
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro and in silico studies of SARS-CoV-2 main protease Mpro inhibitors isolated from Helichrysum bracteatum†
Gehad Abdel Wahab ,
Walaa S. Aboelmaaty,
Mohamed Farid Lahloub and
Amal Sallam*
,
Walaa S. Aboelmaaty,
Mohamed Farid Lahloub and
Amal Sallam*
Pharmacognosy Department, Faculty of Pharmacy, Mansoura University, Mansoura, 35516, Egypt. E-mail: asallam@mans.edu.eg; gehadabdelwahab@mans.edu.eg; algehadalhaq@yahoo.com; walaa_safwat@mhans.edu.eg; walaa_m_s@yahoo.com; mfilah@yahoo.com; Fax: +20502247496; Tel: +201092017949
First published on 22nd June 2022
Abstract
Discovering SARS-CoV-2 inhibitors from natural sources is still a target that has captured the interest of many researchers. In this study, the compounds (1–18) present in the methanolic extract of Helichrysum bracteatum were isolated, identified, and their in vitro inhibitory activities against SARS-CoV-2 main protease (Mpro) was evaluated using fluorescence resonance energy transfer assay (FRET-based assay). Based on 1D and 2D spectroscopic techniques, compounds (1–18) were identified as 24-β-ethyl-cholesta-5(6),22(23),25(26)-triene-3-ol (1), α-amyrin (2), linoleic acid (3), 24-β-ethyl-cholesta-5(6),22(23),25(26)-triene-3-O-β-D-glucoside (4), 1,3-propanediol-2-amino-1-(3′,4′-methylenedioxyphenyl) (5), (−)-(7R,8R,8′R)-acuminatolide (6), (+)-piperitol (7), 5,7,4′-trihydroxy-8,3′-dimethoxy flavanone (8), 5,7,4′-trihydroxy-6-methoxy flavanone (9), 4′,5-dihydroxy-3′,7,8-trimethoxyflavone (10), 5,7-dihydroxy-3′,4′,5′,8-tetramethoxy flavone (11), 1,3-propanediol-2-amino-1-(4′-hydroxy-3′-methoxyphenyl) (12), 3′,5′,5,7-tetrahydroxy-6-methoxyflavanone (13), simplexoside (piperitol-O-β-D-glucoside) (14), pinoresinol monomethyl ether-β-D-glucoside (15), orientin (16), luteolin-3′-O-β-D-glucoside (17), and 3,5-dicaffeoylquinic acid (18). Compounds 6, 12, and 14 showed comparable inhibitory activities against SARS-CoV-2 Mpro with IC50 values of 0.917 ± 0.05, 0.476 ± 0.02, and 0.610 ± 0.03 μM, respectively, compared with the control lopinavir with an IC50 value of 0.225 ± 0.01 μM. The other tested compounds showed considerable inhibitory activities. The molecular docking study for the tested compounds was carried out to correlate their binding modes and affinities for the SARS-CoV-2 Mpro enzyme with the in vitro results. Analyzing the results of the in vitro assay together with the obtained in silico results led to the conclusion that phenylpropanoids, lignans, and flavonoids could be considered suitable drug leads for developing anti-COVID-19 therapeutics. Moreover, the phenylpropanoid skeleton oxygenated at C3, C4 of the phenyl moiety and at C1, C3 of the propane parts constitute an essential core of the SARS-CoV-2 Mpro inhibitors, and thus could be proposed as a scaffold for the design of new anti-COVID-19 drugs.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel strain of the coronavirus group. It emerged in the city of Wuhan, China at the end of 2019, causing an outbreak of unusual viral pneumonia. It caused acute respiratory disease that was named coronavirus disease 2019 (COVID-19) by the WHO on 11 February 2020. Being highly transmissible and pathogenic such that it has spread fast all over the world, WHO defined it on 11 March 2020 as a pandemic, posing an extraordinary threat to global public health.1 To date, there are no generally proven antiviral drugs against SARS-CoV-2, although several clinical trials all over the world are testing several known antiviral drugs.2 Therefore, finding natural, semisynthetic, or synthetic remedies for COVID-19 is the target of many researches now.The main protease (Mpro), papain-like protease (PLpro), and RNA-dependent RNA polymerase (RdRp) of SARS-CoV-2 are considered decisive factors in the infectious route of the virus; they have been reported as important targets for therapeutic strategies.
SARS-CoV-2 main protease (Mpro) or (3-chymotrypsin-like protease 3CLpro) is an enzyme responsible for the proteolysis and release of essential functioning peptides, playing a great role in replication, and thus the life cycle of the virus.2–5 Moreover, Mpro is highly conserved across coronaviruses; thus, inhibiting main proteases is considered an attractive target for the discovery of effective antiviral drugs for the treatment of not only SARS-CoV-2 but also other coronaviruses.6 Although researchers have focused their efforts on studying the potential antiviral properties of plants and their constituents from different classes including flavonoids and lignans as anti-COVID-19 agents through in silico studies targeting Mpro and other enzymes, little has been found concerning the in vitro studies.4–7
The genus Helichrysum belonging to family Asteraceae (Compositae) includes approximately 600 species spread widely all over the world, especially in the Southern Hemisphere; it is also widespread through Eurasia, Australia, and the Mediterranean region.8–13 Several classes of phytoconstituents have been reported in different Helichrysum species, mainly phenolics, lignans, phloroglucinols, pyrones, fatty acids, and terpenoid compounds.11,12 Since ancient times, Helichrysum species are well known for their medicinal properties as diuretic, anti-inflammatory, hepatoprotective, and anti-psoriasis. Also, they have been used in treatment of colds, cough, inflammation, and allergy conditions such as those related to the respiratory tract.11,12
It is reported that several species of genus Helichrysum show antiviral activities against different viruses including coronaviruses or similar viruses. H. arenarium showed antiviral activities against Herpes simplex virus Type-1 (HSV-1) and Para-influenza-3. H. italicum and H. auronitens have antiviral activities against HSV-1, while H. melanacme has antiviral activity against HIV.9,10,13–15 Helichrysetin, a chalcone derivative found within numerous Helichrysum species, was reported to be able to inhibit MERS-CoV 3CLpro.15
H. bracteatum is known as straw flower and is widely cultivated as an ornamental plant.16,17 Previous phytochemical studies have reported the presence of different classes of compounds as flavonoids, lignans, and phenolic acids.17–19
This study is concerned with discovering potential antiviral leads against COVID-19 based on the evaluation of the ability of H. bracteatum methanolic extract, as well as fractions and isolated phytoconstituents to inhibit SARS-CoV-2 main protease (Mpro) through in vitro and in silico studies.
2. Results and discussion
2.1 Characterization of the isolated compounds
Phytochemical investigation of petroleum ether, methylene chloride, and ethyl acetate fractions of the methanolic extract of H. bracteatum leaves resulted in the isolation and structure elucidation of eighteen compounds (1–18) (Fig. 1) including two steroidal compounds (1,4), one pentacyclic triterpene (2), one fatty acid (3), two phenyl propanoid derivatives nitrogenated at C2 (5,12), four lignans (6,7,14,15), seven flavonoids, three flavanones (8,9,13), four flavones (10,11,16,17), and one phenolic acid derivative (18). The proton and carbon values beside the spectra of the isolated compounds are present in the ESI file (Data S1 and Fig. S1–S50†).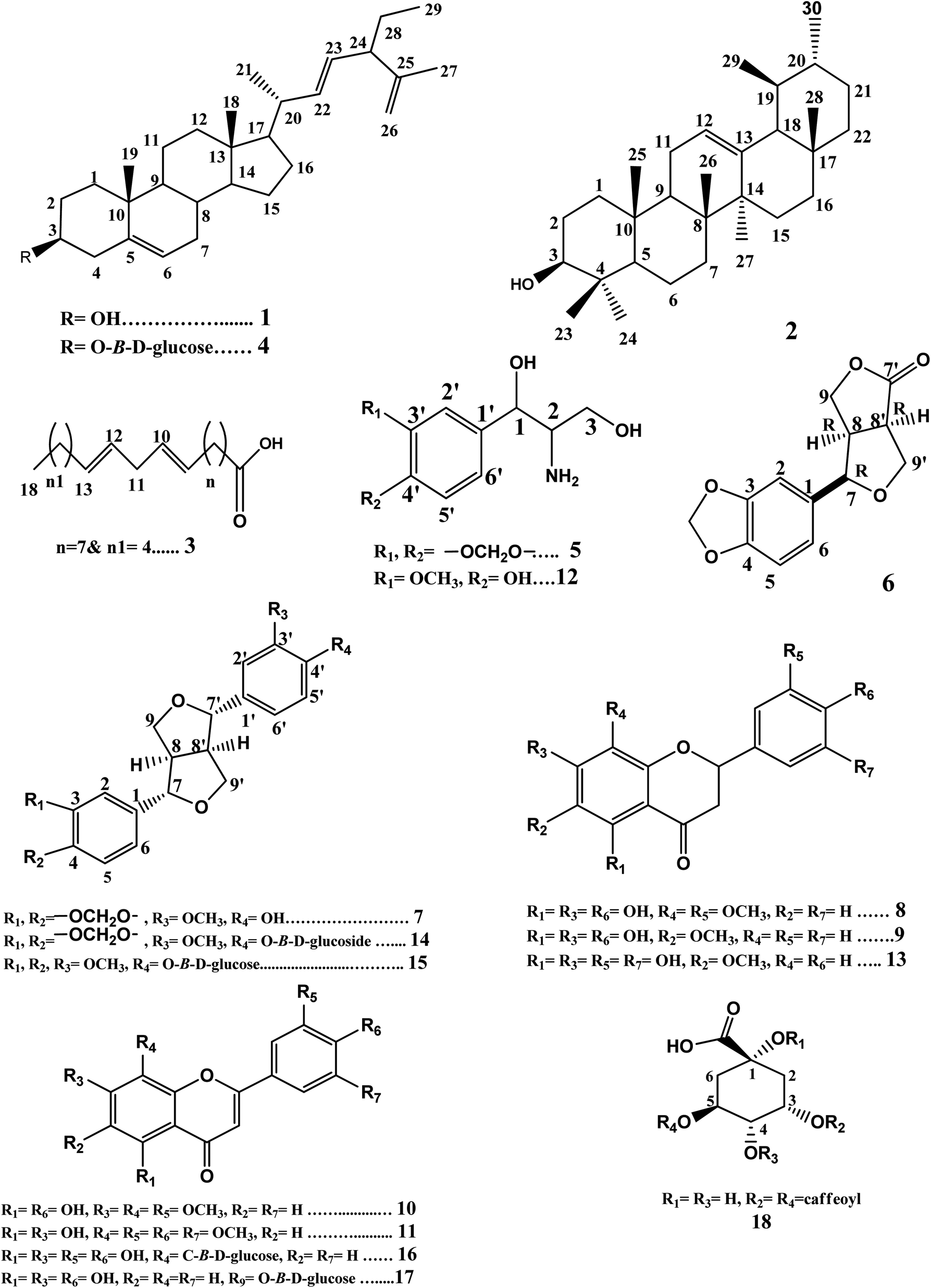 | ||
| Fig. 1 Structures of compounds (1–18). | ||
Compounds (1–4) were isolated from the petroleum ether fraction. They were confirmed from their proton and carbon chemical shift values (Fig. S1–S8†) compared with those reported in the literature. They were identified as 24-β-ethyl-cholesta-5(6),22(23),25(26)-triene-3-ol (1) and its glucoside 24-β-ethyl-cholesta-5(6),22(23),25(26)-triene-3-O-β-D-glucoside (4),20 besides α-amyrin (2),21,22 and linoleic acid (3).23 It is the first time that compounds 1 and 4 have been isolated from family Compositae. It is the first time that compound 2 has been isolated from genus Helichrysum. Compound 3 was previously reported from Helichrysum seed oil.24
