
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible ammonia uptake at room temperature in a robust and tunable metal–organic framework†
Jaechul Lee‡
a,
Younggyu Seo‡a,
Dong Won Kang b,
Seungjae Parka,
Hyunyong Kima,
Jaheon Kimc,
Kimoon Kimade,
Chang Seop Hong*b,
Dae-Woon Lim*fg and
Eunsung Lee*ad
b,
Seungjae Parka,
Hyunyong Kima,
Jaheon Kimc,
Kimoon Kimade,
Chang Seop Hong*b,
Dae-Woon Lim*fg and
Eunsung Lee*ad
aDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), Pohang, 37673, Republic of Korea. E-mail: eslee@postech.ac.kr
bDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: cshong@korea.ac.kr
cDepartment of Chemistry, Soongsil University, Seoul 06978, Republic of Korea
dDivision of Advanced Materials Science, Pohang University of Science and Technology, Pohang 37673, Republic of Korea
eCenter for Self-assembly and Complexity, Institute for Basic Science (IBS), Pohang, 37673, Republic of Korea
fDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa Oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan
gDepartment of Chemistry and Medical Chemistry, Yonsei University, Wonju, 26493, Republic of Korea. E-mail: limdaewoon@yonsei.ac.kr
First published on 8th March 2022
Abstract
Ammonia is useful for the production of fertilizers and chemicals for modern technology, but its high toxicity and corrosiveness are harmful to the environment and human health. Here, we report the recyclable and tunable ammonia adsorption using a robust imidazolium-based MOF (JCM-1) that uptakes 5.7 mmol g−1 of NH3 at 298 K reversibly without structural deformation. Furthermore, a simple substitution of NO3− with Cl− in a post-synthetic manner leads to an increase in the NH3 uptake capacity of JCM-1(Cl−) up to 7.2 mmol g−1.
Introduction
Ammonia is one of the essential chemicals not only for fertilizers in agriculture but also for various industrial applications such as an alternative fuel, refrigerants, and a precursor for numerous nitrogenous compounds. As the second most-produced chemical in the world, over 150 million tons of ammonia is annually produced.1 Such a huge production and extensive usage of NH3 raises concerns about the environment and human health issues because of its toxic and corrosive properties. The United States Department of Labor alerts that exposure only to a small amount of ammonia can damage a human body. In the atmosphere, NH3 can combine with sulfate and nitrate to form fine particulate matter (PM2.5), causing severe particulate air pollution.2–4 In this context, practical methods and materials for the removal of ammonia emitted in the air are highly desirable.Metal–organic frameworks (MOFs) have emerged as a promising material for the capture of toxic chemicals such as NH3,5–19 NOx,20–23 SOx,24–26 and volatile organic compounds27,28 because well-ordered porosity with feasible chemical and structural tunability of pore surfaces enables such applications. Synthesis of MOFs with high structural stability toward corrosive species is a critical issue for real-world applications. In this regard, several MOFs that can take up NH3 under atmospheric condition has been developed recently.5–19,29 Nevertheless, structural instability of adsorbent and high regeneration temperature are still remaining challenges for practical applications.5,7–19 MOFs showing recyclable ammonia adsorption while retaining its porosity are still very rare. The limited stability of MOFs under ammonia conditions is ascribed to the coordinating nature of NH3. That is, NH3 as a good sigma donating ligand can replace the existing ligand in MOFs, leading to framework decomposition.30 Next, strong NH3⋯MOF interactions through open metal sites or acid–base interactions necessitate high regeneration temperature, which in turn needs high thermal stability of MOFs and energy-intensive regeneration processes as well. Thus far, MFM-300(Al) reported by M. Schröder and co-workers in 2018 is a representative MOF that can repeatedly adsorb ammonia with regeneration at room temperature.29 In addition, there have been reported numerous examples for uptaking ammonia using MOFs for the past five years (Table S3†). Nevertheless, it is still challenging to devise robust MOFs that enables reversible uptake of ammonia under mild regeneration conditions.
Recently, we reported a microporous MOF, JCM-1 ([Co(PzIm)(NO3)], H2PzIm = 1,3-bis(3,5-dimethyl-1H-pyrazol-4-yl)-imidazolium).31 JCM-1 has a cationic three-dimensional framework formed by Co(II)–pyrazolate coordination bonds, where imidazolium groups are aligned along the channels containing NO3− ions (Fig. S1†). Since azolates such as pyrazolate, imidazolate, and triazolate have a greater Lewis basicity than common carboxylates, MOFs constructed from azolate linkers are well-known to possess superior durability against polar solvent.32–34 Indeed, JCM-1 has outstanding properties such as high hydrolytic stability, a unique pore structure, and gas separation abilities.31 For its unique gas sorption properties, the imidazolium functionality in JCM-1 plays a crucial role as a mimic of ionic liquid solutions. We also found that some imidazolium-based ionic liquids show high ammonia solubility by weak hydrogen bonds between imidazolium salt and NH3.35–38 Studies on the NH3 adsorption using imidazolium-based MOFs have not been reported so far, which motivated us to investigate the NH3 capture ability of JCM-1 (Fig. 1).
