
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped graphene–poly(hydroxymethylated-3,4-ethylenedioxythiophene) nanocomposite electrochemical sensor for ultrasensitive determination of luteolin†
Shanshan Yua,
Yining Chena,
Ying Yanga,
Yuanyuan Yao *a and
Haijun Song*b
*a and
Haijun Song*b
aJiaxing Key Laboratory of Molecular Recognition and Sensing, College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, PR China. E-mail: yaoyuanyuan2007@163.com
bCollege of Mechanical and Electrical Engineering, Jiaxing University, Jiaxing 314001, PR China. E-mail: songhaijun8837@126.com
First published on 23rd May 2022
Abstract
An ultrasensitive luteolin electrochemical sensor was constructed by co-electropolymerization of nitrogen-doped graphene (N-GR) and hydroxymethylated-3,4-ethylenedioxythiophene (EDOT-MeOH) using cyclic voltammetry (CV). Because of the synergistic effects of the large surface area, superior electrical conductivity, and large amount of chemically active sites of N-GR together with the satisfactory water solubility and high conductivity of poly(hydroxymethylated-3,4-ethylenedioxythiophene) (PEDOT-MeOH), the N-GR–PEDOT-MeOH nanocomposite sensor exhibited high electrochemical sensitivity towards luteolin with a wide linear range of 0.005–10.06 μM and low detection limit of 0.05 nM. Satisfactory reproducibility, selectivity, and stability were exhibited by this electrochemical sensor. Additionally, the proposed sensor was employed for trace-level analysis of luteolin in actual samples of herbal medicines (thyme (Thymus vulgaris L.), honeysuckle (Lonicera japonica Thunb.), and Tibetan Duyiwei (Lamiophlomis rotata (Benth.) Kudo)) with satisfactory results.
1. Introduction
Luteolin (3′,4′,5,7-tetrahydroxyflavone, shown in Scheme 1) is a flavonoid that is abundant in plants and herbal medicines including thyme, green pepper, celery, artichoke, perilla leaf, peppermint, and chamomile tea.1 It is of particular interest in the pharmaceutical and clinical fields because of its anti-oxidative, anti-proliferative, anti-platelet, anti-inflammatory, anti-allergy, anti-viral, anti-bacterial, anti-ulcer, anti-cancer, anti-carcinogenic, cardiovascular protection, and cataract prevention activities.2,3 Thus, it is vital to establish an accurate and effective approach for the determination of luteolin in drug quality control and the clinical field. | ||
| Scheme 1 The modification process for N-GR–PEDOT-MeOH/GCE, and the mechanism for the determination of luteolin in practical samples. | ||
Various reported technologies have been used for the determination of luteolin, for instance, capillary electrophoresis,4 fluorimetry,5 gas chromatography,6 spectrophotometry,7 and high-performance liquid chromatography (HPLC).8 Although these methods are generally highly sensitive and efficient, they are often costly and time consuming as well. Instead, there has been interest in electrochemical sensors because of their advantages of simple operation, time-saving, low-cost, sensitivity, selectivity, and real-time online detection. Furthermore, luteolin is an electroactive compound, and in recent years, chemically modified electrodes for luteolin detection have been developed. Thus far, electrochemical sensors and biosensors based on different modified materials have been reported for the determination of luteolin, such as macroporous carbon,9 carbon nanotubes,10–12 hydroxyapatite,13 graphene and graphene oxide,13–18 polymer,10,13,17 and metals and metal oxides.14–17
Graphene is a true two-dimensional sp2-hybridized material that is used in various applications, including energy storage, nanoelectronics, and optoelectronics, and particularly in electrochemical sensors and biosensors, because of its remarkable electrical conductivity, satisfactory mechanical strength, large specific surface area, high chemical stability, and ease of modification.19,20 The chemical doping of graphene with heteroatoms such as boron, nitrogen, oxygen, sulfur, phosphorus, chlorine, or fluorine provides a promising strategy to effectively modulate and tailor its chemical reactivity and electronic properties.21–23 Among various potential dopants, nitrogen is identified as an extraordinary doping element for graphene due to its five valence electrons and comparable atomic size, which is beneficial for forming strong valence bonds with carbon atoms.23,24 Because of its large surface area, high ratio of surface active groups to volume, satisfactory biocompatibility, superior electrical conductivity, and large amount of chemically active sites, nitrogen-doped graphene (N-GR) has been regarded as a promising sensing material and exhibits satisfactory electrocatalytic ability for thiols, hydrogen peroxide,25 dopamine,26 catechol and hydroquinone,27 pesticides,28,29 ascorbic acid, dopamine, and uric acid,30,31 and nitrite.32
Recently, there have been numerous investigations on nanocomposites of conducting polymers (CPs), graphene, and graphene derivatives. Poly(3,4-ethylenedioxythiophene) (PEDOT) is one of the most successful commercially available CPs today, and it has been considered as a fascinating electrode material because of its satisfactory conductivity, low band gap, extraordinary environmental stability, and biocompatibility.33–35 In comparison to 3,4-ethylenedioxylthiophene (EDOT) monomer, its polar derivative hydroxymethylated-3,4-ethylenedioxylthiophene (EDOT-MeOH) possesses greater water solubility and lower onset oxidation potential. In addition, the polymer film of EDOT-MeOH (PEDOT-MeOH) displays more optimal electrochemical properties and biocompatibility than PEDOT.35–38 Furthermore, our previous works proved that PEDOT-MeOH is a promising electrode material for the establishment of chemo and bio-sensors.35,38 Therefore, combining the merits of N-GR with PEDOT-MeOH may be a good choice for constructing a novel electrochemical sensor with satisfactory electrochemical performance. However, application of nanocomposite based on PEDOT-MeOH and N-GR for the fabrication of a luteolin sensor has not been reported until now.