Compounds 5 and 12 were concluded to be phenyl propanoid derivatives nitrogenated at C2. The careful examination of the 1H-NMR spectrum of compound 5 (Fig. S9†) showed signals at δH 6.78 (br d, 1H, J = 8, H-5′), δH 6.80 (br d, 1H, J = 10.8, H-6′), and δH 6.85 (br s, 1H, H-2′), indicating the possible presence of the tri-substituted phenyl ring. It also showed the signal of an oxygenated methylene that may represent a methylenedioxy group at δH 5.95 (s, 2H), substituting the phenyl ring. Signals representing one oxygenated methine proton at δH 4.72 (d, 1H, J = 3.2, H-1) and one aminomethine proton δH 3.05 (m, 1H, H-2) besides an oxygenated methylene group at δH 4.23 (dd, 1H, J1 = 6.4 & 8.4, H-3a) and δH 3.87 (dd, 1H, J = 6.8 & 2, H-3b) were also observed, indicating the possible presence of the 1,3-dihydroxy-2-amino-propane moiety. The APT spectrum (Fig. S10†) showed signals supporting the previous possibilities. It revealed two oxygenated quaternary aromatic carbon signals at δC 148.0 (C-3′) & 147.1 (C-4′), a quaternary aromatic carbon at δC 135.0 (C-1′), and three aromatic methine carbons at δC 106.5 (C-2′), 108.2 (C-5′), and 119.4 (C-6′). It also showed carbon signals representing oxygenated methylene carbon at δC 101.1, indicating the presence of the methylenedioxy group substituting the phenyl ring. Moreover, it showed signals representing one oxygenated aliphatic methine carbon at δC 85.8 (C-1) and one oxygenated methylene carbon at δC 71.7 (C-3) and one aminomethine carbon at δC 54.3 (C-2), which was shifted upfield than the oxygenated carbons (C-1 & 3), thus confirming the presence of the 1,3-dihydroxy-2-amino-propane moiety. The HMBC spectrum (Fig. S11†) showed a cross peak correlating the proton signal at δH 4.71 (H-1) with the quaternary carbon signals at δC 135.0 (C-1′), 106.5 (C-2′), and 119.4 (C-6′). This indicated that the 1,3-dihydroxy-2-amino-propane moiety substituted the phenyl ring at C-1′. The HMBC cross peaks from the methylenedioxy group δH 5.95 to carbons δC 148.0 (C-3′) & 147.1 (C-4′) confirmed that the methylendioxy group is substituting the phenyl ring at C-3′ & 4′. Compound 5 was identified as 1,3-propanediol-2-amino-1-(3′,4′-methylenedioxyphenyl). It was previously isolated only from Artemisia selengensis F. Compositae;25 also, it was reported as a synthetic compound.26
The careful examination of the 1H-NMR and APT spectra of compound 12 (Fig. S26 and S27†) indicated that it is also a phenyl propanoid derivative nitrogenated at C2. Comparing its 1H-NMR and APT spectra with those of compound 5 revealed that compound 12 was similar to compound 5, but the phenyl ring is substituted with methoxy and hydroxy groups instead of methylenedioxygroup (at C-3′, 4′). Compound 12 was identified as 1,3-propanediol-2-amino-1-(4′-hydroxy-3′-methoxyphenyl).27 It was previously isolated only from Santolina chamaecyparissus F. Compositae and our study reports its detailed chemical shift values for the first time.
Compounds 7, 14, and 15 were found to be furofuranlignans. Comparing the data obtained from 1H-NMR and APT spectra (Fig. S14 and S15†) with that reported in the previous literature and the stereochemistry reported for furofuranlignans,28–30 compound 7 was identified as (+)-piperitol.31,32 Compound 14 was found to be the glucoside of 7; this was revealed by its 1H-NMR and APT spectra (Fig. S40 and S41†), which showed the doublet signal representing the anomeric proton (H-1′′) at 4.88 ppm (1H, d, J = 6.7 Hz) in addition to the carbon signal at δC 100.6 (C-1′′) and the four hydroxylated aliphatic methine carbons at δC 71.6, 77.3, 70.1, 77.4, besides the hydroxylated aliphatic methylene carbon at δC 61.1 (C-6′′), indicating the presence of the hexose sugar glucose. Compound 14 was identified as simplexoside (piperitol-O-β-D-glucoside).33,34 Both compounds 7 & 14 were previously isolated from H. bracteatum aerial parts.17 A careful examination of the 1H-NMR and APT spectra of compounds 14 & 15 (Fig. S40–S43†) revealed that compound 15 is similar to compound 14, except for the presence of two signals at δH 3.76 (6H, s, OCH3-3 & OCH3-4), representing two methoxy groups instead of the signal at 6.00 (2H, s, OCH2O), thus representing the methylenedioxy group in compound 14. Compound 15 was identified as pinoresinol monomethyl ether-β-D-glucoside.35 Compound 15 is isolated from F. Compositae (Asteraceae) for the first time in this study. Comparing the 1H-NMR and APT spectra of compound 6 (Fig. S12 and S13†) with compound 7 revealed that 6 is similar to 7, but it lacks the presence of one aryl group; instead, it showed a carbonyl moiety. This was deduced from the carbonyl group at δC 178.1 corresponding to C-7′. Comparing this data with the previous literature, compound 6 was identified as (−)-(7R,8R,8′R)-acuminatolide.36 To the best of our knowledge, acuminatolide was previously isolated from aerial parts of H. acuminatum.37
Compounds 8, 9, and 13 were found to be flavanones depending on the common characteristic features that appeared in the UV-spectral data besides both 1H-NMR and APT data. The UV-spectra of these three compounds showed two absorption bands, band I in the λmax range of 289–291 nm and band II in the λmax range of 232–236 nm, which appeared to be in agreement with those characteristic for flavanones or dihydroflavonols.38,39 The 1H-NMR spectra of the three compounds (Fig. S16, S19 and S28†) revealed the presence of two double doublet signals characteristic for H-3α and H-3β in the range of δH 2.5–3.01 besides one double of doublet signal corresponding to the oxygenated methine H-2 at δH 5.20. The APT spectra of the three compounds (Fig. S17, S20 and S31†) supported this deduction by showing signals representing carbonyl (C-4) in the range of δC 195.2–197.2, a signal for the methylene group at 42.7 (C-3), besides the signal representing oxygenated methine (C-2) at δc 79.2.40
The UV spectral data in different shift reagents (Table S1†) suggested that 8 has a flavanone structure with free OH groups at positions 5, 7, and 4′.38 The 1H-NMR spectrum (Fig. S16†) showed a signal at δH 5.18 (1H, dd, J = 2.8 & 12.6) representing the oxygenated methine H-2 and pair of double doublets at δH 2.56 (1H, dd, J = 2.8 & 17.2) and δH 2.97 (1H, dd, J = 12.8 & 17.2) representing the methylene protons H-3α and 3β, respectively. These signals are characteristic of the flavanone skeleton. Also, it revealed a singlet signal at δH 5.79 (1H, s, H-6), indicating that ring A has only one free proton. Besides, the signals representing the ABX system in ring B that were obtained at δH 6.97 (1H, d, J = 1.6, H-2′), δH 6.71 (1H, d, J = 8, H-5′), and δH 6.81 (1H, dd, J = 8.2 & 2, H-6′). In addition, it showed two signals of two methoxy groups at δH 3.66 & 3.78. The APT spectrum (Fig. S17†) showed six oxygenated aromatic carbon signals along with a carbonyl signal in the range of δC 130–200 ppm. The presence of a carbonyl signal at δC 195.7 (C-4), an oxygenated methine signal at δC 79.2 (C-2) and a methylene signal at 42.7 (C-3) confirmed the flavanone skeleton.40,41 There were three signals representing the oxygenated aromatic carbons substituted with free hydroxyl groups at δC 159.0, 164.6, & 146.6 representing C-5, 7, & 4′, respectively, which were confirmed previously by UV data; the other two signals were methoxylated, one at δC 147.7 representing C-3′ as ring B showed an ABX system with free OH at C-4′ only. The other one at δC 130.7 represents C-8 rather than C-6 that was not substituted and appeared at δC 96.3. Reviewing previous literature that reported flavanone oxygenated at C-8 while C-6 is free and those reported compounds oxygenated at C-6 while C-8 is free, it could be noticed that when C-8 is oxygenated while C-6 is free, C-6 appears at a chemical shift value that is slightly downfield shifted (δC 96.3 as in compound 8) compared with the chemical shift of C-8 if it is free and C-6 is oxygenated, where C-8 in this case appears slightly shifted upfield41,42 (Table S2†). The data obtained from UV, 1H-NMR, and APT suggested that compound 8 is a flavanone hydroxylated at 5, 7, & 4′ and methoxylated at 8 and 3′. The HMBC spectrum of compound 8 (Fig. S18†) showed cross peaks correlating the methoxy group proton signal at δH 3.66 (H-R4) with the carbon signal at δC 130.7 (C-8); also, it showed cross peaks correlating the methoxy group proton signal at δH 3.78 (H-R5) with the carbon signal at δC 147.7 (C-3′). These data confirmed that both C-8 and 3′ are blocked with OCH3 groups, while C-5, 7, and 4′ are substituted with free hydroxyl groups. Also, it showed cross peaks correlating the methine H-6 at δH 5.79 with the carbon signals at δC 159.0 (C-5), 130.7 (C-8), & 100.7 (C-10), which also supports this hypothesis. The previous data suggests that compound 8 is 5,7,4′-trihydroxy-8,3′-dimethoxy flavanone. Comparing the carbons values obtained from the APT spectrum of compound 8 with previous literature revealed that compound 8 is 5,7,4′-trihydroxy-8,3′-dimethoxy flavanone.41 It worth noting that this is the first study that reports the isolation of compound 8 (5,7,4′-trihydroxy-8,3′-dimethoxy flavanone) from the family Compositae. It was previously isolated from Iris unguicularis43 and our study reports its detailed chemical shift values.
