
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCordyceps sinensis-mediated biotransformation of notoginsenoside R1 into 25-OH-20(S/R)-R2 with elevated cardioprotective effect against DOX-induced cell injury†
Jishuang Liu a,
Yu Xina,
Zhidong Qiua,
Qi Zhanga,
Tianzhu Heb,
Ye Qiu*a and
Weinan Wang*a
a,
Yu Xina,
Zhidong Qiua,
Qi Zhanga,
Tianzhu Heb,
Ye Qiu*a and
Weinan Wang*a
aSchool of pharmaceutical sciences, Changchun University of Chinese Medicine, Changchun, Jilin Province, China. E-mail: cnweinanwang@163.com; ccyeqiu@163.com; Tel: +86-431-86172225 Tel: +86-431-86172211
bSchool of Basic Medical Sciences, Changchun University of Chinese Medicine, Changchun, Jilin Province, China
First published on 28th April 2022
Abstract
Notoginsenoside R1 is a dammarane saponin in Panax notoginseng with promising cardioprotective effects. The bioactivity–structure relationship of such saponins suggested that the presence of a hydroxyl group at C25 could elevate its performance. To fulfill that goal, bioconversion of notoginsenoside R1 was mediated by a biocatalytic system of Cordyceps sinensis that had successfully produced multiple 25-OH derivatives from ginsenoside Re and Rg1. The major metabolic products of notoginsenoside R1 were identified as 25-OH-20(S/R)-R2 via the techniques of HRMS, 13C-NMR, 1H-NMR, HSQC and HMBC. Time-course experiments were designed to monitor the reaction process, establishing a biocatalytic pathway of “R1→20(S/R)-R2→25-OH-20(S/R)-R2”. The bioconversion rate of these 25-OH derivatives added up to 69.87% which greatly precedes the previous report. Afterwards, the effect of these biocatalytic products against doxorubicin-induced cardiotoxicity was evaluated, indicating a significant increase in efficacy after the hydration of the C24–C25 double bond on the dammarane skeleton. In conclusion, the biocatalytic system employed in this paper is able to harvest 25-OH-20(S/R)-R2 in high yield from notoginsenoside R1, which will provide lead compounds or drug candidates to alleviate myocardial injury caused by doxorubicin.
1. Introduction
Notoginsenoside R1 (R1) is a dammarane saponin originated from Panax notoginseng.1,2 It was reported to have potent cardioprotective effects in multiple research cases and has been accepted as a promising drug candidate in the treatment of myocardial ischemia.3–5 According to the studies on the bioactivity–structure relationship of dammarane saponins, the number of sugar moieties and their position, the difference in aglycone types, the position of double bond and hydroxyl groups, as well as the R/S stereoselectivity of C20 profoundly influence their overall efficacy in dealing with cancer, myocardial injury etc., among which the hydroxylation at C25 has been emphasized in the last decade.6 For instance, 25-OH protopanaxadiol (25-OH PPD) could inhibit proliferation, cause cell cycle arrest, and induce apoptosis of ill-defined cells in the setting of pancreatic and prostate cancer. Its efficacy is significantly higher than protopanaxadiol and even, ginsenoside Rg3, an approved anticancer drug clinically applied in China.7,8 However, these 25-OH saponins are usually scarce in natural sources which hinders their scale-up application in drug development and clinical practice.Biotransformation and biocatalysis have gained momentum in recent years, playing increasingly important roles in the chemical modification of natural products and the development of environment-friendly industries. As in the field of saponin biotransformation, great effort has been made on deglycosylation reactions that could convert primary saponins into secondary saponins and/or their aglycones, leveraged by the notion that saponins with less sugar moieties might demonstrate better bioactivity and bioavailability comparing to their prototypes. In contrast, modifications on the aglycone of saponins have drawn less attention and were sometimes recognized as side reactions as revealed in the practice of gastric acid transformation on ginsenosides.9–11 Though some researchers witnessed diversified derivatives of saponins during either in vitro/vivo metabolism or microbial transformation, most of the key enzymes and mechanisms underlying these specific biocatalytic behavior kept in dark.12,13
To achieve 25-OH derivatives from dammarane type saponins, a hydration reaction on the double bonds between C24 and C25 is necessary which could be catalyzed by potential hydratase under mild condition.14,15 Unfortunately, such hydratase for the biocatalysis of dammarane saponins is yet to be discovered. Microorganisms, on the other hand, possess versatile tool kits for almost all types of catalytic reactions. Some of them are promising source of hydratase against complex structures like dammarane saponins, which, upon being properly elicited, could be utilized on the production of their 25-OH derivatives.16,17 Prior to this paper, we unraveled and optimized a fungus-based biocatalytic system as a potent catalyst for the structural modification of multiple ginsenosides, among which the hydration of ginsenoside Rh1 and Rg2 were of great interests.18,19 Herein, we took the advantage of this fungal system to investigate the biocatalytic pathway of R1 with the aim of digging its 25-OH derivatives and substantiate their elevated bioactivity on myocardiocyte protection, thereby providing lead compounds for the development of new drugs against myocardial injury.
2. Material and methods
2.1. Materials and reagents
D-(+)-Glucose, MgSO4, (NH4)2SO4, KH2PO4, FeCl3·6H2O and n-butanol were purchased from Beijing Chem Works. Trapidil (>99%), pyridine-d5 (99.5%), peptone and yeast extract were purchased from Sigma-Aldrich. Doxorubicin (DOX), DMSO (cell culture grade), Trypan Blue Stain solution (0.4%) were purchased from Solarbio. Acetonitrile and methanol were purchased from Thermo Fisher Scientific. Mega BE-C18 (1 g, 6 mL) was purchased from Agilent Technologies. Fetal Bovine Serum, DMEM basic were purchased from Gibco. Cell counting kit-8 (CCK-8) was bought from Invigentech. Penicillin–streptomycin solution (100×) was purchased from Beyotime. PBS (1×) and trypsin–EDTA were purchased from Hyclone. Notoginsenoside R1 and 20(S/R)-R2 with a purity of more than 98.0% (HPLC-UV) were purchased from national institutes for food and drug control (China).2.2. Fungal culture
The fungal strain Cordyceps sinensis (CICC 14017) was obtained from China Center of Industrial Culture Collection. The culture condition was optimized and described previously.18 In short, Cordyceps sinensis was revived from a PDA slant under 30 °C. The slant was then flushed with saline and diluted to 1 × 107 spores per mL to achieve the spore stock. 5% (v/v) of spore stock was transferred to a seed culture of 10 g L−1 peptone, 2 g L−1 yeast extract, 20 g L−1 D-(+)-glucose, 0.5 g L−1 MgSO4, 0.5 g L−1 KH2PO4, and incubated at 28 °C in a thermostat shaker (150 rpm) for 2 days. After that, 5% (v/v) of seed culture was mixed with a reaction medium consisting of 20 g L−1 D-(+)-glucose, 5 g L−1 (NH4)2SO4 and 1 mM Fe3+. The reaction medium experienced a prolonged 4 day-incubation to establish the biocatalytic system.2.3. Cell culture
The rat cardiomyoblast H9C2 cells were purchased from Procell Life Science & Technology Co., Ltd (China). The cell line was cultured in DMEM medium containing 10% FBS and 1% penicillin–streptomycin solution and incubated at 37 °C with 5% CO2. The cells were passaged and harvested for 2–3 days prior to further experiments.2.4. General process for the whole-cell biocatalysis and time-course experiments
Notoginsenoside R1 was dissolved in 50% (v/v) aqueous methanol solution to prepare a substrate stock with the concentration of 5 mg mL−1. Afterwards, 5% (v/v) of substrate stock was disinfected through membrane filtration before mixing with the biocatalytic system. The whole-cell biocatalysis was carried out in a thermostat shaker (28 °C, 150 rpm). Meanwhile 5% (v/v) of disinfected substrate stock was added to a fresh reaction medium and incubated under the same condition as a substrate blank. The culture of Cordyceps sinensis mycelium prepared in the reaction medium with no substrate was treated as a biocatalyst blank. In order to monitor the biocatalytic process, 0.5 mL aliquot was withdrawn from each of the above groups daily and subjected to HPLC-MS analysis.For time-course experiments, the relative amount of the substrates and biocatalytic products was calculated as their individual peak area versus the sum of all detected peak areas.
