
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled synthesis of gold nanorod dimers with end-to-end configurations†
Xuxing Lu ,
Deep Punj and
Michel Orrit*
,
Deep Punj and
Michel Orrit*
Huygens-Kamerlingh Onnes Laboratory, 2300 RA Leiden, Netherlands. E-mail: Orrit@physics.leidenuniv.nl
First published on 5th May 2022
Abstract
End-to-end gold nanorod dimers provide unique plasmonic hotspots with extremely large near-field enhancements in the gaps. Thereby they are beneficial in a wide range of applications, such as enhancing the emissions from ultra-weak emitters. For practical purposes, synthesis of gold nanorod dimers with high yield, especially on the substrates, is essential. Here, we demonstrate two controllable strategies to synthesize gold nanorod dimers based on the self-assembly of gold nanorods, either in bulk solution or on the surface of a glass substrate directly. Both methods can give a high yield of gold nanorod dimers, yet, assembling them directly on the substrate provides more flexibility in controlling the shape and size of each nanorod within the dimer. We also show that these gold nanorod dimers can be used to enhance two-photon-excited fluorescence signals at the single-molecule level.
1 Introduction
Plasmonic coupling between metallic nanoparticles can strengthen the near-field enhancement inside the interparticle gaps,1–10 as well as the local density of photon states,6,11–15 compared to individual nanoparticles. As a consequence, metal nanoparticle aggregates can be applied to enhance ultra-weak signals, such as two-photon-excited (TPE) fluorescence.16 These TPE fluorescence signals are normally too weak to be detected at the single-molecule level, particularly when single nanoparticles are used. Nanoparticle dimers provide single plasmonic hotspots without reducing the overall fluorescence enhancements. Therefore nanoparticle dimer structures can be considered as the best plasmon-coupling systems for single-molecule fluorescence enhancement in the sense that, (i) they provide single sites for fluorescence enhancements, and (ii) the photoluminescence background from the nanoparticles can be reduced compared to multimer aggregates.Among all the metal nanoparticles, chemically synthesised gold nanorods (GNRs) with elongated shape have proven to be excellent building blocks for constructing plasmonic dimers. Compared to spherical nanoparticles, the anisotropy of the GNRs gives rise to much richer flexibility in tailoring the optical responses of the dimer structures. GNRs support two surface plasmon modes along their transversal and longitudinal axes.17,18 The longitudinal plasmon resonance can be tuned from visible to the near-infrared region by varying the aspect ratio.17,19,20 When two GNRs are placed close to each other, the plasmon resonance can be further adjusted by the interparticle gap and by their relative orientation.21–24 End-to-end GNR dimers are of particular interest as they produce higher near-field enhancement than other configurations as a result of the stronger coupling between the longitudinal modes. Preparation of end-to-end GNR dimers with high yield, especially on the substrate, is therefore beneficial for various plasmon-mediated applications.24,25 Towards this aim, various approaches have been developed to link the GNRs in an end-to-end fashion.
DNA origami templates can be used to assemble individual GNRs into well-defined nanoarchitectures,22,26–28 including the end-to-end dimer,29–31 yet this approach requires harsh treatments to both the GNR suspensions and the DNA templates. A more straightforward approach is to link GNRs directly in an end-to-end fashion through electrostatic attraction32,33 or through covalent binding via molecular linkers.34–36 Chemically synthesised GNRs are highly anisotropic in surface reactivity due to their anisotropic structures, hence allow site-specific functionalization on the surfaces. For cetyltrimethylammonium bromide (CTAB)-stabilized GNRs, for instance, the tips are more accessible to dithiol groups as a result of a smaller density of CTAB,37,38 which thus can trigger the end-to-end self-assembly of GNRs in the suspension.17 For most cases, the dithiol ligands can serve not only as linkers, but also as spacers that separate the GNRs. Hence, dithiol ligands with different molecular sizes can be used to control the interparticle gaps of GNR assemblies.34,39–41 Despite the facilitation of end-to-end connections, controlled synthesis of GNR dimers with high yield in bulk suspension remains challenging because it lacks control to stop the dimers from randomly growing into chain-like morphologies in the suspension.34–36,39–43
Here, we describe two new, simple, but efficient strategies to synthesize end-to-end GNR dimers on glass cover slides with the aid of biotinylated streptavidin as molecular linkers. Firstly, we illustrate the assembly in a bulk suspension of GNRs in the presence of the biotinylated streptavidin. The biotinylated streptavidin can bind specifically to the tips of the GNRs in the presence of the surfactant CTAB, hence ensuring the end-to-end assembly of the GNRs. The assembly was monitored by recording the extinction spectra of the assembly suspension over time. A drop of this suspension was later deposited and dried between two glass slides. Secondly, we performed the GNR dimer assembly directly on glass slides. We first immobilized single nanorods on a clean glass slide, and functionalized them with the biotinylated streptavidin specifically on the tips of the each GNR. The molecular linkers functionalized on the immobilized gold nanorods have the chance of adsorbing a second GNR diffusing in the solution, hence forming dimer structures. During the assembly, based on the bottom-up strategy described hereafter, we can separate the inevitable multimer aggregates in the suspensions. Thereby we can get purified end-to-end GNR dimer structures on glass.
