
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous poly-N-vinylcarbazole polymer as a novel matrix for the determination of low molecular weight compounds by MALDI-TOF MS†
Xiu-Ying Chenabc,
Yong-hui Wangb,
Shu-Yue Renb,
Shuang Li b,
Yu Wangb,
Kang Qinb,
Sen Lib,
Dian-Peng Hanb,
Yuan Pengb,
Tie Hanb,
Zhi-Xian Gaob,
Bao-Xiang Gao*a and
Huan-ying Zhou*b
b,
Yu Wangb,
Kang Qinb,
Sen Lib,
Dian-Peng Hanb,
Yuan Pengb,
Tie Hanb,
Zhi-Xian Gaob,
Bao-Xiang Gao*a and
Huan-ying Zhou*b
aKey Laboratory of Medicinal Chemistry and Molecular Diagnosis, College of Chemical and Environmental Sciences, Hebei University, Baoding 071002, China. E-mail: bxgao@hbu.edu.cn
bTianjin Key Laboratory of Risk Assessment and Control Technology for Environment and Food Safety, Tianjin Institute of Environmental and Operational Medicine, Tianjin 300050, China. E-mail: zhouhytj@163.com
cNanpu Development Zone Administrative Examination and Approval Bureau, Tangshan 063305, China
First published on 18th May 2022
Abstract
Traditional matrices for matrix-assisted laser desorption/ionization mass spectrometry (MALDI-TOF MS) are usually crystalline small molecules. The heterogeneous co-crystallization of the analyte and the matrix creates a sweet spot effect and reduces point-to-point reproducibility. In this study, an amorphous poly-N-vinylcarbazole polymer (PVK) was studied as a novel matrix for MALDI-TOF MS to detect various low molecular weight compounds (LMWCs) in the negative ion mode. The PVK achieved excellent matrix action and showed high sensitivity, good salt tolerance, and reproducibility. These results significantly broaden the design rules for new and efficient polymeric MALDI matrices.
Introduction
As a soft ionization technique, matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) has become an essential tool for biomolecular analysis because of the high throughput and low sample consumption.1,2 It has gained a widespread use in the analysis of proteins,3–5 peptides,6,7 oligonucleotides,8 nucleic acids,9,10 metabolites,11 organic polymers,12 and other biological macromolecules. However, the analysis of small molecules using this technique remains challenging for two reasons. On the one hand, the traditional organic small molecule matrix would exhibit a large number of matrix-related peaks in the low-mass region with molecular weight less than 700 Da, which interfere with the detection of low-molecular-weight compounds,13 and on the other hand, traditional organic small molecule compounds are crystalline matrices, which easily undergo matrix-targeted heterogeneous co-crystallization, resulting in a “sweet spot” effect, leading to poor point-to-point reproducibility and the inability to quantitatively analyze according to the signal intensity of the tested substance.14In order to improve the analysis performance of MALDI, different approaches were investigated, such as matrix-free,15 inorganic matrices16,17 and novel organic matrices. These materials usually include metal nanoparticles, carbon nanomaterials, silicon-based nanomaterials, organic framework materials, hybrid materials, or surfaces. Inorganic matrices or surfaces are easy for desorption/ionization and to eliminate matrix-related peak interference problems in low-quality areas, and most of them have low solubility in organic solvents and poor dispersibility in water, leading to an uneven crystallization with the analytes. It would reduce the sensitivity and point-to-point reproducibility. Even more important is that most of these matrices or surfaces can only be analyzed for small molecule compounds in the positive mode. This is related to the fact that its ionization mechanism depends on surface properties, such as pore size or particle size, specific surface area, and nanostructure. In the positive mode, complex polybasic metal ion adducts as well as protonated ions in the spectrum tend to make it difficult to identify, and only a single deprotonated ion is produced in the spectrum in negative ion mode, and the background is relatively clear and easy to interpret.
Organic matrices have advantages, such as lower cost and easier to synthesize, high solubility in organic solvents, stability, and good salt tolerance. In recent years, researchers have developed a variety of organic compounds and ionic liquid matrices, but most of them are only suitable for the detection of small analytical compounds in the positive ion mode, and not many organic compound matrices are suitable for detection in the negative ion mode. Furthermore, achieving point-to-point reproducibility still faces challenges due to the “sweet spot” effect caused by the uneven co-crystallization between the crystalline matrix and the analyte. Recently, Horatz et al.18 synthesized two amorphous copolymers P(TNDIT–FI(C10C8)) and P(TNDIT–FI(C10C8)) as MALDI matrices from 9,9-bis(2-ethylhexyl)fluorene as the starting material for LMWC detection in positive and negative ion modes, respectively, and explored the relationship between matrix crystallinity and the performance as a matrix. The results show that amorphous matrices have obvious advantages in the LMWC signal intensity detection compared to semi-crystalline matrices. It significantly broadens the design rules for new and efficient polymeric MALDI matrices. Inspired by this, we investigated an amorphous polymer, poly(N-vinyl carbazole) (PVK), as a novel matrix for the MALDI-TOF MS detection of diverse LMWC in the negative ion mode.
Amorphous PVK is an aromatic-structured nitrogen-heterocyclic homopolymer with large rigid side-groups and large π-electron branched-chain-based conjugate systems, which endow it with chemical stability and excellent dielectric properties.19,20 PVK is an unconjugated polymer. There are carbazole groups on its backbone and a large conjugated system on the side chain.21 These groups facilitate strong absorption in the ultraviolet spectrum and favor the charge transfer, which can satisfy the fundamental requirements of a MALDI MS matrix. Furthermore, the N atom of carbazole with a lone pair of electrons is an excellent electron-donating group, which facilitates the hole transport. Consequently, it has attracted considerable attention as an electron donor and hole-transporting material for use in electroluminescent devices22,23 and the optoelectronic materials industry.24–26 PVK has good ultraviolet absorption and electron donating ability, which are favorable as the medium of energy absorption and transfer and electron transfer, and has the potential as the MALDI matrix-assisted ionization of LMWC. However, to the best of our knowledge, amorphous PVK has not been reported as a matrix.
