
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of Fe3+ coordination by excess Cl− in alcohol solutions†
Yunika Nomuraa,
Dai Inoueb and
Yutaka Moritomo *abcd
*abcd
aSchool of Science and Engineering, University of Tsukuba, Tsukuba 305-8571, Japan. E-mail: moritomo.yutaka.gf@u.tsukuba.ac.jp
bGraduate School of Pure and Applied Sciences, University of Tsukuba, Tsukuba 305-8571, Japan
cFaculty of Pure and Applied Sciences, University of Tsukuba, Tsukuba 305-8571, Japan
dTsukuba Research Center for Energy Materials Science (TREMS), University of Tsukuba, Tsukuba 305-8571, Japan
First published on 17th June 2022
Abstract
We spectroscopically investigated coordination state of Fe3+ in methanol (MeOH) and ethanol (EtOH) solutions against Cl− concentration ([Cl−]). In both the system, we observed characteristic absorption bands due to the FeCl4 complex at high-[Cl−] region. In the MeOH system, the proportion (r) of [FeCl4]− exhibits a stationary value of 0.2–0.3 in the intermediate region of 10 mM < [Cl−] < 50 mM, which is interpretted in terms of [FeClnL6−n]3−n (n = 1 and 2). In the EtOH system, r steeply increases from 0.1 at [Cl−] = 1.5 mM to 0.7 at [Cl−] = 3.5 mM, indicating direct transformation from [FeL6]3+ to [FeCl4]−. We further found that the coordination change significantly decreases the redox potential of Fe2+/Fe3+.
Introduction
The coordination state around the redox pair in solution has a great influence on the redox potential (V) as well as its temperature coefficient (SEC) because V is equivalent to −ΔG/e, where ΔG and e are the variation in the Gibbs free energy associated with reduction reaction and elementary charge (>0). In a technological point of view, the electrochemical parameters can be used for energy harvesting device, such as liquid thermoelectric cell (LTE).1–6 In this sense, it is scientifically and technologically important to deeply comprehend and control the coordination state of redox pairs in solution. The Fe ion in solution is usually octahedrally coordinated by six solvent molecules (L) forming the FeL6 complex. Inada et al.7 reported that Fe2+ is coordinated by six L in aqueous, methanol (MeOH), ethanol (EtOH), dimethyl sulfoxide (DMSO) solutions. If the aqueous solution contains Cl−, however, it is reported that Fe3+ takes various coordination state,8,9 such as [FeCln(H2O)6−n]n−3 and [FeCl4]−, reflecting a strong interaction between Fe3+ and Cl−. The Fe coordination in aqueous solution containing Cl− is still controversial.Recently, there has been an interest in the electrochemistry of redox pairs not only in aqueous solutions but also in organic solutions. Especially, Inoue et al.10 reported that SEC of Fe2+/Fe3+ in several organic solvents are much higher than SEC in aqueous solution. For example, SEC (= 3.6 mV K−10) of Fe2+/Fe3+ in acetone is much larger than that (= 1.5 mV K−6) in aqueous solution. In addition, Wake et al.11 reported that the dimensionless performance index (ZT) of LTE is much enhanced if aqueous solution of Fe2+/Fe3+ is replaced with acetone solution of Fe2+/Fe3+. In this situation, a comprehension of the coordination state of Fe ions in the organic solution is desired. In general, the formation of FeCl4 in solution is governed by the equilibrium constant (K); K = [FeCl4−]/[Fe3+][Cl−]4 for Fe3+ + 4Cl− ↔ FeCl4−. Then, systematic investigation against Cl− concentration ([Cl−]) at a fixed Fe3+ concentration is effective for comprehension and control of the complex formation. We emphasized that the ultraviolet-visible (UV-vis) absorption spectroscopy is a sensitive probe for complex formation, because Fe3+ complex exhibits characteristic absorption bands in this region.12 In addition, the spectroscopy is sensitive even in a dilute Fe3+ solution of sub mM and is suitable for investigation of organic solution.
