
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-induced polymerization and bandgap-adjustment of TPEPA†
Jun Han‡
a,
Jieshun Cui‡b,
Qunfei Zhenga,
Zhipeng Yana,
Yun Lic,
Jian Chena,
Xiaodong Yaoa,
Guangyang Daia,
Shanmin Wanga,
Ying Liu*a,
Hsing-Lin Wang*b,
Yusheng Zhao ac and
Jinlong Zhu*ac
ac and
Jinlong Zhu*ac
aDepartment of Physics, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: zhujl@sustech.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, China
cAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen 518055, China
First published on 19th April 2022
Abstract
Organic solar cells have become an important development direction in solar cell materials because of their low cost, light weight, and good flexibility. However, the size of their bandgap is difficult to continuously regulate, resulting in a low power conversion efficiency. In this work, an organic molecule TPEPA was synthesized, and its luminescence performance and polymerization under high pressure were studied by performing in situ Raman, IR, fluorescence, and UV-vis spectroscopy. The Raman and IR spectroscopic results show that single bonds (C–H, C–Ph) and long chains (C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C) are more unstable and prone to amorphization under high pressure. At 10 GPa, the TPEPA molecule undergoes a transition of amorphization accompanied by a few polymerizations in the CC bond structure. After holding pressure at 20 GPa for one day and releasing to ambient pressure, the other peaks almost disappeared, while the new peak of C(sp3)–H from the polymerization of the benzene ring was observed, indicating that the irreversible amorphization and polymerization did occur. UV-vis spectra results show that the bandgap is reduced from 2.9 eV to 1.3 eV, which is just in the maximum conversion efficiency bandgap range (1.3–1.4 eV) of p–n junction solar cell materials. This pressure is within the working pressure range of a large volume press, which is favorable in applications of large-scale synthesis. Our strategy may provide a method for the large-scale synthesis of novel organic solar cell materials.
C–C) are more unstable and prone to amorphization under high pressure. At 10 GPa, the TPEPA molecule undergoes a transition of amorphization accompanied by a few polymerizations in the CC bond structure. After holding pressure at 20 GPa for one day and releasing to ambient pressure, the other peaks almost disappeared, while the new peak of C(sp3)–H from the polymerization of the benzene ring was observed, indicating that the irreversible amorphization and polymerization did occur. UV-vis spectra results show that the bandgap is reduced from 2.9 eV to 1.3 eV, which is just in the maximum conversion efficiency bandgap range (1.3–1.4 eV) of p–n junction solar cell materials. This pressure is within the working pressure range of a large volume press, which is favorable in applications of large-scale synthesis. Our strategy may provide a method for the large-scale synthesis of novel organic solar cell materials.
Introduction
In recent years, organic optoelectronic materials have attracted extensive attention in science and industry for their low-cost, lightweight, ultra-thin, and flexible properties.1–5 Organic materials can be precisely designed and synthesized, which can achieve multiple regulations of molecular luminescence properties.6–9 The size of the bandgap is one of the most important factors affecting the performance of an organic solar cell. Whether a material meets the Shockley–Queisser (SQ) limit bandgap range of 1.3–1.4 eV will directly affect the luminescence efficiency and application, which was proposed by Shockley and Queisser in 1961.10,11 As an independent thermodynamic parameter, high pressure can not only tailor the electronic structure but also effectively alter interactions between atoms. This enables fully continuous and controlled modulation of the bandgap to facilitate the synthesis of new materials, even as a meta-stable phase quenched to ambient conditions. For example, organic compounds containing unsaturated bonds can undergo pressure-induced polymerization (PIP) under high pressures.12–21 Similarly, high pressure enables continuous regulation of bandgap size, resulting in continuous changes in sample luminous performance, especially for organic compounds.22–25 Applying pressure on organic optoelectronic materials would profoundly modify electronic orbitals and bonding patterns, thus triggering different reactions according to the difference in the chemical bond energy.26,27Previous studies have found that tris(4-phenylethynyl)-phenyl amine (TPEPA) is a high-performance fluorescent material, and temperature can effectively regulate its bandgaps.28 However, the range of bandgap regulation is limited and does not reach the SQ limit bandgap range.10,11 Therefore, high pressure, another important thermodynamic parameter, is employed to regulate the bandgap of TPEPA, hoping that the bandgap energy can be regulated to 1.3–1.4 eV, and continuous change of fluorescence.