2 Materials and method
2.1 Materials
The following chemicals and materials were used for the synthesis of the GNR dimers: unconjugated streptavidin (SNN1001, Invitrogen), N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio) propionamide (EZ-link HPDP-biotin; Pierce), tris((2-carboxyethyl)phosphine) (TCEP, Pierce), cetyltrimethylammonium bromide (CTAB), CTAB-stabilized gold nanorods with plasmon resonance of 700 nm (NA-40-700, OD-50, Nanoseedz), glass slides (Menzel-Gläser, ϕ = 25 mm).2.2 Methods
Fig. 1 schematically illustrates the two assembly strategies we applied for the synthesis of gold nanorod dimers: (a) assembly in solution, which we can call bulk-assembly process; and (b) step-wise assembly on the surface of the substrate. The processes can be briefly described as follows.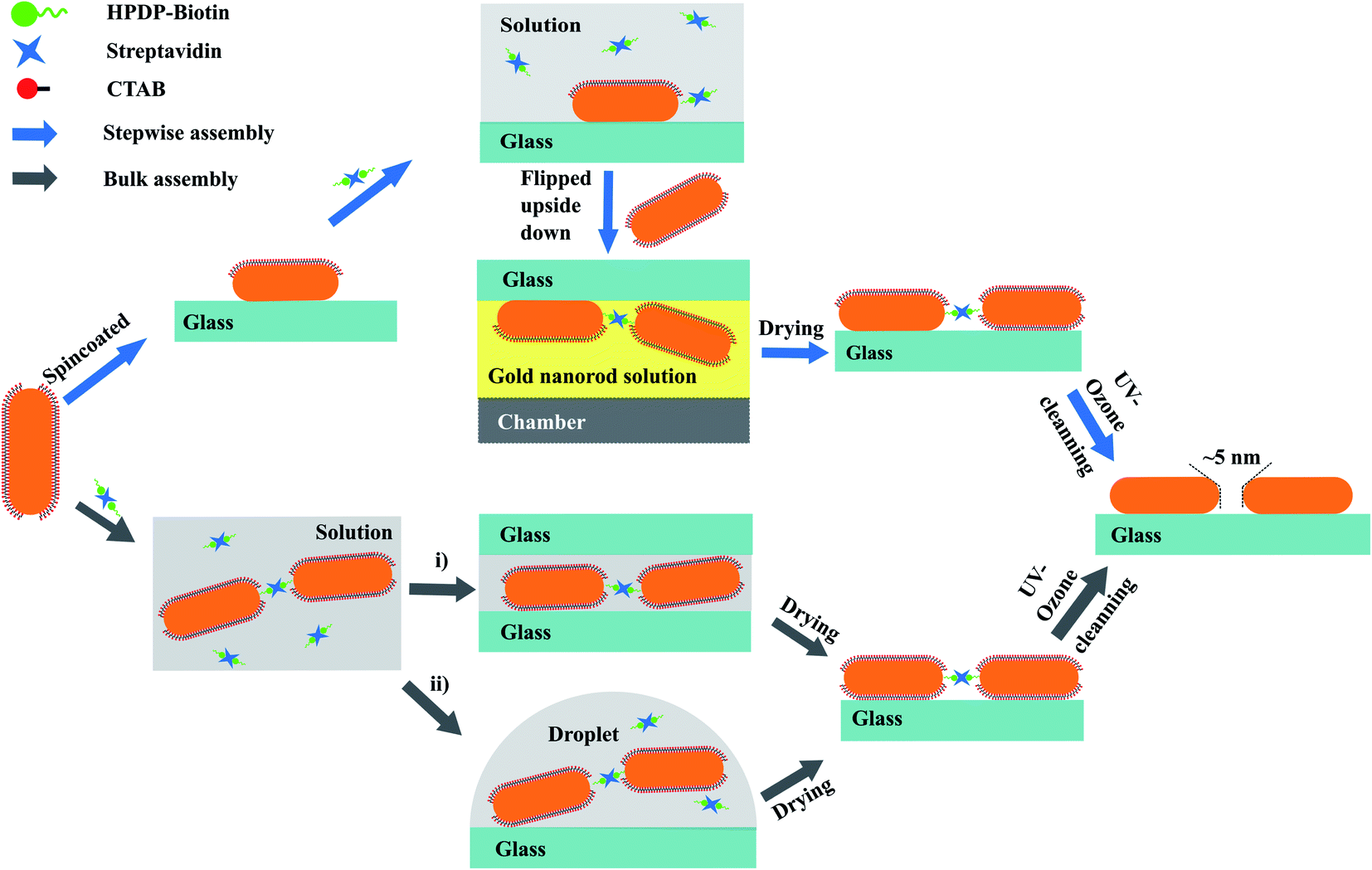 | ||
| Fig. 1 Assembly schemes for two synthesis approaches of gold nanorod dimers. Blue arrows illustrate the step-wise assembly approach where two GNRs of the dimer are immobilized sequentially on the glass surface, by spin-coating and by molecular linking, respectively. Grey arrows show the bulk-assembly approach. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 biotin/TCEP ratio. This reaction was allowed at room temperature for 15 min to break the disulfide bonds in the HPDP-biotin molecules. This process can be verified by monitoring the extinction peak at 343 nm (ref. 44) (see Fig. S1b in ESI†). The mixture solution was added to the streptavidin solution (1 μM) in phosphate-buffered saline solution (PBS, pH = 7.4), with a ratio of 4:1 for biotin/streptavidin. The incubation lasted for 45 min to allow the binding of streptavidin with at least two biotins. The excess of unbound biotin disulfide and TCEP was removed by centrifugation in centrifugal filter devices (Ultra-0.5 10k, Amicon). The residue was dispersed in 100 μL deionized (DI) water.