In this work, we spectroscopically investigated coordination state of Fe3+ against [Cl−] in the MeOH and EtOH solutions. In MeOH solution, with increases in [Cl−], octahedral [FeL6]3+ gradually transforms to tetrahedral [FeCl4]− via mixed [FeClnL6−n]3−n state. In EtOH solution, [FeL6]3+ directly transforms to [FeCl4]− with increase in [Cl−]. The variation of the Fe coordination significantly decreases V of Fe2+/Fe3+.
Experiment
UV-vis absorption
The UV-vis absorption spectra of Fe3+ solutions were investigated with a spectrometer (V750, Jasco) at room temperature. Absorption spectra were obtained by dividing the transmission intensity spectra (I) of the solution by that (I0) without cell. The molar absorption coefficient (ε) was defined by −ln(I/I0)/cd, where c (= 0.5 mM) and d (= 1 cm) are the molar concentration of Fe3+ and thickness of the optical cell, respectively.The solvents investigated were water (H2O), MeOH (FUJIFILM Wako corp.), EtOH (FUJIFILM Wako corp.). The solvents are purchased and used as received. The solutions contain 0.5 mM Fe3+. The [Cl−] value was controlled by dissolving FeCl3·6H2O (FUJIFILM Wako corp.), Fe(ClO4)3·6.9H2O (FUJIFILM Wako corp.), and NaCl (FUJIFILM Wako corp.) in appropriate molar ratios. At several [Cl−] in MeOH and EtOH solutions, we confirmed that the crystal water in solution does not affect the spectra. The intensity and shape of the spectra of anhydrous FeCl3 (FUJIFILM Wako corp.) solutions are essentially the same as that of the corresponding hydrous FeCl3 solution. In aqueous solution, LiCl (FUJIFILM Wako corp.) was used instead of NaCl, because LiCl shows higher solubility in water. We further investigated the solutions containing 0.5 mM Fe(ClO4)3·6.9H2O (FUJIFILM Wako corp.). The ClO4− concentration ([ClO4−]) was controlled by dissolving extra NaClO4 (FUJIFILM Wako corp.).
Variation of redox potential V
We systematically investigated variation in V of Fe2+/Fe3+ against [Cl−]. We define ΔV as Vsample − Vref, where Vsample and Vref are V of the sample and reference cells, respectively. The reference and sample cells were beaker cells which were connected by a salt bridge. The salt bridge was made as follows. NaClO4 (10 g per 100 mL) and agar (4 g per 100 mL) were added to water. Then, the solution was heated, dissolved, poured into a U-shaped tube, and cooled to harden. A Pt electrode was inserted into each cell.The electrolyte in the reference cell was MeOH (or EtOH) solution containing 0.5 mM Fe(ClO4)2·6H2O (FUJIFILM Wako corp.) and 0.5 mM Fe(ClO4)3·6.9H2O. The electrolytes in the sample cell were [Cl−] controlled MeOH (or EtOH) solution containing 0.5 mM Fe2+ and 0.5 mM Fe3+. The [Cl−] value was controlled by dissolving FeCl2·4H2O, FeCl3·6H2O, Fe(ClO4)2·6H2O, Fe(ClO4)3·6.9H2O, and NaCl in appropriate molar ratios. ΔV between the cells were carefully investigated with confirming stabilization of the voltage. At several [Cl−] in MeOH and EtOH solutions, we confirmed that the crystal water in solution does not affect the redox potential. The redox potentials of anhydrous FeCl2 (FUJIFILM Wako corp.)/FeCl3 (nacalai tesque) solutions are the same as that of the corresponding hydrous FeCl2/FeCl3 solutions within the experimental error (<6 mV).