In this work, an organic molecule TPEPA was synthesized. TPEPA is an organic molecule with a typical aromatic conjugate system, consisting of three diphenyl acetylene connected to a nitrogen atom. The crystal structure of TPEPA polycrystalline is monoclinic, the space group is P21/C, and there are 280 atoms in the unit cell.28 The luminescence characteristics and polymerization under high pressure were studied by performing in situ Raman spectroscopy, infrared (IR) spectroscopy, fluorescence spectroscopy, and UV-visible (UV-vis) absorption spectroscopy. Raman and IR spectroscopy were performed to study the structural evolution and main vibration or rotation change processes of the functional groups. The fluorescence spectrum was then conducted to study the evolution of luminescence performance under high pressure. The relationship between luminescence properties and bandgap variation was studied by UV-vis absorption spectrum.
Experimental methods
Sample preparation and characterization
TPEPA sample was synthesized according to the literature28,29 with a slightly changed procedure (Scheme 1). In a 100 mL two-neck flask, tris(4-iodophenyl)amine (3.11 g, 5.0 mmol), phenylacetylene (2.14 g, 21.0 mmol), Pd(PPh3)4 (145 mg, 0.125 mmol) and CuI (152 mg, 0.125 mmol) were loaded under nitrogen. Tetrahydrofuran (THF, 30.0 mL) and triethylamine (TEA, 7.0 mL) were degassed and injected into the flask via cannula under nitrogen. The mixture was then heated at 80 °C under N2 for 10 h. Upon cooling to room temperature, the solvents were removed in vacuo and the residue was purified by column chromatography (silica gel, dichloromethane/hexane as the eluent) to obtain the target molecule TPEPA as a white powder (2.32 g, yield: 85%). This compound was characterized by 1H-NMR, 13C-NMR, and High-Resolution Mass Spectroscopy (HRMS) spectrums (Fig. S1–S3†). 1H-NMR (600 MHz, CDCl3) δ 7.52 (d, J = 7.8 Hz, 6H), 7.44 (d, J = 8.3 Hz, 6H), 7.34 (t, J = 7.5 Hz, 9H), 7.08 (d, J = 8.3 Hz, 6H). 13C-NMR (151 MHz, CDCl3) δ [ppm] = 146.81, 132.95, 131.66, 128.48, 128.26, 124.15, 123.53, 118.11, 89.44, 89.39. HRMS: calculated [M + H]+ 546.22163, found [M + H]+ 546.22057. | ||
| Scheme 1 Synthesis of TPEPA. | ||
In situ high-pressure experiments
We used DAC equipped with a pair of type-IIa diamond anvils with a culet size of 300 μm to generate high pressure. T-301 stainless steel gaskets were pre-indented to about 45 μm in thickness for all experiments. A sample hole with a diameter of ∼150 μm was drilled at the center of the indentation, and ruby balls were used to calibrate the pressure.30The in situ Raman spectra were collected on an Andor spectroscopy system equipped with a He–Ne laser (λ = 638 nm) without a pressure transmission medium. Because TPEPA is as soft as NaCl, which can keep the sample chamber in a quasi-hydrostatic condition. The Raman spectra were recorded with a backscattering configuration, and a 600 lines per mm grating was used. In situ IR spectra were collected on a Bruker VERTEX 70v system with an HYPERION 2000 microscope. Using Globar as a conventional source, the IR spectra were collected in transmission mode with a pressure transmission medium of KBr in the range of 600–4000 cm−1. In situ fluorescence spectra were collected on a Renishaw micro-Raman spectroscopy system with a 325 nm laser without a pressure transmission medium. In situ UV-vis spectra were collected on an Andor spectroscopy system in the range of 350–1000 nm, and silicone oil was used as the pressure transmission medium, which provides quasi hydrostatic conditions up to 10 GPa (ref. 31) and more importantly leaves a clean window for collecting a referenced background at each pressure point.
