
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Construction of sulfur-containing N-vinylimides: N-addition of imides to propargyl sulfonium salts†
Shou-Jie Shen *a,
Le-Mei Wanga,
Guo-Mei Gonga,
Yan-Jiao Wanga,
Jin-Yan Liang*b and
Jun-Wen Wang*a
*a,
Le-Mei Wanga,
Guo-Mei Gonga,
Yan-Jiao Wanga,
Jin-Yan Liang*b and
Jun-Wen Wang*a
aKey Laboratory of Magnetic Molecules, Magnetic Information Materials Ministry of Education, The School of Chemical and Material Science, Shanxi Normal University, Linfen, 041004, China. E-mail: shoujie_shen@outlook.com
bCollege of Life Science, Shanxi Normal University, Linfen, 041004, China
First published on 26th April 2022
Abstract
An N-addition reaction between imides and propargyl sulfonium salts was developed to afford sulfur-containing N-vinylimides with moderate to excellent yields. Under the activation of NaOAc·3H2O, imides could undergo deprotonation and propargyl sulfonium salts could isomerize to allenic sulfonium salts. The N-nucleophilic attack initiates the reaction and gives the desired products. Various imides, including arylimides, aliphatic imides and N-(arylsulfonyl) alkyl acylamides, and even bioactive saccharin, thalidomide and pomalidomide could provide organosulfur N-vinylimides compounds. The simple, mild and metal-free reaction conditions, the broad scope of substrates, gram-scale synthesis and convenient transformation embody the synthetic superiority of this process.
N-vinylimides represent crucial structural motifs due to the importance of these frameworks (Fig. 1), which are the core structure found in biologically active structures,1 functional materials2 and natural products such as the parazoanthines A–E.3 They can also serve as versatile synthetic intermediates in the synthesis of β-2-amino acid derivatives4 and other complex structures.5
 | ||
| Fig. 1 Representative functional N-vinylimides scaffolds. | ||
Due to the importance of these N-vinylimides frameworks, the growing interest in this featured moiety has catalyzed a recent spurt of attention for methodology appropriate for its construction. Conventionally, protocols for the synthesis of this important substrate class included transition-metal-catalyzed en-imidic C(sp2)–N bond formation reactions of imides with vinyl halides, pseudohalides or alkynes. The main strategies involved Cu-catalyzed Chan–Lam–Evans reactions,6 Ru-catalyzed hydroimidation reaction of imide with alkyne7 and Pd-catalyzed oxidative amination of alkenes.8 In 2021, Sandtorv's group reported Cu-catalyzed Chan–Lam–Evans reaction for coupling cyclic imides and alkenylboronic acids by forming C(sp2)–N-bonds, enables the practical and mild preparation of (E)-enimides (Scheme 1a).6a In 2020, Schaub's group reported Ru-phosphine catalyzed hydroimidation reaction of cyclic amides with acetylene under low pressure, affording new method for synthesis of N-vinylimides (Scheme 1b).7a Hull's group reported Pd-catalyzed anti-Markovnikov oxidative amination reaction, alkenes are shown to react with imides in the presence of a palladate catalyst to generate the terminal imide, providing mild and robust complementary routes (Scheme 1c).8a Besides, rarely examples of organo-catalytic conjugate additions of imides to acetylene can also provide methods for the synthesis of N-vinylimides.9
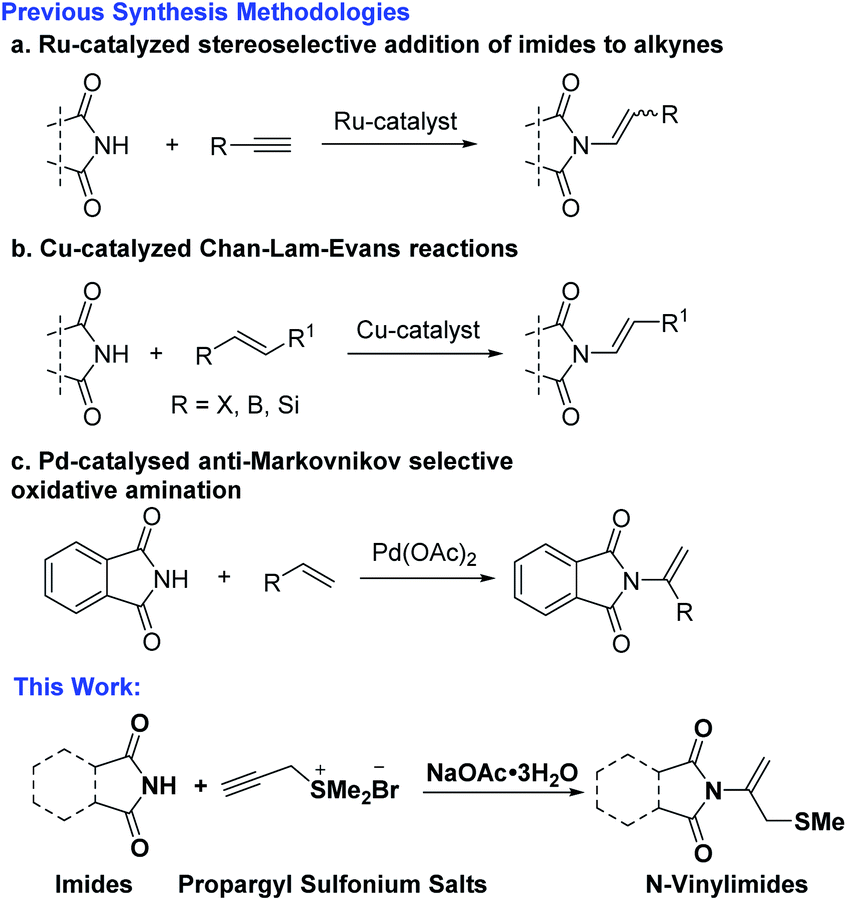 | ||
| Scheme 1 Approaches for vinylation of imides. | ||
The previously reported synthesis strategies mainly involved the use of expensive Ru and Pd-catalysts, otherwise toxic copper catalyst, and the structural limitations imposed to phthalimide and therefore specialized. Considering the limitation in generality, the harsh reaction condition, and the use of metal-catalysis decreased the attractiveness for synthetic applications, the development of new kind of vinylation reagents and their application of building N-vinylimides in a simple, mild, metal-free and efficient manner are highly desirable.
Our earlier work inspired our interest in synthesis of N-vinylimides by employing propargyl sulfonium salts as vinylation reagent. We have been exploring new reaction patterns of sulfonium salts and developed propargyl sulfonium salts involved [3 + 2] annulation/substitution reaction and N-addition/[2,3]-sigmatropic rearrangement reaction in an acyclic model.10 Based on our processive interests on constructing N-functionalized vinylation reaction and exploring the diverse reactive pathway of propargyl sulfonium salts, we herein report the realization of inorganic base promoted N-addition reaction of imides and propargyl sulfonium salts, delivering potential bioactive sulfur-containing N-vinylimides in moderate to excellent yields (Scheme 1).