Comparing the proton and carbon values of compound 9 obtained from 1H-NMR and APT spectra (Fig. S19 and S20†) with those of compound 8 revealed that compound 9 is similar to compound 8 with the presence of the methoxy group at C-6 rather than C-8 (owing to the slightly upfield shifted value of C-8 at δc 94.8 compared to the value of the unsubstituted C-6, which is slightly shifted downfield when unsubstituted at δC 96.3 as in compound 8) and the absence of the methoxy group at C-3′ (Fig. S19, S20 and Table S2†). Thus, compound 9 was identified as 5,7,4′-trihydroxy-6-methoxy flavanone and it is the first time to be isolated from F. Compositae in this study.41,42
The molecular formula of compound 13 was determined to be C16H14O7 from the [M + H]+ peak at m/z 319.24 appearing in the LC-ESI+-MS spectrum (Fig. S36†), which is in agreement with the calculated one at m/z 319.28. The UV spectral data in different shift reagents (Table S1†) indicated that 13 has a flavanone structure with free OH groups at positions 5 and 7.38 Comparing the proton and carbon chemical shift values of ring A in compound 13 obtained from 1H-NMR and APT spectra (Fig. S28–S31, S38 and Table S2†) with those of compound 9 revealed that both compounds show the same substitution patterns in ring A (5,7-dihydroxy & 6-methoxy). The 1H-NMR, APT, & HSQC spectra showed signals characteristic for the flavanone skeleton at δH 5.16 (1H, dd, J = 2.8 & 12.8) representing the oxygenated methine H-2 correlated to δc 79.2, a pair of double doublets at δH 2.60 (1H, dd, J = 2.8 & 17.2), and δH 2.96 (1H, dd, J = 12.8 & 17.2) representing methylene protons H-3α and 3β, respectively, which are correlated to δC 42.7 besides the carbonyl at δC 197.2 (C-4).40,41 The singlet signal at δH 5.87 (1H, s, H-8) correlated to δc 94.8 and a methoxy group at δH/C 3.68/59.6 were also present. Singlet signals representing three protons of ring B at δH 6.68 (2H, s) and δH 6.81 (1H, s) representing H-2′, 4′, & 6′ correlated to δC 117.9, 114.8, & 113.3, respectively, revealed the absence of AB or ABX system in ring B. This suggestion was supported by the HMBC cross peaks from δH 6.68 (H-2′) & 6.81 (H-6′) to the carbon signal at δC 79.2 (C-2) and from δH 6.68 & 6.81 (H-2′& H-6′) to δC 114.8(C-4′) (Fig. S34 and S35†). The APT spectrum (Fig. S31†) showed also six oxygenated aromatic carbon signals along with a carbonyl one in the range of δC 129–200 ppm. The carbon signal at δC 129.0 is methoxylated rather than hydroxylated; this could be detected from the HMBC spectrum (Fig. S34†) that showed cross peaks correlating the methoxyprotons at δH 3.68 with the carbon signal at δc 129.0. The carbon signal representing the methoxylated aromatic carbon at δC 129.0 was assigned to C-6 rather than C-8 that was not substituted and appeared at a slightly upfield chemical shift value (δH 94.3, as C-8 of compound 9) when compared with the chemical shift value of the unsubstituted C-6 that appeared at a slightly downfield shifted value (δH 96.3, as C-6 in compound 8)41,42 (Table S2†). The HMBC spectrum showed cross peaks correlating the signal at δH 5.87 (H-8) with carbon signals at δC 129.0 (C-6), 158.8 (C-9), 159.5 (C-7), & 102.1 (C-10). The hydroxylated carbon signals at δC 155.2 & 159.5 were assigned to C-5 & 7. The remaining oxygenated carbon signals at δC 145.1 & 145.5 were assigned to the hydroxylated carbons in ring B. Lacking an AB or ABX system in ring B and absence of free hydroxyl group at C-4′ (according to UV data) suggested rare substitution by the hydroxyls at C-3′ & C-5′ not the common one at C-3′ & C-4′ in ring B. This suggestion was supported by the HMBC correlation from δH 6.68 (H-2′) to δC 145.1 (C-3′) and from δH 6.81 (H-6′) to δC 145.5 (C-5′) (Fig. S34†). The ESI+-MS fragmentation of compound 13 showed a base peak at m/z 183.61 [M + H]+ corresponding to the fragment (a) 3-(3,5-dihydroxyphenyl)propanoic acid fragment (calculated m/z 183.18), confirming that ring B is disubstituted by two hydroxyl groups (Fig. S37A and B†). The previous data suggested that compound 13 is 5,7,3′,5′-tetrahydroxy-6-methoxy flavanone. Reviewing the current literature, it was found that this study is the first to report the isolation of 5,7,3′,5′-tetrahydroxy-6-methoxy flavanone from F. Compositae (Asteraceae). It was previously isolated only from Salvia plebeian F. Labiatae;44 also, it was reported as a semi-synthetic compound.45 This study is the first that reports detailed data for that rare substituted flavanone at C-3′ & 5′.
The careful examination of the UV, 1H-NMR, and APT spectra of compounds 10 & 11 indicated that they are flavones. They were identified as 4′,5-dihydroxy-3′,7,8-trimethoxyflavone (10) and 5,7-dihydroxy-3′,4′,5′,8-tetramethoxy flavone (11). This was deduced from the UV-spectra of the two compounds that showed two absorption bands, band I in the λmax range of 323–345 nm and band II in the λmax range of 276–278 nm, which appear to be in agreement with those characteristic for flavones.38,39 The 1H NMR spectra of the two compounds (Fig. S21 and S24†) revealed the presence of a singlet signal corresponding to the methine proton H-3 at (δH 6.99 & 6.62 for compounds 10 & 11, respectively). The APT spectra of the two compounds (Fig. S22 and S25†) supported this deduction by showing signals representing carbonyl (C-4) (at δC 182.7 & 182.4 for compounds 10 & 11, respectively) and the signal for the methine carbon C-3 (at δC 103.5 & 105.4 for compounds 10 & 11, respectively).