Results and discussion
In situ Raman spectra of TPEPA up to 20.0 GPa are shown in Fig. 1(a), and no obvious peaks were observed outside this range in Fig. S(4).† The evolution of Raman vibration modes upon increasing pressure was plotted in Fig. 1(b). Careful assignment of different crystal vibrational modes was identified according to the available references of diphenylacetylene in Fig. 1(c).32–36 Before 10.1 GPa, all the peaks exhibit blue-shifted under increasing pressure, indicating that the bond length becomes shorter and the vibration becomes stronger under high pressure. After 10.1 GPa, the peak of CC vibration (2223 cm−1 at 1.2 GPa) splits into a new broader peak. Simultaneously, these two anti-symmetrically coupled C–Ph stretches (a* vibration in Fig. 1(c) ∼1250 cm−1 at 1.2 GPa) are disappeared, indicating that amorphous and polymeric reactions began thereafter. Furthermore, these a* vibrations of C–Ph stretches exhibit the biggest blue shift rate, and their intensities decrease sharply upon the increase of pressure, indicating that the a* vibrations in diphenylacetylene are easy to be compressed and destroyed under high pressure. In addition, the CC bond vibration splits into two peaks and disappears at ∼13 GPa, indicating that it is also slightly prone to react under high pressures. However, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond vibrations on benzene rings (∼1597 cm−1 at 1.2 GPa) and the symmetric C–Ph stretch (1141 cm−1 at 1.2 GPa) are disappeared at the pressure of ∼20 GPa, indicating that the benzene ring and symmetric C–Ph are the relatively stable groups, and less likely to be destroyed under high pressure. Thus, we summarized propose that the reaction of the CC bond occurred firstly in the TPEPA molecule at 10 GPa, which causes anti-symmetrically coupled of C–Ph stretches to be destroyed firstly under high pressure, while symmetrically C–Ph stretches and benzene ring vibration are little affected. Until 20 GPa, completely amorphization occurred in the whole organic molecule crystals. After holding for one day at 20.0 GPa, the sample was decompressed, and no Raman peaks were observed during the whole decompression process. This indicates that most of the samples undergo an irreversible amorphous process, and a few samples might undergo polymerization under high pressure.
C bond vibrations on benzene rings (∼1597 cm−1 at 1.2 GPa) and the symmetric C–Ph stretch (1141 cm−1 at 1.2 GPa) are disappeared at the pressure of ∼20 GPa, indicating that the benzene ring and symmetric C–Ph are the relatively stable groups, and less likely to be destroyed under high pressure. Thus, we summarized propose that the reaction of the CC bond occurred firstly in the TPEPA molecule at 10 GPa, which causes anti-symmetrically coupled of C–Ph stretches to be destroyed firstly under high pressure, while symmetrically C–Ph stretches and benzene ring vibration are little affected. Until 20 GPa, completely amorphization occurred in the whole organic molecule crystals. After holding for one day at 20.0 GPa, the sample was decompressed, and no Raman peaks were observed during the whole decompression process. This indicates that most of the samples undergo an irreversible amorphous process, and a few samples might undergo polymerization under high pressure.