We began our investigation by selecting phthalimide 1a and propargyl sulfonium salt 2a as model substrates (Table 1). When phthalimide 1a (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.) in CH3CN (3.0 mL, c = 0.1 M) were mixed, the reaction mixture was stirred for 10 min at 22 °C and propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred for 6 h continuously, phthalimide 1a was consumed completely and afforded N-addition product 3a with 71% yield (Table 1). Extensive exploration of a range of bases indicated that NaOAc·3H2O was the most efficient to promote the process. Replacement of NaOAc·3H2O with anhydrous NaOAc, Na2CO3, K2CO3, Cs2CO3, KOH and lithium carbonate gave related product with 24–46% yields (Table 1, entries 1–7). Replacement of either stronger inorganic bases such as NaH and KOtBu, or organic base Et3N and DBU could not promote the reaction efficiently (Table 1, entries 8–11, yields from 33 to 40%). Screening of solvents including THF, CHCl3, DCE, DCM, and toluene did not give better yield (Table 1, entries 12–16, yields from trace to 37%). Temperature affected the reaction greatly and the desired product was obtained only in 11% yield when the reaction was conducted at 22 °C (entry 17 in Table 1). As the temperature was risen to 30 and 60 °C, the product 3a was obtained gradually to 23 and 61%, respectively (Table 1, entries 18 and 19). When the temperature was risen to 80 and 90 °C, the reaction efficiency was slightly decreased to 51 and 49%, respectively (entries 20 and 21 in Table 1). We also probed the influence of the ratio of sulfonium salts 2a and NaOAc·3H2O (for reaction details, see ESI†).
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Temp. (°C) | Yieldb (%) |
a Unless otherwise noted, the reactions were performed under air and imide 1a (0.3 mmol, 1.0 equiv.), base (0.45 mmol, 1.5 equiv.) in solvent (3.0 mL, c = 0.1 M) were mixed, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6 h until starting material 1a was fully consumed (monitored by TLC).b Isolated yield. DCE: 1,2-dichloroethane; DCM: dichloromethane.c With the ratio of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a:NaOAc·3H2O = 1:1.5:1.5. :2a:NaOAc·3H2O = 1:1.5:1.5. |
||||
| 1 | NaOAc | CH3CN | 50 | 45 |
| 2 | Na2CO3 | CH3CN | 50 | 42 |
| 3 | K2CO3 | CH3CN | 50 | 46 |
| 4 | Cs2CO3 | CH3CN | 50 | 39 |
| 5 | KOH | CH3CN | 50 | 24 |
| 6 | LiOAc·2H2O | CH3CN | 50 | 33 |
| 7 | LiOAc | CH3CN | 50 | 42 |
| 8 | NaH | CH3CN | 50 | 40 |
| 9 | KOtBu | CH3CN | 50 | 37 |
| 10 | Et3N | CH3CN | 50 | 36 |
| 11 | DBU | CH3CN | 50 | 33 |
| 12 | NaOAc·3H2O | THF | 50 | 37 |
| 13 | NaOAc·3H2O | CHCl3 | 50 | 34 |
| 14 | NaOAc·3H2O | DCE | 50 | 28 |
| 15 | NaOAc·3H2O | DCM | 50 | 35 |
| 16 | NaOAc·3H2O | Toluene | 50 | Trace |
| 17c | NaOAc·3H2O | CH3CN | 22 | 11 |
| 18c | NaOAc·3H2O | CH3CN | 30 | 23 |
| 19c | NaOAc·3H2O | CH3CN | 60 | 61 |
| 20c | NaOAc·3H2O | CH3CN | 80 | 51 |
| 21c | NaOAc·3H2O | CH3CN | 90 | 49 |
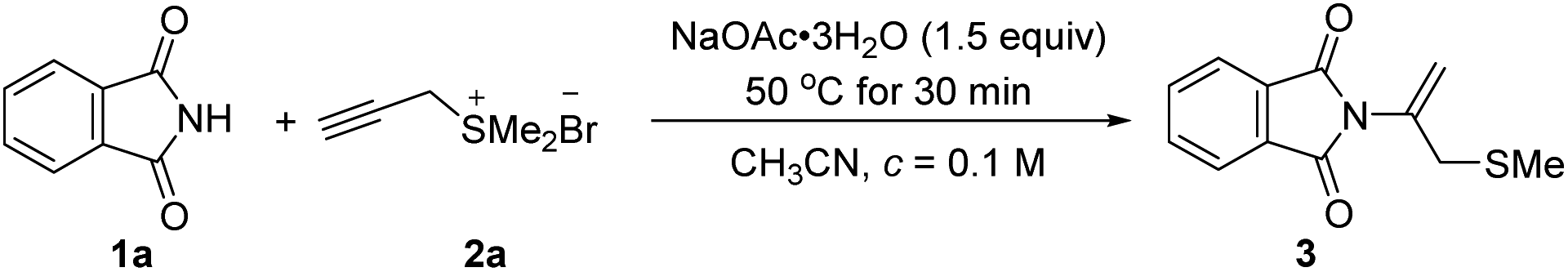
Having established the optimized conditions, we commenced to explore the substrate scope of the reaction. A selection of arylimides and aliphatic imides was next investigated with propargyl sulfonium salt 2a in Scheme 2. Generally, arylimides containing electron-withdrawing group such as tetrachloro-, 4-bromo-, 4-nitro- and 3-nitrophthalimide provided desired N-vinylimides products 3b–3e with moderate yields (Scheme 2, 3b–3e, with yields of 27–52%), probably due to the electron withdrawing effect of substituents. 1,8-Naphthalimide and 2,3-naphthalimide were well-tolerated to provide N-vinylimide products 3f and 3g with 62 and 65% yields, respectively. 3,4-Pyridinedicarboximide could also be engaged in the reaction to obtain 3h with yield of 43%. Subsequently, we went on to evaluate the reactivity of aliphatic imides. Unexpected, maleimide could not provide the desired N-nucleophilic addition product under the optimized conditions with recovering of the starting material. Oppositely, the method was high yielding and tolerable to succinimide and substituted succinimides. Succinimide and substituted succinimides worked well to deliver N-vinylimides products 3j–3o with moderate to excellent yields of 52–93%.11 Continuously, we evaluated the reactive effectiveness of glutarimide and substituted glutarimides. Under optimized conditions, glutarimide and substituted glutarimides could also react with 2a and give desired products 3p, 3q and 3r with yields of 32, 24 and 24%, respectively.