The UV spectral data in different shift reagents suggested that 10 has a flavone structure with free OH groups at positions 5 and 4′.38 Both 1H- and DEPT-Q NMR data also supported the previous conclusion. The 1H-NMR spectrum (Fig. S21†) showed two singlet signals at δH 6.59 (1H, s, H-6) and 6.99 (1H, s, H-3). Also, it showed signals representing aromatic protons of ring B at δH 7.59 (1H, s, H-2′), 7.00 (1H, d, J = 6.7, H-5′), and 7.60 (1H, d, J = 6, H-6′). Besides, it revealed three methoxy groups at δH 3.92, 3.86, & 3.90 and carbon signals at δC (56.9, 61.6, & 56.5) representing R3, R4, & R5. The DEPT-Q spectrum (Fig. S22†) showed seven oxygenated aromatic carbon signals along with the carbonyl signal in the range of δC 125–183 ppm. The presence of a carbonyl signal at δC 182.7 (C-4) and a signal at 103.5 (C-3) confirmed the flavone skeleton.40,41 The two signals at δC 164.3 & 151.4 ppm were assigned to the two ether-linked carbons C-2 and C-9, respectively. The two signals of hydroxylated aromatic carbons at δC 157.1 & 148.5 were assigned to C-5 & 4′, respectively, which was confirmed from the HMBC spectrum (Fig. S23†) that showed cross peaks correlating δH 6.59 (H-6) with both oxygenated carbons at δC 128.9 (C-8) & 104.3 (C-10), and the absence of a cross peak with C-9 (δC 151.4), confirming that H-8 is blocked. The other three methoxylated aromatic carbon signals at δC 158.8, 128.9, & 149.2 were assigned to carbons C-7, 8, & 3′; this was supported by the HMBC cross peaks correlating the methoxy group proton signal at δH 3.92 (OCH3-R3) with the carbon signal at δC 158.8 (C-7). Also, it showed a cross peak correlating the methoxy group proton signal at δH 3.86 (OCH3-R4) with the carbon signal at δC 128.9 (C-8). The third methoxy group at δH 3.90 (OCH3-R5) showed a cross peak with the carbon signal at δC 149.2 (C-3′). The data obtained from UV, 1H, and DEPT-Q spectra revealed that compound 10 is a flavone hydroxylated at position 5 & 4′ and methoxylated at C-7, 8, & 3′. This structure is in agreement with that representing 4′,5-dihydroxy-3′,7,8-trimethoxyflavone. Comparing this data with that reported in the literature, compound 10 could be identified as 4′,5-dihydroxy-3′,7,8-trimethoxyflavone.41 It is the first report for the isolation of 4′,5-dihydroxy-3′,7, 8-trimethoxyflavone from F. Compositae (Asteraceae). It was previously isolated from Ocimum sanctum leaves.46 It was reported as a synthetic compound in a previous study.47 This study reports its detailed proton and carbon chemical shift values for the first time.
The 1H-NMR spectrum of compound 11 (Fig. S24†) showed one signal at δH 7.13 representing the singlet protons (2H-2′, 6′). Besides, it revealed four methoxy groups at δH 4.00 (3H, s, R4), 3.95 (6H, s, R5 & R7), & 3.94 (3H, s, R6). The APT spectrum (Fig. S25†) showed eight oxygenated aromatic carbon signals alongwith the carbonyl signal in the range of δC 125–183 ppm. The data obtained from UV, 1H, and APT spectra revealed that compound 11 is a flavone hydroxylated at positions 5 & 7 and methoxylated at 8, 3′, 4′, and 5′. Comparing proton and carbon values of compound 11 with previous literature, compound 11 could be identified as 5,7-dihydroxy-3′,4′,5′,8-tetramethoxy flavone.41 It is the first time to report the isolation of this compound from family Compositae. It was previously isolated from Dikamali gum,48 which reported it as a semi-synthetic compound;49 our study is the first to report its detailed chemical shift values.
Careful examination of the UV, 1H-NMR, and APT spectra of compounds 16 & 17 indicated that these compounds are glucosidated flavones. They were identified as orientin (16) and luteolin-3′-O-β-D-glucoside (17). The UV spectra of the two compounds showed two absorption bands, band I in the λmax range of 331–346 nm and band II at λmax 271 nm, which appear to be in agreement with those characteristic for flavones.38,39 The 1H-NMR spectra of the two compounds (Fig. S44 and S46†) revealed the presence of the singlet signal corresponding to the methine proton H-3 at (δH 6.68 & 6.52 for compound 16 & 17, respectively). The APT spectra of the two compounds (Fig. S45 and S46†) supported this deduction by showing signals representing carbonyl (C-4) at δC 182.5 for the two compounds and signal for the methine carbon C-3 (at δC 102.9 & 103.7 for compounds 16 & 17, respectively).
These UV-spectral data in different shift reagents suggested the presence of a flavone structure in 16 with free OH groups at positions 5, 7, 3′, and 4′. The doublet at 4.69 ppm (1H, d, J = 9.6 Hz) in addition to the carbon signal at δC 73.9 and the four hydroxylated aliphatic methine carbons at δC 71.2, 79.2, 71.2, 82.5 besides the hydroxylated aliphatic methylene carbon at δC 62.1 indicated the presence of a hexose sugar. The doublet at 4.69 ppm (1H, d, J = 9.6) represents the anomeric proton of the glucose sugar (H-1′′); this coupling constant (J = 9.6) indicates that the sugar is β-linked. The upfield shifted carbon signal at δC 73.9 represents the anomeric carbon of the sugar (C-1′′) was reported for C-linked-β-D-sugar rather than O-linked-β-D-sugar.40 The glucose substitutes compound 16 at C-8 rather than C-6 owing to the slight upfield shifted value of the glycosylated C-8 at δC 105.0 as in compound orientin compared with the value of the glycosylated C-6, which is slightly downfield shifted at δC 108.9 as isoorientin.38,50 Reviewing the current literature, the data obtained from the UV, 1H, and APT spectra of compound 16 are consistent with those reported for orientin.50 This study is the first that reports the isolation of orientin from genus Helichrysum.
The data obtained from UV spectra in different shift reagents (Table S1†), 1H-NMR, APT, and HMBC spectra (Fig. S46–S48†) of compound 17 are consistent with those reported for luteolin-3′-O-β-D-glucoside.38,40 It was previously isolated from H. arenarium flowers.51
The careful examination of both 1H-NMR and APT spectra of 18 (Fig. S49 and S50†) showed that compound 18 contains quinic acid nucleus esterified with the two caffeoyl moieties at C-3 & C-5. Thus, compound 18 was identified as 3,5-dicaffeoylquinic acid (isochlorogenic acid).52 It was previously isolated from H. bracteatum flowers and H. italicum aerial parts.18,53
2.2 Inhibitory activities against SARS-CoV-2 main protease (Mpro or 3CLpro)
Several species of genus Helichrysum were reported to have antiviral activities against coronaviruses and other viruses such as Herpes simplex virus Type-1.10,15 Previous literature reported the ability of helichrysetin, a chalcone derivative isolated from certain Helichrysum species, to inhibit MERS-CoV 3CLpro.15 Several classes of phytoconstituents as flavonoids and lignans are reported in in silico studies as potential anti-COVID-19 agents by inhibiting SARS-CoV-2 main protease (Mpro) and other enzymes involved in the virus life cycle.4,5 These facts encouraged us to evaluate the activity of H. bracteatum leaves methanolic extract, fractions, and the isolated compounds as inhibitors of SARS-CoV-2 Mpro.The methanolic extract exhibited inhibitory activity with IC50 value of 14.47 ± 0.74 μg mL−1. Among the tested four fractions, the ethyl acetate fraction showed the highest inhibitory activity followed by petroleum ether, methylene chloride, and butanol fractions with IC50 values of 2.589, 3.466, 16.05, and 21.9 μg mL−1, respectively, compared with the standard antiviral compound lopinavir with an IC50 value of 0.225 ± 0.01 μM (Table S4 and Fig. S55†).
The isolated compounds (1–18) were evaluated for their inhibitory activities against SARS-CoV-2 Mpro. Compounds 6, 12, and 14 showed comparable inhibitory activities against SARS-COV-2 Mpro with IC50 values of 0.917 ± 0.05, 0.476 ± 0.02, and 0.610 ± 0.03 μM, respectively, compared with the control lopinavir with an IC50 value of 0.225 ± 0.01 μM. Compounds 2, 5, 11, 13, & 18 showed moderate inhibitory activities with IC50 values in the range of 4–8 μM, while compounds 1, 7, 9, 10, 15, & 17 exhibited significant activities with IC50 values in the range of 10–16 μM. Compounds 3, 8, & 16 showed weak activities with IC50 values in the range of 20–28 μM. The lowest activity was reported for compound 4 (IC50 value of 89.99 ± 4.59 μM).
Compounds 12, 14, and 6 showed the highest inhibitory activity. Compounds 6 and 14 are lignans, while compound 12 is a phenyl propanoid derivative nitrogenated at C2. The three compounds share the presence of the phenyl propanoid part oxygenated at C1 & C3 of the propane moiety and C3′ & C4′ of the phenyl moiety. It seems that the oxygenated phenyl propanoid part is crucial for the inhibitory activity, as revealed by the other tested compounds with moderate and significant activities as phenyl propanoid derivative nitrogenated at C2 (5), the flavanone compounds 9 & 13, and the lignan compound 15.
Although ethyl acetate & petroleum ether fractions showed better inhibitory activities than that of the methylene chloride fraction, compounds 12, 14, & 6 that exhibited the highest inhibitory activities were isolated from the methylene chloride fraction. This may be explained by the antagonistic effect of these compounds together and/or with other constituents.
This study is the first that reports the in vitro promising SARS-CoV-2 Mpro inhibitory activities of compounds 12, 14, & 6 besides the moderate activities of compounds 11, 13, & 5 against the Mpro enzyme. Compounds 2 & 18 were reported for their promising inhibitory activity against the Mpro enzyme through the in silico study.54,55 This study is the first study that proves this promising activity through the in vitro assay.