 | ||
| Fig. 1 (a) Selected Raman spectra of TPEPA upon compression and decompression, (b) frequency shifts of Raman modes as a function of pressure. (c) Characteristic region: functional group vibration labeling. | ||
To avoid fluorescence interference, the infrared spectrum is used to obtain more spectral peaks vibration information. The in situ IR measurements were carried out up to 20.5 GPa, as shown in Fig. 2(a). Similar to the Raman results, all the peaks before 10 GPa were blue-shifted and broadened under compression. After 10 GPa, most vibrations of C–H, C–C, and CC bonds were red-shifted and gradually disappeared in Fig. 2(b), indicating that the TPEPA molecule was distorted, so that these vibrations directions were easier to vibrate and rotate. It can be seen from Fig. 2(b) that, the skeleton vibration peak of the benzene ring (in the range of 1400–1600 cm−1) still holds a steady change after 10 GPa, and gradually disappears at 20 GPa, while most C–H, C–Ph, and N–Ph vibration peaks show large redshift after 10 GPa. It indicates that after 10 GPa, the vibration of the benzene ring remains stable and its local chemical environment was still preserved, while these chain structures undergo amorphization and partial polymerization, which is consistent with the results of Raman spectroscopy. At about 20 GPa, most of the vibrational models of the TPEPA molecule disappeared, indicating that the sample was completely amorphous. Meanwhile, it can be seen from Fig. 2(c) that after 10 GPa, the sample showed a dark color, corresponding to the process of amorphization and polymerization. When compressed to 20 GPa and even released to ambient pressure, the color of the sample remains dark yellow, indicating that the amorphization and polymerization process was irreversible. After releasing pressure from holding 20.5 GPa for one day, the peak centered at 2207 cm−1 (CC stretching) disappeared, and a new peak at 2926 cm−1 (C(sp3)–H stretching) was observed in Fig. 2(d), demonstrating that the C–H vibration with sp2 transformed to sp2 + sp3 during polymerization. CC bond in the chain structure might be transformed into single or double-bonded amorphous network polymers (vibration in the range of 1200–1600 cm−1). This C(sp3)–H vibration is likely to arise from the polymerization of the unsaturated CC bond in the benzene ring. Raman spectroscopy showed that the TPEPA molecule was completely amorphous after high pressure, and no spectral peak was detected. While the infrared spectroscopy (region average measurement) showed that most samples were amorphous after compression, there was still an obvious new peak of the polymerized quenched sample.
 | ||
| Fig. 2 (a) Selected infrared absorption spectra of TPEPA upon compression and decompression, (b) frequency shifts of IR peaks as a function of pressure in 680–840 cm−1 and 1200–3150 cm−1, (c) optical microscopic images of TPEPA at different pressures, (d) before and after the high pressure of pure sample by infrared spectrum. | ||
Summarily, Raman and IR results show that single bonds (C–H, C–Ph) and long chains (C–CC–C) are more unstable and prone to amorphization under high pressure. After 10 GPa, amorphization accompanied by partial polymerization in CC bond structure began to take place. After 20 GPa, the irreversible amorphous process of the sample occurs.
To study the influence of the amorphous process on TPEPA luminescence performance, high-pressure in situ fluorescence spectroscopy was performed, as shown in Fig. 3. There are two peaks in the fluorescence spectrum at ∼470 nm and ∼501 nm respectively, and the corresponding sample produces blue and green light when excited. The peak at 470 nm is probably from the electron transition from π* antibonding orbital to π bonding orbital (π* → π) in the vibration of the conjugated π bond from the benzene ring. The peak at 501 nm corresponds to the π* → π electron transition from the carbon–carbon triple bond. The peak at 470 nm red-shifted from 0.2 GPa at 470 nm (cyan) to 10 GPa at 550 nm (green), and the fluorescence intensity was decreasing continuously during compression. Before 2.1 GPa, the fluorescence emission intensity of the benzene ring is weaker than that of the CC bond, and then exceeds that of the CC bond vibration thereafter. This indicates that the benzene ring is more stable, which is consistent with the Raman and IR data results. After 10 GPa, the spectral peaks disappeared basically, and the corresponding sample began to undergo an amorphous and polymerization process. After being pressurized to 20 GPa and held for one day, the spectrum peak did not appear again, and TPEPA was completely amorphous under high pressure. If we want to obtain the fluorescence enhancement phenomenon of TPEPA, we can introduce hydroxyl or groups containing N, and F atoms into the end group of the TPEPA molecule to make it conducive to forming hydrogen bonds under high pressure. Thus, intermolecular vibration can be reduced, the non-radiative transition can be reduced, and fluorescence can be enhanced.37,38 In summary, the π–π stacking interaction in TPEPA was enhanced by pressure, resulting in a significant decrease in fluorescence intensity and irreversible amorphization.