Herein, we constructed a luteolin electrochemical sensor by combining the advantages of N-GR with PEDOT-MeOH. The N-GR–PEDOT-MeOH electrode material was prepared by one-step co-electropolymerization using a cyclic voltammetry (CV) method. Scanning electron microscopy (SEM), Fourier transform infrared (FT-IR) spectrometry, and electrochemical impedance spectroscopy (EIS) were carried out to characterize the N-GR–PEDOT-MeOH nanocomposite. The modified electrode exhibited satisfactory electrocatalytic activity toward luteolin, with wide linear range, satisfactory sensitivity, selectivity, and stability, and low detection limit. We also investigated the analytical applications of a N-GR–PEDOT-MeOH-modified electrode for the trace-level detection of luteolin in samples of thyme (Thymus vulgaris), honeysuckle (Lonicera japonica), and Tibetan Duyiwei (Lamiophlomis rotata (Benth.) Kudo) capsules.
2. Experimental
2.1. Reagents
Luteolin (≥98%) was purchased from J&K Chemical Ltd. (Shanghai, China). EDOT (98%) was obtained from Sigma-Aldrich (USA). EDOT-MeOH was synthesized by our group (Fig. S1, shown in the ESI†). N-GR was purchased from Nanjing XFNANO Materials Tech Co., Ltd. (Nanjing, China). Lithium perchlorate trihydrate (LiClO4) was purchased from Shanghai Chemical Co. Ltd. (Shanghai, China). N,N-Dimethylformamide (DMF) was purchased from Beijing Chemical Industry Co., Ltd. and used after purification. All the other reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). For the supporting electrolyte solution, 0.1 M phosphate-buffered saline (PBS) with various pH values was employed, and was prepared from stock solutions of 0.1 M NaH2PO4, Na2HPO4, H3PO4, and NaOH. A stock solution of luteolin (1 × 10−3 M) in ethanol was prepared and stored at 4 °C. The other solutions were prepared with double-distilled deionized water (DDW).2.2. Apparatus
CV, differential pulse voltammetry (DPV), and EIS were performed using a model CHI660B electrochemical workstation (Chenhua Instrument Company, Shanghai, China). All the measurements were performed under the conventional three-electrode system, with a platinum wire (Φ = 1 mm) as a counter electrode, bare glassy carbon electrode (GCE) (Φ = 3 mm), PEDOT/GCE, PEDOT-MeOH/GCE, or N-GR–PEDOT-MeOH/GCE as the working electrode, and saturated calomel electrode (SCE) as a reference electrode. FT-IR was recorded by a Magna-IR 750 instrument (Nicolet, USA). SEM images were acquired with a JEOL JSM-6700F scanning electron microscope (Tokyo, Japan).2.3. Preparation of modified electrodes
Before modification, each of the bare GCEs was polished with chamois leather containing 0.05 μm γ-alumina slurry until a surface with a mirror-like shine was obtained, followed by successive sonication in DDW, ethanol, DDW for 5 min, and then drying under temperature. The PEDOT/GCE and PEDOT-MeOH/GCE were individually constructed by CV in 10 mM monomers containing 0.1 M LiClO4 with the potential scanning range from −0.8 V to 1.0 V vs. SCE for 10 cycles at 100 mV s−1 (Fig. S2A and B†). The N-GR/GCE-modified electrode was developed by electropolymerization in 1 mg mL−1 N-GR, and the N-GR–PEDOT-MeOH/GCE modified electrode was produced by co-electropolymerization in 10 mM EDOT-MeOH monomer and 1 mg mL−1 N-GR containing 0.1 M LiClO4 under the same conditions (Fig. S2C†). Then, the modified electrodes were repeatedly washed with DDW to remove the electrolyte monomers, dried at room temperature, and stored at 4 °C. The fabrication process is shown in Scheme 1.2.4. Sample preparation
Air-dried T. vulgaris was purchased from a local market. Next, 1.0 g each of T. vulgaris and L. japonica samples were washed with DDW and then dried in an electro-thermostatic blast oven. They were powdered, sifted through a 60-mesh sieve, placed in an Erlenmeyer flask containing 30 mL absolute ethanol, constantly stirred for 12 h, and then diluted to 50 mL volume with absolute ethanol. Next, the extract was filtered and centrifuged. L. rotata capsules were purchased from a drugstore. Two capsules were pounded in a mortar, and the powders were dissolved in 30 mL absolute ethanol. Then, they were filtered and diluted to 50 mL volume with absolute ethanol. All the sample solutions were stored at 4 °C.2.5. Experimental measurements
Five milliliters of 0.1 M PBS (pH 7.0) with a certain concentration of luteolin was transferred into a sealed electrochemical cell using a micro pipette. EIS was carried out with the potential of open circuit potential (OCP), and a frequency range between 100 mHz and 10 kHz, using equal concentrations of [Fe(CN)6]3−/4− redox couple (5 mM), with 0.1 M KCl as the supporting electrolyte. CV was performed from −0.2 V to 0.8 V at a scan rate of 50 mV s−1. DPV was recorded from −0.1 V to 0.6 V, with a potential increase of 0.004 V, amplitude of 0.05 V, pulse width of 0.05 s, and pulse interval of 0.2 s. Prior to each experiment, all detection solutions were deoxygenated by purging with nitrogen for 10 min. For the accumulation of luteolin, the modified electrode was placed in a stirred 0.1 M PBS (pH 7.0) solution containing a specific amount of luteolin for 120 s at 0 V.3. Results and discussion