The equation is presented as below:
| Relative amount (%) = [Ai/(A1 + A2 + … + An)] × 100% |
2.5. Sample preparation and single compound isolation
The biocatalytic products were pretreated with ultrasonication for 10 min and then extracted three times with equivalent volume of water-saturated n-butanol. All the n-butanol extracts were merged and concentrated under reduced pressure at 60 °C. After that, the dryness was dissolved in 1 mL methanol and filtered through a 0.22 μm organic membrane before being analyzed by HPLC-MS.As for single compound isolation, the dryness was suspended in distilled water and subjected to a Solid-phase extraction (SPE) C18 column (1 g, 6 mL). The column was preconditioned with 6 mL of acetonitrile and 12 mL of water. The loading amount of the dryness was 20 mg for each gram of the adsorbent. Then the sample was washed with approximately 30 mL of water and sequentially eluted with 40 mL of 5%, 10%, 15%, 20%, 25%, 30% aqueous acetonitrile. Finally, the effluent was combined and concentrated according to the results of TLC chromatography. The obtained products were named as compound 1 to 4 following the peak order. The purity of each compound was evaluated using HPLC-MS.
2.6. HPLC-MS analysis
Separation of the biocatalytic products on HPLC was accomplished by an Agilent SB-Aq C18 column (250 mm × 4.6 mm, 5 μm) with a flow rate of 1.0 mL min−1. The mobile phase consisted of A (0.1% formic acid in H2O) and B (0.1% formic acid in acetonitrile). The gradient elution procedure was operated as below: 0–30 min 19% B, 30–35 min 19–24% B, 35–60 min 24–40%, 60–75 min 40–90% B and 75–80 min 90% B. The column temperature was set to 30 °C. The injection volume was 10 μL for either qualification or quantification experiments.Qualification of the biocatalytic products was performed on a LCMS-IT-TOF facility (Shimadzu Corporation, Japan) equipped with a dual electrospray ionization (ESI) source. The MS scan was conducted in a m/z range of 400–1000 under negative mode. Specific operating parameter was set as follows: nebulizer gas = 1.5 L min−1, dry gas pressure = 100 kPa, curved desolvation line temperature = 250 °C, heat block temperature = 250 °C, interface voltage = 3.5 kV, detector voltage = 1.7 kV, ion accumulation time for MS = 30 ms. To perform high-accuracy mass measurements, the instrument was calibrated using a standard solution (Shimadzu, Japan) prior to qualification. Data analysis was performed on the LCMSsolutionVer3 software coupled with Accurate Mass Calculator and Formula Predictor (Shimadzu, Japan). Each sample was measured thrice to validate the repeatability of the results.
Quantification experiments were performed with a triple quadrupole mass spectrometer (Shimadzu LC-MS 8040, Japan). The MS spectra was recorded in negative ionization mode. The settings were as follows: dry gas = 15 L min−1, nebulizer gas = 3 L min−1, heat block temperature = 400 °C, DL temperature = 250 °C, interface voltage = 3.5 kV, detector voltage = 1.72 kV, scanning range (m/z) = 300–1300. Data analysis was processed using the Labsolutions LCMS-8040 software Ver. 5 and Profiling Solution Ver. 1.1. The accuracy, stability and repeatability of this LC-MS method had been investigated elsewhere by our research group.20
2.7. Identification of the biocatalytic products
The purified products were evaporated to dryness under vacuum conditions and redissolved in pyridine-d5 before being subjected to NMR analysis. 1H-NMR, 13C-NMR, HSQC and HMBC were performed on a BRUKER AVANCE III-500 instrument with tetramethylsilane (TMS) as the internal standard. 13C-NMR spectra were recorded under attached proton test (APT) mode.2.8. CCK-8 assay for compound cytotoxicity
H9C2 cells were seeded in sextuplicate wells of 96-well plates (5000 cells per well) in a CO2 incubator (5% CO2, 37 °C) for 24 h. Notoginsenoside R1 and the purified biocatalytic products were separately dissolved in DMSO and diluted to different concentrations (0, 10, 40, 70, 100 μg mL−1) as sample solutions. Then the sample solutions were added to the wells, thereby incubating the plates under the same condition for another 18 h. After removal of the sample-containing medium, the wells were refilled with 110 μL CCK-8 solution (Vcomplete medium/VCCK8 = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and incubated for 40 min. Afterwards, absorbance of each well was measured at 450 nm to evaluate the cell viability of each group using a Microplate Reader (Molecular Devices, USA).
:1) and incubated for 40 min. Afterwards, absorbance of each well was measured at 450 nm to evaluate the cell viability of each group using a Microplate Reader (Molecular Devices, USA).
