28 °C. The 16S rDNA gene sequence was amplified by universal primers 27F (5′-AGTTTGATCMTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). PCR amplification reactions were prepared in a 25 μL reaction volume containing 12.5 μL of PCR 2× T5 Mix, 8.5 μL of distilled H2O, 1 μL of 27F primer, 1 μL of 1492R primer, 1 μL DMSO, and 1 μL of DNA template, and was performed under reference conditions. PCR products were sent to the Sangon Biotech for sequencing. The 16S rDNA sequence was submitted to GenBank and blasted on NCBI. The multiple sequences were aligned by Clustal W. Phylogenetic trees were constructed with the MEGA7 software using neighbor-joining method. Bootstrap analysis was used to evaluate the trees topology. Kimura two-parameter model was used for phylogeny construction and evolutionary distances analysis.
Fermentation, isolation, and structure characterization
The ISP2 medium was used for sporulation. Strain MYH522 was cultured with fermentation medium (starch soluble 10.0 g, yeast extract 4.0 g, peptone 2.0 g, CaCO3 1.0 g, Fe2(SO4)3·4H2O (8 g L−1) 5.0 mL, KBr (20 g L−1) 5.0 mL, distilled water 1.0 L, pH 7.0) at 28 °C for 7 days. An 18 L of fermentation broth was extracted with EtOAc for three times. The combined extracts were concentrated to afford a residue (1.54 g), which was subjected to ODS column chromatography, eluted by gradient MeOH–H2O solution (30–100%, v/v) to yield 6 fractions (Fr. 1–6). Fr. 3 was separated by Sephadex LH-20 (MeOH/CH2Cl2, 1:2) to give 5 fractions (Fr. 3.1–3.5). Fr. 3.2 was subjected to semipreparative RP-HPLC (70% MeOH, 2.0 mL min−1) to afford 2 (2.3 mg, tR 15.6 min), 4 (14.9 mg, tR 16.4 min), 3 (2.0 mg, tR 18.5 min), 1 (65.5 mg, tR 20.7 min), and 5 (6.5 mg, tR 27.2 min), respectively.
:1); [α]27.8D −76 (c 0.2, MeOH); IR (film) νmax 3302, 2935, 2873, 1712, 1643, 1444, 1258, 981 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; HRESIMS m/z: 457.3287 [M + H]+ (calcd for C24H45N2O6, 457.3278).:1); [α]28.2D −29 (c 0.2, MeOH); IR (film) νmax 3284, 2923, 2873, 1722, 1641, 1462, 1103, 979 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; HRESIMS m/z: 471.3444 [M + H]+ (calcd for C25H47N2O6, 471.3434).:1); [α]28.6D −29 (c 1.0, MeOH); IR (film) νmax 3294, 2932, 1638, 1443, 1281,1102, 1017, 827 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; HRESIMS m/z: 471.3449 [M + H]+ (calcd for C25H47N2O6, 471.3434).:1); [α]28.9D −9.7 (c 0.2, MeOH); IR (film) νmax 3251, 2922, 1665, 1600, 1495, 1453, 1202, 702 cm−1; 1H and 13C NMR spectroscopic data, see Tables 1 and 2; HRESIMS m/z: 499.3763 [M + H]+ (calcd for C27H51N2O6, 499.3747).Antibacterial activity assays
The antibacterial activity assays were performed against Gram-positive bacteria S. aureus and Gram-negative bacteria E. coli according to the Clinical and Laboratory Standards Institute (CLSI) methods.34 The bacteria were grown in LB medium at 37 °C, 200 rpm for overnight, and were diluted to a concentration of 105 colony forming unit (CFU) mL−1. Tested compounds and kanamycin in dimethyl sulfoxide (DMSO) were 2-fold serially diluted with LB medium to prepare 100 μL of solutions ranging from 128 to 4 μg mL−1, and were added to 96-well microtiter plates with 100 μL of bacterial suspensions in each well. The plates were incubated at 37 °C for 24 h. Optical density was recorded at 630 nm using microplate reader. IC50 values refer the lowest concentration of compounds that cause 50% inhibition of bacterial growth.Antifungal activity assays29
The antifungal activity assays was performed using a broth microdilution methodology adjusted from CLSI M38-A2 and M27-A3 standard. The fungal suspension was applied to 96-well plates at a concentration of 103 CFU mL−1 in RPMI 1640 medium. The tested compounds and fluconazole were serially diluted with final concentrations ranging from 64 to 0.125 μg mL−1. To evaluate the synthetic effect, fluconazole was added into the fungal suspension at a concentration of 8 μg mL−1 before applied to 96-well plates, while the compounds were serially diluted. Plates were incubated for 24 hours at 30 °C. Optical density was measured at 630 nm with microplate reader, and background optical density was removed. The minimum inhibitory concentration (MIC80) is that at which the growth of 80% tested strains is inhibited.Author contributions
Conceptualization and supervision – P. S.; writing – H. Y. and P. S.; investigation and methodology – H. Y., S. C., H. L., and R. W.; review & editing – Y. J. and L. Y.; all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (41876184, 81622044, and 82173867) and National Key Research and Development Program of China (2019YFC0312502 and 2019YFC0312601), Shanghai International Science and Technology Cooperation Project (21430713000), Shanghai Science and Technology Support Project in the Field of Biomedicine Project (19431901300), and Shanghai Pujiang Program (21PJD0081).Notes and references
- J. Bérdy, J. Antibiot., 2005, 58, 1–26 CrossRef PubMed.