3.1. Characterization of modified electrodes
The surface morphologies of composite modified electrode films were investigated by SEM. PEDOT film showed a twisted structure (Fig. 1A), while the PEDOT-MeOH film presented a much more rough, compact, and uniformly twisted morphology (Fig. 1B), which is beneficial for increasing the active surface area and promoting electron transfer. The morphology image of N-GR exhibits a crumpled and overlapped structure (Fig. 1C), which is favorable for enhancing the electroactive area on the electrode surface. As shown in Fig. 1D, N-GR is uniformly deposited on the surface or interior of PEDOT-MeOH film. The surface roughness of N-GR–PEDOT-MeOH film is significantly increased, supplying a greater amount of active surface area and electroactive sites, which contributes to the exchange of analytes between the surface of the electrode and the electrolyte. | ||
| Fig. 1 SEM images of (A) PEDOT, (B) PEDOT-MeOH, (C) N-GR, and (D) N-GR–PEDOT-MeOH films. | ||
FT-IR spectra of (a) PEDOT, (b) PEDOT-MeOH, (c) N-GR, and (d) N-GR–PEDOT-MeOH films were obtained, as shown in Fig. 2A. For the PEDOT and PEDOT-MeOH spectra, the vibrations at 1520, 1473, and 1325 cm−1 were attributed to the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C in the thiophene ring. The peaks at 968, 830, and 680 cm−1 were assigned to the stretching vibrations of C–S–C. The stretching modes of C–O–C in the ethylenedioxy group can be seen at 1195, 1134, 1080, and 1040 cm−1.35,38 For the N-GR spectrum, the oxygen-containing functional groups are nearly removed. The characteristic peaks of N-GR and PEDOT-MeOH were observable for the FT-IR spectrum of N-GR–PEDOT-MeOH, indicating the successful co-electropolymerization of N-GR and EDOT-MeOH.
C and C–C in the thiophene ring. The peaks at 968, 830, and 680 cm−1 were assigned to the stretching vibrations of C–S–C. The stretching modes of C–O–C in the ethylenedioxy group can be seen at 1195, 1134, 1080, and 1040 cm−1.35,38 For the N-GR spectrum, the oxygen-containing functional groups are nearly removed. The characteristic peaks of N-GR and PEDOT-MeOH were observable for the FT-IR spectrum of N-GR–PEDOT-MeOH, indicating the successful co-electropolymerization of N-GR and EDOT-MeOH.
 | ||
| Fig. 2 (A) FT-IR spectra of (a) PEDOT, (b) PEDOT-MeOH, (c) N-GR, and (d) N-GR–PEDOT-MeOH films. (B) Nyquist plots and CVs (inset) of (a) bare GCE, (b) N-GR/GCE, (c) PEDOT/GCE, (d) PEDOT-MeOH/GCE, and (e) N-GR–PEDOT-MeOH/GCE in 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl. (C) Plots of anodic peak current of [Fe(CN)6]3− (1 mM) vs. square root of scan rates for: (a) bare GCE, (b) PEDOT/GCE, (c) PEDOT-MeOH/GCE, (d) N-GR, and (e) N-GR–PEDOT-MeOH/GCE. (D) Raman spectra of (a) N-GR/GCE, (b) PEDOT-MeOH/GCE, and (c) N-GR–PEDOT-MeOH/GCE. | ||
To investigate the electron transfer capability of modified electrodes, EIS was conducted for (a) bare GCE, (b) N-GR/GCE, (c) PEDOT/GCE, (d) PEDOT-MeOH/GCE, and (e) N-GR–PEDOT-MeOH/GCE, as displayed in Fig. 2B. The bare GCE shows a small quasi-semicircle, and the electron transfer resistance (Rct) is 217 Ω. When the surface of the GCE was modified with N-GR, PEDOT, PEDOT-MeOH, or N-GR–PEDOT-MeOH, all the modified electrodes dramatically decreased, nearly to zero. Four straight lines at nearly 45° were obtained, indicating that the electron transfer process of N-GR/GCE, PEDOT/GCE, PEDOT-MeOH/GCE, and N-GR–PEDOT-MeEOH/GCE is a diffusion-limited process.39 In addition, the lower Rct suggests that the synergistic effect of N-GR and PEDOT-MeOH accelerates electron transfer between species in solution and at the electrode interface.
Furthermore, CV scans of (a) GCE, (b) N-GR/GCE, (c) PEDOT/GCE, (d) PEDOT-MeOH/GCE, and (e) N-GR–PEDOT-MeOH/GCE were also carried out in 5 mM [Fe(CN)6]3−/4− containing 0.1 M KCl (Fig. 2B (inset)). All the electrodes show a pair of well-defined anodic and cathodic peaks. Compared with GCE (curve a), the peak-to-peak separation between anodic and cathodic peaks (ΔEp) at N-GR/GCE (75 mV, curve b), PEDOT/GCE (82 mV, curve c), PEDOT-MeOH/GCE (78 mV, curve d), and N-GR–PEDOT-MeOH/GCE (66 mV, curve e) is smaller than that of bare GCE (98 mV, curve a), and the response current intensities are greatly enhanced as compared to the bare GCE. This results in high conductivity of PEDOT and PEDOT-MeOH and a large surface area and superior electrical conductivity for N-GR. The lowest ΔEp of N-GR–PEDOT-MeOH/GCE suggests that the N-GR–PEDOT-MeOH nanocomposite is a satisfactory modified electrode material, which is beneficial for acceleration of the electron transfer between the electrode interface and species in solution.