2.9. Evaluation of the biocatalytic products against DOX-induced cardiomyocyte injury
H9C2 cells were plated in 96-well plates (5000 cells per well) for 24 h before mixing with DOX (0.5 μM) for 4 h in the CO2 incubator (5% CO2, 37 °C) to establish the model group. Thereafter, cells were treated with different concentrations (0, 10, 40, 70, 100 μg mL−1) of R1 and its biocatalytic products for another 18 h as the treatment groups. In parallel, equal amounts of Trapidil were administered to the model and incubated for 18 h as the positive control group. Cell cultures without DOX and any other samples were established as the blank control group. All the groups above were conducted in 6 parallel wells. Surviving cells were measured by CCK-8 assay as described in section 2.8.2.10. Statistical analysis
All assay are repeated at least three times and data presented in this paper is expressed as means ± standard deviation. Statistical analysis was performed using one-way ANOVA with Tukey's post hoc test. The P < 0.05 and P < 0.01 were considered as significant differences between two groups.3. Results and discussion
3.1. Identification of the biocatalytic products
Primarily, biocatalytic products of R1 were analyzed by HPLC-Q-TOF-MS. As shown in Fig. 1, four metabolites that structurally associated with R1 are characterized and named as compounds 1 to 4 in increasing order of retention time. Based on the HRMS data, both formulations of compounds 3 and 4 were calculated as C41H70O13 which are identical to 20(S)- and 20(R)-notoginsenoside R2 (R2) respectively (Table 1).21 The identification results of compound 3 and 4 were further confirmed by aligning their retention time to the standards (Fig. 1). On the other hand, compounds 1 and 2 were reasoned out to be the hydrated derivatives of R2 since their molecular weights are 18 Da higher than compound 3 and 4. Judging from the feature of dammarane skeleton, the only double bond locates between C24–C25 of R2, suggesting compounds 1 and 2 be the C24/25-OH isomers of R2 which needs to be discriminated by NMR techniques.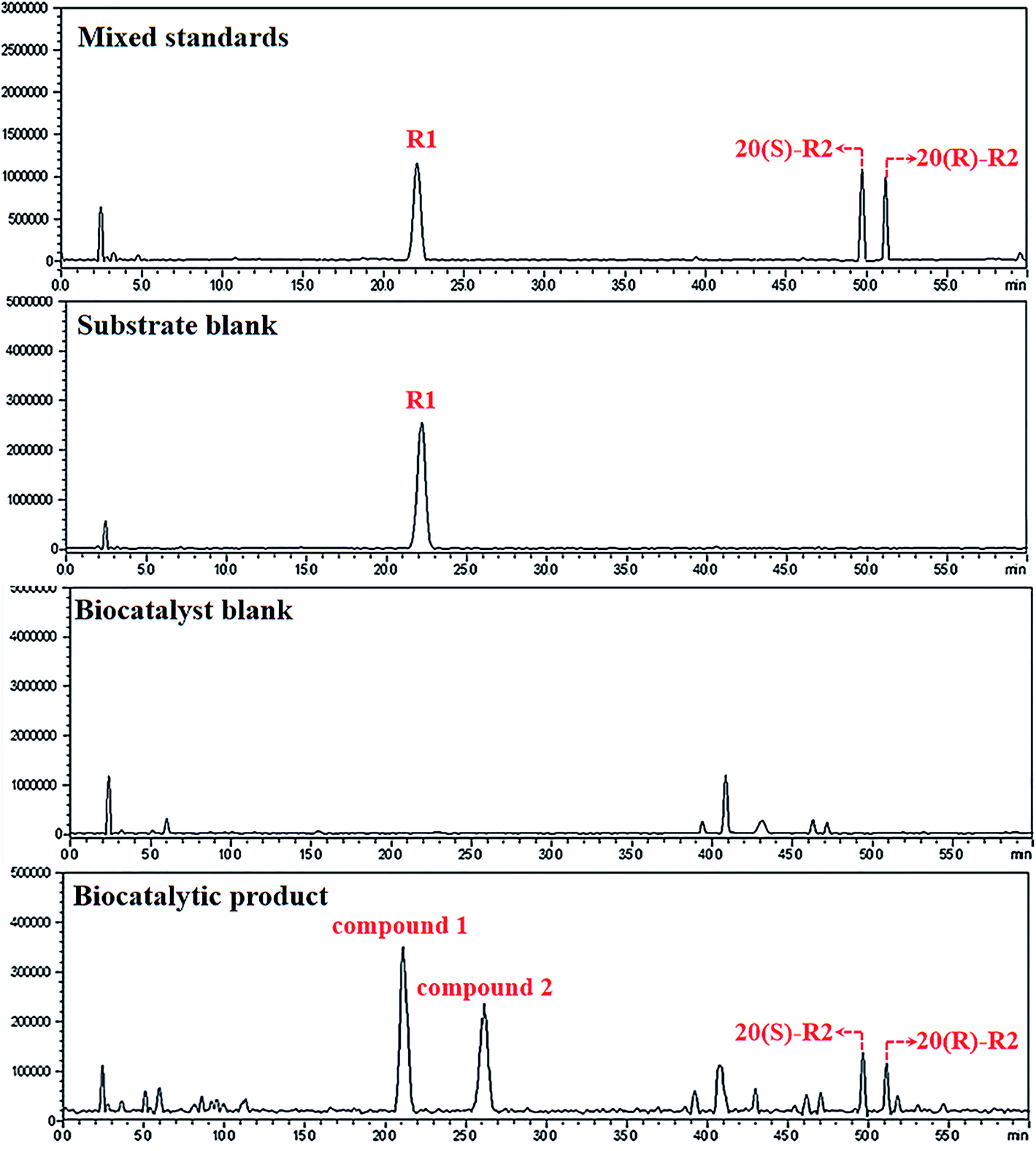 | ||
| Fig. 1 LCMS-IT-TOF analysis of the biocatalytic products. Irrelevant peaks are located and excluded by aligning chromatogram of the products to the standards and different blank groups. | ||
| Peak | tR (min) | [M − H]− (m/z) | Calculate mass errors (ppm) | Calculated formulation |
|---|---|---|---|---|
| 1 | 21.100 | 787.4845 | −0.1395 | C41H72O14 |
| 2 | 26.695 | 787.4845 | −0.1395 | C41H72O14 |
| 3 | 49.904 | 769.4739 | 0.0714 | C41H70O13 |
| 4 | 51.359 | 769.4739 | 0.0714 | C41H70O13 |
To confirm the structures of compound 1 and 2, these two products were isolated and purified from the reaction mixture, and subjected to 1H-NMR, 13C-NMR, HSQC and HMBC analysis.
Compound 1 (26.4 mg, peak purity >95.0%) was obtained as a white powder. The chromatogram for purity assessment were presented in Fig. 1S.† Its 1H-NMR and 13C-NMR spectra show eight methyl signals (δH 0.79, 0.97, 1.21, 1.39, 1.39, 1.39, 1.45, 2.04; δc 16.60, 16.79, 17.35, 17.53, 26.84, 29.78, 30.09, 31.61) which belongs to the dammarane skeleton22,23 (Fig. 3S and 4S†). Unlike the 13C-NMR spectrum of R2, there is one more signal of hydroxylated carbon at 69.62 ppm. HSQC data indicates no cross-peak between proton and carbon at this signal, determining it as a quaternary carbon (Fig. 5S†). Since the olefinic signals of C24 and C25 were absent after microbial transformation of R2, the new hydroxyl group on compound 1 should be located at C25, which was confirmed by its HMBC spectrum that demonstrates strong long-range correlations between this quaternary carbon and H-26, H-27 (Fig. 2 and 6S†). Furthermore, the 13C-NMR data of compound 1 is identical to what was reported for 3β,12β,20(S),25-tetrahydroxydammarane-6-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranoside, thereby determining it as 25-OH-20(S)-R2.24
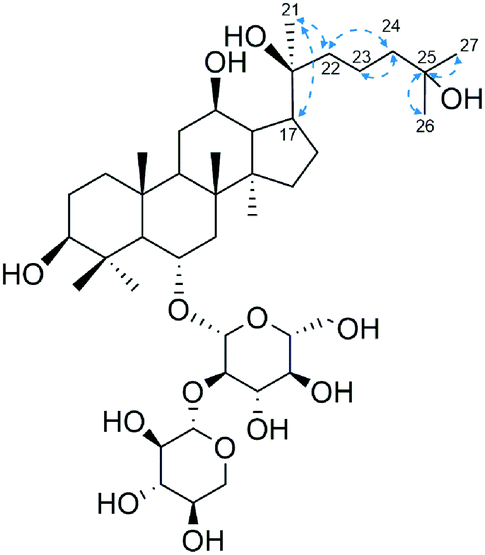 | ||
| Fig. 2 Critical HMBC correlation of compound 1. Blue arrows indicate the correlation between 1H and 13C nuclei. | ||
On the other hand, compound 2 (19.1 mg, peak purity >95.0%) is the isomer of compound 1 and shares common 13C-NMR data with 3β,12β,20(R),25-tetrahydroxydammarane-6-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranoside (Table 2, Fig. 2S and 7S†). Hence, compound 2 is identified as 25-OH-20(R)-R2.