2.3 Molecular docking results
Molecular docking is considered as a tool that can be used for predicting the binding mode of the tested compounds with the targeted enzymes. SARS-CoV-2 main protease (Mpro) is an enzyme responsible for proteolysis and releasing the essential functioning peptides. It is a promising target against SARS-CoV-2 due to its importance in the viral life cycle replication. Thus, Mpro inhibition can better stop the viral replication and recover the symptoms of COVID-19 disease.3–5 The crystallographic structure of Mpro complexed with the co-crystallized ligand (N3) was downloaded from Protein Data Bank (E-mail: https://www.rcsb.org; , code 6LU7). The molecular docking simulation of the isolated tested compounds (1–18) was carried out compared with the co-crystallized inhibitor (N3 ligand) and also with lopinavir (standard) to demonstrate their binding modes and affinities for Mpro (3CLpro), thus explaining their possible inhibitory activities against Mpro. The binding scores and interacting amino acid residues involved in the binding between the tested compounds (1–18) and the Mpro active pockets beside their IC50 values are summarized in Tables 1, S5 and S6.†| Compound (code) | In vitroSARS-COV-2 Mpro IC50 μM | Binding energy (kcal mol−1) (docking score) | Compound (code) | In vitroSARS-COV-2 Mpro IC50 μM | Binding energy (kcal mol−1) (docking score) |
|---|---|---|---|---|---|
| a The standard used Lopinavir. | |||||
| Lopinavira | 0.225 ± 0.01 | −9.61 | |||
| 1 | 12.51 ± 0.64 | −9.99 | 10 | 12.83 ± 0.65 | −13.45 |
| 2 | 4.185 ± 0.21 | −10.29 | 11 | 5.069 ± 0.26 | −12.48 |
| 3 | 20.67 ± 1.05 | −10.39 | 12 | 0.476 ± 0.02 | −10.79 |
| 4 | 89.99 ± 4.59 | −11.92 | 13 | 5.565 ± 0.28 | −12.81 |
| 5 | 8.532 ± 0.43 | −8.97 | 14 | 0.61 ± 0.03 | −12.96 |
| 6 | 0.917 ± 0.05 | −9.39 | 15 | 11.46 ± 0.58 | −11.69 |
| 7 | 16.31 ± 0.83 | −12.34 | 16 | 27.5 ± 1.4 | −14.34 |
| 8 | 27.86 ± 1.42 | −12.69 | 17 | 10.12 ± 0.52 | −15.61 |
| 9 | 11.83 ± 0.6 | −11.49 | 18 | 4.74 ± 0.24 | −16.24 |
Previous studies reported that the amino acid residues involved in the binding of the co-crystallized ligand (N3) with Mpro are Thr 26, Gly 143, Glu 166, and Gln 189 through hydrogen bonding.4,5 Lopinavir, the standard antiviral compound used in this study, showed H-bonding binding with Glu166 & Gln189 amino acid residues of Mpro (Fig. S6†). Compounds sharing nearly the same binding mode of the co-crystallized inhibitor (N3) and/or lopinavir with Mpro will be considered as potential Mpro (3CLpro) inhibitors, which can be used to explain the in vitro results.
The docking results (Tables 1, S5, S6† and Fig. 2a) showed that the interaction of lopinavir (reference drug, IC50 0.225 ± 0.01 μM) shared the N3 ligand in H-bonding binding with Glu166 & Gln189 amino acid residues. The most potent tested compound 12 (IC50 0.476 ± 0.02 μM & binding energy slightly higher than that of lopinavir −10.79 & −9.61 kcal mol−1, respectively) exhibited four H-bonds with Glu 166, Gln 189, Thr 190, & Arg 188 amino acid residues through the free hydroxyl groups in the phenyl moiety and in the propane diol side chain (Fig. 2b). It shared both lopinavir and N3 ligand in H-bonding binding with Glu 166 & Gln 189 amino acid residues. Compound 14 (IC50 0.61 ± 0.03 μM & binding energy −12.96 kcal mol−1 higher than that of lopinavir) exhibited seven H-bonds with Gln 189, Glu 166, Thr 26, Thr 24, Ser 46, & Gly 143 amino acid residues through the free hydroxyl groups of the glucose moiety and the oxygenated phenyl moiety besides the furan moiety (Fig. 2c). It shared both lopinavir and the N3 ligand in H-bonding binding with Gln 189 & Glu 166 amino acid residues; it shared the N3 ligand in binding with Gly 143 and Thr 26 amino acid residues. Compound 6 (IC50 0.917 ± 0.05 μM & binding energy of −9.39 kcal mol−1 nearly similar to that of lopinavir) exhibited two H-bonds with Glu 166 & Ser 144 amino acid residues (Fig. 2d). It shared lopinavir in H-bonding binding with the amino acid residue Glu 166.
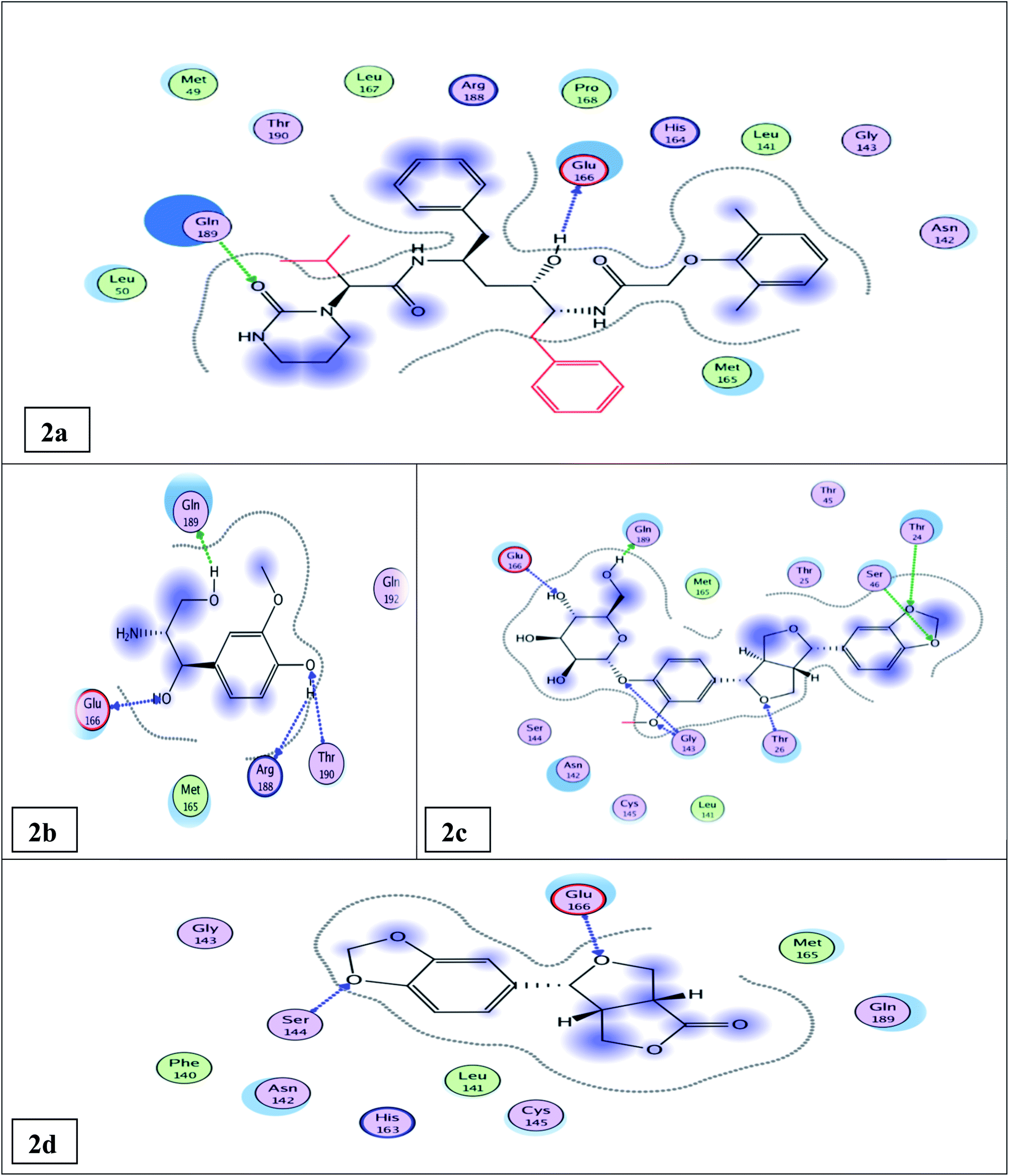 | ||
| Fig. 2 Docking results of compounds 12, 14, 6, & the standard lopinavir in the active site of SARS-CoV-2 Mpro (6LU7). 2D interactions of standard lopinavir (2a). 2D interactions of compound 12 (2b). 2D interactions of compound 14 (2c). 2D interactions of compound 6 (2d). | ||
The other isolated compounds (Tables S5 and S6†) also showed good binding interactions to Mpro active site's crucial amino acid residues, which are very close to that of the N3 ligand and the standard lopinavir drug. All the tested compounds shared both lopinavir and N3 ligand in binding with Glu166 amino acid residue through H-bonding except compounds 3 and 4. Compounds 4, 5, and 18 shared both lopinavir and N3 ligand in binding with Gln189 amino acid residue. The results of the docking scores and different interactions with amino acids of the protein pocket (two-dimensional visualization) are shown in Tables S5 and S6.† The three-dimensional visualization of the docking results and the binding pocket surface mapping were also shown to simulate ligand binding to the Mpro active pocket (Table S7†). Analyzing the docking results of all the tested compounds, compared to the ligand inhibitor N3 of Mpro and lopinavir, represented a good idea about their binding modes and affinities. However, the tested compounds showed variable binding strengths, as discussed above.