To investigate the enhancement in the electrode active surface of different modified electrodes, the redox behavior of [Fe(CN)6]3−/4− was used as a molecular probe. CV was performed for (a) PEDOT/GCE, (b) PEDOT-MeOH/GCE, (c) N-GR, and (d, e) N-GR–PEDOT-MeOH/GCE in 1 mM [Fe(CN)6]3− with the scan rates ranging from 10 mV s−1 to 500 mV s−1 (Fig. S3†). The anodic peak currents (ipa) for 1.0 mM of [Fe(CN)6]3− as a function of the square root of the scan rate (ν1/2) are shown in Fig. 2C. There was a linear relationship between ipa and ν1/2 for all the electrodes. The active electrode area was calculated according to the Randles–Ševčík equation:40
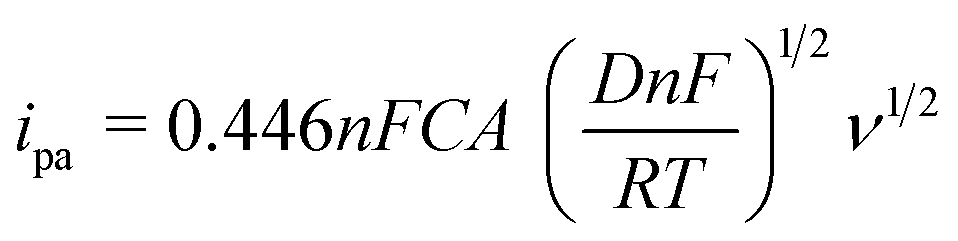
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480C mol−1).
480C mol−1).
The active electrode areas for (a) bare GCE, (b) PEDOT/GCE, (c) PEDOT-MeOH/GCE, (d) N-GR, and (e) N-GR–PEDOT-MeOH/GCE were calculated to be 0.069 cm2, 0.099 cm2, 0.40 cm2, 0.39 cm2, and 0.64 cm2, respectively (n = 1, D = 6.70 × 10−6 cm s−1). All the surface areas of the different modified electrodes were increased, which might be beneficial to increase the electrochemical active sites, and thus enhance the electrochemical response and decrease the detection limit. In addition, N-GR–PEDOT-MeOH/GCE has the highest surface area, which may be due to the synergistic effects of N-GR and PEDOT-MeOH.
The Raman spectra of N-GR, PEDOT-MeOH, and N-GR–PEDOT-MeOH nanomaterials were investigated. As shown in Fig. 2D, a salient characteristic peak at approximately 1590 cm−1 appears in the Raman spectra, corresponding to the G bands of graphene (curve a). High intensity of the D-band was observed at approximately 1350 cm−1 in the N-GR film, which clearly indicates the presence of defects in the graphene layer. These defects are usually generated during nitrogen doping.41 As shown in Fig. 2D (curve b), the Raman spectrum for the PEDOT-MeOH nanomaterial presents bands with excellent correlation to those found in the literature.42,43 The band at 440 cm−1 was assigned to oxyethylene ring deformation, 567 cm−1 is related to oxyethylene ring deformation, 1379 cm−1 to Cβ–Cβ stretching, 1436 cm−1 to symmetric CαCβ (–O) stretching, and 1559 cm−1 to asymmetric CαCβ stretching. For the N-GR–PEDOT-MeOH nanomaterial, the bands assigned to the νCC of the thiophene ring in the region between 1300-1600 cm−1 present a strong enhancement without significant shift, indicating that thiophene rings are perpendicular to the N-GR surface.
3.2. Electrochemical behavior of luteolin
CV was performed for 50 μM luteolin at the (a) bare GCE, (b) N-GR/GCE, (c) PEDOT/GCE, (d) PEDOT-MeOH/GCE, and (e) N-GR–PEDOT-MeOH/GCE in 0.1 M PBS (pH 7.0) with a scan rate of 50 mV s−1, and the results are shown in Fig. 3. All the electrodes exhibit a couple of redox peaks, but the reduction peak currents are slightly lower than the oxidation peak currents, suggesting that the electrochemical reaction of luteolin is a quasi-reversible process. Fig. 3 shows that a relatively poor oxidation peak current is observed on the bare GCE (curve a). However, when the surface of the bare GCE was modified by PEDOT and PEDOT-MeOH, the electrochemical response of luteolin was improved because of the satisfactory conductivity and catalytic activity of PEDOT and PEDOT-MeOH. For N-GR/GCE (curve b), the electrochemical response of luteolin was also improved due to the large surface area of N-GR. Compared to PEDOT/GCE (curve c), PEDOT-MeOH/GCE (curve d) displays a negative shift of peak potential and higher peak height, which may be attributed to the satisfactory electrocatalytic activity of PEDOT-MeOH film. For N-GR–PEDOT-MeOH/GCE (curve e), a remarkable increase in peak current and relatively lower peak potential is observed, which may be attributed to the synergistic effects of large surface area, superior electrical conductivity, and large amount of chemically active sites of N-GR and the satisfactory water solubility and high conductivity of PEDOT-MeOH. Hence, the N-GR–PEDOT-MeOH nanocomposite can be applied as a promising modified electrode material for the electrochemical determination of luteolin. | ||