:10 biotin/TCEP ratio. This reaction was allowed at room temperature for 15 min to break the disulfide bonds in the HPDP-biotin molecules. This process can be verified by monitoring the extinction peak at 343 nm (ref. 44) (see Fig. S1b in ESI†). The mixture solution was added to the streptavidin solution (1 μM) in phosphate-buffered saline solution (PBS, pH = 7.4), with a ratio of 4:1 for biotin/streptavidin. The incubation lasted for 45 min to allow the binding of streptavidin with at least two biotins. The excess of unbound biotin disulfide and TCEP was removed by centrifugation in centrifugal filter devices (Ultra-0.5 10k, Amicon). The residue was dispersed in 100 μL deionized (DI) water.Step 2: tip-specific functionalization of the GNRs: the CTAB solution was replaced with the pretreated biotinylated streptavidin solution (∼200 nM). In the presence of ∼1 mM CTAB, the thiolated linkers can bind specifically at the tip of the GNRs. The tip-specific binding reaction was allowed to proceed for 90 min, and afterward the slide was cleaned gently with de-ionized water to remove any reactant residues on the surface.
Step 3: synthesis of GNR dimers on the glass surface: the glass slide was then flipped and immersed in GNR colloid in an upside-down manner for 12 hours, with the surface containing tip-functionalized GNRs facing the bottom of the sample chamber (see the scheme in Fig. 1). This upside-down strategy can significantly reduce the deposition of GNR monomers or aggregated multimers onto the substrate under the influence of gravity. The thiol-functionalized GNRs on the glass slide provided binding sites for the GNRs, and had the chance to “adsorb” the second GNR to form a GNR dimer through gold–thiol bonds. This step-wise assembly approach allows us to construct high-purity gold nanorod dimers on the substrate, and provides the flexibility of adjusting the shapes or sizes of each particle in the dimers.
(i) Deposition of the GNR assemblies between two glass slides. A small amount of the GNR assembly solution (∼10 μL) was deposited onto a clean cover glass slide, and was immediately covered with a second glass slide. The capillary action between the two slides squeezed the assembled solution and formed a thin layer. Compared to a normal drop-cast method where the solution is free to dry in a 3-dimensional area, we found this sandwich strategy helps in stopping further assembly of GNR due to the drainage of excess gold nanorods available to react at any specific location confined in the 2-dimensional area. This strategy provides a more uniform distribution of GNR assemblies on the surfaces of the glass slides. After deposition, the two slides were left to react and dry for around 60 min and then were separated by immersing them in clean water.
(ii) Drop-coating deposition of the GNR assemblies. As a comparison, a traditional drop-coating approach of depositing the GNR assemblies was also examined in this study. Briefly, a small drop of GNR assembly solution was deposited on a clean glass slide, and blown dry gently with nitrogen gas.
3 Results and discussion
We begin our discussion with the assembly process of GNRs in the bulk suspension. As is well known, the formation of nanoparticle aggregates, or nanoparticle assemblies, can be characterized by the change of their plasmon resonances compared to single particles. The assembly process in bulk solution can be visualized by the evolution of the visible-near-infrared (vis-NIR) extinction spectra of the GNR suspension mixed with the linkers. Specifically, the elongated GNRs can be assembled side-by-side, end-to-side, or end-to-end, depending on the binding sites of the molecular linkers on the surface of each GNR. In the bulk-assembly experiments, the formation of these assemblies can be probed by monitoring the absorption bands corresponding to the transverse or longitudinal SPR of the GNRs: the side-by-side and the end-to-side assemblies will induce a red-shift to the initial transverse SPR band of single GNRs, while the end-to-end assemblies will give rise to a red-shift of the longitudinal SPR band. The red-shifts of the plasmon modes depend on the sizes of the individual GNRs and on the gaps between them. Considering the interparticle gap of about 5 nm, determined by the size of streptavidin,39 