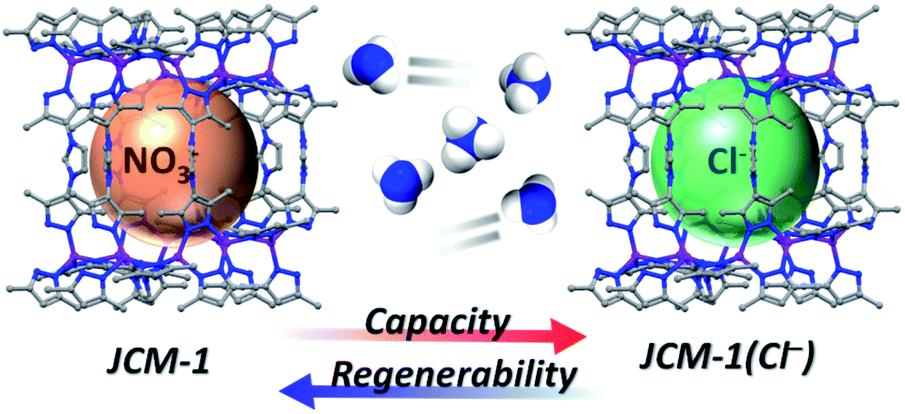 | ||
| Fig. 1 Schematic illustration of ammonia adsorption with JCM-1 and JCM-1(Cl−). | ||
Results and discussion
Synthesis and characterization of JCM-1 and JCM-1(Cl−)
Herein, we report the recyclable and tunable ammonia adsorption using JCM-1, which is the first demonstration of NH3 capture utilizing an imidazolium-based MOF. JCM-1 was synthesized according to the previous procedure by heating a mixture of Co(NO3)2(H2O)6 and the H2PzImCl ligand in N,N-dimethylformamide (details in the ESI†). In the single-crystal X-ray structure of JCM-1, one-dimensional channels are running through the [001] direction with an aperture size of 12.5 × 3.9 Å. The nitrate anion in the channels of JCM-1 can be fully replaced by chloride ion to obtain JCM-1(Cl−) by post-synthetic anion exchange method following the previous procedures. The crystal structure of JCM-1(Cl−) by single-crystal X-ray diffraction analysis showed that the space group of JCM-1(Cl−) was not changed (I41/amd) with almost the same unit cell parameters as JCM-1 (Fig. S2 and Table S1†); the cationic scaffold remained unchanged, and only the anion species in the pore was replaced after ion substitution. When the crystals of JCM-1 were treated with 1 M ammonia solution in THF for 24 hours (Fig. 2a), the crystals remained intact, which secures the chemical stability of JCM-1 toward corrosive ammonia, enabling NH3 gas uptake measurements.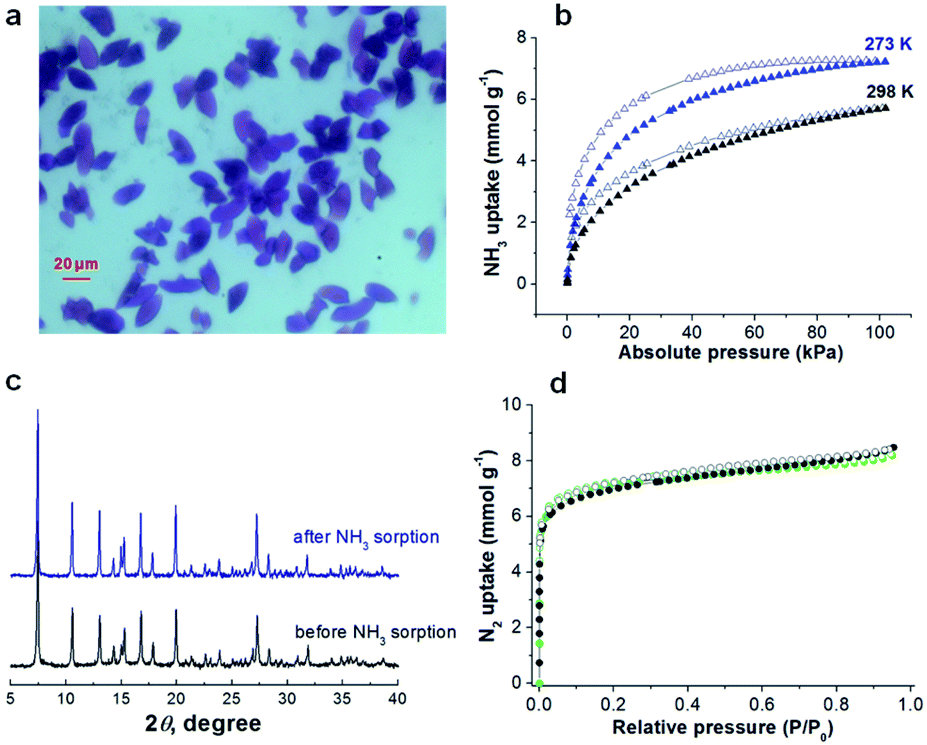 | ||
| Fig. 2 (a) A picture for the crystals of JCM-1 stored in 1 M ammonia THF solution at room temperature for 24 hours. (b) NH3 adsorption isotherms of JCM-1 at 273 K and 298 K. (c) PXRD patterns and (d) N2 adsorption isotherms of JCM-1 before and after ammonia adsorption experiments (black circle, before NH3 sorption; light-green circle, after NH3 sorption). | ||
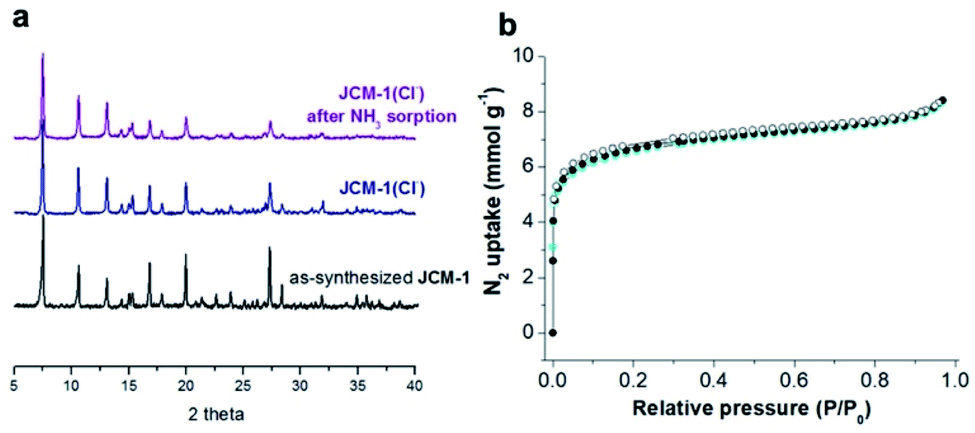 | ||
| Fig. 3 (a) PXRD patterns of as-synthesized JCM-1, JCM-1(Cl−) before NH3 adsorption, and JCM-1(Cl−) after NH3 adsorption experiment. (b) N2 adsorption isotherms of JCM-1(Cl−) before and after ammonia adsorption experiments (black circle: before NH3 sorption, cyan circle: after NH3 sorption). | ||