| Position | 3β,12β,20(S),25-Tetrahydroxydammarane-6-O-β-D-xylopyranosyl-(1→2)-β-D-gluco-pyranoside | 3β,12β,20(R),25-Tetrahydroxydammarane-6-O-β-D-xylopyranosyl-(1→2)-β-D-gluco-pyranoside | Compound 1 | Compound 2 |
|---|---|---|---|---|
| 1 | 39.5 | 39.5 | 39.40 | 39.40 |
| 2 | 27.8 | 27.8 | 27.68 | 27.68 |
| 3 | 78.7 | 78.7 | 78.57 | 78.57 |
| 4 | 40.2 | 40.2 | 40.11 | 40.11 |
| 5 | 61.3 | 61.3 | 61.22 | 61.22 |
| 6 | 79.4 | 79.4 | 79.48 | 79.48 |
| 7 | 45.0 | 45.0 | 44.88 | 44.88 |
| 8 | 41.1 | 41.1 | 40.90 | 40.90 |
| 9 | 50.1 | 50.1 | 49.97 | 49.97 |
| 10 | 39.7 | 39.7 | 39.61 | 39.61 |
| 11 | 32.1 | 32.1 | 31.90 | 31.90 |
| 12 | 71.0 | 70.9 | 70.88 | 70.87 |
| 13 | 48.3 | 48.9 | 48.27 | 48.82 |
| 14 | 51.7 | 51.7 | 51.68 | 51.68 |
| 15 | 31.3 | 31.3 | 31.20 | 31.20 |
| 16 | 26.9 | 26.7 | 26.64 | 26.66 |
| 17 | 54.7 | 50.8 | 54.50 | 50.60 |
| 18 | 17.4 | 17.4 | 17.35 | 17.35 |
| 19 | 17.6 | 17.6 | 17.53 | 17.53 |
| 20 | 73.3 | 73.3 | 73.20 | 73.20 |
| 21 | 27.2 | 22.8 | 27.14 | 22.71 |
| 22 | 36.5 | 44.0 | 36.40 | 43.85 |
| 23 | 19.1 | 18.7 | 19.01 | 18.62 |
| 24 | 45.6 | 45.8 | 45.55 | 45.78 |
| 25 | 69.7 | 69.7 | 69.62 | 69.62 |
| 26 | 29.9 | 29.9 | 29.78 | 29.78 |
| 27 | 30.2 | 30.2 | 30.09 | 30.09 |
| 28 | 31.7 | 31.7 | 31.61 | 31.61 |
| 29 | 16.7 | 16.7 | 16.60 | 16.60 |
| 30 | 16.9 | 17.1 | 16.79 | 16.99 |
|
||||
| Glc | ||||
| 1′ | 103.5 | 103.5 | 103.32 | 103.32 |
| 2′ | 80.3 | 80.3 | 80.19 | 80.19 |
| 3′ | 78.0 | 78.0 | 77.84 | 77.84 |
| 4′ | 71.8 | 71.8 | 71.72 | 71.72 |
| 5′ | 79.5 | 79.5 | 79.44 | 79.44 |
| 6′ | 63.0 | 63.0 | 62.88 | 62.88 |
|
||||
| Xyl | ||||
| 1′′ | 104.9 | 104.9 | 104.79 | 104.79 |
| 2′′ | 75.8 | 75.8 | 75.75 | 75.75 |
| 3′′ | 78.7 | 78.7 | 78.67 | 78.67 |
| 4′′ | 71.2 | 71.2 | 71.16 | 71.16 |
| 5′′ | 67.3 | 67.3 | 67.19 | 67.19 |
3.2. Time-course variation of R1 and its biocatalytic pathway
Several studies had unveiled the metabolic pathways of notoginsenoside R1 either in vitro or in vivo, within which deglycosylation reaction often dominates.24–31 Zhang et al.25 explored the metabolic pathway of R1 in rats using ultra-fast liquid chromatographic-tandem mass spectrometric method. A total of seven metabolites were characterized, including 20(S/R)-R2, 20(S/R)-ginsenoside Rh1, Rg1, F1 and protopanaxatriol. Ruan et al.32 investigated R1 metabolism by gut microbiota on a Caco-2 model. Four metabolites generated through the stepwise pathway of R1 → Rg1 → F1 → PPT → dehydrogenated PPT. Comparing to deglycosylation, the hydration of double bond on dammarane skeleton is more intriguing but with less focus. For instance, 25-OH R2 was found in the biotransformation products of R1 by Absidia coerulea. However, there were more than ten compounds collected from the metabolites of R1, within which 25-OH-R2 only took a minor fraction.24 On the basis of a zebrafish model, trace amount of 25-OH-R1 was found in vivo after administering R1 to the research object, with R2 and multiple other deglycosylated ginsenosides being the main metabolites.27 As in the case of Cordyceps sinensis, there was few literature about its catalytic characteristics toward natural products. Within these limited resources, Cordyceps sinensis demonstrated catalytic features of oxidation, hydrogenation, hydrolyzation as well as hydration but on substrates other than ginsenosides.33–35In our study, the hydrated products obviously overwhelmed the deglycosylated products and there are two reasonable pathways for the bioconversion of R1 into 25-OH-20(S/R)-R2 (Fig. 3). One is the hydrolysis of the glucosyl group at C-20 prior to the hydration of the double bond between C24–C25. The other is a beforehand hydration of the double bond to produce 25-OH-R1 followed by the hydration of its glucosyl group at C-20, ending in the epimers of 25-OH-20(S/R)-R2.