Results and discussion
Overall feature of spectra
Fig. 1(a) shows molar absorption coefficient (ε) spectra of Fe(ClO4)3 dissolved in H2O, MeOH, and EtOH. Thick and thin curves represent for the spectra without and with excess ClO4−, respectively. In all solutions, the spectra with excess ClO4− are the same as those without excess ClO4−. This observation indicates that the Fe3+ complex is stable even with excess ClO4−. In MeOH and EtOH solutions, the spectra exhibit two absorption bands at 360 nm and 260 nm. We ascribed the spectral feature to formation of [FeL6]3+. We calculated the absorption spectra of [Fe(MeOH)6]3+ cluster with Gaussian 16W program13 (Fig. S1†). The calculated spectrum shows two-band structure at 190 nm and 370 nm due to the ligand to metal charge transfer (LMCT) transition and qualitatively reproduces the observed spectra [Fig. 1(a)]. In aqueous solution, traces of absorption bands are discernible at 300 nm, which is ascribed to [Fe(OH)(H2O)5]2+.14,15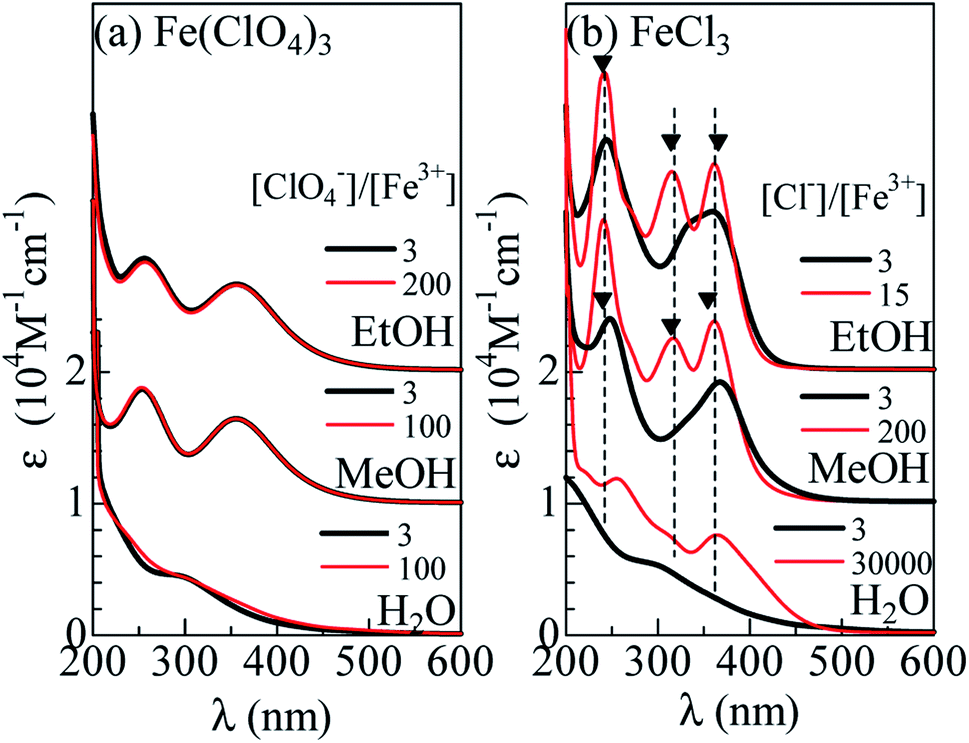 | ||
| Fig. 1 Molar absorption coefficient (ε) spectra of (a) Fe(ClO4)3 and (b) FeCl3 dissolved in H2O, methanol (MeOH) and ethanol (EtOH). Thin curves (a) and (b) are the spectra with excess ClO4− and Cl−, respectively. Filled triangles in (b) represent the absorption bands due to [FeCl4]−. | ||
Fig. 1(b) shows the ε spectra of FeCl3 dissolved in H2O, MeOH, and EtOH. Thick curves represent for the spectra without excess Cl−. The spectrum of the MeOH and EtOH solutions without excess Cl− still exhibit two-band structure at 250 nm and 370 nm. The spectral weight of the 260 nm band is higher than that of the Fe(ClO4)3 solutions [(a)]. We ascribed the spectral feature to formation of [FeClnL6−n]3−n (n = 1 and 2), because the oscillator strength (f) of an electron transfer from Cl to Fe3+ is larger than that from O to Fe3+. We calculated the absorption spectra of [FeClMeOH5]2+, trans [FeCl2MeOH4]+, and cis [FeCl2MeOH4]+ clusters with Gaussian 16W program13 (Fig. S1†). The total oscillator strength (ftot) of transitions forming the higher energy band is larger in the Cl-substituted clusters than ftot (= 0.42) in [FeMeOH6]3+; ftot is 0.56 in [FeClMeOH5]2+, 0.73 in trans [FeCl2MeOH4]+, and 0.74 in cis [FeCl2MeOH4]+.The spectral shape of the FeCl3 aqueous solution without excess Cl− is the same as that of the Fe(ClO4)3 aqueous solution, indicating that the Fe coordination state remains [Fe(OH)(H2O)5]2+.14,15
Surprisingly, addition of excess Cl− completely changes the spectra. Thin curves in Fig. 1(b) represent for the spectra with excess Cl−. In the MeOH and EtOH solutions, excess Cl− causes sharp absorption bands at 362, 318, and 242 nm, as indicated by filled triangles. We emphasize that the spectral profile of the MeOH solution is the same as that of the EtOH solution. The sameness of the two spectra indicates that Fe3+ is not coordinated by L, but Cl−. In Fig. 2, we compared the spectra of the MeOH solution containing 0.5 mM Fe3+ with excess at [Cl−] = 101.5 mM with that of nitromethane solution of [N(C2H5)4][FeCl4].3 Concerning to the lower-lying two absorption bands, the spectral profiles are essentially the same. This clearly indicates formation of [FeCl4]− in the MeOH and EtOH solutions with excess Cl−. On the other hands, the spectrum of aqueous solution at [Cl−]/[Fe3+] = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 exhibits two-band structure. The spectral profile is consistent with the spectra at [Cl−] = 15 M reported by Liu et al.,12 who ascribed the spectral feature to [FeCl3(H2O)3] and/or [FeCl4]−. We emphasize that the 260 nm band is located at higher wavelength side than the 242 nm band due to [FeCl4]−, as indicated by broken straight lines. This clearly indicates that [FeCl4]− is not the dominant species even at the highest [Cl−].