The total NH3 uptake amounts of JCM-1 at 1 bar were 7.9 and 5.7 mmol g−1 at 273 K and 298 K, respectively (Fig. 2b), which are comparable with other NH3 adsorbing MOFs.5–19,29 Notably, JCM-1 surpasses other materials in terms of framework stability upon regeneration, which are the critical properties of energy-efficient NH3 adsorbent materials. Although a substantial number of MOFs were tested for ammonia adsorption thus far, they suffer from structural deformation after exposure to corrosive NH3 gas.5,7–19,39,41 Many of the MOFs show decreased porosity with the degradation of crystallinity during repeated NH3 adsorption–desorption processes, resulting in peak broadening in powder X-ray diffraction (PXRD) patterns. In contrast, JCM-1 could maintain the initial crystallinity and the porosity after NH3 adsorption experiments, which was confirmed by the almost unchanged PXRD patterns and N2 sorption isotherms (Fig. 2c and d).
Ammonia adsorption of JCM-1 and JCM-1(Cl−)
Unlike other MOFs capturing ammonia, JCM-1 has no open metal sites and no acid functional groups for strong interaction with NH3. Instead, it possesses imidazolium functional groups on the pore surfaces to form weak hydrogen bonds with NH3. The imidazolium protons and counter anions in JCM-1 can act respectively as a proton donor and a proton acceptor, enabling cooperative hydrogen bonding with NH3. This weak but effective intermolecular interaction seems to facilitate the re-activation of JCM-1 after repeated NH3 adsorption measurements. We carried out cycle experiments (Fig. 4a and c), and observed that the sample could be fully evacuated at room temperature under dynamic vacuum. The NH3 uptake capacity was well retained over five cycles, showing the facile regenerability of JCM-1 under mild conditions.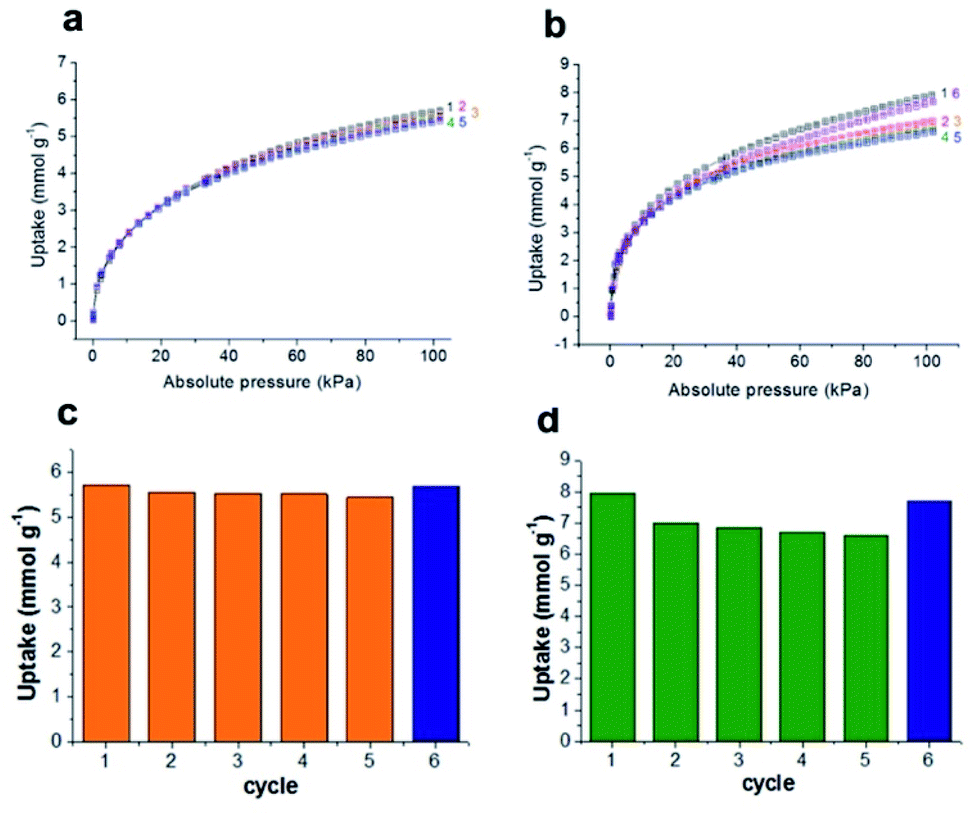 | ||
| Fig. 4 Adsorption isotherms of (a) repeated NH3 adsorption cycles in JCM-1 and (b) JCM-1(Cl−) at ambient conditions (298 K and 1 bar) uptake capacity of (c) repeated NH3 adsorption cycles in JCM-1 and (d) JCM-1(Cl−) at ambient conditions (298 K and 1 bar). | ||
The NH3 uptake capacity and sorption affinity of JCM-1 could be also modulated by replacing NO3− with Cl− in a post-synthetic manner, where JCM-1 was soaked in a methanol solution of LiCl for a week to give anion-exchanged JCM-1(Cl−). Although the effectiveness of anion inserted into a MOF for modulating ammonia uptake was reported by Binaeian,15 the exchange of counter anions in the cationic framework was investigated for ammonia uptake in our case. The NH3 uptake capacity of JCM-1(Cl−) was increased, although its N2 adsorption behavior was almost the