- W. Fenical and P. R. Jensen, Nat. Chem. Biol., 2006, 2, 666–673 CrossRef CAS PubMed.
- P. R. Jensen, B. S. Moore and W. Fenical, Nat. Prod. Rep., 2015, 32, 738–751 RSC.
- P. Manivasagan, K.-H. Kang, K. Sivakumar, E. C. Y. Li-Chan, H.-M. Oh and S.-K. Kim, Environ. Toxicol. Pharmacol., 2014, 38, 172–188 CrossRef CAS PubMed.
- S. Qi, M. Gui, H. Li, C. Yu, H. Li, Z. Zeng and P. Sun, Chem. Biodiversity, 2020, 17, e2000024 CrossRef CAS PubMed.
- R. Subramani and D. Sipkema, Mar. Drugs, 2019, 17, 249 CrossRef CAS PubMed.
- K. Tiwari and R. K. Gupta, Crit. Rev. Biotechnol., 2012, 32, 108–132 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang and J. Ruan, Int. J. Syst. Bacteriol., 1998, 48, 411–422 CrossRef PubMed.
- V. R. Hegde, M. G. Patel, V. P. Gullo, A. K. Ganguly, O. Sarre, M. S. Puar and A. T. McPhail, J. Am. Chem. Soc., 1990, 112, 6403–6405 CrossRef CAS.
- N. Naruse, T. Tsuno, Y. Sawada, M. Konishi and T. Oki, J. Antibiot., 1991, 44, 741–755 CrossRef CAS PubMed.
- V. R. Hegde, M. G. Patel, V. P. Gullo, A. C. Horan, A. H. King, F. Gentile, G. H. Wagman, M. S. Puar and D. Loebenberg, J. Antibiot., 1993, 46, 1109–1115 CrossRef CAS PubMed.
- V. R. Hegde, M. G. Patel, A. C. Horan, A. H. King, F. Gentile, M. S. Puar and D. Loebenberg, J. Antibiot., 1998, 51, 464–670 CrossRef CAS PubMed.
- R. Cooper, I. Truumees, R. Yarborough, D. Loebenberg, J. Marquez, A. Horan, M. Patel, V. Gullo, M. Puar and B. Pramanik, J. Antibiot., 1992, 45, 633–638 CrossRef CAS PubMed.
- V. Hegde, M. Patel, A. Horan, V. Gullo, J. Marquez, I. Gunnarsson, F. Gentile, D. Loebenberg, A. King, M. Puar and B. Pramanik, J. Antibiot., 1992, 45, 624–632 CrossRef CAS PubMed.
- V. R. Hegde, M. G. Patel, V. P. Gullo and M. S. Puar, J. Chem. Soc., Chem. Commun., 1991, 810–812 RSC.
- S. Ayers, D. L. Zink, J. S. Powell, C. M. Brown, A. Grund, O. Genilloud, O. Salazar, D. Thompson and S. B. Singh, J. Antibiot., 2008, 61, 59–62 CrossRef CAS PubMed.