000 exhibits two-band structure. The spectral profile is consistent with the spectra at [Cl−] = 15 M reported by Liu et al.,12 who ascribed the spectral feature to [FeCl3(H2O)3] and/or [FeCl4]−. We emphasize that the 260 nm band is located at higher wavelength side than the 242 nm band due to [FeCl4]−, as indicated by broken straight lines. This clearly indicates that [FeCl4]− is not the dominant species even at the highest [Cl−].
 | ||
| Fig. 2 Molar absorption coefficient (ε) spectra of MeOH solution containing 0.5 mM Fe3+ at [Cl−] = 101.5 mM. Red symbols show the ε spectra of nitromethane solution of [N(C2H5)4][FeCl4] (cited from ref. 8). | ||
Spectral change against [Cl−]
Now, let us investigate in detail how the ε spectrum changes with increase in [Cl−]. Fig. 3(a) shows the ε spectra of the MeOH solution containing 0.5 mM Fe3+ against [Cl−]. At [Cl−] = 0.0 mM, the spectrum exhibits two broad absorption bands at 360 nm and 260 nm, which are due to an electron transfer from L to Fe3+ within the FeL6 complex. At [Cl−] = 1.5 mM, the spectra still show two-band structure, but the spectral weight of the higher energy band is much higher than those at [Cl−] = 0.0 mM. As discussed in the previous subsection, the spectral change is interpreted in terms of the formation of [FeClnL6−n]3−n (n = 1 and 2). At [Cl−] = 11.5 mM, trace of an additional absorption band is discernible, as indicated by an open triangle. Its spectral weight gradually increases as [Cl−] increases. At [Cl−] = 71.5 mM, the spectrum shows characteristic three band structure due to FeCl4− (Fig. 2). The spectra remain unchanged in the [Cl−] region above 71.5 mM. This means that all Fe3+ form the [FeCl4]− complex, because the equilibrium does not move with increase in [Cl−]. Hereafter, we will call the absorption band at 318 nm as “FeCl4 band”. A detailed investigation revealed three isosbestic points at 225 nm, 255 nm, and 385 nm above [Cl−] = 11.5 mM [Fig. S2(a)†]. The isosbestic point reflects the reaction between octahedral ([FeClnL6−n]3−n) to tetrahedral ([FeCl4]−) complexes with increase in [Cl−]. | ||
| Fig. 3 Molar absorption coefficient (ε) spectra of (a) MeOH and (b) EtOH solution containing 0.5 mM Fe3+ against Cl− concentration ([Cl−]). Open triangles indicate FeCl4 band (see text). | ||
A similar [Cl−]-dependent spectral change is observed in the EtOH solution [Fig. 3(b)]. At [Cl−] = 0.0 and 0.9 mM, the spectra exhibit two-band structure. At [Cl−] = 2.5 mM, an additional absorption band appears as indicated by an open triangle. Its spectral weight steeply increases as [Cl−] increases. At [Cl−] = 4.5 mM, the spectrum shows characteristic three band structure due to FeCl4 (Fig. 2). The spectra remain unchanged in the [Cl−] region above 4.5 mM, indicating that all Fe3+ forms the FeCl4 complex. We found three isosbestic points at 225 nm, 260 nm, and 385 nm above [Cl−] = 0.9 mM [Fig. S2(b)†]. The isosbestic point reflects the reaction between octahedral ([FeClnL6−n]3−n) to tetrahedral ([FeCl4]−) complexes with increase in [Cl−].