same (Fig. 3). Because of high porosity of JCM-1 and the weaker interaction with nitrogen compared to ammonia, the anion exchange effect on the nitrogen adsorption properties of JCM-1 and JCM-1(Cl−) is less pronounced compared to the ammonia adsorption. At 1 bar, JCM-1(Cl−) adsorbed 11.7 and 7.2 mmol g−1 of NH3 at 273 K and 298 K, respectively, which increased by 48% (273 K) and 26% (298 K), compared with JCM-1 (Fig. 5). The crystallinity and porosity of JCM-1(Cl−) were well retained after NH3 adsorption, as confirmed by PXRD and N2 adsorption data (Fig. 3). In the cycling experiment, JCM-1(Cl−) showed gradually decreasing NH3 capacity after the first cycle (Fig. 4b and d), which means that the sample was not fully activated at room temperature. Activation of the sample after the 5th cycle at an elevated temperature of 358 K for 5 h regenerated JCM-1(Cl−) and the initial NH3 uptake capacity of was almost recovered (Fig. 4d).
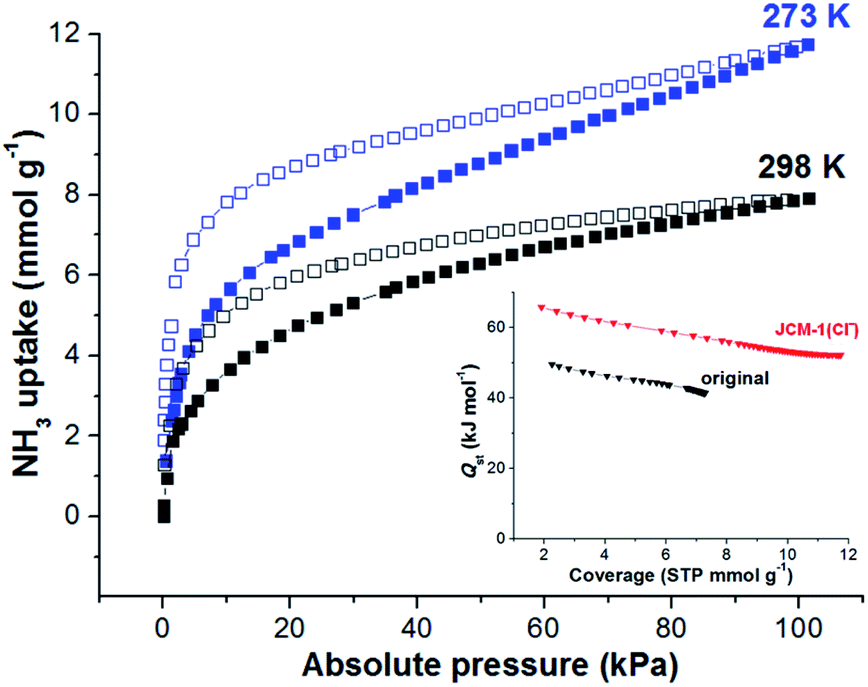 | ||
| Fig. 5 NH3 adsorption isotherms of JCM-1(Cl−) at 273 K and 298 K. Inset: isosteric heat of sorption (Qst) of NH3 in JCM-1 and JCM-1(Cl−). | ||
The isosteric heats (Qst) of adsorption for NH3 within JCM-1 or JCM-1(Cl−) was calculated by employing the Virial method.41 Because of the hysteresis in the NH3 adsorption isotherms, we calculated the Qst values using the desorption curves, which is also valid for explaining the change in the regeneration temperatures.42–44 The Qst values for NH3 desorption in JCM-1(Cl−) were higher by ca. 20 kJ mol−1 than JCM-1 in all coverages (inset in Fig. 5). The higher Qst value of JCM-1(Cl−) supports the reason for the increased regeneration temperature after ion substitution. The enhanced NH3 affinity of JCM-1(Cl−) could be confirmed using density functional theory (DFT) calculations. For DFT calculations, the appropriate structural models of each framework were prepared using the crystal structures of JCM-1 and JCM-1(Cl−). The initial coordinate of NH3 was loaded into each model structure, followed by further geometry optimization (see the ESI†). The NH3 molecule showed stronger interaction with the imidazolium-Cl− pair than the imidazolium-NO3− in the optimized structures, as the interaction enthalpies of JCM-1 and JCM-1(Cl−) with NH3 molecule were calculated to be 20.8 kJ mol−1 and 50.0 kJ mol−1, respectively (Fig. S3 and S4†), which is qualitatively following the experimental observations.