Here, we report a novel amorphous PVK matrix. The characteristics of the amorphous matrix were evaluated by testing multiple targets, including amino acids, fatty acids, nucleic acid base, bisphenols, quinolones and other small molecules. The PVK matrix has shown high sensitivity, good salt tolerance and reproducibility for the detection of LMWC in the negative ion mode. Furthermore, the successful achievement of small molecule quantification of enrofloxacin in milk samples indicates that the amorphous PVK matrix has the potential that can be generalized to the analysis of other metabolic small molecules and complex samples.
Experimental materials and methods
PVK were purchased from J&K Chemicals (Beijing). Histidine (His), phenylalanine (Phe), methionine (Met), threonine (Thr), adenine (A), uracil (U), thymine (T), palmitic acid (C16), stearic acid (C18), bisphenol A (BPA), bisphenol B (BPB), bisphenol F (BPF), bisphenol S (BPS), lomefloxacin (LOM), fleroxacin (FLE), norfloxacin (NOR), ciprofloxacin (CIP), sarafloxacin hydrochloride (SAR), enrofloxacin (ENR), enrofloxacin-D5 (ENR-D5), and tetrahydrofuran (THF) were bought from Sigma-Aldrich (St. Louis, MO, USA). Poly(N-vinyl carbazole) (PVK) and N-vinyl carbazole (VK) poly-n-vinylcarbazole and N-vinylcarbazole were purchased from Beijing Lingwei Chemical Reagent Co., Ltd. Dichloromethane (DCM) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Acetonitrile and methanol were bought from Fisher Scientific (Fair Lawn, New Jersey, USA). Deionized water was purified using a Direct-Q system (Millipore, Bedford, MA, USA). Except for PVK, other reagents were not further purified.Thermogravimetric analysis (TGA) was conducted on a TGA/DSC 1 (Mettler Toledo, Switzerland). X-Ray diffraction (XRD) patterns of the samples were recorded using a SmartLab 9 automated multipurpose X-ray diffractometer (Rigaku, Japan) adopting Cu Kα radiation (λ = 1.54056 Å). Fourier-transform infrared (FT-IR) spectra were recorded using a Nicolet 380 FT-IR spectrometer (Thermo Fisher Scientific, USA) employing KBr as the background. Raman spectra were obtained from a Renishaw inVia confocal Raman microscope. X-ray photoelectron spectroscopy (XPS) was performed using an ESCALAB 250Xi X-ray photoelectron spectrometer (Thermo Fisher Scientific, USA). Ultraviolet visible (UV VIS) spectra: 2.0 mg PVK was dissolved in a 1 mL dichloromethane solvent, and dichloromethane was used as the reference, which was tested by an UV2600 (Golden Island, Japan) double beam UV spectrophotometer.
Purification of the PVK matrix
PVK was purified by the recrystallization method. The recrystallization purification process of poly(N-vinylcarbazole) uses its different solubility in different solvents for purification. PVK is easy to dissolve in tetrahydrofuran and difficult to dissolve in methanol. When methanol is added to the tetrahydrofuran solution of PVK, there will be precipitation. The resulting precipitation needs to be centrifuged to remove the supernatant. The specific operation steps were as follows: initially, 2.0 g PVK was dissolved with 100 mL of THF. Then, 200 mL of methanol was added. There was a precipitate produced, and the resulting precipitate was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min. The steps were repeated for five times. The purified PVK were dried at 80 °C for 24 h and stored at room temperature.
000 rpm for 15 min. The steps were repeated for five times. The purified PVK were dried at 80 °C for 24 h and stored at room temperature.
Preparation of matrices and low molecular weight standard compounds
2 mg PVK was dispersed in dichloromethane to form the matrix solution (2 mg mL−1). His, Phe, Met, and Thr were dispersed in water. A, U, and T were dispersed in boiling water (100 °C). C16, C18, BPA, BPB, BPF, BPS, LOM, FLE, NOR, CIP, SAR, and ENR were dissolved in methanol to prepare a solution with a concentration of 1 mg mL−1. NOR was dissolved in acetonitrile to prepare a solution with a concentration of 1 mg mL−1. All standard store solutions were kept at 4 °C.Sample preparation
200 μL milk sample was mixed with 1.6 mL of 1% formic acid acetonitrile and vortexed for 10 min. Then, the mixed solution was centrifuged at 10000 rpm min−1 for 10 min at room temperature. The supernatant was purged using nitrogen. The dry residue was reconstituted in 200 μL of acetonitrile for the MALDI-TOF MS analysis.
MALDI-TOF MS analysis
The analyte solution was mixed with the matrix solution at a ratio of 1:1 (v/v). 2 μL mixture was dropped on the target plate and dried at room temperature. The mass spectrometric detection was performed on a MALDI micro MX mass spectrometer (Waters/Micromass, Manchester, UK) equipped with a nitrogen laser (337 nm). The laser intensity was set at 71%. The raw data were analysed using MassLynx v4.1.