FeCl4 formation against [Cl−]
The solution system investigated contains multiple complex species, such as, [FeL6]3+, [FeClnL6−n]3−n (n = 1 and 2), [FeCl4]−. It is difficult to unambiguously decompose the spectrum into the respective components because there is no quantitative information on the spectra due to [FeClnL6−n]3−n. Fortunately, we know the spectrum of [FeIIICl4]−. In addition, the FeCl4 band at 318 nm is well separated from the absorption bands due to other complexes. In the following, we focused our attention on the FeCl4 formation against [Cl−]. We evaluated the intensities and peak positions (λp) of the FeCl4 band by least-squares fitting with three Gaussian functions (Fig. S3 and S4†). The intensities were normalized by the value at [Cl−] = 101.5 mM (7.5 mM) for the MeOH (EtOH) solutions, where all Fe3+ is considered to form the FeCl4 complex. Then, the proportion (r) of the FeCl4 complex is the same value as the normalized intensity (I).Fig. 4 shows I (upper panel) and λp (middle panel) against [Cl−] in (a) MeOH and (b) EtOH solutions. For convenience of explanation, we define regions I, II, and III as regions where r (= I) < 0.2, 0.2 < r < 0.7, and r > 0.7, respectively. The region I and III are dominated by the [FeL6]3+ and [FeCl4]− complexes, respectively. In (a) MeOH system, r exhibits a stationary value of 0.2–0.3 in the region of 10 mM < [Cl−] < 50 mM (region II). The fact that r remains 0.2–0.3 indicates existence of a dominant species other than [FeCl4]− and [FeL6]3+, that is, [FeClnL6−n]3−n (n = 1 and 2). In (b) EtOH, r steeply increases from 0.5 at [Cl−] = 1 mM to 0.7 at [Cl−] = 2.5 mM. The steep increase in r indicates that FeL6 almost directly transfers to FeCl4 with increases in [Cl−].
 | ||
| Fig. 4 (a) Normalized intensity (I; upper panel) and wavelength (λp; middle panel) at the peak of the FeCl4 band, and variation (ΔV; bottom panel) in V of Fe2+/Fe3+ in MeOH solution against [Cl−]. The intensity was normalized by the value at [Cl−] = 101.5 mM. (b) I (upper panel) and λp (middle penal), and ΔV (bottom panel) in EtOH solution against [Cl−]. The intensity was normalized by the value at [Cl−] = 7.5 mM. For convenience of explanation, regions I, II, and III are defined (see text). | ||
The difference in the complex formation between the MeOH and EtOH solutions is probably reflects the difference in solubility of Cl−. The solubility (= 6 mM) of NaCl in EtOH is much smaller than that (= 100 mM) in MeOH. In EtOH system, Cl− in solution is unstable and tend to form the [FeCl4]− complex if Fe3+ exits in solvent. The [Cl−] value (= 2.0 mM) at the boundary to region III is nearly the same value (= 2.0 mM) as required for all Fe3+ to become FeCl4. In MeOH system, Cl− in solution is stabler than in EtOH. This causes the stable [FeL6]3+ in small [Cl−] region (<10 mM; region I) and [FeClnL6−n]3−n formation in the intermediate [Cl−] region (10 mM < [Cl−] < 50 mM; region II). In aqueous solution where Cl− is much more stabler than in alcohol solutions, no trace of [FeCl4]− is observed even at [Cl−] = 15 M [Fig. 1(b)]. Thus, our investigation revealed the solvent dependence in complex formation against [Cl−].