Mechanism of ammonia adsorption on JCM-1 and JCM-1(Cl−)
A simple interpretation of the above observation is that the adsorbed NH3 molecule interacts more strongly with Cl− than NO3− ions. Contrary to this expectation, DFT calculations revealed a more delicate interplay between NH3 and the imidazolium–anion pairs. Before NH3 loading, NO3− or Cl− is engaged in hydrogen bonding with the imidazolium in the framework. The distance between NO3− and imidazolium proton is 1.93 Å, whereas the distance between Cl− and imidazolium proton is 2.21 Å (Fig. S5†). Upon adsorption, NH3 molecules have to compete with the anions to form hydrogen bonding with the imidazolium proton. As expected, in the optimized structures of the NH3-loaded JCM-1 and JCM-1(Cl−), the distances between the anion and imidazolium proton increased (Fig. 5). On the other hand, a weak hydrogen bond between an NH3 molecule and the imidazolium proton was detected. In the NH3@JCM-1, the distance between the nitrogen atom of NH3 and the imidazolium proton was 2.83 Å (Fig. 6a). Interestingly, in the NH3@JCM-1(Cl−), the distance was 2.51 Å, which is significantly shorter than that of JCM-1 and explains why JCM-1(Cl−) exhibits the enhanced NH3 affinity (Fig. 6b). As above mentioned, this interaction is similar to that for imidazolium-based ionic liquids having high ammonia solubility.35–38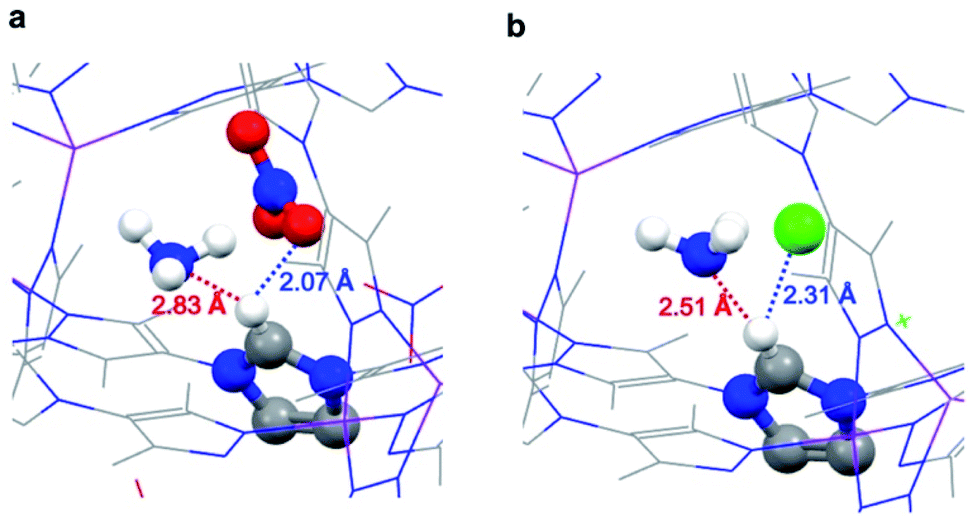 | ||
| Fig. 6 NH3-loaded optimized structures of (a) JCM-1 and (b) JCM-1(Cl−). | ||
The interaction between NH3 and the imidazolium group on the pore surface was experimentally observed by IR spectroscopy (Fig. S6†). We measured the IR spectra of the frameworks before and after exposure to NH3 gas. In both structures, imidazolium C–H stretching bands45 (2900–3100 cm−1) were slightly broadened and blue-shifted after exposure to the NH3 atmosphere.40,46 The band-broadening and band-shift are indicative of the formation of the weak hydrogen bond between imidazolium and the NH3 molecule, which is coincident with the DFT calculation results.
Dynamic breakthrough curves were obtained under dry and humid conditions (0% relative humidity (RH) and 80% RH) at a feed concentration of 1000 ppm NH3 and a temperature of 298 K. We observed the increase in the adsorption amount of ammonia in JCM-1(Cl−) in both humid and wet conditions, which shows the same tendency with the adsorption isotherms, while JCM-1 adsorbs only dry ammonia (Fig. S7–S10†).