 | ||
| Scheme 2 Scope of imidesa. aUnless otherwise noted, the reactions were performed under air and imide 1 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.) in CH3CN (3.0 mL, c = 0.1 M) were mixed, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6 h until starting material 1 was fully consumed (monitored by TLC). bIsolated yield. | ||
Surprising reaction appeared when we explored the reaction of tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione 1s and 1t (Scheme 3).12 Under the optimized conditions, 1s worked well with propargyl sulfonium salt 2a to deliver the corresponding product 3s with yield of 65%. Under the same conditions, compound 1t gave the desired N-vinylimide product 3t with yield of 35%, meanwhile with the unexpected N-vinylimide product 3i with yield of 16%, which unavailable in the reaction of maleimide with propargyl sulfonium salt 2a, probably due to the retro-Diels–Alder reaction of 1t with generation of maleimide intermediate.
 | ||
| Scheme 3 Scope of imides. | ||
The reaction performance could also be adapted to N-(arylsulfonyl) alkyl acylamides (Scheme 4). The method smoothly transferred electron-deficient aryl sulfonyl acylamides to form N-vinylimide products such as 5a–5e in moderate yields. In contrast, when N-(arylsulfonyl) aryl acylamides 5i and N-(arylacyl) alkyl acylamides 5j–5l were involved, the reaction was sluggish and no desired N-vinylimide products could be obtained probably due to its low nucleophilicity (Scheme 4, 5i–5l).
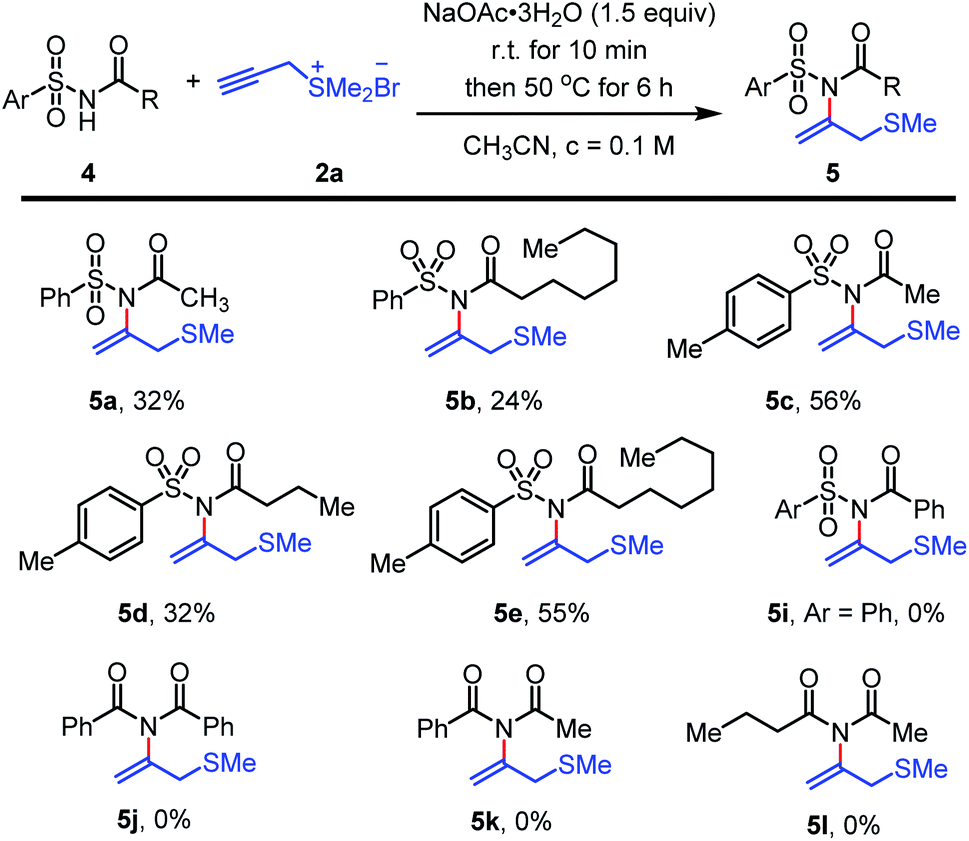 | ||
| Scheme 4 Scope of aryl sulfonyl amides and carbonimidesa, aUnless otherwise noted, the reactions were performed under air and imide 4 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.) in CH3CN (3.0 mL, c = 0.1 M) were mixed, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6 h until starting material 4 was fully consumed (monitored by TLC). bIsolated yield. | ||
To further broaden the scope of the reaction, other representative propargyl sulfonium salts were also investigated (Scheme 5). Trimethylsilyl contained propargyl sulfonium salt 2b could be applied to the reaction and the desilylation product 3a was obtained with a yield of 63%. The method was high yielding and tolerable to diverse bioactive molecules, such as saccharin, thalidomide and pomalidomide. Saccharin derivatives have been reported as good hCAs inhibitors,13 and thalidomide and pomalidomide belongs to an important class of molecules known as immunomodulatory imide drugs (IMiDs).14 We found that under optimized conditions, saccharin, thalidomide and pomalidomide were also compatible with propargyl sulfonium salt 2a and provided the corresponding products 7, 9 and 11 with 52, 87, and 80% yields, respectively (Scheme 5).
 | ||
| Scheme 5 Scope of propargyl sulfonium salts and bioactive molecules. | ||
To demonstrate the synthetic utility of this protocol, we performed the gram-scale operation using phthalimide 1a (1.01 g, 6.8 mmol) and propargyl sulfonium salt 2a (1.5 equiv.) as the representative substrates under the optimized conditions, providing the related product 3a (1.03 g) with 65% yield (Scheme 6). The typical transformation was also conducted by oxidation of compound 3a with m-chloro peroxybenzoic acid (3.0 equiv.) and sulfonyl product 12 was obtained with 94% yield.
 | ||
| Scheme 6 Gram-scale synthesis and further transformation. | ||
According to the previous reports on α-alkylidene pyrazolinones and propargyl sulfonium ylides,10b,c a possible mechanism is proposed to account for the formation of N-vinylimides 3 (Scheme 7). Under the activation of inorganic base NaOAc·3H2O, the imides 1 may undergo deprotonation to form intermediate I and propargyl sulfonium salt 2a can isomerize to allenic sulfonium salts II. The N-nucleophilic attack of I to allenic sulfonium salts II initiates the reaction and gives intermediate III. Subsequently, protonation of the species III and release of MeBr provided the desired product 3.
 | ||
| Scheme 7 Plausible reaction mechanism. | ||
In summary, we have developed NaOAc·3H2O promoted N-addition reaction between imides and propargyl sulfonium salts, delivering potentially bioactive N-vinylimides in moderate to excellent yields. Various imides, including arylimides, aliphatic imides and N-(arylsulfonyl) alkyl acylamides, even bioactive saccharin, thalidomide and pomalidomide could tolerate and function to provide organosulfur N-vinylimides compounds. Gram-scale synthesis and convenient transformations are also furnished. The simple, mild, metal-free and efficient reaction condition, the broad scope of substrates, gram-scale synthesis and convenient transformation embody the synthetic superiority of this reaction process.