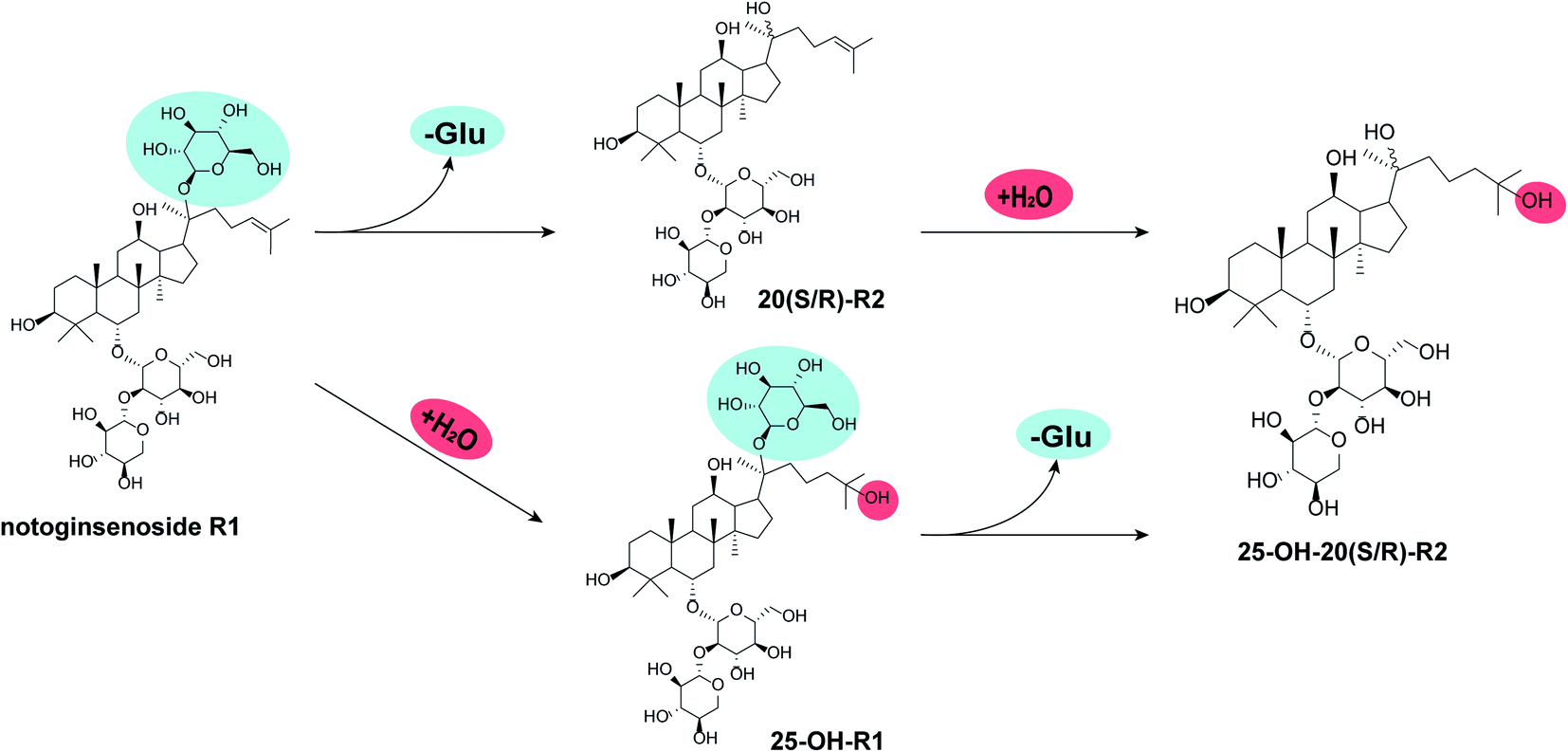 | ||
| Fig. 3 Potential biocatalytic pathways for R1. “-Glu” indicates the loss of glucosyl group while “+H2O” indicates the hydration of C24–C25 double bond. | ||
Herein, a time-course experiment was designed to clarify the pathway by HPLC-MS. As shown in Fig. 4 and 5, R1 is consumed completely after four days of biotransformation. The mixture of 20(S/R)-R2 emerged and peaked in the first two days before their peak area started to go down. Since then, 25-OH-20(S/R)-R2 gradually accumulated and reached their equilibrium amount on the 6th day where the reaction finished. Through the whole process, no signal of 25-OH-R1 (m/z [M − H]− = 995) was ever observed, thereby rejecting the route that starts with the hydration of R1. Accordingly, the biocatalytic pathway of R1 is determined as “R1→20(S/R)-R2→25-OH-20(S/R)-R2”. The relative conversion rate of 25-OH-20(S/R)-R2 and 20(S/R)-R2 was calculated as 69.87% and 12.53% respectively, which makes our catalytic system more advantageous than the previous reports that firstly found 25-OH-20(S/R)-R2 within a repertoire of reaction products mediated by Absidia coerulea.24
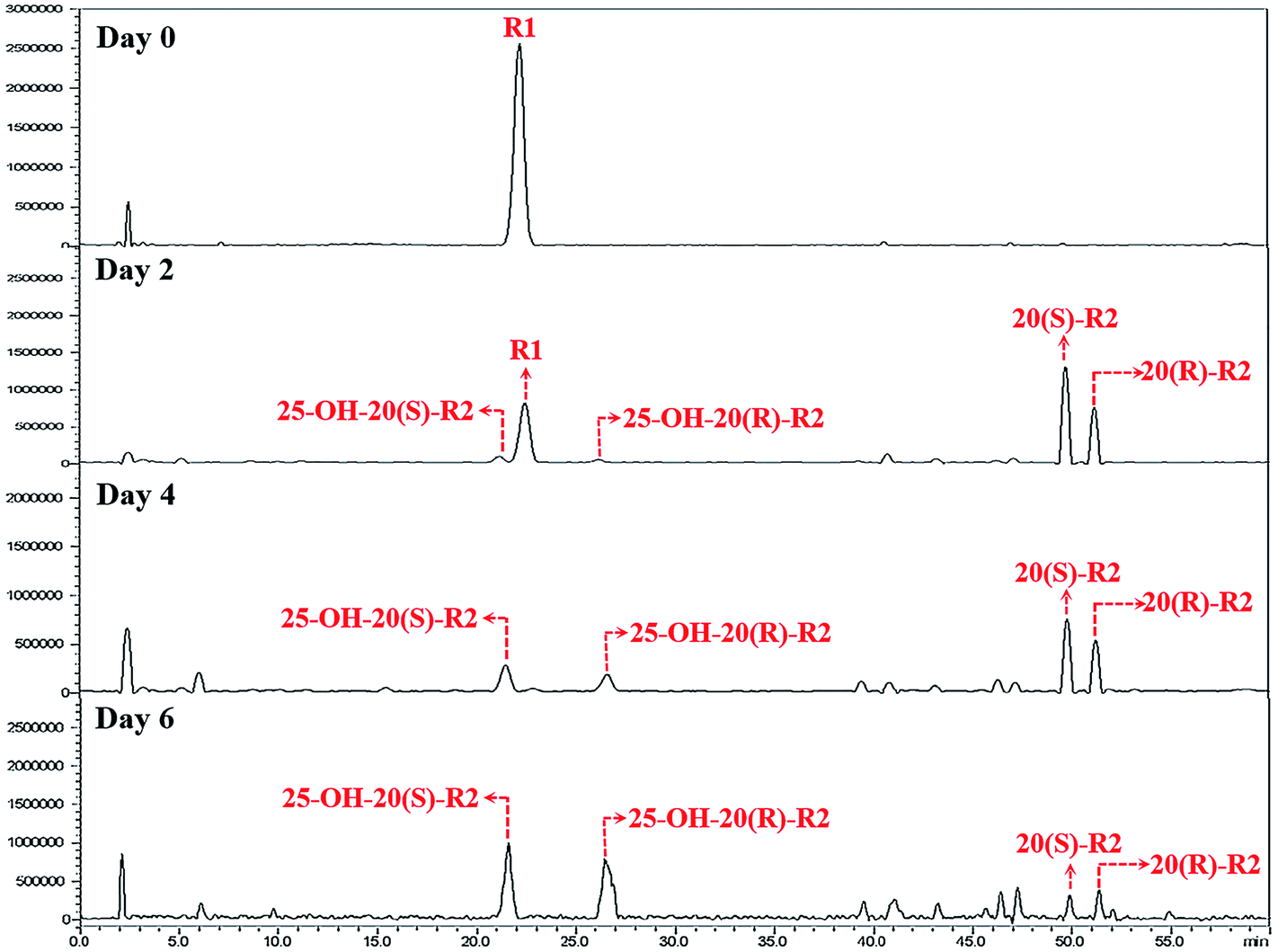 | ||
| Fig. 4 HPLC-MS chromatograms of time-course experiments. These chromatograms are recorded in auto-scan mode in order to reveal all potential impurities in the biocatalytic products. | ||
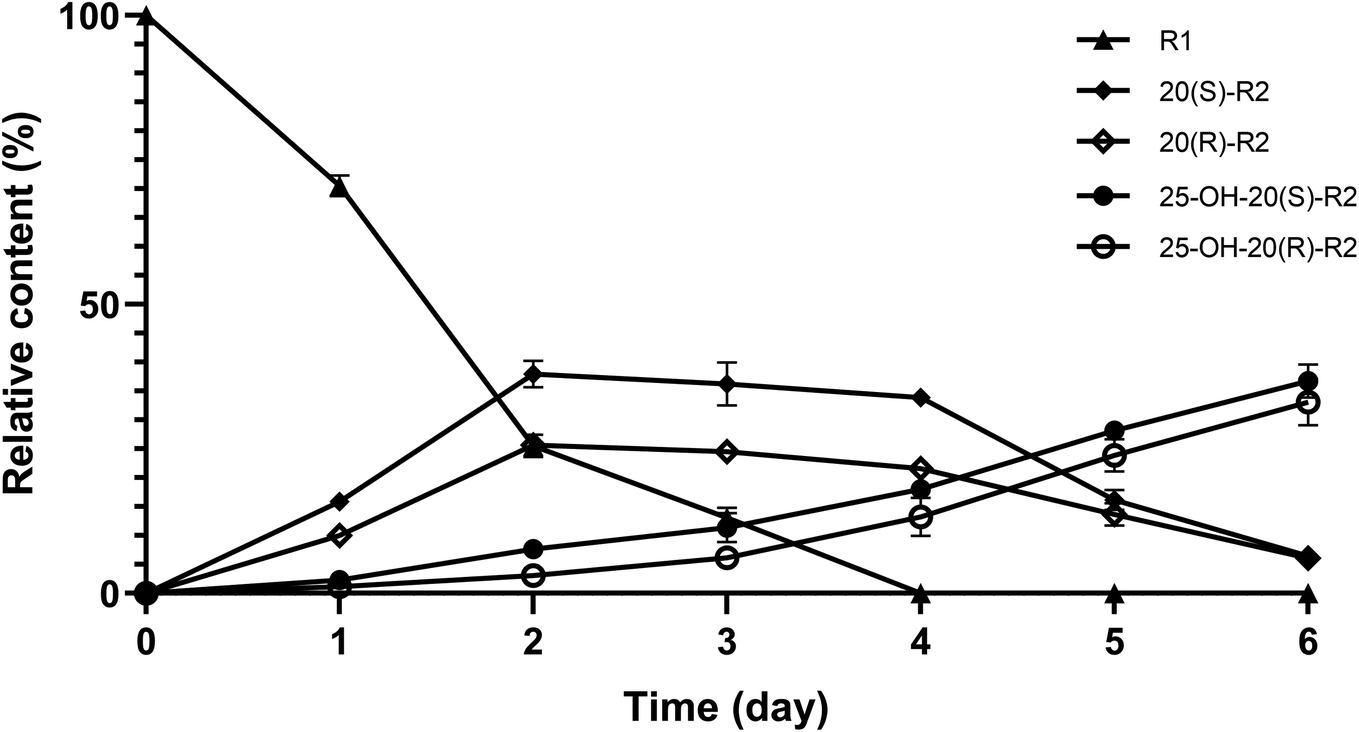 | ||
| Fig. 5 Variation in relative content of R1 and its biocatalytic products during time-course experiments. Data expressed as mean ± standard deviation. | ||
In the setting of microbial transformation, the modification of compounds mostly depend on various enzymes from the host microorganisms.36 Based on the established pathway and the structure of R1, we speculated that the glucosyl moiety at C-20 might barricade the double bond from getting close to the functional domain of the target enzymes which prevents the hydration of R1. In contrast, it would be easier to hydrate the C24–C25 double bond on R2 because the glucosyl moiety at C-20 was removed.
3.3. Effect of the biocatalytic products against doxorubicin-induced cardiomyocyte injury