and the average sizes 40 nm × 97 nm of the GNRs, we expect a red-shift of about 100 nm for the longitudinal SPR of the end-to-end and a red-shift of about 30 nm for the transverse SPR of the side-by-side dimer, compared to the modes of single GNRs (see Fig. S2 in ESI†). Fig. 2a shows the evolution of the vis-NIR extinction spectra of the GNR suspension after the addition of the pretreated dithiolated streptavidin-biotin compounds. The spectra were acquired every 2 min at room temperature. After adding the molecular linkers, the extinction peak, corresponding to longitudinal SPR band of isolated GNRs, decreased gradually in intensity without shift of its spectral position, while the red-side shoulder became broader, indicating the formation of the end-to-end assemblies of GNRs in the solution. After a few minutes, we observed a small tail at the longer-wavelength side of the longitudinal plasmon band, which could be assigned to the increased content of the end-to-end GNR chains over time. From the spectra, we conclude that the fraction of the longer chain assemblies (e.g. the assemblies with more than three GNRs) in the solution is low, as the extinction at the end of the longer-wavelength tail is relatively small compared to the extinction at the red-side shoulder at the wavelength of around 800 nm (which corresponds to the SPR of end-to-end dimer from simulations, see Fig. S2†). The ratio of short-chain assemblies (e.g. GNR dimers) can be further controlled by adjusting the assembly time and the stoichiometric ratio between GNR and the molecular linker.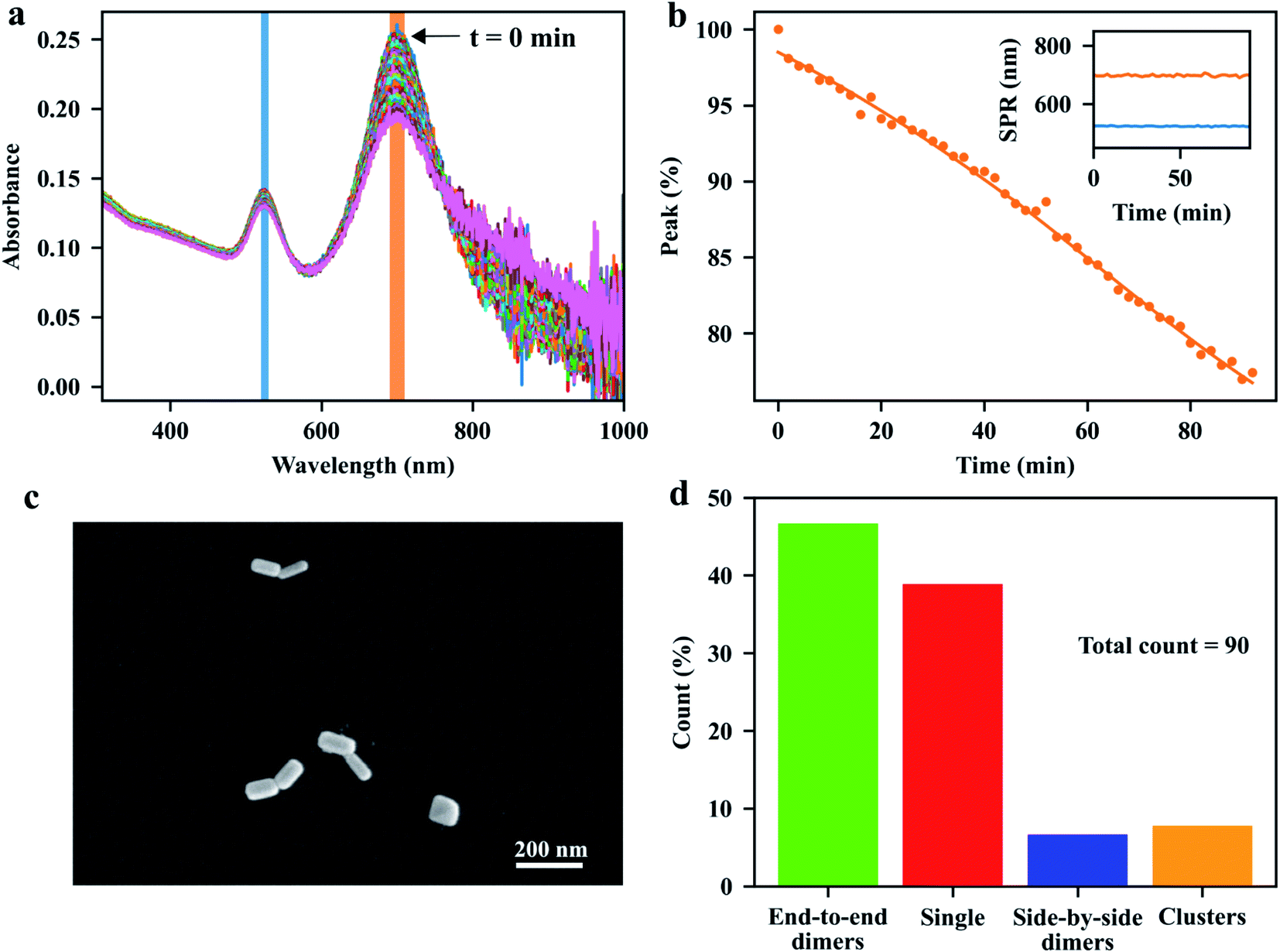 | ||
| Fig. 2 Bulk assembly experiment. (a) Spectral change during the assembly process. (b) Percentage decrease in the peak value of spectra as time progresses. The orange shaded area shown in (a) is used to calculate the normalized values. Inset: the resonance wavelengths of the transverse (blue) and longitudinal (orange) plasmon modes. (c) SEM image of typical gold nanorod dimers on glass slide deposited by using “two-slide” strategy. (d) The statistics percentages of end-to-end dimers (green), single GNRs (red), side-by-side dimers (blue), and clusters (orange). The statistics was done by counting 90 nanoparticles. | ||
From Fig. 2a, we do not observe any noticeable change in the shape of the transverse SPR band except for a reduction of the intensity, which indicates that the GNRs assembled in the side-by-side configuration were rare in the solution. Here, the facilitation of end-to-end GNR assembly can be attributed to the rigid structure of the streptavidin and of the streptavidin-biotin complexes. The relative large size of streptavidin makes it more difficult for the short thiol-end groups of the compounds to bind with the gold surfaces on the sides of the GNRs, which are coated with a CTAB layer, favoring the binding of the linkers to the GNR's tips, where the CTAB density is lower. We therefore propose that the pre-treatments of the molecular linkers mentioned earlier make them more robust to assemble the GNRs into end-to-end configurations. This statement can be confirmed by a control experiment, where we added the solutions of streptavidin, biotin and TCEP into the GNR suspension simultaneously to trigger the GNR assembly, instead of using the pre-treated solution that contained only the streptavidin-biotin compounds. Fig. S3† shows the time-dependent extinction spectra of GNRs after the addition of the mixture of streptavidin, HPDP-biotin and TCEP, with the stoichiometric ratio of 1:4:8. As shown in the plot, the transverse SPR band was red-shifted slightly as time evolved, accompanied by an increase of the extinction at the dip of the spectra between the two SPR peaks. Hence it was evident that the GNRs could also be assembled side-by-side in the presence of free thiolated biotin and unconjugated streptavidin. Additionally, the assembly rate was faster than the assembly process with only streptavidin-biotin compounds. More control experiments were performed using: (i) biotin and TCEP, (ii) streptavidin and TCEP and (iii) streptavidin-biotin linker molecules. As is shown in Fig. S4,† we see that biotin-only or streptavidin-only yields negligible spectral change as time goes, compared to the case with streptavidin-biotin linker molecules.