- S. Ayers, D. L. Zink, K. Mohn, J. S. Powell, C. M. Brown, T. Murphy, A. Grund, O. Genilloud, O. Salazar, D. Thompson and S. B. Singh, J. Nat. Prod., 2007, 70, 1371–1373 CrossRef CAS PubMed.
- A. S. Leutou, I. Yang, T. C. Le, D. Hahn, K. M. Lim, S. J. Nam and W. Fenical, J. Antibiot., 2018, 71, 609–612 CrossRef CAS PubMed.
- M. Costa, P. Zúñiga, A. M. Peñalver, M. Thorsteinsdottir, M. Pérez, L. M. Cañedo and C. Cuevas, Nat. Prod. Commun., 2017, 12, 679–682 CrossRef PubMed.
- N. Naruse, O. Tenmyo, K. Kawano, K. Tomita, N. Ohgusa, T. Miyaki, M. Konishi and T. Oki, J. Antibiot., 1991, 44, 733–740 CrossRef CAS PubMed.
- K. Tomita, N. Oda, Y. Hoshino, N. Ohkusa and H. Chikazawa, J. Antibiot., 1991, 44, 940–948 CrossRef CAS PubMed.
- E. Llàcer, F. Urpí and J. Vilarrasa, Org. Lett., 2009, 11, 3198–3201 CrossRef PubMed.
- Y. G. Suh, S. A. Kim, J. K. Jung, D. Y. Shin, K. H. Min, B. A. Koo and H. S. Kim, Angew. Chem., Int. Ed. Engl., 1999, 38, 3545–3547 CrossRef CAS PubMed.
- M. Martín, G. Mas, F. Urpí and J. Vilarrasa, Angew. Chem., Int. Ed. Engl., 1999, 38, 3086–3089 CrossRef.
- P. Sun, F.-Y. Cai, G. Lauro, H. Tang, L. Su, H.-L. Wang, H. H. Li, A. Mándi, T. Kurtán, R. Riccio, G. Bifulco and W. Zhang, J. Nat. Prod., 2019, 82, 1264–1273 CrossRef CAS PubMed.
- J. Gong, P. Sun, N. Jiang, R. Riccio, G. Lauro, G. Bifulco, T.-J. Li, W. H. Gerwick and W. Zhang, Org. Lett., 2014, 16, 2224–2227 CrossRef CAS PubMed.
- P. Sun, D.-X. Xu, A. Mándi, T. Kurtán, T.-J. Li, B. Schulz and W. Zhang, J. Org. Chem., 2013, 78, 7030–7047 CrossRef CAS PubMed.
- N. Naruse, M. Konishi, T. Oki, Y. Inouye and H. Kakisawa, J. Antibiot., 1991, 44, 756–761 CrossRef CAS PubMed.
- H. Quan, Y. Y. Cao, Z. Xu, J. X. Zhao, P. H. Gao, X. F. Qin and Y. Y. Jiang, Antimicrob. Agents Chemother., 2006, 50, 1096–1099 CrossRef CAS PubMed.
- T. Y. Lin, L. S. Borketey, G. Prasad, S. A. Waters and N. A. Schnarr, ACS Synth. Biol., 2013, 2, 635–642 CrossRef CAS PubMed.
- A. Miyanaga, Y. Hayakawa, M. Numakura, J. Hashimoto, K. Teruya, T. Hirano, K. Shin-Ya, F. Kudo and T. Eguchi, Biosci., Biotechnol., Biochem., 2016, 80, 935–941 CrossRef CAS PubMed.
- F. Del Vecchio, H. Petkovic, S. G. Kendrew, L. Low, B. Wilkinson, R. Lill, J. Cortes, B. A. Rudd, J. Staunton and P. F. Leadlay, J. Ind. Microbiol. Biotechnol., 2003, 30, 489–494 CrossRef CAS PubMed.
- C. D. Reeves, S. Murli, G. W. Ashley, M. Piagentini, C. R. Hutchinson and R. McDaniel, Biochemistry, 2001, 40, 15464–15470 CrossRef CAS PubMed.
- J. H. Rex and M. J. Ferraro, in Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition, Clinical and Laboratory Standards Institute, Wayne, PA, 2012, vol. 32, 07–A0907–A0907–A09 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1, S2 and Fig. S1, S2–S5 (MS and NMR spectra of 2–5). See https://doi.org/10.1039/d2ra01701f |
| This journal is © The Royal Society of Chemistry 2022 |