2.4 Structure activity relationship study
Observing the structure activity relationships of the isolated compounds depending on their binding affinities and binding modes to the Mpro pocket and comparing it with their in vitro inhibitory activity results against the Mpro enzyme compared to lopinavir can give an insight into the characteristic features for the compounds that can be considered as leads for designing anti-COVID-19 drugs. In this study, generally, compounds 12, 14, and 6 achieved nearly the same inhibitory activities against the Mpro enzyme compared with lopinavir; they shared the presence of the phenyl propanoid part oxygenated at C1 & C3 of the propane moiety and C3’ & C4’ of the phenyl moiety, which seemed to be potential sources for developing anti-COVID-19 drugs (Table S5†).Compounds 5 and 12, the two phenyl propanoid derivatives nitrogenated at C2 and shared both N3 ligand and lopinavir in binding with both Gln189 and Glu166 amino acid residues, showed different in vitro inhibitory activities. This may be attributed to the free hydroxyl group located at C-4′ in compound 12 that increased its binding affinities compared to 5, where this free phenolic hydroxyl binds by two hydrogen bonds with Thr190 & Arg188 amino acid residues, while the methylenedioxy group in compound 5 binds by only one hydrogen bond with Gln192 (Tables S5 and S6†). This may explain the results of the in vitro inhibitory activity assay where compound 12 showed higher inhibitory activity than 5 (IC50 value of 0.476 ± 0.02 and 8.532 ± 0.43 μM, respectively) (Fig. S51†).
A careful study of the two lignans 7 and its glucoside 14 revealed that the glucose moiety of compound 14 increased the binding affinities of this compound compared to its aglycone 7 as the glucose moiety in compound 14 shared the N3 ligand and the standard lopinavir in binding by two H-bonds with both Gln189 and Glu166 amino acid residues; in addition, it shared the N3 ligand only in binding by two H-bonds with Gly143 amino acid residue, while the aglycone 7 shared the N3 inhibitor ligand and lopinavir in binding with only Glu 166 amino acid residue (Tables S5 and S6†). This is in agreement with the results of the in vitro inhibitory activity assay where compound 14 showed an IC50 value of 0.61 ± 0.03 and compound 7 showed an IC50 value of 16.31 ± 0.83 (Tables S5 and S6†) (Fig. S53†).
Comparing the binding affinities of the two lignans 14 and 15, the methylenedioxy group in 14 increased its binding affinities compared with compound 15 that showed two methoxy groups at C-3 & 4 instead of the methylenedioxy group in compound 14. The methylenedioxy group in compound 14 is bound by the hydrogen bond with two amino acids (Thr 24 & Ser 46), while one of the two methoxy groups in compound 15 is bound by only one hydrogen bond with Thr 26. This matches with the results of the inhibitory activity assay where compound 14 showed an IC50 value of 0.61 ± 0.03 μM and compound 15 showed an IC50 value of 11.46 ± 0.58 μM (Tables S5 and S6†) (Fig. S54†).
In the steroidal compound 4, the glucose moiety decreased the activity of this compound compared to its aglycone 1, where compound 4 showed an IC50 value of 89.99 ± 4.59 μM, while 1 showed an IC50 value of 12.51 ± 0.64 μM. This revealed that the blocking of the hydroxyl group at the C-3 of sterol decreased the inhibitory activity against Mpro (3CLpro) (Tables S5 and S6†) (Fig. S52†).
3. Experimental
3.1 Reagents and apparatus
A UV-visible spectrophotometer (Shimadzu 1601 PC, model TCC-240A, Japan) was employed. ESI-HPLC-Mass, TSQ Quantum Access MAX triple stage quadrupole mass spectrometer equipped with an electrospray ionization (ESI) was operated in the positive ionization mode, Thermo Scientific, New York, USA and Accela U-HPLC system using Accela 1250 quaternary pump and Accela open autosampler (operated at 25 °C) New York, USA. Nuclear Magnetic Resonance spectra (1H-NMR, APT, DEPT-Q, HMBC, and HSQC) using TMS as an internal standard were recorded on a Bruker AV-400 spectrometer at 400 MHz for 1H and 100 MHz for 13C NMR. Compounds were dissolved in CDCl3, CD3OD, or DMSO-d6. Chemical shifts were given in ppm with a TMS as an internal standard. Column chromatography was performed on silica gel 60 (Merck, Germany) and thin-layer chromatography on precoated silica gel 60 GF254 (20 × 20 cm × 0.2 mm thick) on an aluminum sheet (Merck, Germany), RP-C18 (Merk, Germany), and Sephadex LH 20 (Pharmacia, USA). Spots were visualized by exposure to vanillin sulfuric spraying reagent.For the FRET-based activity assay, 3CL Protease (SARS-CoV-2) Assay Kit (Catalog #79955-1) was used to measure the main protease activity. The kit comes in a convenient 96-well format with purified main protease, fluorogenic substrate, and main protease assay buffer for 100 enzyme reactions. Also, lopinavir was included as a positive control. In addition, microtiter plate-reading fluorimeter was used to measure the fluorescence intensity.
(6042 Cornerstone Court West, Ste. BSan Diego CA 92121, Email: E-mail: info@bpsbioscience.com).
3.2 Preparation of the plant material
The aerial parts of H. bracteatum ornamental plant were collected from El-Orman Garden, Cairo, Egypt in June 2019. The plant identity was confirmed by Associate Prof. Dr Mahmoud Makram Qassem, Department of Vegetables & Floriculture, Faculty of Agriculture, Mansoura University, Egypt. Voucher specimens were coded as Hb-1-2019 and kept in Pharmacognosy Department, Faculty of Pharmacy, Mansoura University, Egypt.3.3 Extraction and isolation
The leaves of the plant were separated, air dried in shade, and then powdered. The dried powdered leaves (2.5 kg) were extracted with 70% hydro-alcoholic methanol (6 × 10 L) by maceration. The collected methanolic extracts were evaporated under reduced pressure to give (444 g) of a dark green viscous residue. The dried methanolic extract was dissolved in the least amount of methanol, diluted with 1000 mL distilled water, fractionated using solvents of increasing polarities such as petroleum ether (12 × 500 mL), methylene chloride (10 × 500 mL), ethyl acetate (9 × 500 mL), and finally with n-butanol (5 × 300 mL). The solvent, in each case, was evaporated to dryness under reduced pressure giving petroleum ether extract (115 g), methylene chloride extract (150 g), ethyl acetate extract (19 g), and n-butanol extract (17 g). The petroleum ether extract (70 g) was subjected to normal silica gel column chromatography, eluted with petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (100:0) till (0:100), and then ethyl acetate:methanol (100:0) till (0:100) to give two groups, namely, group 1 and group 2. Group 1, eluted with petroleum ether:ethyl acetate (90:10), when left for crystallization, precipitated a white powder (compound 1, 14 mg), while the supernatant was collected, dried, and then re-chromatographed over normal silica gel column using petroleum ether:ethyl acetate (100:0) till (0:100) to give two subgroups 1A and 1B; subgroup 1A (fractions 28–43), eluted with petroleum ether:ethyl acetate (98:2), was also re-chromatographed over a normal silica gel column using petroleum ether:ethyl acetate (100:0) till (0:100) and yielded sub-fractions (41–49), eluted with petroleum ether:ethyl acetate (88:12), which produced compound 2, 5 mg, while subgroup 1B (fractions 81–103) was further re-chromatographed over normal silica gel column using petroleum ether:methylene chloride (100:0) till (0:100), yielded sub-fractions (103–109), eluted with methylene chloride (100%), produced compound 3, 5 mg. Group 2, eluted with petroleum ether:ethyl acetate (20:80), was re-chromatographed over a normal silica gel column using methylene chloride:methanol (100:0 till 0:100), sub-fractions (35–49), eluted with methylene chloride:methanol (96:4), when left for crystallization, white substance precipitated (compound 4, 86 mg).
:ethyl acetate (100:0) till (0:100), and then ethyl acetate:methanol (100:0) till (0:100) to give two groups, namely, group 1 and group 2. Group 1, eluted with petroleum ether:ethyl acetate (90:10), when left for crystallization, precipitated a white powder (compound 1, 14 mg), while the supernatant was collected, dried, and then re-chromatographed over normal silica gel column using petroleum ether:ethyl acetate (100:0) till (0:100) to give two subgroups 1A and 1B; subgroup 1A (fractions 28–43), eluted with petroleum ether:ethyl acetate (98:2), was also re-chromatographed over a normal silica gel column using petroleum ether:ethyl acetate (100:0) till (0:100) and yielded sub-fractions (41–49), eluted with petroleum ether:ethyl acetate (88:12), which produced compound 2, 5 mg, while subgroup 1B (fractions 81–103) was further re-chromatographed over normal silica gel column using petroleum ether:methylene chloride (100:0) till (0:100), yielded sub-fractions (103–109), eluted with methylene chloride (100%), produced compound 3, 5 mg. Group 2, eluted with petroleum ether:ethyl acetate (20:80), was re-chromatographed over a normal silica gel column using methylene chloride:methanol (100:0 till 0:100), sub-fractions (35–49), eluted with methylene chloride:methanol (96:4), when left for crystallization, white substance precipitated (compound 4, 86 mg).