Results and discussion
Characterization of PVK as a MALDI matrix
The most fundamental requirement required for a MALDI matrix is the good UV absorbing ability in the wavelength range emitted by the laser light source. Commonly used laser sources mainly include a nitrogen laser (337 nm), triple neodymium-doped yttrium fluoride lithium laser (349 nm) and triple or quadruple neodymium-doped yttrium aluminum garnet laser (355 nm or 266 nm).27 As shown in Fig. 1A, the PVK had strong UV absorption ranges from 250 to 370 nm, which included all wavelengths of UV laser sources. It indicated that the matrix had the ability to strongly absorb laser energy and transfer it to analyte, which met the essential requirements of a MALDI matrix. The XRD pattern of PVK is shown in Fig. 1B. The broad peak in the 2θ region (“*” in Fig. 1B) of 15–25° is attributed to the amorphous structure of PVK,28 which indicated that PVK is not crystalline. Fourier-transform infrared (FT-IR)29 spectroscopy is shown in Fig. S1.† 1600–1580 cm−1 and 1500–1450 cm−1 were the stretching vibration peak of the benzene ring, which formed the π-conjugated structure and was beneficial to the analyte desorption/ionization. We also recorded the Raman spectra of PVK to identify its vibrational modes. Fig. S2† shows the Raman spectra and vibrational frequencies of the PVK molecule. The structure of the PVK molecule can be divided into three parts: the long polyethylene chain, the o-disubstituted benzene ring, and the heteropentacycle (Table S1, ESI†). Thermogravimetric analysis (TGA) was used to further evaluate the stability of PVK under the detection. It can be seen from Fig. 1C that the decomposition of polymer PVK underwent two processes: the weight loss at ∼130 °C was caused by the evaporation of residual reagents in the product, the thermal decomposition occurred at around 458 °C PVK, and the decomposition was complete at 500 °C. It indicated that PVK had good thermal stability. The LDI MS spectrum of PVK was detected ranging from 0 to 1000 Da. As shown in Fig. 1D, a [VK–CHCH2–H]− ion peak appeared at m/z 165 in the negative mode. In the positive mode, the complex polybasic adducts as well as protonated ions appeared (Fig. S3†). The background of PVK in the negative mode was relatively clear compared with that in the positive mode.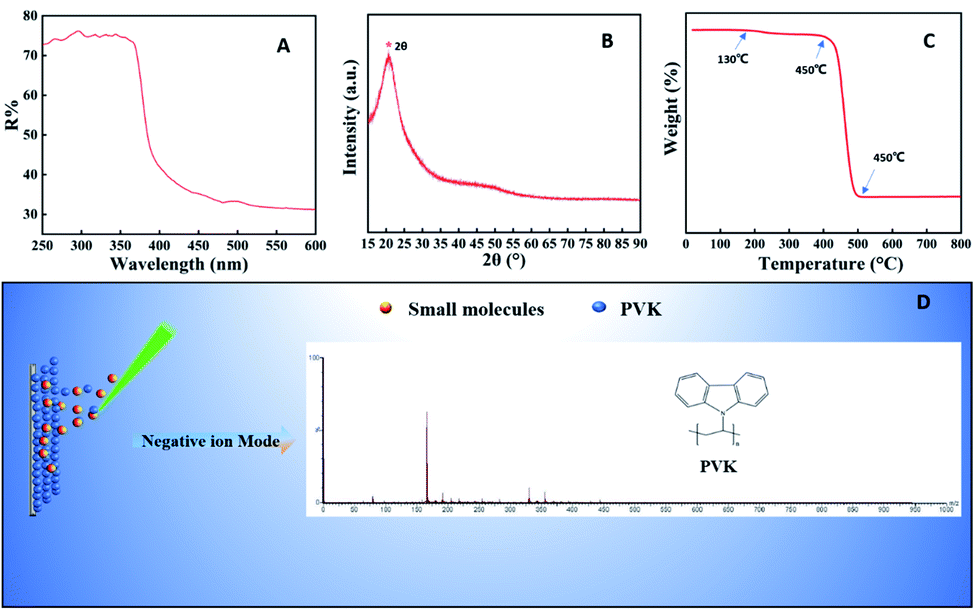 | ||
| Fig. 1 Characterization of PVK. (A) UV-Vis spectra of PVK. (B) XRD spectra of PVK. (C) Thermogram of PVK. (D) LDI MS spectrum of PVK in the negative ion mode. | ||
Optimization of experimental conditions
The concentration of the matrix affects the uniformity of crystallization between the matrix and the sample, and also affects the dispersion of the sample, which will ultimately affect the sensitivity of analysis and the quality of peak. It is also very important to select the appropriate matrix concentration. To confirm whether matrix-assisted function was affected by different matrix concentrations, matrix concentration optimization was performed by mixing equal volumes of 1 mg mL−1 each of A, U, His, and Thr as targets with 0.2, 0.5, 1.0, 2.0, 5.0, and 10.0 mg mL−1 matrix PVK, respectively, and taking 2 μL of the target plate to measure the mass spectrum, taking a bar chart made of the absolute signal