The GNR assemblies were examined via SEM measurements after deposition onto a clean glass slide. We applied the two-slide-sandwich strategy mentioned in the method section to deposit the GNR assemblies onto the glass slides. Compared to other methods, such as the drop-coating or spin-coating deposition approaches, our strategy is very simple, yet, very effective in the distribution of the assemblies on the glass surface, and can maintain the configurations of the assemblies during the deposition process. A typical SEM image of GNR dimers with well-defined end-to-end configuration is shown in Fig. 2c. More images taken from the same sample (see Fig. S6–S8 in ESI†) indicate that our approaches are very efficient in preparing end-to-end GNR dimers in the substrate. As shown in Fig. 2d, we obtained a ratio of 47% for the end-to-end dimers from the total count of 90 particles. More comparisons of different deposition approaches can be found in the ESI.†
So far we have discussed the bulk assembly approach to synthesize the end-to-end GNR dimers in GNR suspension. Our result shows that suitable treatments of the thiolated streptavidin-biotin complex facilitate the assembly of the GNRs into end-to-end configurations, which had earlier been proved by K. K. Caswell et al.39 Yet assembly of GNRs in bulk suspension normally ends up with long chains of GNRs, and the separation of the GNR dimers from the monomers and multimers could be an issue to prepare the sample with GNR dimers on the substrate with high yields.
Inspired by the extraordinarily high affinity of streptavidin for biotin and by the chemical stability of the complex, we developed a step-wise assembly approach to synthesize GNR dimers directly on a glass surface. Briefly, the first GNR of the dimer was first immobilized on the glass surface, and later combined with the second GNR through the thiol–gold bond with the aid of the thiolated streptavidin-biotin compound functionalized at the tip of the first GNR. Similar step-wise assembly approach had been reported to create gold nanoparticle dimers with high yield by using the alkanedithiol (SH(CH2)nSH) or double-stranded DNA as the molecular linkers.48–51 So far, however, this strategy has only be exploited to the assembly of spherical gold nanoparticles with small sizes (∼20 nm), and special functionalizations of the glass surface were needed. Here, we show that the step-wise assembly method can also be applied to fabricate GNR dimers on the substrate. By using the flipped upside-down strategy shown in Fig. 1, where the targeted GNRs were in the solution below the glass surface with tip-functionalized GNRs, we can prevent precipitation of the monomers or sedimentation of the inevitable aggregated multimers on the substrate, hence selectively bind dimer structures onto the glass surface.
Fig. 3 shows a representative SEM image of the GNR dimers assembled using the step-wise upside-down approach. As shown in the SEM image, we got 8 GNR dimers with end-to-end configurations (noted with the green dashed circles) out of the 15 nanoparticles in the area. We define an end-to-end assembled dimer as a dimer in which the contact does not involve any cylindrical side of the component nanorods. Meanwhile, we also see 3 particles with GNRs aggregated in side-by-side configurations. Interestingly, we don't see any end-to-end assemblies in this area with more than three GNRs in the arrangement, which confirms that our method is effective in the fabrication of end-to-end GNR assemblies of dimers and very short chains (with very low probability of clusters of 3 GNRs or more).
 | ||
| Fig. 3 SEM image of the assembly sample prepared by using the step-wise assembly method. We see a total of 8 end-to-end GNR dimers out of 15 nanoparticles (green dashed circles) in this area. The blue and red circles represent the side-by-side assemblies and single GNRs, respectively. Below shows the zoomed in configurations of the assemblies. | ||
Several factors could affect the efficiency of the fabrication of end-to-end dimers on the substrate by using our step-wise upside-down strategy: (i) the number of the active binding sites for the targeted GNRs at the tips of the individual GNRs on the glass; (ii) the denaturation of the dithiolated streptavidin-biotin complex during the assembly process; (iii) the surface conditions of the glass, such as the surface roughness or the surface charge of the glass, which may influence the sticking of the second GNRs on the glass surface; (iv) after taking away the glass from the GNR suspension after the assembly, there might also be some GNR suspension left on the glass surface, which might stick on the glass or even aggregate into clusters during the drying process.
We examined the SEM images in 12 different areas recorded on three different samples (including the area shown in Fig. 3) using the same method of step-wise assembly. Shown in Fig. S9 and S10,† we see that the ratios of end-to-end GNR dimers can be very different in different areas. The non-uniform distribution of GNR dimers on the glass surface might be due to the non-uniform surface conditions of the glasses, which might affect the tip functionalization of each GNR, or might affect the binding efficiency of the linker with the second GNR. The random distribution of the GNR residues on the glass surface after the assembly might also affect the proportion of GNR dimers in different areas, as they will add more single GNRs on the surface. Nonetheless, we got a total of 50 end-to-end GNR dimers out of the 140 particles in all areas, which yields a ratio of 36% for end-to-end GNR dimers, as can be seen in Fig. 4 (green bar).