Methylene chloride extract (120 g) was subjected to normal silica gel column chromatography, elution was carried out using petroleum ether:ethyl acetate (100:0) till (0:100) then ethyl acetate-methanol (100:0) till (0:100) to give seven groups 1–7. Group 1 (fractions 81–97), eluted with petroleum ether:ethyl acetate (88:12), was re-chromatographed over a normal silica gel column using petroleum ether:ethyl acetate (94:6) to yield sub-fractions (33–39), when left for crystallization, precipitated a white substance (compound 5; 13 mg). Group 2 (fractions 139–155), eluted with petroleum ether:ethyl acetate (79:21), was re-chromatographed over normal silica gel column using petroleum ether:methylene chloride (60:40 till 0:100) for elution; two subgroups were obtained, namely, 2A and 2B. Subgroup 2A (fractions 65–80), eluted with petroleum ether:methylene chloride (95:5), when left for crystallization, precipitated a white needle substance (compound 6; 7 mg). Subgroup 2B (fractions 121–125), eluted with methylene chloride (100%), was re-chromatographed over normal silica gel column using petroleum ether: methylene chloride (50:50 till 0:100) for elution giving fractions (30–52), eluted with petroleum ether:methylene chloride (10:90), when left for crystallization, a white substance was precipitated (compound 7; 1 g). Group 3 (fractions 168–175), eluted with petroleum ether:ethyl acetate (76:24), was re-chromatographed over normal a silica gel column using methylene chloride:methanol (99:1) using isocratic elution, yielded two subgroups 3A and 3B. Subgroup 3A (fraction 9) was re-chromatographed over normal silica gel preparative TLC using methylene chloride:methanol (98:2), when left for crystallization, precipitated a yellowish white substance (compound 8; 7 mg). Subgroup 3B (fractions 50–80), when left for crystallization, precipitated a yellowish white substance (compound 9; 33 mg). Group 4 (fractions 180–200), eluted with petroleum ether:ethyl acetate (73:27), when left for crystallization, precipitated a yellow substance (compound 10; 20 mg). Group 5 (fractions 208–230), eluted with petroleum ether:ethyl acetate (70:30), was re-chromatographed over normal silica gel column using methylene chloride:methanol (100:0 till 98:2) to elute fractions (36–39), when left for crystallization, precipitated yellow substance (compound 11; 6 mg). Group 6 (fractions 243–276), eluted with petroleum ether:ethyl acetate (67:33), was re-chromatographed over normal silica gel column using petroleum ether:methylene chloride (5:95 till 0:100) then methylene chloride:methanol (100:0 till 0:100) yielded two subgroups 6A and 6B. Subgroup 6A (fractions 60–75), eluted with methylene chloride (100%), when left for crystallization, precipitated colorless needles (compound 12; 10 mg), while subgroup 6B (fractions 119–131), eluted with methylene chloride:methanol (98:2), when left for crystallization, a yellow powder precipitated (compound 13, 6 mg). Group 7 (fractions 360–375), eluted with ethyl acetate:methanol (95:5), was re-chromatographed over a normal silica gel column using methylene chloride:methanol (100:0 till 0:100) and produced two subgroups 7A and 7B. Subgroup 7A (fractions 31–41), eluted with methylene chloride:methanol (96:4), when left for crystallization, a white powder precipitated (compound 14, 1.5 g). Subgroup 7B (fractions 42–50), also eluted with methylene chloride:methanol (96:4), was re-chromatographed over normal silica gel preparative TLC using methylene chloride:methanol (90:10), when left for crystallization, a white substance precipitated (compound 15, 5 mg).
The ethyl acetate extract (15 g) was subjected to normal silica gel column chromatography, eluting with ethyl acetate:methanol (100:0) till (0:100) to give two groups: group 1 and group 2. Group 1 (fractions 26–39), eluted with ethyl acetate: methanol (95:5), when left for crystallization, precipitated a yellow substance (compound 16, 65 mg). Group 2 (fractions 40–45), also eluted with ethyl acetate:methanol (95:5), was re-chromatographed over a Sephadex LH 20 column using methanol (100%), yielded two subgroups 2A and 2B. Subgroup 2A (fractions 28–37), which was re-chromatographed over normal silica gel column using methylene chloride:methanol (100:0 till 0:100) to give sub-fractions (50–100), eluted with methylene chloride:methanol (85:15), when left for crystallization, precipitated a yellow powder (compound 17, 5 mg). Subgroup 2B (fractions 43–70) was re-chromatographed over reversed silica gel column RP-C18 using water:methanol (100:0 till 0:100), yielded fractions (17–23), eluted with water:methanol (95:5), when left for crystallization, a yellow substance was precipitated (compound 18, 7 mg).
3.4 FRET-based activity assay (Fluorescence Resonance Energy Transfer assay)
The inhibitory activity against the SARS-CoV-2 main protease (Mpro or 3CLpro) assay was carried out based on the FRET-based activity assay.56 The principle of the assay depends on the C-terminal of the peptide substrate being linked to a fluorophore (Edans) and the N-terminal has a fluorescence quencher (Dabcyl) that quenches the fluorescence signal of Edans. Thus, the peptide substrate exhibits low fluorescence because the fluorescence intensity of Edans in the C-terminal is quenched by the Dabcyl in the N-terminal of the substrate. When the Mpro hydrolyzes the substrate, it yields two fragments: non-fluorescent Dabcyl fragment and highly florescent Edans fragment. Consequently, an increase in the fluorescence signal proportional to the protease activity occurs. Main protease (Mpro) inhibitor causes the inhibition of fluorescent fragment release, and thus decrease intensity of the fluorescence signal. Fluorescence intensity is measured with a fluorescent microtiter plate reader capable of reading excitation/emission = 360/460 nm.573.5 Molecular modelling simulation study
The binding affinities of the isolated compounds 1–18 to SARS-CoV-2 main protease (Mpro) pocket amino acids residues were predicted by carrying out a docking experiment for them and comparing their results with the co-crystallized ligand (N3 inhibitor)4 and the standard lopinavir. Lopinavir was previously reported for its promising inhibitory activity against SARS-COV-2 Mpro enzyme through in silico computational study58 and it was also reported for inhibiting SARS-CoV-2 replication in vitro study.594. Conclusion
The methanolic extract, petroleum ether, methylene chloride, and ethyl acetate fractions of H. bracteatum leaves besides eighteen isolated and identified compounds (1–18) were in vitro evaluated for their inhibitory activities against SARS-CoV-2 main protease (Mpro) using fluorescence resonance energy transfer assay (FRET-based assay). The tested isolated compounds (1–18) included phenylpropanoid derivatives nitrogenated at C-2, lignans, and rare flavonoids. The methanolic extract and fractions exhibited promising inhibitory activities. Compounds 6, 12, and 14 showed comparable inhibitory activities against SARS-COV-2 Mpro with IC50 values of 0.917 ± 0.05, 0.476 ± 0.02, and 0.610 ± 0.03 μM, respectively, compared with the control lopinavir with an IC50 value of 0.225 ± 0.01 μM. The other tested compounds showed significant inhibitory activities. Thus, the methanolic extract of H. bracteatum leaves and the isolated phenylpropanoid derivatives, lignans, followed by flavonoids, could be considered promising natural SARS-COV-2 Mpro inhibitors. The molecular docking study for the tested compounds was carried out to correlate their binding modes and affinities for Mpro enzyme with the in vitro results. Combining the results of the in vitro and in silico studies led us to suggest the structural basis of potential inhibitors targeting SARS-COV-2 Mpro. It could be concluded that phenylpropanoids skeleton oxygenated at C3, C4 of the phenyl moiety and at C1, C3 of the propane part is the essential core of the SARS-COV-2 Mpro inhibitors and could be considered during the isolation of natural compounds, semi-synthesis, or synthesis of effective anti-COVID-19 drugs.Conflicts of interest
We declare that we have no conflict of interest.Acknowledgements
The authors would like to acknowledge assistant lecturers Mohamed A. Sabry and Eman T. Warda, for helping in molecular docking study.References
- F. Wu, S. Zhao, B. Yu, Y. Chen, W. Wang, Z. Song, Y. Hu, Z. Tao, J. Tian, Y. Pei, M. Yuan, Y. Zhang, F. Dai, Y. Liu, Q. Wang, J. Zheng, L. Xu, E. C. Holmes and Y. Zhang, Nature, 2020, 579, 265–284 CrossRef CAS PubMed.
- B. Hu, H. Guo, P. Zhou and Z. Sh, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed.
- J. Huang, G. Tao, J. Liu, J. Cai, Z. Huang and J. Chen, Front. Pharmacol., 2020, 11, 588508 CrossRef CAS PubMed.
- A. A. Zaki, A. A. Al-Karmalawy, Y. A. El-Amier and A. Ashour, New J. Chem., 2020, 44, 16752–16758 RSC.
- A. E. Allam, Y. Amen, A. Ashour, H. K. Assaf, H. A. Hassan, I. M. Abdel-Rahman, A. M. Sayed and K. Shimizu, RSC Adv., 2021, 11, 22398 RSC.
- J. O. Ogidigo, E. A. Iwuchukwu, C. U. Ibeji, O. Okpalefe and M. E. S. Soliman, J. Biomol. Struct. Dyn., 2022, 40(5), 2284–2301 CrossRef CAS PubMed.
- A. Zrig, Pharm. Chem. J., 2022, 55(10), 1080–1084 CrossRef CAS PubMed.
- A. M. Kakam, K. Franke, J. C. Ndom, E. Dongo, T. N. Mpondo and L. A. Wessjohann, Biochem. Syst. Ecol., 2011, 39, 166–167 CrossRef.
- D. A. Viegas, A. Palmeira-de-Oliveira, L. Salgueiro, J. Martinez-de-Oliveira and R. Palmeira-de-Oliveira, J. Ethnopharmacol., 2014, 151, 54–65 CrossRef PubMed.
- I. Kutluk, M. Aslanb, I. E. Orhan and B. Özçelik, S. Afr. J. Bot., 2018, 119, 252–257 CrossRef CAS.