intensity. The optimal concentration of the matrix was 2.0 mg mL−1, which was tested at 2.0 mg mL−1 PVK in subsequent articles (Fig. 2A). The solvent needs to effectively regulate analyte–matrix interactions to increase or decrease the MALDI ion signals.30–32 The mass spectra of His and A exhibited weaker signals when the matrix was dissolved in THF than DCM (Fig. S4 and S5†). The absolute signal intensity of A at m/z 135([M − H]−) with DCM as the matrix solvent was two times higher than that of THF. The absolute signal intensity of His at m/z 154([M − H]−) with DCM as the matrix solvent was three times higher than that of THF. The degree of crystalline homogeneity of the matrix and sample influenced the reproducibility. The spotting method is a key factor, which could influence the uniform co-crystallization of the matrix and the sample. In this study, three spotting methods, including the sample first technique, the matrix first technique, and the dried-droplet technique (a mixture of the sample and matrix techniques), were compared. The results showed that the dried-droplet technique was more conducive than the other two spotting methods (Fig. S6 and S7†).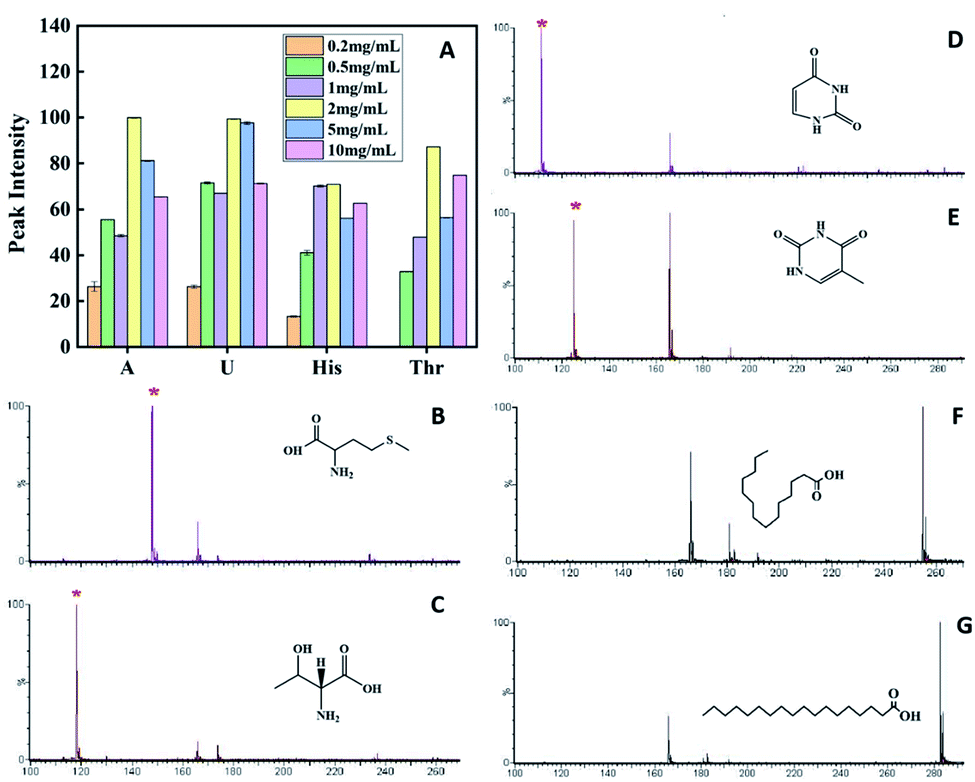 | ||
| Fig. 2 Comparison of the MALDI-TOF MS spectrum intensity of adenine, uracil, histidine and threonine with different concentrations of the matrix (A). MALDI-TOF MS spectra of Met (B), Thr (C), U (D), T (E), palmitic acid (F) and stearic acid (G) with amorphous PVK as the matrix, respectively. | ||
Analysis of multiple low molecular weight compounds
The crystal matrix 9-AA has been reported to be effective for LMWC testing in the negative ion mode.33 A comparative analysis was performed with different matrices, including 9-AA, VK, and PVK. A carboxylic acid mixture containing ascorbic acid (Vc, MW 176.12) and citric acid (CA, MW 192.14) was employed to test small molecules. The intensity of the [M − H]− peak with crystal 9-AA and VK as the matrix was lower than that with amorphous PVK, proving the superiority of amorphous PVK as a matrix (Fig. S8†).Amino acids, nucleic acid bases, and fatty acids are important biological components, which play important roles in various life activities. To validate the broad applicability of amorphous PVK as a matrix, multiple small bioactive molecules were analyzed. As shown on Fig. 2B–G and S9,† the clear deprotonated peaks belonging to [M − H]− (marked with *) were observed with Phe, MW 165.19; Met, MW 149.21; Thr, MW 119.11; A, MW 135.13; U, MW 112.09; T, MW 126.11; C18, MW 284.48; and C16, MW 256.42; respectively.
BPs are a kind of typical endocrine disruptors that have adverse effects on the human immune system.34 In order to further verify the applicability of amorphous PVK as a matrix, BPA, MW 228.29; BPB, MW242.32; BPF, MW200.24; and BPS, MW 250.27 were used as analytes. As shown on Fig. 3A–D, the [M − H]− peak and [M − CH4–H]− fragmentation peaks of BPA appeared at m/z 225.89, m/z 209.86. The peaks at m/z 239.90 and m/z 209.87 belonged to BPB corresponding to [M − H]− and [M − C2H6–H]−. For BPF, the peak at m/z 197.90 belonged to its dehydrogenated rearrangement ion. The peak at m/z 248.58 for BPS corresponded to the [M − H]− ion. Fig. S10† demonstrates the fragmentation pathways of BPs.