Experimental
General information
All reactions were performed in oven-dried or flame-dried round-bottom flasks and vials. Stainless steel syringes and cannula were used to transfer air- and moisture-sensitive liquids. Flash chromatography was performed using silica gel 60 (230–400 mesh) from Aladdin.General procedure for substrates
To a flame-dried sealable 3-dram vial equipped with a stir bar was added imides 1 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), subsequently treated CH3CN (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10–30 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6–12 h until imides 1 was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 3.Characterization data for the products
:1 to 25:1) as colorless oil (33 mg, 71% yield). IR νmax (neat)/cm−1: 3465, 3437, 2925, 2848, 1768, 1711, 1635, 1460, 1375, 1087, 717; 1H NMR (600 MHz, CDCl3): δ 7.82 (dd, J = 0, 0.6 Hz, 2H), 7.69 (dd, J = 0, 0.6 Hz, 2H), 5.39 (s, 1H), 5.24 (s, 1H), 3.53 (s, 2H), 1.97 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 167.1, 134.3, 134.0, 131.7, 123.7, 116.8, 37.1, 11.5. HRMS (ESI, m/z): calcd for C12H12NO2S+, [M + H]+, 234.0583, found 234.0585.:1 to 35:1) as colorless oil (20 mg, 27% yield). IR νmax (neat)/cm−1: 3452, 3437, 2974, 2925, 2855, 1719, 1648, 1388, 1368, 1157, 1116, 730; 1H NMR (600 MHz, CDCl3): δ 5.53 (s, 1H), 5.35 (s, 1H), 3.57 (s, 2H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 162.3, 140.5, 133.4, 130.1, 127.2, 117.9, 36.9, 14.6. HRMS (ESI, m/z): calcd for C12H8Cl4NO2S+, [M + H]+, 369.9024, found 369.9025.:1 to 25:1) as colorless oil (32 mg, 51% yield). IR νmax (neat)/cm−1: 3465, 3445, 2961, 2910, 2848, 1753, 1712, 1648, 1375, 1087, 892; 1H NMR (600 MHz, CDCl3): δ 8.02 (s, 1H), 7.89 (dd, J = 0.6, 7.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 5.47 (s, 1H), 5.30 (s, 1H), 3.57 (s, 2H), 2.02 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 166.3, 165.7, 137.3, 133.8, 133.3, 130.2, 129.2, 127.0, 125.0, 117.0, 37.0, 14.5. HRMS (ESI, m/z): calcd for C12H11BrNO2S+, [M + H]+, 311.9688, found 311.9687.:1 to 5:1) as colorless oil (29 mg, 52% yield). IR νmax (neat)/cm−1: 3465, 3437, 3108, 2939, 2910, 2856, 1782, 1720, 1648, 1537, 1375, 1339, 1087, 926, 722; 1H NMR (600 MHz, CDCl3): δ 8.73 (d, J = 1.8 Hz, 1H), 8.66 (dd, J = 1.8, 7.8 Hz, 1H), 8.11 (d, J = 7.8 Hz, 1H), 5.54 (s, 1H), 5.38 (s, 1H), 3.60 (s, 2H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 165.0, 164.7, 151.9, 136.0, 133.6, 133.1, 129.5, 124.9, 119.1, 117.5, 36.9, 14.5. HRMS (ESI, m/z): calcd for C12H11N2O4S+, [M + H]+, 279.0434, found 279.0436.:1 to 6:1) as colorless oil (23 mg, 42% yield). IR νmax (neat)/cm−1: 3470, 3430, 3111, 2942, 2915, 2858, 1789, 1727, 1653, 1543, 1381, 1343, 1092, 933, 726; 1H NMR (600 MHz, CDCl3): δ 8.16 (t, J = 9.0 Hz, 2H), 7.97 (t, J = 7.8 Hz, 1H), 5.54 (s, 1H), 5.37 (s, 1H), 3.58 (s, 2H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 164.6, 161.6, 145.4, 135.6, 133.7, 133.5, 128.8, 127.4, 123.3, 117.8, 36.9, 14.5. HRMS (ESI, m/z): calcd for C12H11N2O4S+, [M + H]+, 279.0434, found 279.0437.:1 to 4:1) as colorless oil (35 mg, 62% yield). IR νmax (neat)/cm−1: 3430, 2910, 2897, 1704, 1663, 1593, 1347, 1242, 899, 779; 1H NMR (600 MHz, CDCl3): δ 8.63 (d, J = 7.2 Hz, 2H), 8.25 (d, J = 8.4 Hz, 2H), 7.78 (t, J = 7.8 Hz, 2H), 5.75 (s, 1H), 5.38 (s, 1H), 3.57 (s, 2H), 2.23 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 163.7, 137.6, 134.2, 131.7, 131.5, 128.5, 126.9, 122.7, 118.4, 38.0, 15.6. HRMS (ESI, m/z): calcd for C16H14NO2S+, [M + H]+, 284.0740, found 284.0742.:1 to 1:1) as colorless oil (37 mg, 65% yield). IR νmax (neat)/cm−1: 3458, 3424, 2967, 2925, 2856, 1768, 1712, 1368, 1095, 758; 1H NMR (600 MHz, CDCl3): δ 8.41 (s, 2H), 8.09 (dd, J = 3.6, 6.0 Hz, 2H), 7.73 (dd, J = 3.6, 6.0 Hz, 2H), 5.54 (s, 1H), 5.40 (s, 1H), 3.66 (s, 2H), 2.09 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 167.0, 135.6, 134.2, 130.3, 129.3, 127.4, 125.2, 117.0, 37.1, 14.6. HRMS (ESI, m/z): calcd for C16H14NO2S+, [M + H]+, 284.0740, found 284.0744.:1 to 5:1) as colorless oil (20 mg, 43% yield). IR νmax (neat)/cm−1: 3437, 2918, 2848, 1720, 1648, 1607, 1417, 1362, 1095, 892; 1H NMR (400 MHz, CDCl3): δ 9.23 (s, 1H), 9.13 (d, J = 3.0 Hz, 1H), 7.82 (d, J = 4.8 Hz, 1H), 5.53 (s, 1H), 5.36 (s, 1H), 3.59 (s, 2H), 2.