- M. Leonardi, S. Giovanelli, K. E. Ambryszewska, B. Ruffoni, C. Cervelli, L. Pistelli, G. Flamini and L. Pistelli, Biochem. Syst. Ecol., 2018, 79, 15–20 CrossRef CAS.
- B. Najar, C. Cervelli, B. Ferri, P. L. Cioni and L. Pistelli, S. Afr. J. Bot., 2019, 124, 178–187 CrossRef CAS.
- J. Werner, W. Ebrahim, F. C. Özkaya, A. Mándi, T. Kurtán, M. El-Neketi, Z. Liu and P. Proksch, Fitoterapia, 2019, 133, 80–84 CrossRef CAS PubMed.
- G. Appendino, M. Ottino, N. Marquez, F. Bianchi, A. Giana, M. Ballero, O. Sterner, B. L. Fiebich and E. Munoz, J. Nat. Prod., 2007, 70, 608–612 CrossRef CAS PubMed.
- S. Verma, D. Twilley, T. Esmear, C. B. Oosthuizen, A. Reid, M. Nel and N. Lall, Front. Pharmacol., 2020, 11, 561334 CrossRef CAS PubMed.
- L. Bailey and the staff of the Bailey hortorium, Manual of cultivated plants, Macmillan Publishing Co. Inc., New York, 5th edn, 1975, pp. 318–320 Search PubMed.
- W. Kisiel, Planta Med., 1980, 38(3), 285–287 CrossRef CAS.
- R. Gunasegaran, A. Ubeda, M. J. Alcaraz, R. Jayaprakasam and A. G. Nair Ramachandran, Pharmazie, 1993, 48(3), 230–231 CAS.
- H. Liu, H. He, X. Yang, M. Chen and X. Hao, Nat. Prod. Res. Dev., 2007, 19(3), 423–426 CAS.
- S. G. Leitao, M. A. C. Kaplan, F. D. Monache, T. Akii-Iisa and T. Tamura, Phytochemistry, 1992, 31(8), 2813–2817 CrossRef CAS.
- V. D. Ebajo Jr, C. Shen and C. Y. Ragasa, J. Appl. Pharm. Sci., 2015, 5(4), 33–39 Search PubMed.
- S. B. Mahato and A. P. Kundu, Phytochemistry, 1994, 37, 1517–1575 CrossRef CAS.
- M. E. D. Pietro, A. Mannu and A. Mele, Processes, 2020, 8, 410 CrossRef CAS.
- R. G. Powell, C. R. Smith and I. A. Wolff, J. Am. Oil Chem. Soc., 1965, 42(3), 165–169 CrossRef CAS.
- A. R. Kim, H. J. Ko, M. A. Chowdhury, Y. Chang and E. Woo, Arch. Pharmacal Res., 2015, 38(6), 1059–1065 CrossRef CAS PubMed.
- S. E. Cellitti, D. H. Jones, L. Lagpacan, X. Hao, Q. Zhang, H. Hu, S. M. Brittain, A. Brinker, J. Caldwell, B. Bursulaya, G. Spraggon, A. Brock, Y. Ryu, T. Uno, P. G. Schultz and B. H. Geierstanger, J. Am. Chem. Soc., 2008, 130, 9268–9281 CrossRef CAS PubMed.
- F. Labeda, M. Masullob, A. Cerullib, F. Benayachea, S. Benayachea and S. Piacenteb, Nat. Prod. Commun., 2017, 12, 1605–1608 Search PubMed.
- W. D. Macrare and G. H. N. Towers, Phytochemistry, 1985, 24, 561–566 CrossRef.
- M. Chiba, S. Hisada, S. Nishibe and H. Thieme, Phytochemistry, 1980, 19, 335–336 CrossRef CAS.
- H. J. Lee, S. M. Seo, O. K. Lee, H. J. Jo, H. Y. Kang, D. H. Choi, K. H. Paik and M. Khan, Helv. Chim. Acta, 2008, 9, 2361–2366 CrossRef.
- T. Lida, Y. Noro and K. Ito, Phytochemistry, 1983, 22(1), 211–213 CrossRef.
- N. Takaku, D. Choi, K. Mikame, T. Okunishi, S. Suzuki, H. Ohashi, T. Umezawa and M. Shimada, J. Wood Sci., 2001, 47, 476–482 CrossRef CAS.
- S. W. Yoo, J. S. Kim, S. S. Kang, K. H. Son, H. W. Chang, H. P. Kim, K. Bae and C. Lee, Arch. Pharmacal Res., 2002, 25(6), 824–830 CrossRef CAS PubMed.
- M. Kim, H. T. Moon, D. G. Lee and E. Woo, Arch. Pharmacal Res., 2007, 30(4), 425–430 CrossRef CAS PubMed.
- V. Atabakia, J. Pourahmad and T. Hosseinabadia, S. Afr. J. Bot., 2021, 137, 399–405 CrossRef.
- R. Saladino, C. Fiani, C. Crestini, D. S. Argyropoulos, S. Marini and M. Coletta, J. Nat. Prod., 2007, 70, 39–42 CrossRef CAS PubMed.
- J. Jakupovic, V. P. Pathak, F. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1987, 26(3), 803–807 CrossRef CAS.
- T. Mabry, K. Markham and M. Thomas, The systematic identification of flavonoids, Springer-Verlag, Berlin, Heidelberg and New York, 1970 Search PubMed.
- K. R. Markham, Techniques of Flavonoid Identification, Biological Technique Series, Academic Press Inc., London and New York, 1982 Search PubMed.
- J. B. Harborne and T. J. Mabry, The Flavonoids: Advances in Research, Champan and Hall Ltd, 1982 Search PubMed.
- P. Agrawal, R. Thakur and M. Bansal, Carbon-13NMR of flavonoids, Elsevier Science Publishing Company Inc., 1989 Search PubMed.
- S. Takagi, M. Yamaki and K. Inoue, Yakugaku Zasshi, 1980, 100(12), 1220–1224 CrossRef CAS PubMed.
- A.-u. -Rahman, S. Hareem, M. I. Choudhary, B. Sener, A. Abbaskhan, H. Siddiqui, S. Anjum, I. Orhan, I. Gurbuz and F. Ayanoglud, Heterocycles, 2010, 82(1), 813–824 CrossRef.
- J. N. Lee, C. M. Lee, G. T. Lee and G. G. Lee, Repub. Korean Kongkae Taeho Kongbo, KR 2012078097 A 20120710, 2012 Search PubMed.
- J. N. Lee, C. M. Lee, G. T. Lee and G. G. Lee, Repub. Korean Kongkae Taeho Kongbo, KR2012078095 A 20120710, 2012 Search PubMed.
- A. Suzuki, O. Shirota, K. Mori, S. Sekita, H. Fuchino, A. Takano and M. Kuroyanagi, Chem. Pharm. Bull., 2009, 57(3), 245–251 CrossRef CAS PubMed.
- D. K. Bhardwaj, A. K. Gupta, R. K. Jain and G. C. Sharma, Curr. Sci., 1978, 47(12), 424–425 CAS.
- M. Krishnamurti, T. R. Seshadri and N. D. Sharma, Indian J. Chem., 1971, 9(2), 189–190 CAS.
- J. Jang, H. P. Kim and H. Park, Arch. Pharmacal Res., 2005, 28(8), 877–884 CrossRef CAS PubMed.
- J. Yang, Y. S. Kwon and M. J. Kim, Arabian J. Chem., 2015, 8, 407–413 CrossRef CAS.
- T. Morikawa, L. Wang, S. Nakamura, K. Ninomiya, E. Yokoyama, H. Matsuda, O. Muraoka, L. Wu and M. Yoshikawa, Chem. Pharm. Bull., 2009, 57(4), 361–367 CrossRef CAS PubMed.
- L. Verotta, L. Belvisi, V. Bertacche and M. C. Loi, Nat. Prod. Commun., 2008, 3(12), 2037–2042 CrossRef CAS.
- B. D'Abrosca, E. Buommino, G. D'Angelo, L. Coretti, M. Scognamiglio, V. Severino, S. Pacifico, G. Donnarumma and A. Fiorentino, Bioorg. Med. Chem., 2013, 21, 7038–7046 CrossRef PubMed.
- M. C. Ali, A. J. Nur, S. S. Khatun, R. Dash, M. M. Rahman and M. M. Karim, J. Adv. Biotechnol. Exp Ther., 2020, 3(4), 57–67 CrossRef.
- S. Shah, D. Chaple, S. Arora, S. Yende, C. Mehta and U. Nayak, J. Biomol. Struct. Dyn., 2021, 1–10 Search PubMed.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, Chembiochem, 2020, 21(5), 730–738 CrossRef CAS PubMed.
- W. Zhu, X. M. Miao, C. Z. Chen, H. Guo, M. Shen, X. Hu, P. Shinn, C. Klumpp-Thomas, S. G. Michael and W. Zheng, ACS Pharmacol. Transl. Sci., 2020, 3(5), 1008–1016 CrossRef CAS PubMed.
- K. A. Peele, C. P. Durthi, T. Srihansa, S. Krupanidhi, A. V. Sai, D. J. Babu, M. Indira, A. R. Reddy and T. C. Venkateswarulu, Inform. Med. Unlocked, 2020, 19, 100345 CrossRef PubMed.
- K. Choy, A. Y. Wong, P. Kaewpreedee, S. F. Sia, D. Chen, K. P. Y. Hui, D. K. W. Chu, M. C. W. Chan, P. P. Cheung, X. Huang, M. Peiris and H. Yen, Antiviral Res., 2020, 178, 104786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01213h |
| This journal is © The Royal Society of Chemistry 2022 |