| Fig. 3 CV scans of 50 μM luteolin at (a) the bare GCE, (b) N-GR/GCE, (c) PEDOT/GCE, (d) PEDOT-MeOH/GCE, and (e) N-GR–PEDOT-MeOH/GCE in 0.1 M PBS (pH 7.0) with a scan rate of 50 mV s−1. | ||
3.3. Influence of pH value
The effect of pH value on the electrochemical determination of luteolin at N-GR–PEDOT-MeOH/GCE was investigated by CV with different pH values (3, 4, 5, 6, 7, 8, 9) at a scan rate of 50 mV s−1. As shown in Fig. 4A, there is a negative shift in the peak potential as the pH value increases, which reveals that protons participate in the electrochemical reaction of luteolin. Fig. 4B shows the relationship of oxidation peak current and peak potential with various pH values. The oxidation peak currents of luteolin at N-GR–PEDOT-MeOH/GCE achieve the maximum at pH 7.0. Thus, pH 7.0 was chosen for the electrochemical determination of luteolin. Furthermore, the relationship between oxidation peak potential (Epa) and pH was also investigated. Epa shifts negatively with increasing pH values and changes linearly with pH values. The regression equation is Epa (V) = −0.0567 pH + 0.692 (R2 = 0.9925). The slope (56.7.8 mV pH−1) is similar to the theoretical value (58.5 mV pH−1), suggesting that the protons and electrons involved in the electrochemical redox process of luteolin are equal.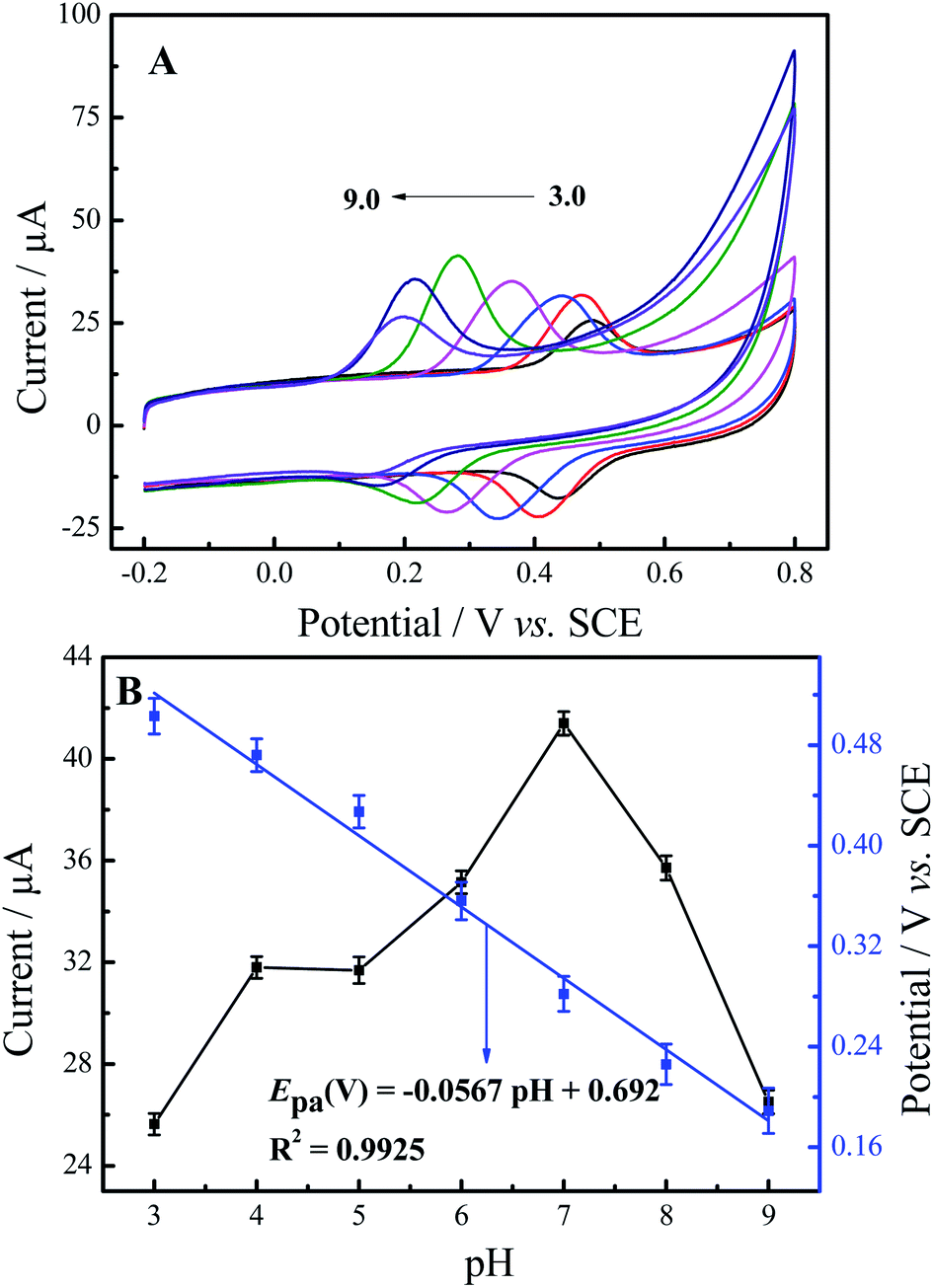 | ||
| Fig. 4 (A) DPVs of 50 μM luteolin at N-GR–PEDOT-MeOH/GCE with different pH values (pH: 3, 4, 5, 6, 7, 8, 9) at the scan rate of 50 mV s−1. (B) Effect of pH values on the anodic peak currents (black line) and anodic peak potentials (blue line). | ||
3.4. Effect of the modifier amounts
The effect of the amount of N-GR was investigated by CV with different concentrations of N-GR ranging from 0 mg mL−1 to 1.5 mg mL−1. As shown in Fig. S4A,† the current response of luteolin gradually increased with the increasing N-GR concentration until it reached a maximum value at 1.0 mg mL−1 due to the catalytic activity of the nanocomposite electrode surface. However, the anodic peak current rapidly decreased when the loading amount increased, which could be attributed to the hindrance of the thickness of the modifier that prevented the electron transfer between the aqueous solution and the interior of the films. Thus, 1.0 mg mL−1 of N-GR was the optimized value for the N-GR–PEDOT-MeOH/GCE-modified electrode.3.5. Influence of accumulation time and potential
Because the electrochemical reaction of luteolin is an adsorption-controlled process,9–21 the accumulation time and accumulation potential may enhance the adsorption of luteolin on the electrode surface and improve the current response of luteolin, while also effectively increasing the sensitivity of the N-GR–PEDOT-MeOH-modified electrode.The influence of accumulation time on the oxidation peak current of luteolin at N-GR–PEDOT-MeOH/GCE using CV at a scan rate of 50 mV s−1 is shown in Fig. 4SB.† The N-GR–PEDOT-MeOH/GCE was dipped into 50 μM luteolin with different accumulation times. With the increase in accumulation time, the oxidation peak current growth was rapid, and it plateaued at 120 s. With further increases in the accumulation time, the oxidation peak current slightly decreased. Therefore, 120 s was chosen as the accumulation time. Because luteolin is not charged, it is not influenced by potential. Thus, the open circuit potential was performed as accumulation potential throughout the accumulation step. Therefore, 120 s accumulation time together with 0 V accumulation potential were selected as parameters throughout the accumulation process.