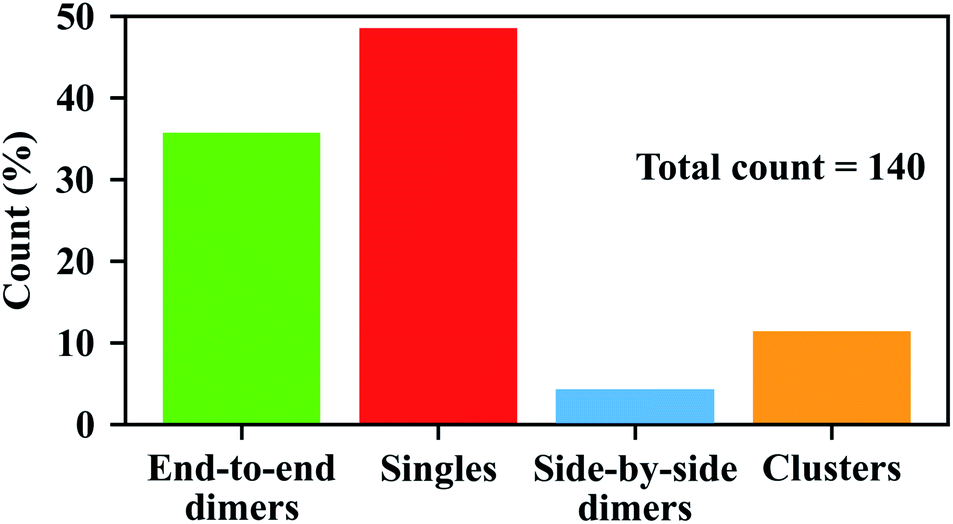 | ||
| Fig. 4 Yields of the nanoparticles with different configurations. The statistics was done by counting 140 nanoparticles from different areas of three samples, all of which were prepared by using the step-wise assembly approach. | ||
The most attractive property of the end-to-end GNR dimers stems from the strong plasmon coupling between the GNRs, which may not only strengthen near-field enhancement but also dramatically increase the local density of photon states (LDOS) in the plasmonic hotspot, compared to a single GNR. Therefore, end-to-end GNR dimers can be used to enhance ultra-weak signals. As an illustration, we applied our GNR dimers to enhance the single-molecule fluorescence from an organic dye, ATTO 610, under two-photon excitation. The experimental setup is shown in the Fig. S11.† Spectral and power dependence measurements of ATTO 610 dye are shown in Fig. S12.† Two-photon-excited fluorescence is a nonlinear optical process where the excitation of the molecule is due to the absorption of two identical photons. As a consequence of the extremely low two-photon absorption cross sections of most organic molecules, detection of two-photon-excited fluorescence from the molecules is very difficult, especially at the single-molecule level. By using a single GNR, our group have earlier reported very strong two-photon absorption enhancement with the enhancement factor up to 105, which allowed us to detect the two-photon-excited photoluminescence from single colloidal quantum dots (Qdots).52 Even such a high enhancement, however, is not large enough to distinguish the enhanced two-photon-excited fluorescence of single organic molecules from the background (here ATTO 610, see orange line in Fig. 5b), as this dye has a much lower two-photon absorption cross-section than the Qdots. Compared to a single GNR, end-to-end GNR dimers can further strengthen the enhancement factor of two-photon excitation by a factor of 103 (see Fig. S13†), for an interparticle gap of 5 nm. With such high enhancements, we successfully detected the enhanced two-photon-excited fluorescence from single ATTO 610 molecules, as demonstrated by the bursts in the luminescence time trace (blue line) in Fig. 5b. Fig. 5a shows the typical photoluminescence spectra of an end-to-end GNR dimer (blue) and of a single GNR (orange). Both structures have their plasmon resonance wavelength at around 760 nm. The insets show their SEM images, respectively. Fig. 5b shows the photoluminescence time traces from the end-to-end GNR dimer (blue) and from the single GNR (orange) immersed in a solution of ATTO 610 with the molecular concentration of 30 nm, respectively. We believe these enhanced fluorescent bursts are coming from the two-photon-excitation of ATTO 610. Further study on this topic is being reported elsewhere since it will be beyond the scope of the work reported here.
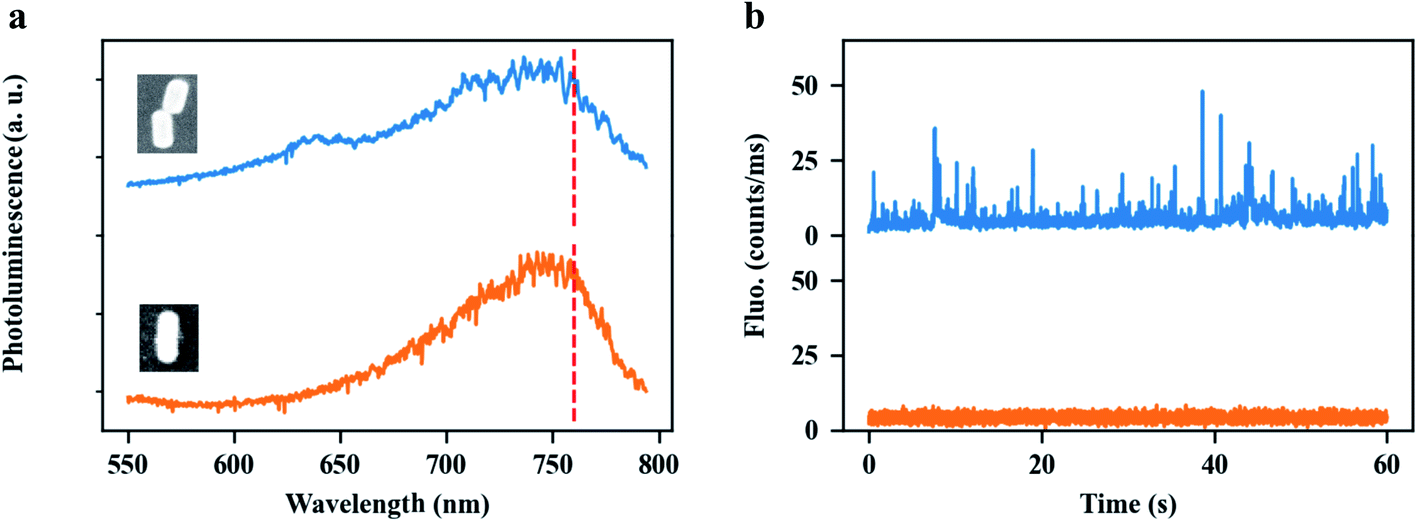 | ||
| Fig. 5 Plasmonically-enhanced single-molecule fluorescence under two-photon-excitation. (a) The typical photoluminescence spectra of an end-to-end GNR dimer (blue) and of a single GNR (orange). Both structures have the plasmon resonance wavelength at around 760 nm. The insets show their SEM images, respectively. (b) Photoluminescence time traces from the end-to-end GNR dimer (blue) and from the single GNR (orange) immersed in a solution of ATTO 610 with the molecular concentration of 30 nM, respectively. | ||
4 Conclusions