Conclusions
In conclusion, we report JCM-1 which can adsorb a high amount of NH3 at an ambient condition without structural deformation. JCM-1 is recyclable even at room temperature through the weak interaction between NH3 and imidazolium functionalities by mimicking ionic liquids. Furthermore, we demonstrated the control of the NH3 sorption property of JCM-1 by simple ion substitution in a post-synthetic manner. After exchanging the anion from nitrate to chloride, the affinity of MOF toward NH3 was enhanced resulting in the increase in the uptake capacity while the regenerability was slightly decreased. This work suggests a new potential application of imidazolium-based MOFs for ammonia capture, as well as the effect of the anion species for ammonia adsorption in ionic porous materials.Experimental
Materials and methods
All the reagents and solvents were commercially available and used as supplied without further purification. Elemental analysis (EA) for C, H, and N were carried out using Flash 2000 (Thermo Fisher Scientific Inc.). FT-IR measurements were processed in Bruker ALPHA FT-IR spectrometer. Single crystal X-ray diffraction (SXRD) analysis was carried out at synchrotron radiation of 2D-SMC and 6D-C&S UNIST-PAL (crystallography) at the Pohang Accelerator Laboratory (PAL, Korea). The powder XRD diffraction patterns were obtained on a Rigaku Smartlab system equipped with Cu sealed tube. Thermogravimetric analysis (TGA) were measured under N2 atmosphere with a PerkinElmer Pyris. All gas sorption isotherms were measured with BELSORP-max volumetric adsorption equipment. Purified compounds were further dried under high vacuum (0.01–0.05 Torr).MOF synthesis
JCM-1 ([Co(PzIm)(NO3)], H2PzIm = 1,3-bis(3,5-dimethyl-1H-pyrazol-4-yl)-1H-imidazol-3-ium) and JCM-1(Cl−) ([Co(PzIm)(Cl)]) was synthesized following our previously reported methods.31X-ray crystallographic analysis
Single crystal X-ray diffraction (SXRD) analysis was carried out at the synchrotron radiation of 2D-SMC at the Pohang Accelerator Laboratory (PAL, Korea). The SXRD patterns of crystals coated with paratone oil were collected at 100 K. Using Olex2,47 the structures were solved by ShelXT48 using Intrinsic Phasing. The crystal structure of JCM-1(Cl−) was refined by ShelXL49 using least squares minimization. CCDC 1912377 contains the structural information for JCM-1(Cl−).Gas sorption experiments
Isosteric heat of sorption
The isosteric heat of sorption Qst was calculated using the virial type expression to fit the isotherms of 273 K and 298 K.50where P is the pressure of the gas phase at equilibrium (bar), N is the adsorbed amount per mass of adsorbent (mmol g−1), T is the absolute temperature, ai and bi are virial coefficients, and m and n are numbers of coefficients used to describe the isotherms.
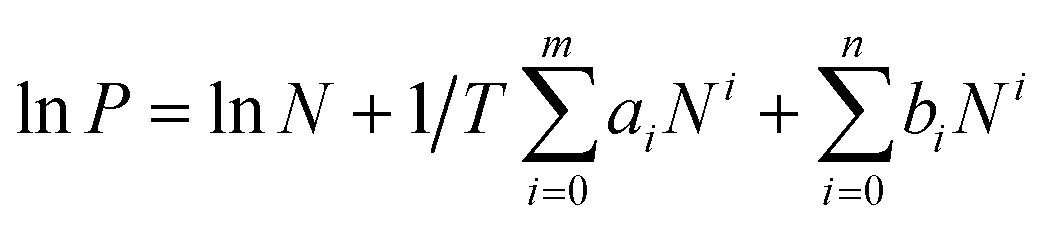
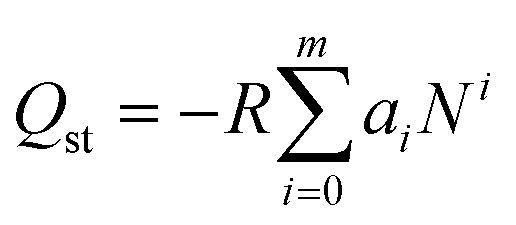
Then, with the virial coefficient (ai) and the following equation, the coverage-dependent isosteric heat of sorption was calculated. R is the universal gas constant.