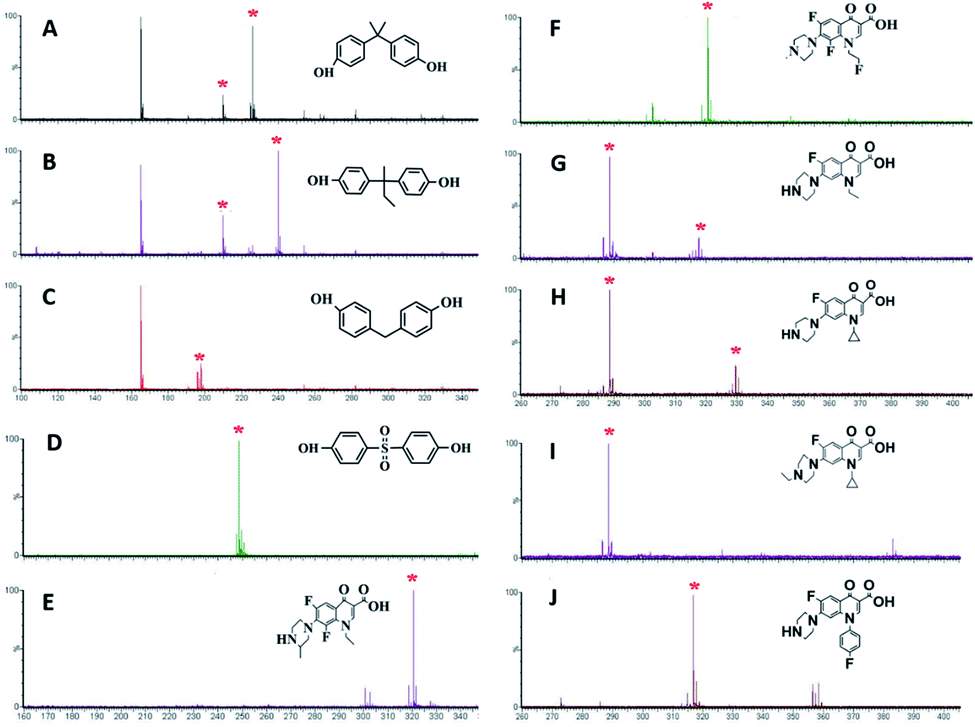 | ||
| Fig. 3 MALDI-TOF MS spectra of (A–D) BPs (BPA, BPB, BPF, BPS) and (E–J) quinolones (LOM, FLE, NOR, CIP, SAR an ENR (J)) with PVK as the matrix. | ||
Quinolones are a group of synthetic broad-spectrum antibiotics, mainly including LOM, MW 351.35; FLE, MW 369.34; NOR, MW 319.33; CIP, MW 331.34; SAR, MW 421.83; and ENR, MW 359.40. Quinolones are relatively broad-spectrum antibiotics used in human and veterinary medicines.35,36 Some potential hazards to humans have been reported owing to the promiscuity of quinolone antibiotics.37,38 As displayed in Fig. 3E–J, six quinolones could be detected using amorphous PVK as the matrix. The fragmentation products were [LOM–C2H5]− (m/z 320.53) for LOM, [FLE–C2H4F]− (m/z 322.34) for FLE, [NOR–H]− (m/z 318.33) and [NOR–C2H5]− (m/z 290.33) for NOR, [CIP–H]− (m/z 330.341) and [CIP–C3H5]− (m/z 290.34) for CIP, [SAR–H]− (m/z 384.33) and [SAR–C6H4F]− (m/z 290.33) for SAR, and [ENR–H]− (m/z 358.4) and [ENR–C3H5]− (m/z 318.78) for ENR. Fig. S11† shows the cleavage pathway of quinolones.39 From the structure and mass spectrometric analysis of antibiotics, we could speculate that the carbon–nitrogen bond could be readily destroyed by the laser energy, and the remaining negative ions could be detected by MALDI-TOF MS. The limits of detection (LOD) were estimated at the threefold signal-to-noise ratio (S/N = 3). The LODs of the analytes including Vc, CA, Phe, Met, Thr, A, U, T, C16, C18, BPA, BPB, BPF, BPS, LOM, FLE, NOR, CIP, SAR, and ENR were determined in the range of 0.0001–0.01 mg mL−1. The detailed LODs for various analytes are shown in Table S2.†
Salt tolerance and stability of the PVK matrix
Numerous samples generally have high salt concentrations, and excessive salt concentration in samples would have inhibitory effects on the signal of the MALDI-TOF MS spectrum. The salt tolerance of amorphous PVK as the matrix was investigated using histidine as a representative analyte. As shown in Fig. 4A, when the concentration of the salt solution increased gradually, the target signal peaks did not disappear and the signal intensity did not weaken significantly. Those results showed that the amorphous PVK matrix possesses high salt tolerance. The stability of the matrix is also one of the main requirements for MALDI-TOF MS. The stability of the matrix was further confirmed by detecting His using an amorphous PVK matrix exposed for 1, 7, 14, and 28 days. Mass spectrometry showed that amorphous PVK could be stably preserved for 28 days with insignificant target signal intensity changes (Fig. 4B). This indicated that amorphous PVK is an extremely stable matrix, which encourages in-depth investigation of its analytical performance.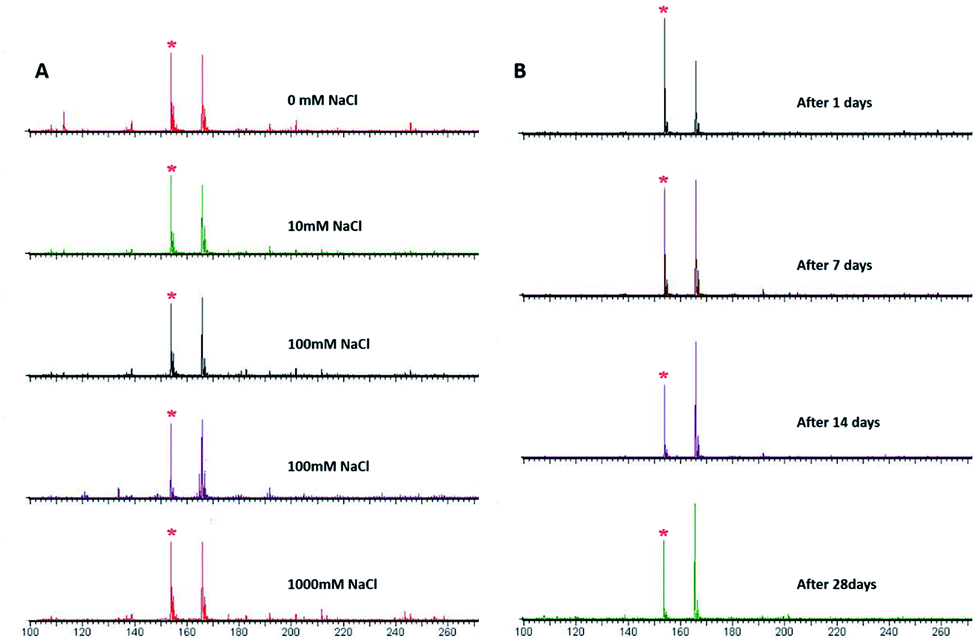 | ||
| Fig. 4 MALDI-TOF MS spectrum of His with matrix of PVK (A) in 0–1000 mM NaCl and (B) after it was stored for 1, 7, 14, and 28 days (* [His–H]−). | ||
Analysis of complex samples