DOX is a famous antibiotic that has been clinically used as potent anti-cancer drug.37,38 Like other chemotherapy medicines, DOX can cause serious side effects to the patients, especially when high dose is administered which brings significant dose-dependent cardiotoxicity.39,40 Hence the development of adjuvant that could tackle or ameliorate DOX-induced cardiotoxicity has been a hot topic within the field of drug discovery. Several dammarane-type saponins, such as ginsenoside Rb1, Rg1 and Rg3, were reported with cardioprotective activity.41–43But these candidates have been suffering from low aqueous solubility and oral bioavailability. Although the hydroxylation at C25 might increase the hydrophilicity of the dammarane structure and improve the efficacy as 25-OH protopanaxatriol (25-OH PPT), one of the aglycones of dammarane-type saponins, performed better than PPT against H2O2-induced cell injury on H9C2 model, its contribution to the glycosides against DOX-induced cardiotoxicity is yet to be proved.44
As in this study, we set up a DOX-induced cell injury model on H9C2 cells to evaluate the therapeutic effect of R1 and its biocatalytic products. Primarily, a cytotoxicity test was conducted on these compounds to clarify the potential influence of their innate feature on the viability of target cells. It turns out from Fig. 6 that none of the biocatalytic products cause apparent cytotoxicity on H9C2 cells in the range of 10–100 μg mL−1, whereas a high concentration of R1 (100 μg mL−1) decreased cell viability (P < 0.05). This finding may explain the inadequacy of R1 in dealing with DOX-induced cell injury at a high concentration, as illustrated in Fig. 7. In contrast, both 20(S/R)-R2 and their hydrated derivatives significantly increase the survival rate of H9C2 cells compared to the model group. Intriguingly, the therapeutic effect significantly improved after the hydration of C24–C25 double bond and the presence of a hydroxy group at C25. At the dose of 100 μg mL−1, the cell viability of 25-OH-20(S)-R2 and 25-OH-20(R)-R2 groups are 14.06% and 10.98% higher than the results brought by 20(S)-R2 and 20(R)-R2, respectively. Besides, the stereostructure of the C-20 hydroxyl was found to influence the performance of these compounds as well. In contrast to the 20(R)-groups, 20(S)-R2 and 25-OH-20(S)-R2 gained 7.70% and 4.62% higher viability at 100 μg mL−1, respectively. Taken together, 25-OH-20(S)-R2 was selected as the most potent protective agent against DOX-induced cardiomyocyte injury among all the biocatalytic products and demonstrated comparable efficacy to the positive control group.