 | ||
| Fig. 3 Fluorescence spectra of TPEPA under different pressures, (a) before 5.6 GPa, (b) after 5.6 GPa. | ||
Finally, to study the effect of high pressure on the accompanying regulation of bandgap during the amorphous process of TPEPA. In situ UV-vis spectra were performed to demonstrate the bandgap evolution as a function of pressure, and the bandgap was fitted according to the Kubelka–Munk function and Tauc plot,39–41 as shown in Fig. 4. It can be seen that this unique peak is the result of the transition of π orbital electrons to π* antibonding in the conjugated system of the benzene ring and CC bond, which can be expressed as π → π*. With the increase of pressure, this peak red-shifts and the corresponding bandgap gradually decreased from 2.9 eV at 0.2 GPa to 1.3 eV at 20.4 GPa. In addition, an obvious mutation occurred at 10 GPa, corresponding to the changes of Raman, IR, and fluorescence spectra, which means an irreversible amorphization began at this pressure point. This pressure is within the working range of a large volume press, which is conducive to large-scale synthesis application. After holding pressure for one day, the bandgap remains at 1.3 eV after dropping to ambient pressure, which is in the maximum conversion efficiency bandgap range (1.3–1.4 eV) of p–n junction solar cell materials.10,11
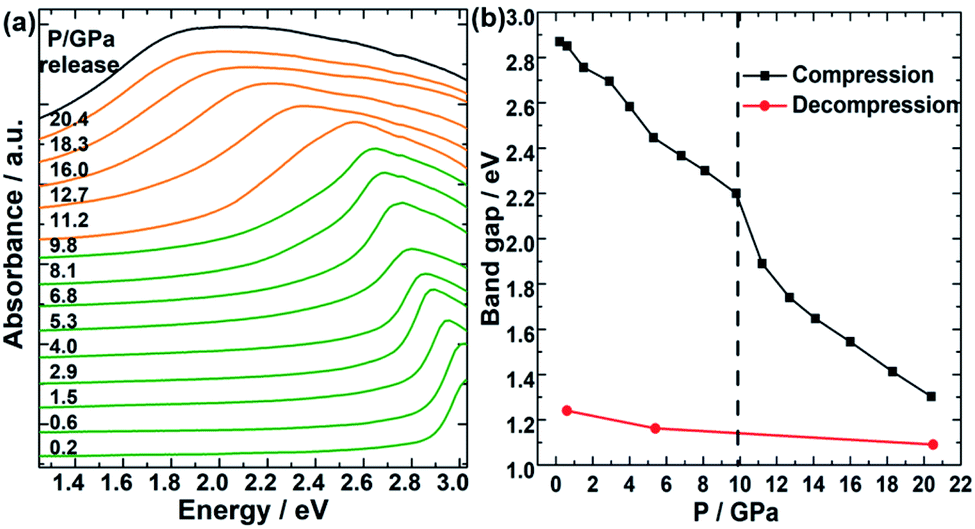 | ||
| Fig. 4 (a) Selected in situ UV-vis patterns of TPEPA under compression. (b) The bandgap of the TPEPA is under high pressure. | ||
Conclusions
In summary, an organic molecule TPEPA was synthesized, and its amorphization and polymerization were studied under high pressure, using in situ Raman, IR, fluorescence, and UV-vis spectroscopy. The Raman and IR results show that the CC bond is the first bond to start the amorphization and polymerization, and C–H (sp2) bond vibration transforms to C–H (sp2 + sp3) bond vibration due to the polymerization of the benzene ring after high pressure, which means that an amorphous state with partially irreversible polymerization has occurred. The polymerization of unsaturated organic compounds under high pressure will irreversibly generate new products so that the properties obtained under high pressure can be partially retained. The fluorescence spectrum peak of π* → π electron transition was red-shifted and gradually disappeared from 470 nm (cyan) at 0.2 GPa to 550 nm (green) at 10 GPa, which means that TPEPA was completely amorphous under high pressure. The UV-vis spectroscopy shows that the bandgap is reduced from 2.9 eV to 1.3 eV, which is in the maximum conversion efficiency bandgap range (1.3–1.4 eV) of the p–n junction solar cell materials. Our researches show that TPEPA can provide a reference for the large-scale synthesis of novel organic solar cell materials.