DFT calculations
Both geometry optimizations and frequency calculations were performed using Gaussian 09 using B3LYP/6-31G(d,p) basis set.51 In the geometry optimization, the default tight convergence in the SCF cycle was used without any orbital symmetry constraints. For the calculation of MOFs, model structures were generated with appropriate pore structure based on crystallography data. Two-layer ONIOM method52 was used with B3LYP/6-31G(d,p) basis set for high level, and molecular mechanics using UFF force field53 for low level. Coordinates of the rigid cationic framework of MOF structures (JCM-1 and JCM-1(Cl−)) were fixed according to the crystallography data, while the geometry of anions (NO3− or Cl−) and ammonia molecule were optimized during the calculation. High level model systems in ONIOM calculation were selected as one imidazolium and its counter anions (NO3− or Cl−) with an ammonia molecule. The interaction enthalpies of the framework with the guest molecule were calculated from the difference in total energies for the model structure (Hinteraction = HMOF + Hguest − HMOF with guest).Breakthrough experiments
Breakthrough experiments were carried out with a BELCAT-II equipped with a BELMASS mass spectrometer. The pelletized sample was packed in the breakthrough column of 0.5 cm in width and 1.0 cm in height and activated at 120 °C for 10 h in a He flow. The dry ammonia breakthrough experiments were performed in 1000 ppm NH3 gas balanced with He at 25 °C. In the case of wet ammonia breakthrough experiments, the sample was pre-saturated with humid (80% RH) He gas for 5 h at 25 °C prior to each measurement. And then, 1000 ppm NH3 gas balanced with humid (80% RH) He was introduced to the sample-packed column for 18 h at 25 °C. The regeneration between consecutive ammonia breakthrough cycles was different to the degassing procedure. The sample was activated with 25 °C for 10 h in a He flow. The effluent gas signal was continuously monitored by the BELMASS mass spectrometer. The amount of adsorbed NH3 was calculated by the integration of the breakthrough curve with ChemMaster program. The total gas flow was set at 30 sccm in all cases.In the case of dry ammonia breakthrough experiments, the amount of adsorbed ammonia in the breakthrough curve was calculated through the ChemMaster program by integrating the hatched area which is the gap between the elute curves of the saturated ammonia in the blank and the sample. The weight of glass beads in blank test (or the weight of the sample in breakthrough curve) was measured by comparing empty column weight and weight of column with beads (or sample) after activation. The calibration factor (count per mmol) of dry 1000 ppm breakthrough test was calculated with the ChemMaster program by choosing the flat region of blank test after NH3 fully was eluted.
In the case of wet ammonia breakthrough experiments, the ammonia breakthrough curves after pre-humidification for 5 h were depicted. The amount of adsorbed ammonia in the breakthrough curve at 80% RH and 1000 ppm was calculated by the ChemMaster program by integrating the hatched area which is the gap between the elute curves of saturated ammonia in the blank and the sample. The weight of glass beads in the blank test (or the weight of the sample in the breakthrough) was measured by comparing empty column weight and weight of column with beads (or sample) after activation. The calibration factor (count per mmol) of the breakthrough test at 80% RH and 1000 ppm was calculated by choosing the flat region of the blank test after NH3 was fully eluted. It should be noted that the ammonia breakthrough curve at dry 1000 ppm and that at 80% RH and 1000 ppm were not compatible in retention time because two have distinct blank breakthrough curves and so the calibration factors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Institute for Basic Science (IBS) [IBSR007-D1]. National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP: Ministry of Science, ICT and Future Planning) (No. NRF-2016H1A2A1908350 – Global PhD Fellowship Program). The X-ray crystallography analysis was performed at the Pohang Accelerator Laboratory (PLS-II BL2D SMC and 6D C&S Unist-PAL beamline). D.-W Lim acknowledges the support from the ACCEL program, Japan Science and Technology Agency (JST), JPMJAC1501. We thank Prof. S. S. Park for helpful discussion.Notes and references
- NITROGEN (FIXED)—ammonia, U.S. Geological Survey, Mineral Commodity Summaries, 2018, pp. 116–117, https://minerals.usgs.gov/minerals/pubs/commodity/nitrogen/mcs-2018-nitro.pdf Search PubMed.
- M. Sharma, S. Kishore, S. N. Tripathi and S. N. Behera, J. Atmos. Chem., 2007, 58, 1–17 CrossRef CAS.
- L. Gong, R. Lewicki, R. J. Griffin, F. K. Tittel, C. R. Lonsdale, R. G. Stevens, J. R. Pierce, Q. G. J. Malloy, S. A. Travis, L. M. Bobmanuel, B. L. Lefer and J. H. Flynn, Atmos. Environ., 2013, 77, 893–900 CrossRef CAS.
- Y. Wu, B. Gu, J. W. Erisman, S. Reis, Y. Fang, X. Lu and X. Zhang, Environ. Pollut., 2016, 218, 86–94 CrossRef CAS PubMed.
- D. Britt, D. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623–11627 CrossRef CAS PubMed.
- O. T. Wilcox, A. Fateeva, A. P. Katsoulidis, M. W. Smith, C. A. Stone and M. J. Rosseinsky, Chem. Commun., 2015, 51, 14989–14991 RSC.
- M. J. Katz, A. J. Howarth, P. Z. Moghadam, J. B. DeCoste, R. Q. Snurr, J. T. Hupp and O. K. Farha, Dalton Trans., 2016, 45, 4150–4153 RSC.
- Y. Chen, X. Zhang, K. Ma, Z. Chen, X. Wang, J. Knapp, S. Alayoglu, F. Wang, Q. Xia, Z. Li, T. Islamoglu and O. K. Farha, ACS Appl. Nano Mater., 2019, 2, 6098–6102 CrossRef CAS.
- S. Moribe, Z. Chen, S. Alayoglu, Z. H. Syed, T. Islamoglu and O. K. Farha, ACS Mater. Lett., 2019, 1, 476–480 CrossRef CAS.
- Y. Zhang, X. Zhang, Z. Chen, K. Ichi Otake, G. W. Peterson, Y. Chen, X. Wang, L. R. Redfern, S. Goswami, P. Li, T. Islamoglu, B. Wang and O. K. Farha, ChemSusChem, 2020, 13, 1710–1714 CrossRef CAS PubMed.
- D. W. Kim, D. W. Kang, M. Kang, J.-H. Lee, J. H. Choe, Y. S. Chae, D. S. Choi, H. Yun and C. S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22531–22536 CrossRef CAS PubMed.