3.6. Influence of scan rates
Fig. 5A shows the effect of different scan rates (10–500 mV s−1 in 0.1 M PBS (pH 7.0)) on the electrochemical behaviors of 50 μM luteolin at N-GR–PEDOT-MeOH/GCE. As shown in Fig. 5B, with the increase in scan rates from 10 to 500 mV s−1, the oxidation peak currents (Ipa) and reduction peak currents (Ipc) linearly grow with the scan rates (ν). The regression equations are Ipa (μA) = 0.357 ν + 14.497 (R2 = 0.988) and Ipc (μA) = −0.269 ν– 3.208 (R2 = 0.997) (ν in mV s−1), indicating that the electrochemical reaction of luteolin is an adsorption-controlled process. Plots of Epa and reduction peak potential (Epc) against the Napierian logarithm of the scan rate (lnν) also demonstrate a linear relationship over the scan rate range of 10–500 mV s−1 (Fig. 5C), with the regression equations Epa (V) = 0.021 ln ν – 1.314 (R2 = 0.991) and Epc (V) = −0.021 lnν – 1.614 (R2 = 0.991) (ν in mV s−1).
 | ||
| Fig. 5 (A) CV scans of 50 μM luteolin at N-GR–PEDOT-MeOH/GCE with different scan rates (10, 20, 30, 50, 70, 100, 150, 200, 250, 300, 400, and 500 mV s−1) in 0.1 M PBS (pH 7.0). (B) The plot of redox peak currents vs. scan rates, and (C) the Napierian logarithm of scan rates. | ||
According to Laviron's equation:39
 | (1) |
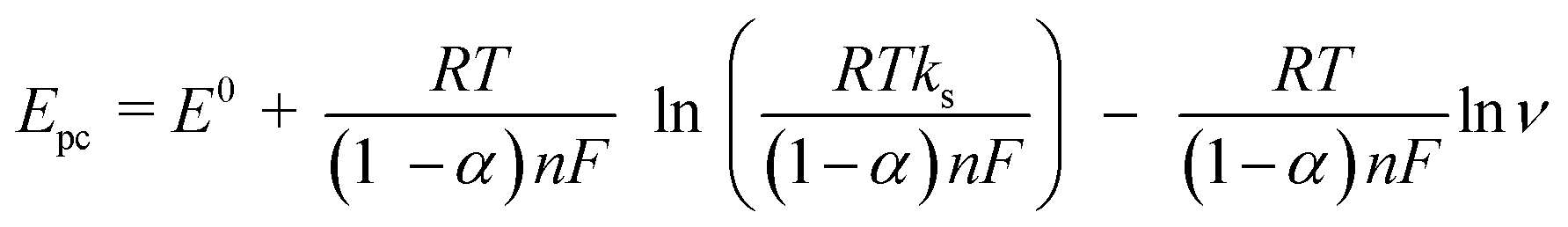 | (2) |
3.7. Detection of luteolin
On account of excellent sensitivity and higher selectivity of DPV as compared to CV, DPV was employed for the measurement of trace amounts of luteolin. Fig. 6A shows the DPVs of different luteolin concentrations at N-GR–PEDOT-MeOH/GCE. Fig. 6B shows that the peak currents are proportional to luteolin concentrations covering two linear ranges, from 0.005 to 0.01 μM and 0.01 to 10.06 μM. The linearization equations are Ip,1 (μA) = 9.45 + 13.56C (μM) (R2 = 0.989) and Ip,2 (μA) = 11.60 + 2.48C (μM) (R2 = 0.992), respectively. The detection limit was calculated to be 0.05 nM (S/N = 3), which is lower than that of most reported electrodes (Table S1†). Moreover, the proposed modified electrode also exhibits short accumulation time, wide linear range, and high sensitivity for the determination of luteolin. | ||
| Fig. 6 (A) DPVs for luteolin at N-GR–PEDOT-MeOH/GCE; luteolin concentration (from bottom to top: 0.005, 0.015, 0.03, 0.06, 0.11, 0.26, 0.56, 1.06, 2.06, 5.06, 10.06 μM). (B) The calibration plot of Ip (μA) vs. concentration of luteolin in 0.1 M PBS (pH 7.0). | ||
3.8. Repeatability, stability, and selectivity
The repeatability of N-GR–PEDOT-MeOH/GCE for the electrochemical analysis of luteolin was investigated. After the 20th successive measurement with the same modified electrode, the relative standard deviation (RSD) was obtained as 0.96% (Fig. S5†). Five different N-GR–PEDOT-MeOH/GCE-modified electrodes were then applied to individually measure luteolin, and the RSD was 2.5%, indicating satisfactory operation stability and reproducibility of N-GR–PEDOT-MeOH/GCE. The long-term stability of N-GR–PEDOT-MeOH/GCE was also studied by storing the modified electrode in a 4 °C refrigerator for 30 days and measuring the same luteolin solution every two days. One month later, the current response still remained at 89.5% of its initial response, indicating the satisfactory long-term stability of N-GR–PEDOT-MeOH/GCE.To demonstrate the selectivity of N-GR–PEDOT-MeOH/GCE for the detection of luteolin, interference studies were carried out in the