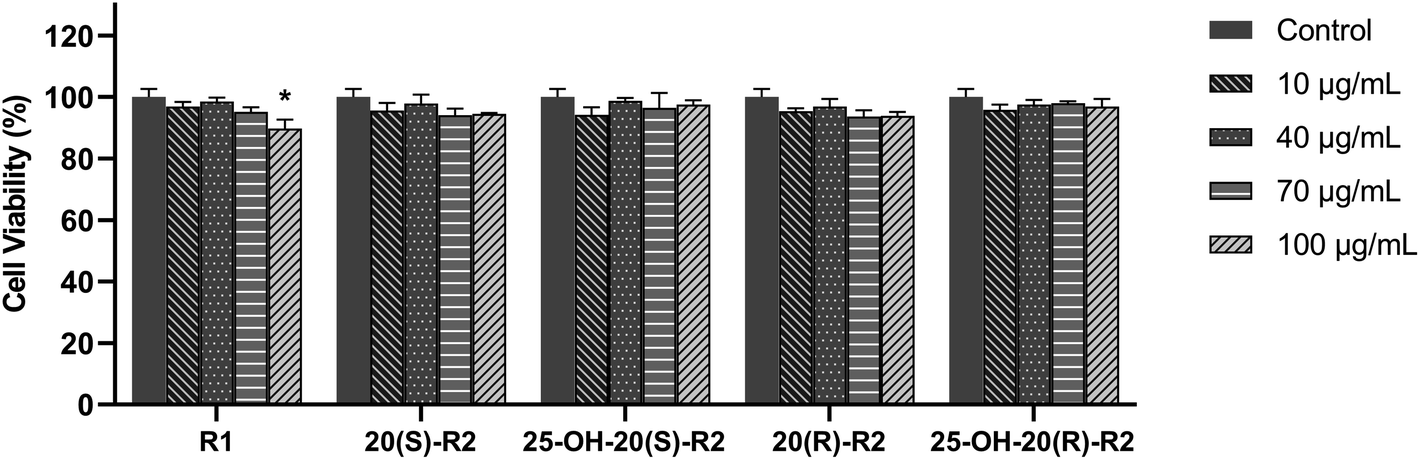 | ||
| Fig. 6 Cytotoxicity test of R1 and its biocatalytic products. Data expressed as mean ± standard deviation. * Indicates a significant difference at the P < 0.05 level between control and treatment groups. | ||
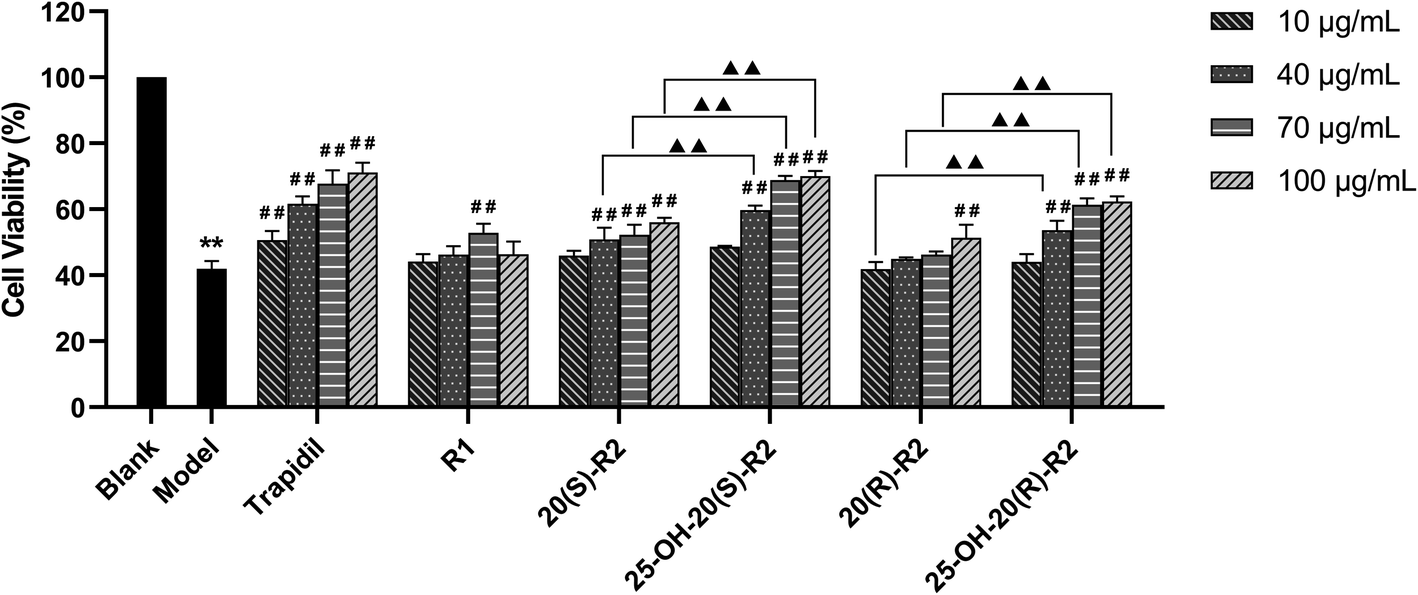 | ||
| Fig. 7 Effect of R1 and its biocatalytic products on the viability of DOX-treated H9C2 cells. Data expressed as mean ± standard deviation. ** Indicates a significant difference at the P < 0.01 level between blank and model group. ## Indicates a significant difference at the P < 0.01 level between treatment and model group. ▲▲ Indicates a significant difference at the P < 0.01 level between 20(S/R)-R2 and their 25-OH derivatives. | ||
4. Conclusions
In summary, the present study enabled notoginsenoside R1 transformation to four metabolites with unnoticeable side products by Cordyceps sinensis. The compounds were characterized as 20(S)-R2, 20(R)-R2, 25-OH-20(S)-R2 and 25-OH-20(R)-R2 using HR-MS/MS and two-dimensional NMR techniques. The biocatalytic pathway of R1 was established by time-course experiments, revealing a preference for an intriguing hydration reaction over deglycosylation which makes the 25-OH derivatives dominant products after the biotransformation. Noteworthy, it provides a mild and regioselective method to obtain 25-OH-20(S/R)-R2 with elevated protective effect against DOX-induced cardiomyocyte injury. It suggests the hydroxylation of C25 on dammarane-type saponins being a promising chemical modification strategy for drug development in the field of heart diseases.Author contributions
J. L. wrote the manuscript and did most of the works. W. W. and Y. Q. designed the framework of the work and polish the manuscript. Y. X. assisted with the experiments. Z. Q. conceived the idea. Q. Z. and T. H. gleaned the material and helped with literature and data processing. All authors contributed to the article and approved the submitted version.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by Jilin Province Science and Technology Department (20210204004YY) and Education Department of Jilin Province (JJKH20210994KJ).References
- S. S. Guo, X. Z. Xi and J. Li, Pharmazie, 2019, 74, 641–647 CAS.
- S. Zhang, C. Chen, W. Lu and L. Wei, Phytother. Res., 2018, 32, 2155–2163 CrossRef CAS PubMed.
- Q. Tong, P. C. Zhu, Z. Zhuang, L. H. Deng, Z. H. Wang, H. Zeng, G. Q. Zheng and Y. Wang, Front. Pharmacol., 2019, 10, 1204 CrossRef CAS PubMed.
- B. Sun, J. Xiao, X. B. Sun and Y. Wu, Br. J. Pharmacol., 2013, 168, 1758–1770 CrossRef CAS PubMed.
- X. Yang, X. Xiong, H. Wang and J. Wang, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 204840 Search PubMed.
- S. A. Nag, J. Qin, W. Wang, M. H. Wang, H. Wang and R. Zhang, Front. Pharmacol., 2012, 3, 25 Search PubMed.
- W. Wang, Y. Zhao, E. R. Rayburn, D. L. Hill, H. Wang and R. Zhang, Cancer Chemother. Pharmacol., 2007, 59, 589–601 CrossRef CAS PubMed.