Coordination effect on V
Now, let us investigate the correlation between the Fe ion coordination and V of Fe2+/Fe3+. The bottom panel of Fig. 4 shows ΔV (= Vsample − Vref) of Fe2+/Fe3+ in (a) MeOH and (b) EtOH solutions. The electrolyte in the reference cell was MeOH (or EtOH) solution containing 0.5 mM Fe2+ and 0.5 mM Fe3+ at [Cl−] = 0.0 mM, where the FeL6 complex is dominated. We must confess that we have no detailed information on the complex state of Fe2+ against [Cl−]. Like Fe3+, the complex state is considered to change from [FeL6]2+, [FeClnL6−n]2−n, to [FeCl4]2− with increase in [Cl−]. In the following argument, we assume that the redox reaction of the complex occurs without substitution of the coordinated molecules/ions.In (a) MeOH system, ΔV is −0.16 V at the entrance of region II. The variation in V between [FeL6]2+/[FeL6]3+ (region I) and [FeClnL6−n]2−n/[FeClnL6−n]3−n (region II) is well explained in terms of the crystal field splitting. We note that six d electrons in the octahedral [FeL6]2+ and [FeClnL6−n]2−n takes high-spin (HS) configuration, similarly to the case of [Fe(H2O)6]2+.16,17 The crystal field from Cl− is weaker than that from oxygen in L. Then, V of [FeCl4]2−/[FeCl4]− is expected to be lower, reflecting narrower crystal field splitting. In (b) EtOH system, ΔV is −0.24 V at the entrance of region III. Variation of V between [FeL6]2+/[FeL6]3+ (region I) and [FeCl4]2−/[FeCl4]− (region III) is also explained in terms of the crystal filed splitting. We note that six d electrons take the HS configuration in tetrahedral [FeCl4]2−, because the crystal field splitting (= 10 Dq) in octahedral complex is larger than that (= 4.45 Dq) in the tetrahedral complex.18 V of [FeCl4]2−/[FeCl4]− is expected to be lower, reflecting narrower ligand field splitting.
Conclusions
We spectroscopically investigated variation of the Fe ion coordination against [Cl−] in MeOH and EtOH solutions. In MeOH solution, with increases in [Cl−], octahedral [FeL6]3+ gradually transforms to tetrahedral [FeCl4]− via mixed [FeClnL6−n]3−n (n = 1 and 2). In EtOH solution, [FeL6]3+ directly transforms to [FeCl4]− with increase in [Cl−]. We found that the coordination change significantly decreases V of Fe2+/Fe3+ and interpreted the observation in terms of the ligand field splitting.Author contributions
Y. N. performed spectroscopic and electrochemical experiment. D. I. have supported the experiment and analysis. D. I further performed quantum chemistry calculation. Y. M. made the experimental plan and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Murata Science Foundation, and joint research with Taisei Rotec Corporation.Notes and references
- T. Ikeshoji, Bull. Chem. Soc. Jpn., 2000, 60, 1505 CrossRef CAS; T. Kim, J. S. Lee, G. Lee, H. Yoon, J. Yoon, T. J. Kangd and Y. H. Kim, Nano Energy, 2017, 31, 160 CrossRef; H. Zhou, T. Yamada and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 10502 CrossRef PubMed; I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985 CrossRef; Y. Mua and T. I. Quickenden, J. Electrochem. Soc., 1996, 143, 2558 CrossRef; J. Kawamura, M. Shimoji and H. Hoshino, J. Phys. Soc., 1981, 50, 194 CrossRef; A. Schiraldi, E. Pezzati and P. Baldini, J. Phys. Chem., 1985, 89, 1528 CrossRef; M. Sindhuja, B. Lohith, V. Sudha, G. R. Manjunath and S. Harinipriya, Mater. Res. Express, 2017, 4, 075513 CrossRef.