Due to the irregular and inhomogeneous crystallization of samples and matrix, it is challenging to quantitatively analyze small molecular compounds with traditional matrices. The quantitative performance of the amorphous PVK matrix was further verified with ENR as a model analyte. Enrofloxacin-D5 (ENR-D5, 1 mg mL−1) was used as an internal standard. Fig. 5A shows the standard curve constructed for ENR. A linear relationship was obtained between the relative peak intensity of the ENR(I[ENR–C3H5]−/I[ENR-D5–C3H5]−) and the relative concentration of ENR(CENR/CENR-D5) from 0.003125 to 0.8 mg mL−1. Correspondingly, the regression equation was y = 1.3526x + 0.0139 (R2 = 0.9905). To further validate the feasibility of our established MALDI MS-based quantitative assay, spiked milk samples with a concentration of 0.1 mg mL−1 enrofloxacin were examined (Fig. 5B). The relative standard deviations between and within groups were within 5% with good precision, recovery of 101.5%, and good reproducibility. Fig. 5B shows that the MS peaks of the substrate of the real sample has slight influence on the target analysis.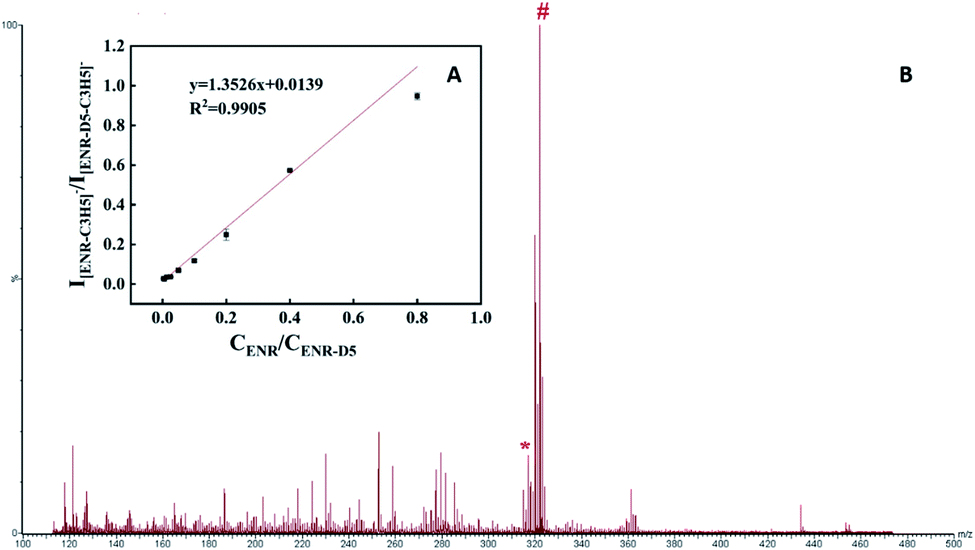 | ||
| Fig. 5 (A) The calibration curve that represents the dependence of the intensity of the [ENR–C3H5]−/[ENR-D5–C3H5]− on the ENR/ENR-D5 concentration in the range of 0.003125–0.8 mg mL; (B) MALDI mass spectrum of a positive milk sample spiked with 0.1 mg mL−1 enrofloxacin using PVK as matrix detection (* [ENR–C3H5]−, # [ENR-D5–C3H5]−). | ||
Ionization mechanism of PVK as a matrix
The excellent properties of the amorphous PVK matrix aroused our curiosity about the influence of the amorphous PVK structure and composition on its ionization efficiency. π-Conjugated structures play a crucial role in assisted-desorption ionization processes. Because it mainly depends on the conjugate structure to absorb and transfer laser energy.40,41 As shown in Fig. 6A, there are two matching peaks in the C1s XPS high-resolution spectrum, which are 284.78 and 285.90 eV,respectively. The latter peak was attributed to the sp2-bonded carbon (C1s binding energy in C–N of the PVK side-chain carbazole ring), and its appearance proved the presence of a series of π-conjugated composition. Simultaneously, a peak of 400.5 eV was observed in the N1s XPS spectrum (Fig. 6B). The results show that the nitrogen atom in the amorphous PVK structural formula existed in the form of pyridinic nitrogen. Because the pyridinic nitrogen atom has a pair of lone pair electrons in its conjugated structure, it has a Lewis base action,42 which can easily capture protons from analytes. Therefore, the pyridinic nitrogen atom in the amorphous PVK structure was considered to accelerate the electron transfer process and promote the increase in the number of negative ions in the negative-ion mode.43 This can be used to explain why the peak intensity obtained by PVK as the matrix was stronger than that obtained using VK as the matrix in the testing of LMWC.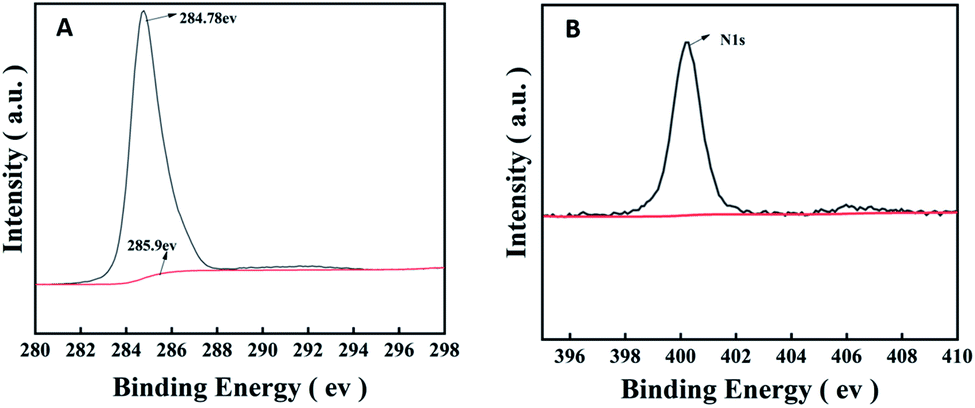 | ||