- W. Wang, E. R. Rayburn, M. Hao, Y. Zhao, D. L. Hill, R. Zhang and H. Wang, The Prostate, 2008, 68, 809–819 CrossRef CAS PubMed.
- H. Hasegawa, J. Pharmacol. Sci., 2004, 95, 153–157 CrossRef CAS PubMed.
- J. R. Wang, L. F. Yau, R. Zhang, Y. Xia, J. Ma, H. M. Ho, P. Hu, M. Hu, L. Liu and Z. H. Jiang, J. Agric. Food Chem., 2014, 62, 2558–2573 CrossRef CAS PubMed.
- H. Dong, L. P. Bai, V. K. W. Wong, H. Zhou, J. R. Wang, Y. Liu, Z. H. Jiang and L. Liu, Molecules, 2011, 16, 10619–10630 CrossRef PubMed.
- M. M. Zheng, F. X. Xu, Y. J. Li, X. Z. Xi, X. W. Cui, C. C. Han and X. L. Zhang, BioMed Res. Int., 2017, 2017, 8601027 Search PubMed.
- S. E. Park, C. S. Na, S. A. Yoo, S. H. Seo and H. S. Son, J. Ginseng Res., 2017, 41, 36–42 CrossRef PubMed.
- M. Engleder, T. Pavkov-Keller, A. Emmerstorfer, A. Hromic, S. Schrempf, G. Steinkellner, T. Wriessnegger, E. Leitner, G. A. Strohmeier and I. Kaluzna, ChemBioChem, 2015, 16, 1730 CrossRef CAS PubMed.
- A. Izumi, D. Rea, T. Adachi, S. Unzai, S. Y. Park, D. I. Roper and J. R. Tame, J. Mol. Biol., 2007, 370, 899–911 CrossRef CAS PubMed.
- T. Kumano, E. Fujiki, Y. Hashimoto and M. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9087–9092 CrossRef CAS PubMed.
- S. M. Thomas and S. Kavitha, Int. J. Sci. Eng. Technol. Res, 2015, 4, 5778–5781 Search PubMed.
- W. Wang, J. Liu, Y. Xin, T. He, Y. Qiu, M. Qu, Y. Song and Z. Qiu, New J. Chem., 2020, 44, 14005–14014 RSC.
- X. Sui, J. Liu, Y. Xin, M. Qu, Y. Qiu, T. He, H. Luo, W. Wang and Z. Qiu, Bioorg. Med. Chem. Lett., 2020, 30, 127504 CrossRef CAS PubMed.
- F. Guo, H. H. Liu, C. Y. Wang, Z. D. Qiu and W. N. Wang, Science Technology and Engineering, 2017, 17, 199–203 Search PubMed.
- D. Q. Dou, J. Ren, Y. Chen, Y. P. Pei and Y. J. Chen, Zhongguo Zhongyao Zazhi, 2003, 28, 522–524 CAS.
- J. G. Cho, M. K. Lee, J. W. Lee, H. J. Park, D. Y. Lee, Y. H. Lee, D. C. Yang and N. I. Baek, J. Ginseng Res., 2010, 34, 113–121 CrossRef CAS.
- R. Teng, H. Li, J. Chen, D. Wang, Y. He and C. Yang, Magn. Reson. Chem., 2002, 40, 483–488 CrossRef CAS.
- G. Chen, M. Yang, Z. Lu, J. Zhang, H. Huang, Y. Liang, S. Guan, Y. Song, L. Wu and D. A. Guo, J. Nat. Prod., 2007, 70, 1203–1206 CrossRef CAS PubMed.
- S. Zhang, Z. Ju, H. Guan, L. Yu, Z. Wang and Y. Zhao, Biomed. Chromatogr., 2019, 33, e4670 CAS.
- J. Q. Ruan, W. I. Leong, R. Yan and Y. T. Wang, J. Agric. Food Chem., 2010, 58, 5770–5776 CrossRef CAS PubMed.
- Y. Wei, P. Li, H. Fan, Y. Peng, W. Liu, C. Wang, L. Shu and X. Jia, Molecules, 2011, 16, 6621–6633 CrossRef CAS PubMed.
- R. Zhang, N. Li, S. Xu, X. Han, C. Li, X. Wei, Y. Liu, T. Tu, X. Tang and J. Zhou, J. Agric. Food Chem., 2019, 67, 3220–3228 CrossRef CAS PubMed.
- K. C. Shin, M. J. Seo and D. K. Oh, Biotechnol. Lett., 2014, 36, 2275–2281 CrossRef CAS PubMed.
- Q. Li, T. Wu, Z. Qi, L. Zhao, J. Pei and F. Tang, BMC Biotechnol., 2018, 18, 1–11 CrossRef.
- D. Bretagne, A. Pâris, R. de Vaumas, P. Lafite and R. Daniellou, Biochimie, 2021, 181, 34–41 CrossRef CAS PubMed.
- J. Q. Ruan, W. I. Leong, R. Yan and Y. T. Wang, J. Agric. Food Chem., 2010, 58, 5770–5776 CrossRef CAS PubMed.
- D. Wang, M. Xu, H. T. Zhu, K. K. Chen, Y. J. Zhang and C. R. Yang, Bioorg. Med. Chem. Lett., 2007, 17, 3195–3197 CrossRef CAS PubMed.
- A. Mira, W. Alkhiary, Q. Zhu, T. Nakagawa, H. B. Tran, Y. M. Amen and K. Shimizu, Chem. Biodiversity, 2016, 13, 1307–1315 CrossRef CAS PubMed.
- A. Mira, M. A. Sabry, K. Shimizu and F. M. Abdel Bar, Med. Chem. Res., 2020, 29, 759–766 CrossRef CAS.
- A. Geraldi, BIO Integr., 2020, 1, 15–24 CrossRef.
- L. Zhu and M. Lin, Anti-Cancer Agents Med. Chem., 2021, 21, 2466–2477 CrossRef PubMed.
- S. Sritharan and N. Sivalingam, Life sciences, 2021, 278, 119527 CrossRef CAS PubMed.
- E. Christidi and L. R. Brunham, Cell Death Dis., 2021, 12, 1–15 CrossRef PubMed.
- P. S. Rawat, A. Jaiswal, A. Khurana, J. S. Bhatti and U. Navik, Biomed. Pharmacother., 2021, 139, 111708 CrossRef CAS PubMed.
- L. Li, J. Ni, M. Li, J. Chen, L. Han, Y. Zhu, D. Kong, J. Mao, Y. Wang and B. Zhang, Drug delivery, 2017, 24, 1617–1630 CrossRef CAS PubMed.
- Z. M. Xu, C. B. Li, Q. L. Liu, P. Li and H. Yang, Int. J. Mol. Sci., 2018, 19, 3658 CrossRef PubMed.
- L. F. Li, Z. C. Ma, Y. G. Wang, X. L. Tang, H. L. Tan, C. R. Xiao and Y. Gao, China Journal of Chinese Materia Medica, 2017, 42, 1365–1369 Search PubMed.
- Z. Wang, G. Su, Z. Zhang, H. Dong, Y. Wang, H. Zhao, Y. Zhao and Q. Sun, Biomed. Pharmacother., 2018, 99, 33–42 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01470j |
| This journal is © The Royal Society of Chemistry 2022 |