- B. Yu, J. Duan, H. Cong, W. Xie, R. Liu, X. Ahuang, H. Wang, B. Qi, M. Xu and L. Wan, Science, 2020, 370, 342 CrossRef CAS PubMed.
- J. H. Kim, J. H. Lee, E. E. Palen, M.-S. Suh, H. H. Lee and R. J. Kang, Sci. Rep., 2019, 9, 8706 CrossRef PubMed.
- J. Duan, G. Feng, B. Yu, J. Li, M. Chen, P. Yang, J. Feng, K. Liu and J. Zhou, Nat. Commun., 2018, 9, 5146 CrossRef PubMed.
- S. W. Lee, Y. Yang, H.-W. Lee, H. Ghasemi, D. Kraemer, G. Chen and Y. Cui, Nat. Commun., 2014, 5, 3942 CrossRef CAS PubMed.
- M. A. Buckingham, F. Marken and L. Aldous, Sustainable Energy Fuels, 2018, 2, 20717 RSC.
- Y. Inada, H. Hayashi, K. Sugimoto and S. Funahashi, J. Phys. Chem., 1999, 103, 1401 CrossRef CAS.
- K. Asakura, M. Nomura and H. Kuroda, Bull. Chem. Soc. Jpn., 1985, 58, 1543 CrossRef CAS.
- D. L. Wertz and M. L. Steele, Inorg. Chem., 1981, 19, 1652 CrossRef CAS; D. L. Wertz and M. D. Lutter, Inorg. Chem., 1961, 20, 3118 CrossRef; G. W. Brady, J. Chem. Phys., 1958, 29, 1361 CrossRef; G. L. Standley and R. F. Kruh, J. Chem. Phys., 1961, 34, 1450 CrossRef; G. W. Brady, M. B. Robin and J. Varimbi, Inorg. Chem., 1964, 3, 1168 CrossRef; M. D. Lind, J. Chem. Phys., 1967, 46, 2010 CrossRef; M. Magini, J. Chem. Phys., 1979, 71, 4255 CrossRef; M. Magini, J. Chem. Phys., 1982, 76, 1111 CrossRef.
- D. Inoue, H. Niwa, H. Nitani and Y. Moritomo, J. Phys. Soc. Jpn., 2011, 90, 033602 CrossRef.
- A. Wake, D. Inoue and Y. Moritomo, Appl. Phys. Express, 2022, 15, 054002 CrossRef.
- W. Liu, B. Etschmann, J. Brugger, L. Spiccia, G. Foran and B. McInnes, Chem. Geol., 2006, 326, 231 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. C. A. Loures, M. A. K. Alcântara, H. J. I. Filho, A. C. S. C. Teixeira, F. T. Silva, T. C. B. Paiva and G. R. L. Samanamud, Int. Rev. Chem. Eng., 2013, 5, 102 Search PubMed.
- R. C. Turner and K. E. Miles, Can. J. Chem., 1957, 35, 1002 CrossRef CAS; H. J. Benkelberg and P. Warneck, J. Phys. Chem., 1995, 99, 5214 CrossRef; R. A. Dandorth and B. Kohler, Chem. Phys. Lett., 2017, 683, 315 CrossRef.
- O. G. Holmes and D. S. McClure, J. Chem. Phys., 1957, 26, 1686 CrossRef CAS.
- D. Harris, G. H. Loew and A. Komornicki, J. Phys. Chem. A, 1997, 101, 3959 CrossRef CAS.
- B. N. Figgis and M. A. Hitchman, Ligand field theory and its application, Wiley-VCH, New York, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01522f |
| This journal is © The Royal Society of Chemistry 2022 |