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 165.9, 165.6, 155.8, 145.3, 139.0, 133.6, 125.6, 117.6, 117.1, 37.0, 14.5. HRMS (ESI, m/z): calcd for C11H11N2O2S+, [M + H]+, 235.0536, found 235.0537.:1 to 1:1) as colorless oil (18 mg, 35% yield), 3i was also detected in the reaction colorless oil (6 mg, 16% yield). IR νmax (neat)/cm−1: 3475, 3434, 2928, 2848, 1707, 1638, 1624, 1391, 1378, 1198; 1H NMR (600 MHz, CDCl3): δ 6.77 (s, 2H), 5.37 (s, 1H), 5.22 (s, 1H), 3.50 (s, 2H), 2.01 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 169.3, 134.1, 133.4, 116.1, 36.9, 14.4. HRMS (ESI, m/z): calcd for C8H10NO2S+, [M + H]+, 184.0427, found 184.0429.:1 to 4:1) as colorless oil (34 mg, 92% yield). IR νmax (neat)/cm−1: 3473, 3430, 2925, 1704, 1635, 1622, 1388, 1375, 1193; 1H NMR (600 MHz, CDCl3): δ 5.45 (s, 1H), 5.22 (s, 1H), 3.47 (s, 2H), 2.82 (s, 4H), 2.03 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 175.9, 134.4, 117.2, 36.4, 28.3, 14.6. HRMS (ESI, m/z): calcd for C8H12NO2S+, [M + H]+, 186.0583, found 186.0584.:1 to 2:1) as colorless oil (39 mg, 86% yield). IR νmax (neat)/cm−1: 3478, 3435, 2929, 1710, 1637, 1652, 1381, 1370, 1198, 630; 1H NMR (600 MHz, CDCl3): δ 5.42 (s, 1H), 5.19 (s, 1H), 3.46 (s, 2H), 2.61 (ABQ, J = 18.6 Hz, 2H), 2.01 (s, 3H), 1.83–1.77 (m, 1H), 1.68–1.62 (m, 1H), 1.36 (s, 3H), 0.95 (t, J = 7.8 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 181.7, 174.9, 134.3, 117.0, 44.2, 40.5, 36.4, 31.0, 24.1, 14.5, 8.7. HRMS (ESI, m/z): calcd for C11H18NO2S+, [M + H]+, 228.1053, found 228.1055.:1 to 5:1) as colorless oil (38 mg, 79% yield). IR νmax (neat)/cm−1: 3457, 3437, 2972, 2919, 1714, 1645, 1375, 1193, 684, 605; 1H NMR (600 MHz, CDCl3): δ 5.40 (s, 1H), 5.19 (s, 1H), 3.49 (s, 2H), 2.95 (s, 2H), 2.03 (s, 3H), 1.90 (s, 4H), 1.49 (d, J = 2.4 Hz, 4H); 13C NMR (150 MHz, CDCl3): δ 178.5, 134.3, 116.7, 40.1, 36.5, 23.9, 21.9, 14.5. HRMS (ESI, m/z): calcd for C12H18NO2S+, [M + H]+, 240.1053, found 240.1054.:1 to 5:1) as colorless oil (44 mg, 93% yield). IR νmax (neat)/cm−1: 3452, 3437, 2961, 2925, 2848, 1712, 1648, 1375, 1208, 694; 1H NMR (600 MHz, CDCl3): δ 5.93 (s, 2H), 5.39 (s, 1H), 5.15 (s, 1H), 3.41 (s, 2H), 3.15 (t, J = 3.0 Hz, 2H), 2.61 (d, J = 15.6 Hz, 2H), 2.29 (d, J = 13.8 Hz, 2H), 1.98 (s, 3H); 13C NMR (600 MHz, CDCl3): δ 179.0, 134.4, 117.0, 39.0, 36.3, 23.5, 14.5. HRMS (ESI, m/z): calcd for C12H16NO2S+, [M + H]+, 238.0896, found 238.0898.:1 to 8:1) as colorless oil (41 mg, 82% yield). IR νmax (neat)/cm−1: 3458, 3437, 2982, 2918, 1704, 1648, 1375, 1355, 1193, 604; 1H NMR (600 MHz, CDCl3): δ 6.20 (s, 2H), 5.35 (s, 1H), 5.05 (s, 1H), 3.45 (s, 2H), 3.35 (t, J = 1.2 Hz, 2H), 3.32 (s, 2H), 2.01 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 176.6, 134.7, 134.3, 117.1, 52.1, 45.8, 45.2, 36.4, 14.5. HRMS (ESI, m/z): calcd for C13H16NO2S+, [M + H]+, 250.0896, found 250.0898.:1 to 4:1) as colorless oil (38 mg, 52% yield). IR νmax (neat)/cm−1: 3458, 3429, 2961, 2925, 1768, 1712, 1648, 1473, 1375, 1208, 766; 1H NMR (600 MHz, CDCl3): δ 7.42 (dd, J = 3.6, 5.4 Hz, 2H), 7.32 (dd, J = 3.0, 5.4 Hz, 2H), 7.21 (dd, J = 3.0, 5.4 Hz, 2H), 7.18 (dd, J = 3.0, 5.4 Hz, 2H), 5.18 (s, 1H), 4.83 (s, 2H), 4.27 (s, 1H), 3.31 (s, 2H), 3.03 (s, 2H), 1.94 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 175.7, 141.3, 138.7, 134.2, 127.0, 126.7, 125.0, 124.2, 116.9, 46.9, 45.7, 36.0, 14.6. HRMS (ESI, m/z): calcd for C22H20NO2S+, [M + H]+, 362.1209, found 362.1212.:1 to 5:1) as colorless oil (13 mg, 32% yield). IR νmax (neat)/cm−1: 3465, 3360, 2988, 2925, 2904, 1671, 1586, 1285, 1213, 1102, 779; 1H NMR (600 MHz, CDCl3): δ 5.53 (s, 1H), 5.11 (s, 1H), 3.32 (s, 2H), 2.73 (t, J = 6.6 Hz, 4H), 2.09 (s, 3H), 2.05–2.00 (m, 2H); 13C NMR (150 MHz, CDCl3): δ 171.9, 137.0, 118.1, 37.8, 32.9, 17.2, 15.4. HRMS (ESI, m/z): calcd for C9H14NO2S+, [M + H]+, 200.0740, found 200.0743.:1 to 4:1) as colorless oil (12 mg, 24% yield). IR νmax (neat)/cm−1: 3465, 3430, 2975, 2920, 2894, 1736, 1673, 1486, 1281, 1233, 1132, 734; 1H NMR (600 MHz, CDCl3): δ 5.53 (s, 1H), 5.11 (s, 1H), 3.36 (s, 2H), 2.84 (dd, J = 4.2, 16.8 Hz, 2H), 2.39 (dd, J = 10.2, 16.8 Hz, 2H), 2.27–2.23 (m, 1H), 2.09 (s, 3H), 1.72–1.68 (m, 1H), 1.28 (dd, J = 7.8, 15.0 Hz, 2H), 0.94 (s, 3H), 0.93 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 171.7, 137.0, 118.2, 44.0, 39.2, 37.9, 27.1, 24.8, 22.5, 15.4. HRMS (ESI, m/z): calcd for C13H22NO2S+, [M + H]+, 256.1366, found 256.1369.:1 to 5:1) as colorless oil (11 mg, 24% yield). IR νmax (neat)/cm−1: 3465, 3437, 2961, 2918, 2856, 1725, 1676, 1355, 1270, 1242, 1131, 625; 1H NMR (600 MHz, CDCl3): δ 5.52 (s, 1H), 5.10 (s, 1H), 3.36 (s, 2H), 2.59 (s, 4H), 2.10 (s, 3H), 1.18 (s, 6H); 13C NMR (150 MHz, CDCl3): δ 171.4, 136.9, 118.2, 46.5, 37.9, 29.3, 27.8, 15.3. HRMS (ESI, m/z): calcd for C11H18NO2S+, [M + H]+, 