absence and presence of 10-fold concentrations of citric acid (CA), ascorbic acid (AA), glucose, lysine, urea, and mannitol, and 100-fold concentrations of inorganic species, including Na+, K+, Mg2+, Ca2+, Ni2+, Zr2+, Al3+, Fe3+, SO42−, PO43−, NO32−, and CO22−. Less than 5% (i.e., 96.8–103.6%) of luteolin peak current was observed. In addition, some natural flavonoids with o-dihydroxy moieties in the B ring and phenolic acids potentially present with luteolin in plants were also investigated as interfering compounds, such as cynaroside (luteolin 7-glucoside), quercetin, diosmetin, myricetin, baicalein, and morin. As shown in Fig. S6,† without abundant flavonoid interfering components (no more than the same concentration), the sensor based on N-GR–PEDOT-MeOH/GCE possesses satisfactory anti-interference ability for luteolin.
3.9. Determination of luteolin in herbal medicines and capsule samples
To evaluate the practical application of N-GR–PEDOT-MeOH/GCE for the detection of luteolin, the standard addition method was employed to analyze real samples of T. vulgaris, L. japonica, and L. rotata. The recoveries and RSD are listed in Table 1. The formula for the samples recovery rate:| Samples | Added (μM) | Found (μM) | Recovery (%) | RSD (%) | |
|---|---|---|---|---|---|
| Thymus vulgaris | 1 | 0 | 4.38 | — | 3.26 |
| 2 | 0.01 | 4.43 | 100.91 | 2.34 | |
| 3 | 10 | 14.13 | 98.26 | 2.78 | |
| Lonicera japonica | 1 | 0 | 7.63 | — | 3.02 |
| 2 | 0.01 | 7.55 | 98.82 | 2.64 | |
| 3 | 10 | 17.73 | 100.53 | 1.89 | |
| Lamiophlomis rotata | 1 | 0 | 4.02 | — | 3.18 |
| 2 | 0.01 | 4.06 | 100.75 | 2.75 | |
| 3 | 10 | 13.94 | 99.43 | 2.12 | |
The average recoveries are in the range of 98.26–100.91, and the RSD is less than 3.5%. Satisfactory results indicate that N-GR–PEDOT-MeOH/GCE is viable for the practical detection of luteolin in Chinese herbal medicines and pharmaceutical formulations.
4. Conclusion
We constructed a sensitive and selective electrochemical sensor for the determination of luteolin by combining the merits of large surface area, superior electrical conductivity, a large amount of chemically active sites from N-GR, and the satisfactory water solubility and high conductivity of PEDOT-MeOH. N-GR–PEDOT-MeOH/GCE exhibited satisfactory electrocatalytic activity in response to luteolin, with wide linear range, rapid response, satisfactory sensitivity, selectivity and stability, and low detection limit. Moreover, the composite modified electrode was successfully employed for detection of luteolin in real samples of T. vulgaris, L. japonica, and L. rotata capsules with extraordinary accuracy and precision. Thus, this sensor provides a promising platform for the detection of flavonoids in Chinese herbal medicines and pharmaceutical formulations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are given for the financial support from the National Natural Science Foundation of China (No. 22074054) and the Natural Science Foundation of Zhejiang Province (No. LQ20B050002, LQ20E030015).References
- M. Liu, F. Tang, Q. Liu, J. Xiao, H. Cao and X. Chen, Nat. Prod. Res., 2018, 34, 2490–2494 CrossRef PubMed.
- N. K. Peters, J. W. Frost and S. R. Long, Science, 1986, 233, 977–980 CrossRef CAS PubMed.
- M. Lopez-Lazaro, Mini-Rev. Med. Chem., 2009, 9, 31–59 CrossRef CAS PubMed.
- S. J. Sheng, L. Y. Zhang and G. Chen, Food Chem., 2014, 145, 555–561 CrossRef CAS PubMed.
- G. Favaro, C. Clementi, A. Romani and V. Vickackaite, J. Fluorescenc., 2007, 17, 707–714 CrossRef CAS PubMed.
- S. Liu, Y. Zhu, N. Liu, D. Fan, M. Wang and Y. Zhao, J. Agric. Food Chem., 2021, 69, 1057–1067 CrossRef CAS PubMed.
- I. Baranowska and D. Rarog, Talanta, 2001, 55, 209–212 CrossRef CAS.