In conclusion, we have demonstrated two different approaches for the fabrication of end-to-end GNR dimers based on the self-assembly of GNRs. Our results showed that pre-conjugation of streptavidin with thiolated-biotin as molecular linkers facilitates GNR assembly into end-to-end dimers in the suspension. In this work, we illustrated a simple but very efficient way of depositing the GNR assemblies onto glass slide by sandwiching a small amount of GNR assembly between two glass slides. This approach helps us to deposit the GNR assemblies uniformly onto the glass slides, and maintain the configurations of the GNR assemblies during the deposition process.We further developed a step-wise assembly method to synthesize GNR dimers directly on the glass. In this method, the two GNRs of the dimer were immobilized sequentially on the glass surface. Briefly, the first GNR of the dimer was immobilized by spin-coating. The second GNR was captured from the suspension with the aid of the molecular linker. By applying a simple upside-down flipping strategy, where the targeted GNRs were in the solution below the glass surface with tip-functionalized GNRs, we ended up with a high yield of end-to-end GNR dimers. Our step-wise assembly method can be readily applied to different nanoparticles, providing the flexibility of controlling the shapes, sizes or materials of each nanoparticle in the assembly.
Finally we demonstrated enhancement by our end-to-end GNR dimer by detecting bursts of two-photon-excited fluorescence emitted by single ATTO 610 molecules in the dimer gap. The simulations show that the enhancement can be 104× higher than the enhancement by a single GNR.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from NWO, The Netherlands Organization for Scientific Research (Grant ECHO). X. L. acknowledges a PhD Grant from the China Scholarship Council.Notes and references
- H. Xu, J. Aizpurua, M. Käll and P. Apell, Phys. Rev. E, 2000, 62, 4318–4324 CrossRef CAS PubMed.
- A. Kinkhabwala, Z. Yu, S. Fan, Y. Avlasevich, K. Müllen and W. E. Moerner, Nat. Photonics, 2009, 3, 654–657 CrossRef CAS.
- T. Kang, S. Hong, Y. Choi and L. P. Lee, Small, 2010, 6, 2649–2652 CrossRef CAS PubMed.
- J. C. Fraire, L. A. Pérez and E. A. Coronado, ACS Nano, 2012, 6, 3441–3452 CrossRef CAS PubMed.
- X. Lan, Z. Chen, X. Lu, G. Dai, W. Ni and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 10423–10427 CrossRef CAS PubMed.
- P. M. R. Paulo, D. Botequim, A. Jóskowiak, S. Martins, D. M. F. Prazeres, P. Zijlstra and S. M. B. Costa, J. Phys. Chem. C, 2018, 122, 10971–10980 CrossRef CAS.
- L. Tie, M. Focsan, J. Bosson, C. Tira, A. Campu, A. Vulpoi and S. Astilean, Mater. Res. Express, 2019, 6, 095038 CrossRef CAS.
- J. Langer and L. M. Liz-Marzán, et al., ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- M. Blanco-Formoso, N. Pazos-Perez and R. A. Alvarez-Puebla, Nanoscale, 2020, 12, 14948–14956 RSC.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121(21), 13342–13453 CrossRef CAS PubMed.
- M. Ringler, A. Schwemer, M. Wunderlich, A. Nichtl, K. Kürzinger, T. A. Klar and J. Feldmann, Phys. Rev. Lett., 2008, 100, 203002 CrossRef CAS PubMed.
- M. P. Busson, B. Rolly, B. Stout, N. Bonod and S. Bidault, Nat. Commun., 2012, 3, 962 CrossRef PubMed.
- Z. Zhang, P. Yang, H. Xu and H. Zheng, J. Appl. Phys., 2013, 113, 033102 CrossRef.
- A. Szenes, B. Bánhelyi, T. Csendes, G. Szabó and M. Csete, Plasmonics, 2018, 13, 1977–1985 CrossRef CAS.
- I. Kaminska, J. Bohlen, S. Mackowski, P. Tinnefeld and G. P. Acuna, ACS Nano, 2018, 12, 1650–1655 CrossRef CAS PubMed.
- O. S. Ojambati, R. Chikkaraddy, W. M. Deacon, J. Huang, D. Wright and J. J. Baumberg, Nano Lett., 2020, 20, 4653–4658 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- P. Zijlstra, P. M. R. Paulo and M. Orrit, Nat. Nanotechnol., 2012, 7, 379–382 CrossRef CAS PubMed.
- W. Ni, X. Kou, Z. Yang and J. Wang, ACS Nano, 2008, 2, 677–686 CrossRef CAS PubMed.
- S. E. Lohse and C. J. Murphy, Chem. Mater., 2013, 25, 1250–1261 CrossRef CAS.
- A. M. Funston, C. Novo, T. J. Davis and P. Mulvaney, Nano Lett., 2009, 9, 1651–1658 CrossRef CAS PubMed.
- X. Lan, Z. Chen, G. Dai, X. Lu, W. Ni and Q. Wang, J. Am. Chem. Soc., 2013, 135, 11441–11444 CrossRef CAS PubMed.