228.1053, found 228.1056.:1 to 2:1) as colorless oil (33 mg, 65% yield). IR νmax (neat)/cm−1: 3465, 3424, 2918, 2910, 1776, 1712, 1648, 1375, 1193, 871; 1H NMR (600 MHz, CDCl3): δ 6.55 (s, 2H), 5.44 (s, 1H), 5.34 (s, 1H), 5.21 (s, 1H), 3.42 (s, 2H), 2.94 (s, 2H), 2.03 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 175.0, 136.6, 134.2, 117.3, 47.4, 36.4, 14.7. HRMS (ESI, m/z): calcd for C12H14NO3S+, [M + H]+, 252.0689, found 252.0692.:1 to 1:1) as colorless oil (18 mg, 35% yield), 3i was also detected in the reaction colorless oil (6 mg, 16% yield). IR νmax (neat)/cm−1: 3456, 2443, 3031, 2984, 2908, 1762, 1701, 1654, 1372, 1153, 1132, 905, 865, 706, 618; 1H NMR (600 MHz, CDCl3): δ 6.51 (s, 2H), 5.39–5.38 (m, 2H), 5.36 (s, 1H), 5.06 (s, 1H), 3.61 (dd, J = 1.8, 3.6 Hz, 2H), 3.33 (s, 2H), 2.01 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 173.7, 134.8, 134.0, 117.3, 79.7, 46.0, 36.3, 14.5. HRMS (ESI, m/z): calcd for C12H14NO3S+, [M + H]+, 252.0689, found 252.0687.General procedure B: to a flame-dried sealable 3-dram vial equipped with a stir bar was added imides 4 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), subsequently treated CH3CN (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6–12 h until imides 4 was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 5.
:1 to 5:1) as colorless oil (18 mg, 32% yield). IR νmax (neat)/cm−1: 3478, 2430, 2925, 2918, 1704, 1635, 1362, 1339, 1249, 1172, 1080, 722, 623; 1H NMR (600 MHz, CDCl3): δ 8.06 (d, J = 7.8 Hz, 2H), 7.66 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 5.72 (s, 1H), 5.26 (s, 1H), 3.54 (s, 2H), 2.23 (s, 3H), 2.20 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 169.9, 141.1, 138.7, 133.9, 129.0, 128.7, 120.6, 39.8, 23.8, 15.9. HRMS (ESI, m/z): calcd for C12H16NO3S2+, [M + H]+, 286.0566, found 286.0569.:1 to 18:1) as colorless oil (18 mg, 24% yield). IR νmax (neat)/cm−1: 3474, 2454, 2925, 2895, 1705, 1635, 1352, 1339, 1270, 1172, 1080, 823, 722, 623; 1H NMR (600 MHz, CDCl3): δ 8.07 (d, J = 8.4 Hz, 2H), 7.65 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.8 Hz, 2H), 5.74 (s, 1H), 5.26 (s, 1H), 3.54 (s, 2H), 2.46 (t, J = 7.8 Hz, 2H), 2.01 (s, 3H), 1.56 (dd, J = 7.2, 13.8 Hz, 2H), 1.27–1.23 (m, 8H), 0.86 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 172.7, 140.6, 139.0, 133.8, 129.0, 128.7, 120.6, 39.9, 35.4, 31.5, 28.9, 28.8, 24.5, 22.5, 16.0, 14.0. HRMS (ESI, m/z): calcd for C18H28NO3S2+, [M + H]+, 370.1505, found 370.1506.:1 to 4:1) as colorless oil (34 mg, 56% yield). IR νmax (neat)/cm−1: 3445, 3416, 2954, 2925, 2869, 1691, 1642, 1339, 1265, 1157, 1136, 1087, 941, 799, 681, 617; 1H NMR (600 MHz, CDCl3): δ 7.95 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 5.71 (s, 1H), 5.25 (s, 1H), 3.53 (s, 2H), 2.46 (s, 3H), 2.22 (s, 3H), 2.20 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 169.9, 145.1, 141.2, 135.8, 130.0, 129.3, 129.1, 127.3, 120.4, 39.8, 23.9, 21.7, 15.9. HRMS (ESI, m/z): calcd for C13H18NO3S2+, [M + H]+, 300.0723, found 300.0726.:1 to 4:1) as colorless oil (21 mg, 32% yield). IR νmax (neat)/cm−1: 3445, 2954, 2925, 2869, 1699, 1642, 1339, 1270, 1172, 1157, 1087, 807, 681, 617; 1H NMR (600 MHz, CDCl3): δ 7.95 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 5.73 (s, 1H), 5.25 (s, 1H), 3.53 (s, 2H), 2.45 (t, J = 6.6 Hz, 2H), 2.21 (s, 3H), 2.07 (s, 3H), 1.61 (dd, J = 7.2, 15.0 Hz, 2H), 0.87 (t, J = 7.8 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 172.5, 145.0, 140.7, 136.0, 129.3, 129.1, 120.5, 39.9, 37.2, 21.7, 18.0, 16.0, 13.5. HRMS (ESI, m/z): calcd for C15H22NO3S2+, [M + H]+, 328.1036, found 328.1038.:1 to 4:1) as colorless oil (42 mg, 55% yield). IR νmax (neat)/cm−1: 3474, 2454, 2923, 2895, 1705, 1635, 1352, 1339, 1270, 1172, 1080, 813, 728, 625; 1H NMR (600 MHz, CDCl3): δ 7.94 (d, J = 7.8 Hz, 2H), 7.33 (d, J = 7.8 Hz, 2H), 5.72 (s, 1H), 5.25 (s, 1H), 3.52 (s, 2H), 2.45 (t, J = 7.2 Hz, 5H), 2.20 (s, 3H), 1.55 (t, J = 7.2 Hz, 2H), 1.28–1.22 (m, 8H), 0.86 (t, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 172.7, 144.9, 140.6, 136.0, 129.3, 129.0, 120.5, 39.9, 35.3, 31.5, 28.9, 28.8, 24.5, 22.5, 21.6, 15.9, 14.0. HRMS (ESI, m/z): calcd for C19H30NO3S2+, [M + H]+, 384.1662, found 384.1665.To a flame-dried sealable 3-dram vial equipped with a stir bar was added imides 1a (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), subsequently treated CH3CN (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2b (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 6 h until imide 1a was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 3a.