- E. Binaeian, Y. Li, H.-A. Tayebi and D. Yuan, J. Hazard. Mater., 2021, 416, 125933 CrossRef CAS PubMed.
- X. Han, W. Lu, Y. Chen, I. Silva, J. Li, L. Lin, W. Li, A. M. Sheveleva, H. G. W. Godfrey, Z. Lu, F. Tuna, E. J. L. Mcinnes, Y. Cheng, L. L. Daemen, L. J. M. McPherson, S. J. Teat, M. D. Frogley, S. Rudic, P. Manuel, A. J. Ramirez-cuesta, S. Yang and M. Schröder, J. Am. Chem. Soc., 2021, 143, 3153–3161 CrossRef CAS PubMed.
- C. Marsh, X. Han, J. Li, Z. Lu, S. P. Argent, I. da Silva, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. P. Thompson, A. J. Blake, S. Yang and M. Schröder, J. Am. Chem. Soc., 2021, 143, 6586–6592 CrossRef CAS PubMed.
- M. K. Matikolaei and E. Binaeian, ACS Appl. Mater. Interfaces, 2021, 13, 27159–27168 CrossRef PubMed.
- L. Li, Y. Wang, J. Yang, Y. Chen and J. Li, ChemPlusChem, 2016, 81, 222–228 CrossRef CAS PubMed.
- A. J. Rieth and M. Dinca, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef CAS PubMed.
- A. J. Rieth, Y. Tulchinsky and M. Dinca, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef CAS PubMed.
- K. Vikrant, V. Kumar, K.-H. Kim and D. Kukkar, J. Mater. Chem. A, 2017, 5, 22877–22896 RSC.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- G. W. Peterson, J. J. Mahle, J. B. DeCoste, W. O. Gordon and J. A. Rossin, Angew. Chem., Int. Ed., 2016, 55, 6235–6238 CrossRef CAS PubMed.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- J. Yang, B. Du, J. Liu, R. Krishna, F. Zhang, W. Zhou, Y. Wang, J. Li and B. Chen, Chem. Commun., 2018, 54, 14061–14064 RSC.
- M. Savage, Y. Cheng, T. L. Easun, J. E. Eyley, S. P. Argent, M. R. Warren, W. Lewis, C. Murray, C. C. Tang, M. D. Frogley, R. T. Murden, M. J. Benham, A. N. Fitch, A. J. Blake, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Adv. Mater., 2016, 28, 8705–8711 CrossRef CAS PubMed.
- L. M. Rodriguez-Albelo, E. Lopez-Maya, S. Hamad, A. R. Ruiz-Salvador, S. Calero and J. A. Navarro, Nat. Commun., 2017, 8, 14457 CrossRef CAS PubMed.
- J. H. Carter, X. Han, F. Y. Moreau, I. Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. H. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695–5727 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- H. G. W. Godfrey, I. da Silva, L. Briggs, J. H. Carter, C. G. Morris, M. Savage, T. L. Easun, P. Manuel, C. A. Murray, C. C. Tang, M. D. Frogley, G. Cinque, S. Yang and M. Schröder, Angew. Chem., Int. Ed., 2018, 57, 14778–14781 CrossRef CAS PubMed.
- C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2010, 20, 111–118 CrossRef CAS.
- J. Lee, C. Y. Chuah, J. Kim, Y. Kim, N. Ko, Y. Seo, K. Kim, T. H. Bae and E. Lee, Angew. Chem., Int. Ed., 2018, 57, 7869–7873 CrossRef CAS PubMed.
- J. G. Duan, W. Q. Jin and S. Kitagawa, Coord. Chem. Rev., 2017, 332, 48–74 CrossRef CAS.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- A. Yokozeki and M. B. Shiflett, Appl. Energy, 2007, 84, 1258–1273 CrossRef CAS.
- A. Yokozeki and M. B. Shiflett, Ind. Eng. Chem. Res., 2007, 46, 1605–1610 CrossRef CAS.
- G. Li, Q. Zhou, X. Zhang, L. Wang, S. Zhang and J. Li, Fluid Phase Equilib., 2010, 297, 34–39 CrossRef CAS.
- K. N. Ruckart, Y. Zhang, W. M. Reichert, G. W. Peterson and T. G. Ind, Ind. Eng. Chem. Res., 2016, 55, 12191–12204 CrossRef CAS.
- T. Kajiwara, M. Higuchi, D. Watanabe, H. Higashimura, T. Yamada and H. Kitagawa, Chem.–Eur. J., 2014, 20, 15611–15617 CrossRef CAS PubMed.
- D. Saha and S. Deng, J. Colloid Interface Sci., 2010, 348, 615–620 CrossRef CAS PubMed.
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197–3204 RSC.
- K. J. Chang and O. Talu, Appl. Therm. Eng., 1996, 16, 359–374 CrossRef CAS.
- D. Ma, J. Zhang, J. Bai and H. Zhang, Nat. Resour., 2014, 5, 782–794 Search PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- S. Cha, M. Ao, W. Sung, B. Moon, B. Ahlstrom, P. Johansson, Y. Ouchi and D. Kim, Phys. Chem. Chem. Phys., 2014, 16, 9591–9601 RSC.
- J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620–4632 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197 RSC.
- G. M. J. Frischet al., Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 461–462, 1–21 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1912377. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra01270g |
| ‡ These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2022 |