Author contributions
Jun Han, Ying Liu, and Jinlong Zhu designed the research, analyzed the data, and wrote the paper; Jun Han and Qunfei Zheng did the experiments; Jieshun Cui and Hsing-Lin Wang synthesized the sample; Jian Chen, and Shanmin Wang, and Yusheng Zhao provide some equipments; Zhipeng Yan, Yun Li, Xiaodong Yao, and Guangyang Dai help analyze the data with software. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the open funding of Xi'an Modern Chemistry Research Institute (Grants No. SYJJ200303), the National Natural Science Foundation of China grant 11904281, and the Major Science and Technology Infrastructure Project of Material Genome Big-science Facilities Platform supported by Municipal Development and Reform Commission of Shenzhen. J. Cui also gratefully acknowledges financial support from the Shenzhen Basic Research Program (JCYJ20190809144215761).Notes and references
- N. Keller and T. Bein, Optoelectronic processes in covalent organic frameworks, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- J. Yang, M. Fang and Z. Li, Organic luminescent materials: the concentration on aggregates from aggregation-induced emission, Aggregate, 2020, 1, 6–18 CrossRef.
- Y. F. Liu, J. Feng, Y. G. Bi, D. Yin and H. B. Sun, Recent Developments in Flexible Organic Light-Emitting Devices, Adv. Mater. Technol., 2019, 4, 1800371 CrossRef.
- J. Sun, J. Wu, X. Tong, F. Lin, Y. Wang and Z. M. Wang, Organic/Inorganic Metal Halide Perovskite Optoelectronic Devices beyond Solar Cells, Adv. Sci., 2018, 5, 1700780 CrossRef PubMed.
- Q. Li and Z. Li, Molecular packing: another key point for the performance of organic and polymeric optoelectronic materials, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- Y. Xiong, Q. Liao, Z. P. Huang, X. Huang, C. Ke, H. T. Zhu, C. Y. Dong, H. S. Wang, K. Xi, P. Zhan, F. Xu and Y. Q. Lu, Ultrahigh responsivity photodetectors of 2D covalent organic frameworks integrated on graphene, Adv. Mater., 2020, 32, 1907242 CrossRef CAS PubMed.
- J. D. Servaites, M. A. Ratner and T. J. Marks, Organic solar cells: a new look at traditional models, Energy Environ. Sci., 2011, 4, 4410–4422 RSC.
- W. Zhao, Z. He and B. Z. Tang, Room-temperature phosphorescence from organic aggregates, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- R. Xue, J. Zhang, Y. Li and Y. Li, Organic Solar Cell Materials toward Commercialization, Small, 2018, 14, 1801793 CrossRef PubMed.
- S. Rühle, Tabulated values of the Shockley–Queisser limit for single junction solar cells, Sol. Energy, 2016, 130, 139–147 CrossRef.
- W. Shockley and H. J. Queisser, Detailed Balance Limit of Efficiency of p-n Junction Solar Cells, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- W. J. Evans, M. J. Lipp, C. S. Yoo, H. Cynn, J. L. Herberg, R. S. Maxwell and M. F. Nicol, Pressure-Induced Polymerization of Carbon Monoxide: Disproportionation and Synthesis of an Energetic Lactonic Polymer, Chem. Mater., 2006, 18, 2520–2531 CrossRef CAS.