- M. Rahimi, S. Bahar, R. Heydari and S. M. Amininasab, Microchem. J., 2019, 148, 433–441 CrossRef CAS.
- M. Y. Cao, X. D. Yin, X. J. Bo and L. P. Guo, J. Electroanal. Chem., 2018, 824, 153–160 CrossRef CAS.
- Q. Zeng, J. Chen, F. Gao, X. Tu, Y. Qian, Y. Yu, L. Lu and W. Wang, Synthetic Met., 2021, 271, 116620 CrossRef CAS.
- Q. Xu, S. Chen, J. Xu, X. Duan, D. Lu, L. Q. Tian, X. Zhang, Y. Cai, X. Lu, L. Rao and Y. Yu, J. Electroanal. Chem., 2021, 880, 114765 CrossRef CAS.
- X. Si, L. Deng, Y. Wang, M. Han and Y. Ding, Microchem. J., 2022, 174, 106864 CrossRef CAS.
- P. Pang, Y. Liu, Y. Zhang, Y. Gao and Q. Hu, Sens. Actuators, B, 2014, 194, 397–403 CrossRef CAS.
- X. Hou, W. Wu, F. Zhao, W. Xie and Q. Yang, Microchim. Acta, 2021, 188, 86 CrossRef CAS PubMed.
- Q. Huang, X. Lin, C. Lin, Y. Zhang, H. Zhang, S. Hu, C. Wei and Q. X. Tong, Anal. Methods, 2016, 8, 6347–6352 RSC.
- X. Niu, Y. Huang, W. Zhang, L. Yan, L. Wang, Z. Li and W. Sun, J. Electroanal. Chem., 2021, 880, 114832 CrossRef CAS.
- L. Wu, Y. Gao, J. Xu, L. Lu and T. Nie, Electroanal, 2014, 26, 2207–2215 CrossRef CAS.
- H. Ibrahim and Y. Temerk, Sens. Actuators, B, 2015, 206, 744–752 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283–2307 RSC.
- Z. Yao, H. G. Nie, Z. Yang, X. M. Zhou, Z. Liu and S. M. Huang, Chem. Commun., 2012, 48, 1027–1029 RSC.
- T. O. Wehling, K. S. Novoselov, S. V. Morozov, E. E. Vdovin, M. I. Katsnelson, A. K. Geim and A. I. Lichtenstein, Nano Lett., 2008, 8, 173–177 CrossRef CAS PubMed.
- N. Talukder, Y. Wang, B. B. Nunna and E. S. Lee, Carbon, 2021, 185, 198–214 CrossRef CAS.
- L. L. Zhang, X. Zhao, H. X. Ji, M. D. Stoller, L. F. Lai, S. Murali, S. McDonnell, B. Cleveger, R. M. Wallace and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 9618–9625 RSC.
- Y. Liu, Y. Chen, J. Deng and J. Wang, Appl. Catal., B, 2021, 297, 120407 CrossRef CAS.
- X. Nie, P. Deng, H. Wang and Y. Tang, RSC Adv., 2021, 11, 37544–37551 RSC.
- Q. Rao, F. X. Hua, L. Y. Gan, C. Guo, Y. Liu, C. Zhang, C. Chen, H. B. Yang and C. M. Li, Electrochim. Acta, 2022, 402, 139427 CrossRef CAS.
- L. Yu, J. Chang, X. Zhuang, H. Li, T. Hou and F. Li, Anal. Chem., 2022, 94, 3669–3676 CrossRef CAS PubMed.
- S. Bilal, A. J. Sami, A. Hayat and M. F. Rehman, Bioelectrochem, 2022, 144, 107999 CrossRef CAS PubMed.
- M. Saraf, K. Natarajan, A. K. Saini and S. M. Mobin, Dalton Trans., 2017, 46, 15848–15858 RSC.
- S. Thareja and A. Kumar, J. Phys. Chem. C, 2021, 125, 24837–24848 CrossRef CAS.
- M. Saraf, R. Rajak and S. M. Mobin, J. Mater. Chem. A, 2016, 4, 16432–16445 RSC.
- B. L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef.
- A. Elschner, S. Kirchmeyer, W. Lvenich, U. Merker and K. Reuter, PEDOT: Principles and applications of an intrinsically conductive polymer, CRC Press, London, 2010 Search PubMed.
- Y. Y. Yao, L. Zhang, Z. F. Wang, J. K. Xu and Y. P. Wen, Anal. Chim. Acta, 2014, 25, 505–510 CAS.
- A. Lima, P. Schottland, S. Sadki and C. Chevrot, Synthetic Met., 1998, 93, 33–41 CrossRef CAS.
- Z. Zhang, G. Tian, X. Duan, H. Chen and D. H. K. Richie, ACS Appl. Bio Mater., 2021, 4, 5556–5565 CrossRef CAS PubMed.
- Y. Y. Yao, L. Zhang, Z. F. Wang, J. K. Xu and Y. P. Wen, Chinese Chem. Lett., 2014, 25, 505–510 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- J. A. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2000 Search PubMed.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed.
- A. Schaarschmidt, A. A. Farah, A. Aby and A. S. Helmy, J. Phys. Chem. B, 2009, 113, 9352 CrossRef CAS PubMed.
- L. D. Sappia, E. Piccinini, W. Marmisollé, N. Santilli, E. Maza, S. Moya, F. Battaglini, R. E. Madrid and O. Azzaroni, Adv. Mater. Interfaces, 2017, 4, 1700502 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01669a |
| This journal is © The Royal Society of Chemistry 2022 |