| Fig. 6 (A) The C1s and (B) N1s spectra of PVK. | ||
Conclusions
In summary, amorphous polymer PVK was used as an effective matrix in the MALDI-TOF MS detection for LMWC in the negative ion mode. The amorphous PVK achieved an excellent matrix action, and showed high sensitivity, good salt tolerance, and reproducibility. The quantification of ENR in milk samples was successfully achieved by the internal standard method. These results significantly broaden the design rules for new and efficient polymeric MALDI matrices.Author contributions
Conceptualization, Huanying Zhou, Baoxiang Gao and Zhixian Gao; data curation, Huanying Zhou and Xiuying Chen; formal analysis, Xiuying Chen and Yonghui Wang; funding acquisition, Huanying Zhou; investigation, Huanying Zhou, Tie Han and Baoxiang Gao; methodology, Zhixian Gao; project administration, Huanying Zhou and Zhixian Gao; resources, Yonghui Wang, Shuyue Ren, and Shuang Li; software, Kang Qin, Sen Li and Dianpeng Han; supervision, Tie Han and Baoxiang Gao; visualization, Shuyue Ren, Shuang Li, Yu Wang,Yuan Peng and Dianpeng Han; writing – original draft, Xiuying Chen; writing – review & editing, Xiuying Chen, Huanying Zhou and Baoxiang Gao.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFF0211301).Notes and references
- M. Karas, U. Bahr, A. Ingendoh and F. Hillenkamp, Angew. Chem., Int. Ed. Engl., 1989, 28(6), 760–761 CrossRef.
- B. O. Keller and L. Li, J. Am. Soc. Mass Spectrom., 2001, 12, 1055–1063 CrossRef CAS.
- M. L. Reyzer and R. M. Caprioli, J. Proteome Res., 2005, 4(4), 1138–1142 CrossRef CAS PubMed.
- A. Matta, R. Ralhan, L. V. Desouza and K. W. MichaelSiu, Mass Spectrom. Rev., 2010, 29(6), 945–961 CrossRef CAS.
- M. Sassi, S. Arena and A. Scaloni, J. Agric. Food Chem., 2015, 63(27), 6157–6171 CrossRef CAS.
- T. G. Kleno, C. M. Andreasen, H. Kjeldal, L. R. Leonardsen, T. N. Krogh, P. F. Nielsen, M. V. Sørensen and O. N. Jensen, Anal. Chem., 2004, 76(13), 3576–3583 CrossRef CAS PubMed.
- L. A. Marzilli, T. R. Golden, R. J. Cotter and A. S. Woods, J. Am. Soc. Mass Spectrom., 2000, 11(11), 1000–1008 CrossRef CAS PubMed.
- W. Y. Chen and Y. C. Chen, Anal. Chem., 2007, 79(21), 8061–8066 CrossRef CAS PubMed.
- O. Bauer, A. Guerasimova, S. Sauer, S. Thamm, M. Steinfath, R. Herwig, M. Janitz, H. Lehrach and U. Radelof, Rapid Commun. Mass Spectrom., 2004, 18(16), 1821–1829 CrossRef CAS PubMed.
- F. Song, Rapid Commun. Mass Spectrom., 2003, 17(15), 1802–1807 CrossRef CAS PubMed.
- J. N. Wang, S. L. Qiu, S. M. Chen, C. Q. Xiong, H. H. Liu, J. Y. Wang, N. Zhang, J. Hou, Q. He and Z. X. Nie, Anal. Chem., 2014, 87(1), 422–430 CrossRef PubMed.
- K. Kim, A. Hasneen, H. J. Paik and T. Chang, Polymer, 2013, 54(22), 6133–6139 CrossRef CAS.
- A. Mandal, M. Singha, P. S. Addy and A. Basak, Mass Spectrom. Rev., 2019, 38(1), 1–19 CrossRef PubMed.
- S. M. Weidner and J. Falkenhagen, Rapid Commun. Mass Spectrom., 2009, 23, 653–660 CrossRef CAS PubMed.