- X. Lu, J. Wu, Q. Zhu, J. Zhao, Q. Wang, L. Zhan and W. Ni, Nanoscale, 2014, 6, 14244–14253 RSC.
- M. A. Beuwer and P. Zijlstra, J. Chem. Phys., 2021, 155, 044701 CrossRef CAS PubMed.
- C. Li, S. Li, A. Qu, H. Kuang, L. Xu and C. Xu, Adv. Funct. Mater., 2020, 30, 2001451 CrossRef CAS.
- X. Lan, X. Lu, C. Shen, Y. Ke, W. Ni and Q. Wang, J. Am. Chem. Soc., 2015, 137, 457–462 CrossRef CAS PubMed.
- P. Zhan, P. K. Dutta, P. Wang, G. Song, M. Dai, S.-X. Zhao, Z.-G. Wang, P. Yin, W. Zhang, B. Ding and Y. Ke, ACS Nano, 2017, 11, 1172–1179 CrossRef CAS PubMed.
- X. Lan, T. Liu, Z. Wang, A. O. Govorov, H. Yan and Y. Liu, J. Am. Chem. Soc., 2018, 140, 11763–11770 CrossRef CAS PubMed.
- Y. Liu, Y. Liu and Y. Shen, Int. J. Intell. Robot. Appl., 2018, 2, 445–453 CrossRef.
- L. A. McCarthy, K. W. Smith, X. Lan, S. A. H. Jebeli, L. Bursi, A. Alabastri, W.-S. Chang, P. Nordlander and S. Link, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 16143–16148 CrossRef CAS PubMed.
- Z. Zhao, X. Chen, J. Zuo, A. Basiri, S. Choi, Y. Yao, Y. Liu and C. Wang, Nano Res., 2021, 15(2), 1327–1337 CrossRef.
- S. M. H. Abtahi, N. D. Burrows, F. A. Idesis, C. J. Murphy, N. B. Saleh and P. J. Vikesland, Langmuir, 2017, 33, 1486–1495 CrossRef CAS PubMed.
- A. Kar, V. Thambi, D. Paital, G. Joshi and S. Khatua, Langmuir, 2020, 36, 9894–9899 CrossRef CAS PubMed.
- P. K. Sudeep, S. T. S. Joseph and K. G. Thomas, J. Am. Chem. Soc., 2005, 127, 6516–6517 CrossRef CAS PubMed.
- Z. Sun, W. Ni, Z. Yang, X. Kou, L. Li and J. Wang, Small, 2008, 4, 1287–1292 CrossRef CAS PubMed.
- W. Ni, R. A. Mosquera, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2010, 1, 1181–1185 CrossRef CAS.
- P. Zijlstra, P. M. R. Paulo, K. Yu, Q.-H. Xu and M. Orrit, Angew. Chem., Int. Ed., 2012, 51, 8352–8355 CrossRef CAS PubMed.
- P. M. R. Paulo, P. Zijlstra, M. Orrit, E. Garcia-Fernandez, T. C. S. Pace, A. S. Viana and S. M. B. Costa, Langmuir, 2017, 33, 6503–6510 CrossRef CAS PubMed.
- K. K. Caswell, J. N. Wilson, U. H. F. Bunz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CrossRef CAS PubMed.
- J. Liu, C. Kan, Y. Li, H. Xu, Y. Ni and D. Shi, Appl. Phys. Lett., 2014, 104, 253105 CrossRef.
- Y. Xu, X. Wang and X. Ma, Dyes Pigm., 2017, 144, 168–172 CrossRef CAS.
- Y. Zhu, H. Kuang, L. Xu, W. Ma, C. Peng, Y. Hua, L. Wang and C. Xu, J. Mater. Chem., 2012, 22, 2387–2391 RSC.
- A. F. Stewart, A. Lee, A. Ahmed, S. Ip, E. Kumacheva and G. C. Walker, ACS Nano, 2014, 8, 5462–5467 CrossRef CAS PubMed.
- H. Chen, H. Zou, H. J. Paholak, M. Ito, W. Qian, Y. Che and D. Sun, Polym. Chem., 2014, 5, 2768–2773 RSC.
- A. Movsesyan, S. Marguet, A. Muravitskaya, J. Beal, P. M. Adam and A. L. Baudrion, J. Opt. Soc. Am. Opt Image Sci. Vis., 2019, 36, C78–C84 CrossRef CAS PubMed.
- H. Le-The, R. M. Tiggelaar, E. Berenschot, A. van den Berg, N. Tas and J. C. T. Eijkel, ACS Nano, 2019, 13, 6782–6789 CrossRef CAS PubMed.
- R. Marie, A. B. Dahlin, J. O. Tegenfeldt and F. Hook, Biointerphases, 2007, 2, 49–55 CrossRef CAS PubMed.
- H. Cha, J. H. Yoon and S. Yoon, ACS Nano, 2014, 8, 8554–8563 CrossRef CAS PubMed.
- D. Lee and S. Yoon, J. Phys. Chem. C, 2015, 119, 7873–7882 CrossRef CAS.
- H. Cha, D. Lee, J. H. Yoon and S. Yoon, J. Colloid Interface Sci., 2016, 464, 18–24 CrossRef CAS PubMed.
- J. Li, T.-S. Deng, X. Liu, J. A. Dolan, N. F. Scherer and P. F. Nealey, Nano Lett., 2019, 19, 4314–4320 CrossRef CAS PubMed.
- W. Zhang, M. Caldarola, X. Lu and M. Orrit, ACS Photonics, 2018, 5, 2960–2968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01288j |
| This journal is © The Royal Society of Chemistry 2022 |