- C. S. Yoo and M. Nicol, Kinetics of a pressure-induced polymerization reaction of cyanogen, J. Phys. Chem., 1986, 90, 6732–6736 CrossRef CAS.
- C. S. Sundar, P. C. Sahu, V. S. Sastry, G. V. N. Rao, V. Sridharan, M. Premila, A. Bharathi, Y. Hariharan, T. S. Radhakrishnan, D. V. S. Muthu and A. K. Sood, Pressure-induced polymerization of fullerenes: a comparative study of C60 and C70, Phys. Rev. B, 1996, 53, 8180–8183 CrossRef CAS PubMed.
- M. J. Lipp, W. J. Evans, B. J. Baer and C. S. Yoo, High-energy-density extended CO solid, Nat. Mater., 2005, 4, 211–215 CrossRef CAS PubMed.
- D. Chelazzi, M. Ceppatelli, M. Santoro, R. Bini and V. Schettino, Pressure-Induced Polymerization in Solid Ethylene, J. Phys. Chem. B, 2005, 109, 21658–21663 CrossRef CAS PubMed.
- A. Friedrichx, I. E. Collings, K. F. Dziubek, S. Fanetti, K. Radacki, J. Ruiz-Fuertes, J. Pellicer-Porres, M. Hanfland, D. Sieh, R. Bini, S. J. Clark and T. B. Marder, Pressure-Induced Polymerization of Polycyclic Arene–Perfluoroarene Cocrystals: Single Crystal X-ray Diffraction Studies, Reaction Kinetics, and Design of Columnar Hydrofluorocarbons, J. Am. Chem. Soc., 2020, 142, 18907–18923 CrossRef PubMed.
- J. Han, X. Tang, Y. Wang, Y. Wang, Y. Han, X. Lin, X. Dong, H. H. Lee, H. Zheng, K. Li and H. K. Mao, Pressure-Induced Polymerization of Monosodium Acetylide: A Radical Reaction Initiated Topochemically, J. Phys. Chem. C, 2019, 123, 30746–30753 CrossRef CAS.
- H. Zheng, L. Wang, K. Li, Y. Yang, Y. Wang, J. Wu, X. Dong, C.-H. Wang, C. A. Tulk, J. J. Molaison, I. N. Ivanov, M. Feygenson, W. Yang, M. Guthrie, Y. Zhao, H.-K. Mao and C. Jin, Pressure induced polymerization of acetylide anions in CaC2 and 107 fold enhancement of electrical conductivity, Chem. Sci., 2017, 8, 298–304 RSC.
- W. S. Tang and T. A. Strobel, Pressure-induced solid-state polymerization of optically-tunable diphenyl-substituted diacetylene, ACS Appl. Polym. Mater., 2019, 1, 3286–3294 CrossRef CAS.
- M. D. Ward, W. S. Tang, L. Zhu, D. Popov, G. D. Cody and T. A. Strobel, Controlled Single-Crystalline Polymerization of C10H8·C10F8 under Pressure, Macromolecules, 2019, 52, 7557–7563 CrossRef CAS.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Piezochromic Luminescence Based on the Molecular Aggregation of 9,10-Bis((E)-2-(pyrid-2-yl)vinyl)anthracene, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed.
- C. Liu, G. Xiao, M. Yang, B. Zou and Z. J. Zhang, Mechanofluorochromic Carbon Nanodots: Controllable Pressure-Triggered Blue-and Red-Shifted Photoluminescence, Angew. Chem., Int. Ed., 2018, 57, 1893 CrossRef CAS PubMed.
- P. McMillan, New materials from high-pressure experiments, Nat. Mater., 2002, 1, 19–25 CrossRef CAS PubMed.
- D. Kim, S. Stefanoski, O. Kurakevych and T. A. Strobel, Synthesis of an open-framework allotrope of silicon, Nat. Mater., 2015, 14, 169–173 CrossRef CAS PubMed.