To a flame-dried sealable 3-dram vial equipped with a stir bar was added saccharin 6 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), anhydrous DMF (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 12 h until saccharin 6 was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 7.
:1 to 4:1) as colorless oil (28 mg, 52% yield). IR νmax (neat)/cm−1: 3452, 3093, 3072, 3038, 2925, 1733, 1607, 1473, 1319, 1172, 982, 751, 591; 1H NMR (600 MHz, CDCl3): δ 7.91 (dd, J = 3.0, 5.4 Hz, 2H), 7.77 (dd, J = 2.4, 5.4 Hz, 2H), 5.49 (s, 1H), 5.33 (s, 1H), 3.61 (s, 2H), 2.06 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 158.0, 137.7, 135.0, 134.4, 133.5, 126.8, 125.5, 121.1, 117.2, 36.9, 14.8. HRMS (ESI, m/z): calcd for C11H12NO3S2+, [M + H]+, 270.0253, found 270.0255.To a flame-dried sealable 3-dram vial equipped with a stir bar was added thalidomide 8 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), subsequently treated CH3CN (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 12 h until thalidomide 8 was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 9.
:1 to 1:1) as colorless oil (60 mg, 87% yield). IR νmax (neat)/cm−1: 3445, 3407, 2961, 2925, 2848, 1720, 1684, 1388, 1262, 1193, 1116, 722; 1H NMR (600 MHz, CDCl3): δ 7.90 (dd, J = 3.6, 5.4 Hz, 2H), 7.79–7.78 (m, 2H), 5.57 (s, 1H), 5.23 (s, 1H), 5.99 (dd, J = 6.0, 12.6 Hz, 1H), 3.33 (dd, J = 15.0, 22.8 Hz, 2H), 3.04 (dd, J = 2.4, 13.2 Hz, 1H), 2.90–2.87 (m, 2H), 2.20–2.17 (m, 1H), 2.08 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 170.3, 167.9, 167.3, 136.9, 134.4, 131.8, 123.7, 118.4, 50.1, 37.5, 32.1, 22.1, 15.3. HRMS (ESI, m/z): calcd for C17H17N2O4S+, [M + H]+, 345.0904, found 345.0905.To a flame-dried sealable 3-dram vial equipped with a stir bar was added pomalidomide 10 (0.3 mmol, 1.0 equiv.), NaOAc·3H2O (0.45 mmol, 1.5 equiv.), subsequently treated DMF (3 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (0.45 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 12 h until pomalidomide 10 was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 11.
:1 to 1:1) as colorless oil (58 mg, 80% yield). IR νmax (neat)/cm−1: 3432, 3397, 2958, 2925, 2851, 1725, 1681, 1378, 1256, 1191, 1107, 718; 1H NMR (600 MHz, CDCl3): δ 7.43 (t, J = 7.8 Hz, 1H), 7.16 (d, J = 7.2 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 5.57 (s, 1H), 5.28 (s, 2H), 5.23 (s, 1H), 5.31 (dd, J = 4.8, 12.0 Hz, 1H), 3.33 (dd, J = 15.0, 21.6 Hz, 2H), 3.06–2.98 (m, 1H), 2.90–2.82 (m, 2H), 2.16–2.15 (m, 1H), 2.09 (s, 3H); 13C NMR (150 MHz, CDCl3): δ 170.5, 168.9, 168.3, 167.5, 145.6, 136.9, 135.5, 132.3, 121.4, 118.4, 113.1, 110.8, 49.6, 37.5, 32.1, 22.1, 15.3. HRMS (ESI, m/z): calcd for C17H18N3O4S+, [M + H]+, 360.1013, found 360.1016.To a flame-dried 100 mL round bottom flask equipped with a stir bar was added phthalimide 1a (1.01 g, 6.8 mmol, 1.0 equiv.), NaOAc·3H2O (1.39 g, 10.2 mmol, 1.5 equiv.), subsequently treated CH3CN (68 mL, c = 0.1 M) was added to vial via syringe, the reaction mixture was stirred for 10 min at 22 °C. Then propargyl sulfonium salt 2a (1.85 g, 10.2 mmol, 1.5 equiv.) was added in one portion. The reaction was stirred at 50 °C for 10 h until phthalimide 1a was fully consumed (monitored by TLC). The organic solvent was removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 3 with yield of 65% (1.03 g).
To a flame-dried sealable 2-dram vial equipped with a stir bar was added phthalimide 3a (50 mg, 0.2 mmol, 1.0 equiv.), Na2CO3 (42.4 mg, 0.4 mmol, 2.0 equiv.) and CH2Cl2 (3 mL, c = 0.2 M). After stirred at 0 °C for 2 min, m-CPBA (103.5 mg, 0.6 mmol, 75%, 3.0 equiv.) was added in portions to the mixture. The reaction mixture was kept stirring for 10 min at 0 °C until 3a was fully consumed (monitored by TLC). The reaction was quenched with aqueous Na2CO3 and extracted with CH2Cl2 (5 mL × 3). The combined organic solvent was dried with anhydrous Na2SO4, removed under reduced pressure and purified through column chromatography (eluent: petroleum ether and EtOAc) to afford the desired product 12 (50 mg, 94%).
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (2019-047), and the 1331 Engineering of Shanxi Province. The instruments were performed using the Analytical Test Center of Shanxi Normal University, Linfen, China.Notes and references
- (a) B. Métayer, A. Angeli, A. Mingot, K. Jouvin, G. Evano, C. T. Supuran and S. Thibaudeau, J. Enzyme Inhib. Med. Chem., 2018, 33, 804 CrossRef PubMed; (b) H. Uhr, C. Boie, H. Rieck, B.-W. Krueger, U. Heinemann, R. Market, M. Vaupel, M. Kugler, K. Stenzel, U. Wachendorffff-Neumann, A. Mauler-Machnik, K.-H. Kuck, P. Loesel and S.-I. Narabu, Preparation of Aryloxydithiazole and Aryloxydithiazine Dioxides as Pesticides and Fungicides, Patent DE19918294, 2000; (c) A. D. Brown, M. E. Bunnage, C. A. L. Lane, R. A. Lewthwaite, P. A. Glossop, K. James and D. A. Price, Preparation of Formylamino Hydroxyphenyl Phenylacetamide Derivatives as B2 Adrenoceptor Agonists, US Pat., US20050215590-A1, 2005 Search PubMed; (d) D. L. Kirkpatrick and M. Indarte, Preparation of N-substituted Thiazolecarboxamides for Inhibiting CNKSR1, US Pat., US20160304533-A1, 2016 Search PubMed; (e) S. L. Zada, K. D. Green, S. K. Shrestha, I. M. Herzog, S. Garneau-Tsodikova and M. Fridman, ACS Infect. Dis., 2018, 4, 1121 CrossRef PubMed.
- (a) T. Ogawa and H. Yamada, Preparation of N-Styrylphthalimide Derivatives as Nonlinear Organic Optical Materials, Patent JP08176107, 1996; (b) S. Otsu, K. Ofuku and N. Kagawa, Semiconductor for Photoelectric Conversion Material, Photoelectric Converter, and Photoelectrochemical Cell, Patent JP2005019124, 2003.
- N. Cachet, G. Genta-Jouve, E. L. Regalado, R. Mokrini, P. Amade, G. Culioli and O. P. Thomas, J. Nat. Prod., 2009, 72, 1612 CrossRef CAS PubMed.
- J. Dai, W. Ren, W. Chang, P. Zhang and Y. Shi, Org. Chem. Front., 2017, 4, 297 RSC.
- (a) E. Alacid and C. Najera, Adv. Synth. Catal., 2008, 350, 1316 CrossRef CAS; (b) W. Susanto, C. Y. Chu, W. J. Ang, T. C. Chou, L. C. Lo and Y. Lam, J. Org. Chem., 2012, 77, 2729 CrossRef CAS PubMed; (c) F. De Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 9009 CrossRef CAS PubMed; (d) J. Szudkowska-Frątczak, G. Hreczycho and P. Pawluć, Org. Chem. Front., 2015, 2, 730 RSC; (e) E. Landagaray, M. Ettaoussi, M. Rami, J. A. Boutin, D. H. Caignard, P. Delagrange, P. Melynyk, P. Berthelot and S. Yous, Eur. J. Med. Chem., 2017, 127, 621 CrossRef CAS PubMed; (f) S. K. Murphy, M. Zeng and S. B. Herzon, Science, 2017, 356, 956 CrossRef CAS PubMed; (g) P. Kramer, J. Schönfeld, M. Bolte and G. Manolikakes, Org. Lett., 2018, 20, 178 CrossRef CAS PubMed.
- For selected references of Cu-catalyzed Chan-Lam-Evans reactions, see: (a) L. N. Berntsen, T. N. Solvi, K. Sørnes, D. S. Wragg and A. H. Sandtorv, Chem. Commun., 2021, 57, 11851 RSC; (b) L. Steemers, L. Wijsman and J. H. Van Maarseveen, Adv. Synth. Catal., 2018, 360, 4241 CrossRef CAS; (c) P. Arsenyan, A. Petrenko, E. Paegle and S. Belyakov, Mendeleev Commun., 2011, 21, 326 CrossRef CAS; (d) R. G. R. Bacon and A. Karim, J. Chem. Soc., Perkin Trans. 1, 1973, 278 RSC; (e) O. Takuji, K. Toshiyuki, H. Kazuo and S. Hitomi, Chem. Lett., 1991, 20, 1443 CrossRef.
- (a) E. Semina, P. Tuzina, F. Bienewald, A. S. K. Hashmi and T. Schaub, Chem. Commun., 2020, 56, 5977 RSC; (b) M. Arndt, K. S. M. Salih, A. Fromm, L. J. Goossen, F. Menges and G. Niedner-Schatteburg, J. Am. Chem. Soc., 2011, 133, 7428 CrossRef CAS PubMed; (c) A. E. Buba, M. Arndt and L. J. Gooßen, J. Organomet. Chem., 2011, 696, 170 CrossRef CAS; (d) L. J. Goossen, M. Arndt, K. S. M. Salih, A. Fromm, F. Menges and G. Niedner-Schatteburg, J. Org. Chem., 2006, 71, 9506 CrossRef CAS PubMed.
- (a) D. G. Kohler, S. N. Gockel, J. L. Kennemur, P. J. Waller and K. L. Hull, Nat. Chem., 2018, 10, 333 CrossRef CAS PubMed; (b) M. M. Rogers, V. Kotov, J. Chatwichien and S. S. Stahl, Org. Lett., 2007, 9, 4331 CrossRef CAS PubMed; (c) J. L. Brice, J. E. Harang, V. I. Timokhin, N. R. Anastasi and S. S. Stahl, J. Am. Chem. Soc., 2005, 127, 2868 CrossRef CAS PubMed; (d) L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737 CrossRef CAS PubMed.
- (a) E. Petit, L. Bosch, J. Font, L. Mola, A. M. Costa and J. Vilarrasa, J. Org. Chem., 2014, 79, 8826 CrossRef CAS PubMed; (b) A. Ramazani, N. Noshiranzadeh and B. Mohammadi, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 539 CrossRef CAS.
- (a) S. Shen, Y. Yang, J. Duan, Z. Jia and J. Liang, Org. Biomol. Chem., 2018, 16, 1068 RSC; (b) S. J. Shen, X. L. Du, X. L. Xu, Y. H. Wu, M. G. Zhao and J. Y. Liang, J. Org. Chem., 2019, 84, 12520 CrossRef CAS PubMed; (c) S. J. Shen, X. L. Du, X. L. Xu, Y. H. Wu, M. G. Zhao and J. Y. Liang, RSC Adv., 2019, 9, 34912 RSC.
- For the synthesis of compound 1o, see selected reference: S. Bova, S. Saponara, A. Rampa, S. Gobbi, L. Cima, F. Fusi, G. Sgaragli, M. Cavalli, C. De los Rios, J. Striessnig and A. Bisi, J. Med. Chem., 2009, 52, 1259 CrossRef CAS PubMed.
- For the Synthesis of Compound 1s and 1t, see selected references: (a) S. H. Frayne, R. M. Stolz and B. H. Northrop, Org. Biomol. Chem., 2019, 17, 7878 RSC; (b) H. Kwart and I. Burchuk, J. Am. Chem. Soc., 1952, 74, 3094 CrossRef CAS.
- M. D'Ascenzio, D. Secci, S. Carradori, S. Zara, P. Guglielmi, R. Cirilli, M. Pierini, G. Poli, T. Tuccinardi, A. Angeli and C. T. Supuran, J. Med. Chem., 2020, 63, 2470 CrossRef PubMed.
- (a) S. Blanco, A. Macario and J. C. López, Phys. Chem. Chem. Phys., 2021, 23, 13705 RSC; (b) D. K. Brownsey, B. C. Rowley, E. Gorobets, B. S. Gelfand and D. J. Derksen, Chem. Sci., 2021, 12, 4519 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01117d |
| This journal is © The Royal Society of Chemistry 2022 |