- D. S. Peterson, Mass Spectrom. Rev., 2007, 26, 19–34 CrossRef CAS PubMed.
- J. M. Chitanda, H. X. Zhang, E. Pahl, R. W. Purves and E. A. Anas, J. Am. Soc. Mass Spectrom., 2016, 27, 1686–1693 CrossRef CAS PubMed.
- L. Muller, K. Baldwin, D. C. Barbacci, S. N. Jackson, A. Roux, C. D. Balaban, B. E. Brinson, M. I. McCully, E. K. Lewis, J. A. Schultz and A. S. Woods, J. Am. Soc. Mass Spectrom., 2017, 28(8), 1716–1728 CrossRef CAS PubMed.
- K. Horatz, K. Ditte, T. Prenveille, K. N. Zhang, D. Jehnichen, A. Kiriy, B. Voit and F. Lissel, ChemPlusChem, 2019, 84(9), 1338–1345 CrossRef CAS.
- K. R. Thomas, J. T. Lin, Y. T. Tao and C. W. Ko, J. Am. Chem. Soc., 2001, 123, 9404–9411 CrossRef CAS PubMed.
- J. L. Hua, W. Zhang, J. D. Luo, J. G. Qin, Y. C. Shen, Y. Zhang and Z. H. Lu, J. Chem. Research (S), 2001, 10, 418–420 CrossRef.
- Z. A. Li, G. Yu, Y. Q. Liu, C. Ye, J. G. Qin and Z. Li, Macromolecules, 2009, 42, 6463–6472 CrossRef CAS.
- B. Hu, Z. Yang and F. E. Karasz, J. Appl. Phys., 1994, 26, 2419–2422 CrossRef.
- D. D. Gebler, Y. Z. Wang, D. K. Fu, T. M. Swager and A. J. Epstein, J. Chem. Phys., 1998, 108, 7842–7848 CrossRef CAS.
- O. B. Pierre, R. Agnesand and T. Sandrine, J. Phys. Chem. B, 2012, 16(2), 802 Search PubMed.
- R. C. Penwell, B. N. Ganguly and T. W. Smith, Journal of Polymer Science Macromolecular Reviews, 1978, 13(1), 63–160 CrossRef CAS.
- G. Safoula, S. Touihrib, J. C. Bern&de, M. Jamali, C. Rabiller, P. Molinied and K. Napo, Polymer, 1999, 40(2), 531 CrossRef CAS.
- H. Ibrahim, K. Jurcic, J. S.-H. Wang, S. N. Whitehead and K. K.-C. Yeung, Anal. Chem., 2017, 89, 12828–12836 CrossRef CAS PubMed.
- Y. Z. Li, M. L. Wang and L. Z. Gao, Chem. Res. Appl., 2014, 26(4), 577 CAS.
- X. L. Wei, J. Li, Q. Zhang, H. Q. Ma and J. Zhu, Journal of Chong Qing University of Arts and Sciences, 2016, 35(2), 120 Search PubMed (in Chinese)..
- S. L. Cohen and B. T. Chait, Anal. Chem., 1996, 68, 31–37 CrossRef CAS PubMed.
- G. Sun, K. Yang, Z. Zhao, S. Guan, X. Han and R. W. Gross, Anal. Chem., 2008, 80, 7576–7585 CrossRef CAS PubMed.
- Y. K. Tzeng, Z. Q. Zhu and H. C. Chang, J. Mass Spectrom., 2009, 44, 375–383 CrossRef CAS PubMed.
- A. Amantonico, J. Y. Oh, J. Sobek, M. Heinemann and R. Zenobi, Angew. Chem., Int. Ed. Engl., 2008, 47, 5382–5385 CrossRef CAS PubMed.
- C. Y. Liao, F. Liu, Y. Guo, H. B. Moon, H. Nakata, Q. Wu and K. Kannan, Environ. Sci. Technol., 2012, 46, 9138–9145 CrossRef CAS PubMed.
- D. Sharma, A. Nagpal, Y. B. Pakade and J. K. Katnoria, Talanta, 2010, 82, 1077–1089 CrossRef CAS PubMed.
- I. Kempf, F. Gesbert, M. Guittet, G. Bennejean and A. C. Cooper, Res. Vet. Sci., 1992, 53, 257–259 CrossRef CAS PubMed.
- Z. F. Fu, Y. Liu, L. Wang and Y. H. Wang, Chromatographia, 2009, 69, 1101–1105 CrossRef CAS.
- H. J. Cho, H. Yi, S. M. Cho, D. G. Lee, K. Cho, A. M. A. El-Aty, J. Shim, S. H. Lee, J. Y. Jeong and H. C. Shin, J. Sep. Sci., 2010, 33, 1034–1043 CrossRef CAS PubMed.
- Y. R. Ma, X. L. Zhang, T. Zeng, D. Cao, Z. Zhou, W. H. Li, H. Y. Niu and Y. Q. Cai, ACS Appl. Mater. Interfaces, 2013, 5, 1024–1030 CrossRef CAS.
- X. L. Dong, J. S. Cheng, J. H. Li and Y. S. Wang, Anal. Chem., 2010, 82, 6208–6214 CrossRef CAS PubMed.
- Q. Liu, M. T. Cheng and G. B. Jiang, Chem.–Eur. J., 2013, 19, 5561–5565 CrossRef CAS PubMed.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni, G. F. Tantardini and J. Nakamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035436 CrossRef.
- Q. H. Min, X. X. Zhang, X. Q. Chen, S. Y. Li and J. J. Zhu, Anal. Chem., 2014, 86, 9122–9130 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01602h |
| This journal is © The Royal Society of Chemistry 2022 |