- J. Aiyasami, Diamond anvil cell and high-pressure physical investigations, Rev. Mod. Phys., 1983, 55, 65 CrossRef.
- H. K. Mao, X. J. Chen, Y. Ding, B. Li and L. Wang, Solids, liquids, and gases under high pressure, Rev. Mod. Phys., 2018, 90, 015007 CrossRef CAS.
- X. Li, J. Cui, Q. Ba, Z. Zhang, S. Chen, G. Yin, Y. Wang, B. Li, G. Xiang, K. S. Kim, H. Xu, Z. Zhang and H. L. Wang, Multiphotoluminescence from a Triphenylamine Derivative and Its Application in White Organic Light-Emitting Diodes Based on a Single Emissive Layer, Adv. Mater., 2019, 31, 1900613 CrossRef PubMed.
- R. Misra, R. Maragani, P. Gautam and S. M. Mobin, Tetracyanoethylene substituted triphenylamine analogues, Tetrahedron Lett., 2014, 55, 7102–7105 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, Calibration of the ruby pressure gauge to 800 kbar under quasi-hydrostatic conditions, J. Geophys. Res.: Solid Earth, 1986, 91, 4673–4676 CrossRef CAS.
- S. Klotz, J. C. Chervin, P. Munsch and G. L. Marchand, Hydrostatic limits of 11 pressure transmitting media, J. Phys. D., 2009, 42, 075413 CrossRef.
- H. Hiura and H. Takahashi, Time-resolved resonance Raman spectroscopy of diphenylacetylene: structures and dynamics of the lowest excited triplet state, radical cation, and radical anion, J. Phys. Chem., 1992, 96, 8909–8915 CrossRef CAS.
- A. Shimojima and H. Takahashi, Ab initio study of geometries and force fields of diphenylacetylene in the ground state, radical cation, radical anion, and lowest excited triplet state, J. Phys. Chem., 1993, 97, 9103–9112 CrossRef CAS.
- G. Baranović, L. Colombo and D. Skare, A valence force field for phenylalkynes: part II. Fundamental frequencies of phenylacetylene and tolane and molecular conformation of tolane in solution, J. Mol. Struct., 1986, 147, 275–300 CrossRef.
- N. C. Craig and S. V. Krasnoshchekov, Vibrational spectroscopy of tolane; Coriolis coupling between Raman-active modes of g symmetry, Mol. Phys., 2019, 117, 1059–1068 CrossRef CAS.
- M. Flock, L. Bosse, D. Kaiser, B. Engels and I. Fischer, A time-resolved photoelectron imaging study on isolated tolane: observation of the biradicalic 1Au state, Phys. Chem. Cheml. Phys., 2019, 21, 13157–13164 RSC.
- S. Zhang, Y. Dai, S. Luo, Y. Gao, N. Gao, K. Wang, B. Zou, B. Yang and Y. Ma, Rehybridization of Nitrogen Atom Induced Photoluminescence Enhancement under Pressure Stimulation, Adv. Funct. Mater., 2017, 27, 1602276 CrossRef.
- Y. Gu, K. Wang, Y. Dai, G. Xiao, Y. Ma, Y. Qiao and B. Zou, Pressure-induced emission enhancement of carbazole: the restriction of intramolecular vibration, J. Phys. Chem. Lett., 2017, 8, 4191–4196 CrossRef CAS PubMed.
- A. L. Companion, Theory and applications of diffuse reflectance spectroscopy, Developments in Applied Spectroscopy, Springer, Boston, MA, 1965. pp. 221–234 Search PubMed.
- P. Makuła, M. Pacia and W. Macyk, How To Correctly Determine the Band Gap Energy of Modified Semiconductor Photocatalysts Based on UV-vis Spectra, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- P. Kubelka and F. Z. Munk, An article on optics of paint layers, Z. Tech. Phys., 1931, 12, 259–274 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01144a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |