
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe concise synthesis and resolution of planar chiral [2.2]paracyclophane oxazolines by C–H activation†
Shashank Tewari ,
Maulik N. Mungalpara,
Suraj Patel and
Gareth J. Rowlands*
,
Maulik N. Mungalpara,
Suraj Patel and
Gareth J. Rowlands*
School of Natural Sciences, Massey University, Private Bag 11 222, Palmerston North, 4442, New Zealand. E-mail: g.j.rowlands@massey.ac.nz
First published on 17th March 2022
Abstract
Planar chiral [2.2]paracyclophanes are resolved through the direct C–H arylation of enantiopure oxazolines, providing a convenient route to ligands and chiral materials. Preliminary results show that hydrolysis followed by decarboxylative phosphorylation leads to enantiopure [2.2]paracyclophane derivatives that are otherwise challenging to prepare.
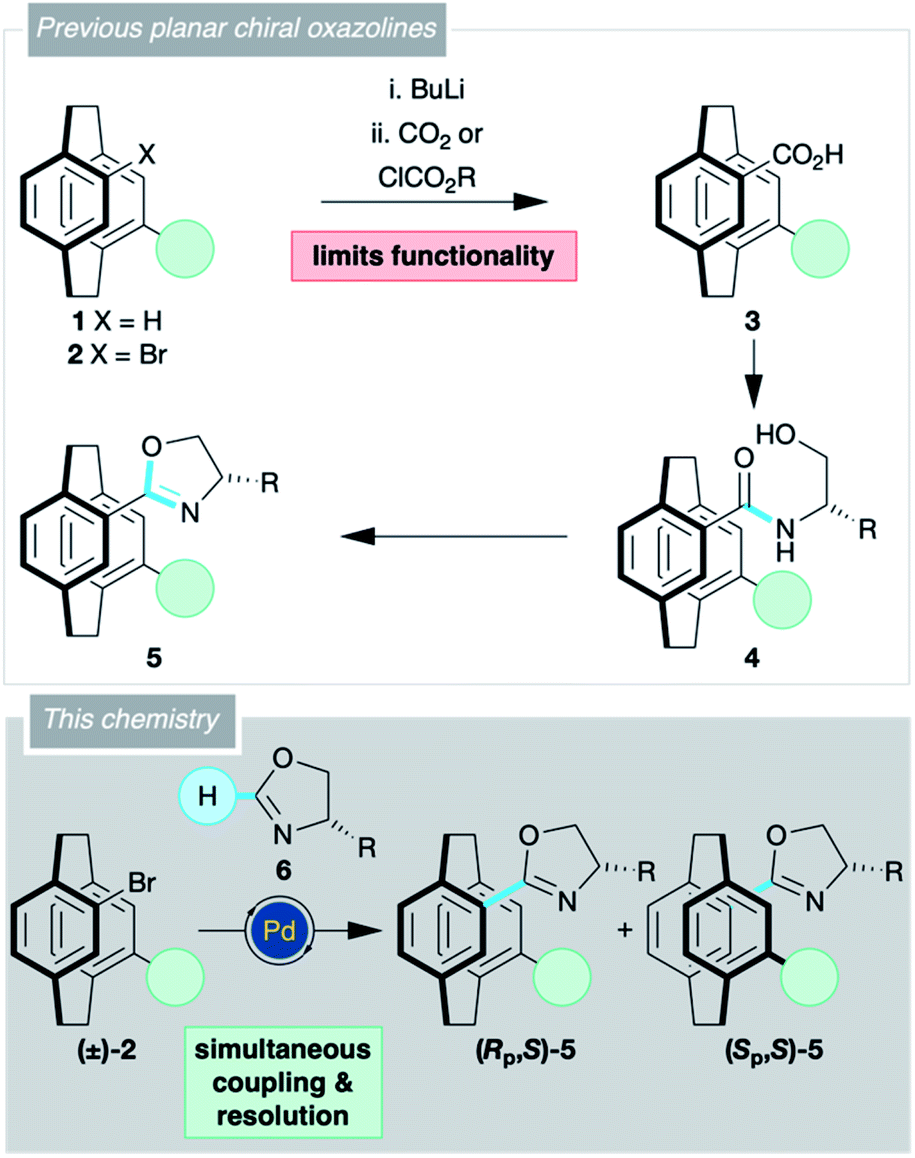
[2.2]Paracyclophane 1 (X = H; Scheme 1) has garnered considerable attention as the core for planar chiral pre-ligands,1,2 catalysts,3,4 bioactive entities5,6 and chiroptical materials.7,8 The rigidity of the [2.2]paracyclophane provides an almost unrivalled opportunity to control the relative spatial position of various functional groups through a series of regioisomers.4,9,10 The major roadblock hampering progress in [2.2]paracyclophane chemistry is the resolution of planar chirality, and there are only a limited number of commonly used enantiopure precursors, some of which are hard to obtain on a large scale.11,12
 | ||
| Scheme 1 Previous routes to oxazolines and proposed chemistry. | ||
Oxazolines form a host of ‘privileged’ pre-ligands,13–15 and it is no surprise that numerous oxazoline-substituted [2.2]paracyclophanes 5 have shown potential in asymmetric catalysis.16–20 The oxazoline moiety aids the synthesis of these compounds, either by permitting resolution of the planar chirality,16–19,21,22 or by directing functionalisation of the cyclophane backbone.16,19,21,23 Every one of these oxazoline-containing [2.2]paracyclophanes 5 was synthesised from a bromo derivative 2 via the carboxylic acid 3, then the amide 4, and finally cyclisation (Scheme 1). This adds steps to the synthesis and limits the functionality found in the molecule as the acid is invariably formed by halogen–metal exchange.
Direct addition of an oxazoline 6 would allow simultaneous functionalisation and resolution of the planar chirality of [2.2]paracyclophane. Such a strategy would permit a more concise synthesis of [2.2]paracyclophane derivatives with a wider range of substituents. Previously, we have used C–H activation chemistry to synthesise planar chiral N-oxides,24,25 and believed that the oxazoline C–H activation chemistry of Ackermann26,27 and Lu28 could be applied to [2.2]paracyclophane. Herein, we report the successful realisation of this strategy. Planar chiral oxazolines are accessed in a single step furnishing molecules that can be used as pre-ligands or as precursors to carboxylic acids that can be further derivatised by traditional means or decarboxylative couplings.
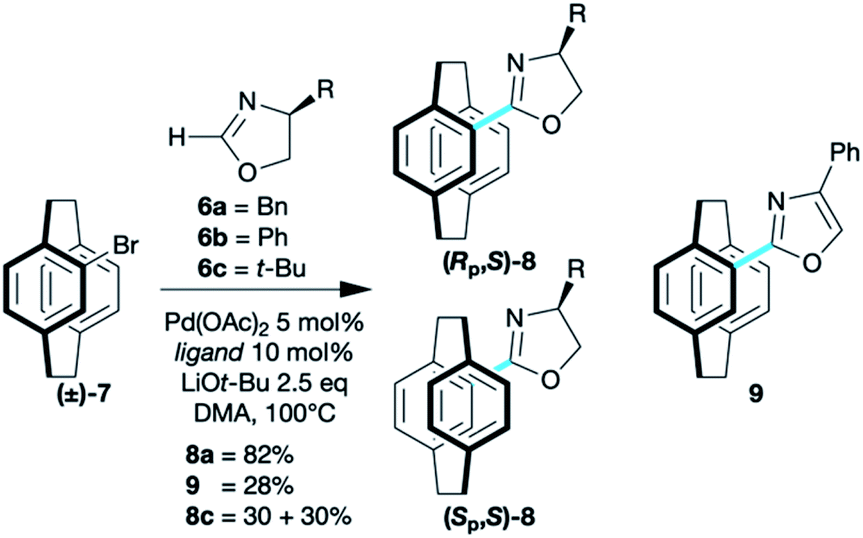
The coupling of oxazolines 6 and 4-bromo[2.2]paracyclophane 7 required little optimisation with the chemistry of Ackermann26 transferring to paracyclophane without incident. A range of (heteroatom-substituted) secondary phosphine pre-ligands [(HA)SPO] were screened, and di-tert-butylphosphine oxide (t-Bu2SPO) gave a catalyst that showed complete conversion of simple [2.2]paracyclophane derivatives, such as 7 (8a 82%; 8c 60% Scheme 2). More complex derivatives required the di-1-adamantylphosphine oxide (Ad2SPO) otherwise returned unreacted starting material (see below).
 | ||
| Scheme 2 Synthesis of 4-oxazolinyl[2.2]paracyclophanes 8a and 8c. | ||
Benzyloxazoline 8a was formed as a mixture of diastereoisomers that are separable by column chromatography, but the similarity of the Rf values make resolution extremely challenging, with only the fractions at the beginning and end of a collection containing pure diastereoisomers. This led to screening a range of oxazolines. All coupled but with less satisfactory yields. The phenyloxazoline 6b appears to couple with concomitant aromatisation furnishing oxazole 9. For mono(oxazolines) 8, tert-butyl derivative 6c gave the best results in terms of resolution of planar chirality, with the diastereoisomers 8c being readily separable.21
Next we established the scope of coupling reaction. Both the pseudo-para- (4,16)- (11a) and pseudo-ortho- (4,12)- bis(oxazolines) 11b were readily prepared from the corresponding dibromo[2.2]paracyclophanes (Table 1). The pseudo-para isomer, 4,16-bis(4′-benzyloxazolin-2′-yl)[2.2]paracyclophane 11a, is formed as a single stereoisomer due to its symmetry. The diastereoisomers of the more useful pseudo-ortho benzyl-substituted oxazoline 11b were readily resolved,17,20,29 but only one diastereoisomer of the tert-butyl derivative 11c could be isolated. The other diastereoisomer co-ran with the two diastereomers of the product of protodebromination (8c). The synthesis of pseudo-ortho-bis(benzyloxazoline) 11b has been performed on >1 g scale. The yields are lower but the separation easier. If a single equivalent of 2H-oxazoline 6a is used in the coupling, it is possible to isolate the bromo mono(oxazoline) 11d, but the reaction is non-selective, with a mixture of mono-along and bis(oxazoline) always formed.16,18,30
| a Conditions A: 6 (1.1 eq), Pd(OAc)2 (5 mol%), (t-Bu)2P(O)H (10 mol%), LiOt-Bu (2.5 eq); conditions B: 6 (2.2 eq), Pd(OAc)2 (10 mol%), (t-Bu)2P(O)H (20 mol%), LiOt-Bu (5.0 eq); conditions C: 6 (4.4 eq), Pd(OAc)2 (40 mol%), (t-Bu)2P(O)H (80 mol%), LiOt-Bu (10.0 eq); conditions D: 6 (1.1 eq), Pd(OAc)2 (5 mol%), (Ad)2P(O)H (10 mol%), LiOt-Bu (2.5 eq); conditions E: 6 (4.4 eq), Pd(OAc)2 (40 mol%), (Ad)2P(O)H (80 mol%), LiOt-Bu (10.0 eq); conditions F: 6 (2.2 eq), Pd(OAc)2 (10 mol%), (Ad)2P(O)H (20 mol%), LiOt-Bu (5.0 eq).b Only single diastereoisomer shown in table. Diastereoisomers in red cannot be separated. Diastereoisomers in blue are partially resolved.c Yield of separate diastereomers (diastereomer1)%/(diastereomer2)%.d Combined yield of all stereoisomers (includes mixed fractions). |
|---|
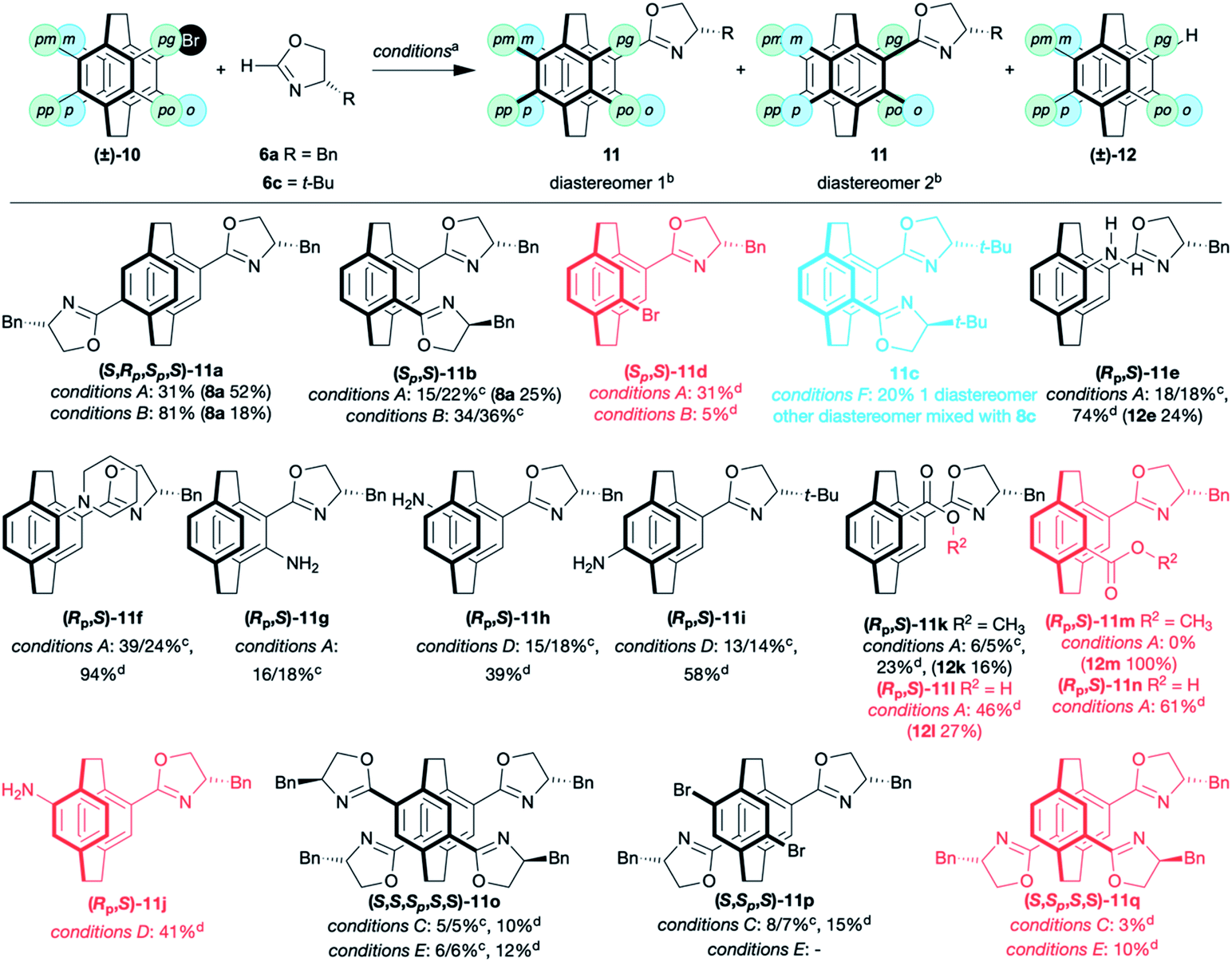 |
As is frequently observed with [2.2]paracyclophane derivatives, each regioisomer behaves differently, and it is hard to predict the influence of the substituents.31,32 Using t-Bu2SPO as pre-ligand, both primary and tertiary pseudo-gem amines gave the desired oxazolines as separable diastereoisomers 11e and 11f. The normally unreactive ortho substituted [2.2]paracyclophane derivative, 4-amino-5-bromo[2.2]paracyclophane,33 appeared to give the amino-oxazoline 11g as a pair of separable diastereomers in 34% yield, although these compounds proved to be unstable. All other amino[2.2]paracyclophanes gave unsatisfactory yields. The pseudo-ortho-amino-bromo[2.2]paracyclophane failed to couple and only furnished the product of protodebromination. On changing to Ad2SPO, the meta 11h and pseudo-para 11i, were synthesised as separable diastereomeric amino oxazolines in moderate to good yields while the pseudo-meta-amino-bromo [2.2]paracyclophane gave 11j as an inseparable mixture of diastereomers. It is unclear why this ligand is superior with these amino-bromo[2.2]paracyclophanes. Nitro-substituted [2.2]paracyclophanes (pseudo-gem and pseudo-meta) are unreactive. Esters proved problematic, giving low yields (11k and 11m) and/or the product of protodebromination (12). We assume that the strongly basic conditions are incompatible with this functionality. This is supported by the fact the free acid reacts in good to moderate yields, 44% for pseudo-gem (11l) and 61% for pseudo-ortho (11n) (Table 1).
To push the limits of this coupling, we synthesised the 4,7,12,15-tetra(4′-benzyloxazolin-2′-yl)[2.2]paracyclophane 11o from tetrabromo[2.2]paracyclophane.34 Initial reaction with the t-Bu2SPO ligand gave a complex mixture of regio- and stereoisomers. The resolved tetra(oxazoline) ligand 11o was isolated in 10%. In addition, the para-bis(oxazoline)-dibromo[2.2]paracyclophane 11p was isolated as separable diastereoisomers along with an unidentified regioisomer of the diastereomeric bis(oxazoline)-dibromo[2.2]paracyclophanes (not included in the yield). Finally, the two diastereoisomers of the protodebromo tris(oxazoline) 11q were isolated. Altering the ligand to Ad2SPO simplified the mixture and gave just the tetra(oxazoline) 11o and tris(oxazoline) 11q.
There are numerous reports describing the utility of [2.2]paracyclophane oxazolines. They have been used as pre-ligands,16,18–20,29 or have directed further functionalisation of the [2.2]paracyclophane by either bromination16,23 or metalation.19,21 We believe straightforward elaboration of the amine or acid functionality will deliver modular pre-ligands, but here we want to increase the versatility of the oxazoline moiety and present preliminary results on the hydrolysis and decarboxylative phosphorylation of the oxazolines.
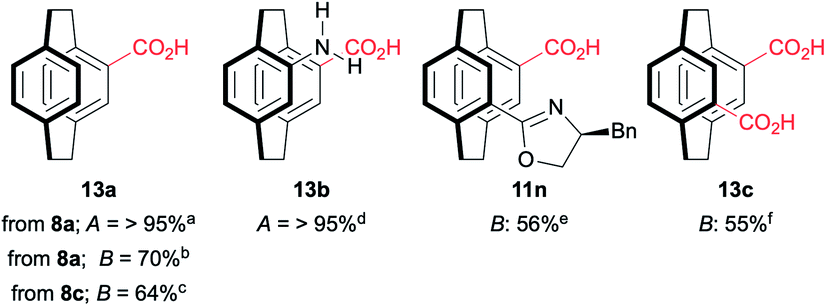
Oxazolines are robust, and hydrolysis is not straightforward. The hydrolysis of 11e by heating to reflux in 6 M HCl has been reported to give 13b in good yield (Table 2),18 and these conditions worked for mono(benzyloxazoline) 8a but failed to hydrolyse the mono(tert-butyloxazoline) 8c or the bis(oxazoline) 11b. More forcing conditions using concentrated sulfuric acid in dioxane at 110 °C lead to the clean hydrolysis of mono(oxazolines) while the pseudo-ortho bis(oxazoline) gives a mixture of the 4-acid-12-oxazoline 11n and the desired 4,12-diacid 13c. Fortunately, the products are easily separated based on solubility and recycling 11n permits good overall yields of 13c to be achieved. No sign of racemisation can be detected by optical rotation. While the conditions are harsh, the rapid synthesis of the oxazolines with concomitant resolution still make this methodology attractive if conducted early in a synthetic sequence.
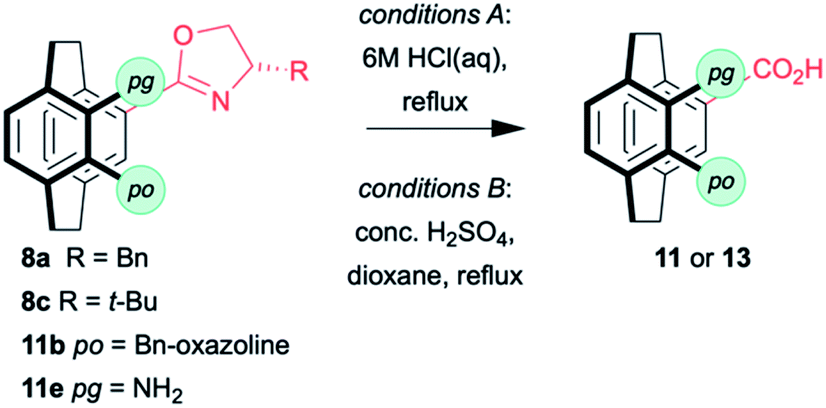
It is possible to subject the enantiomerically enriched 4-carboxylic acid 13a to a decarboxylative phosphorylation to give the phosphine oxide 14 in 8–20% (along with 50% unreacted starting material with no erosion of enantiopurity (Scheme 3).35–37 So far, we have been unable to react the pseudo-ortho-diacid under the same conditions. Although the preliminary results are low yielding, they demonstrate that it is possible to isolate enantiomerically enriched phosphine oxide in just four steps. Only our own sulfoxide chemistry38,39 is as efficient and that methodology was limited by the need for a sulfoxide–lithium exchange with tert-butyllithium.
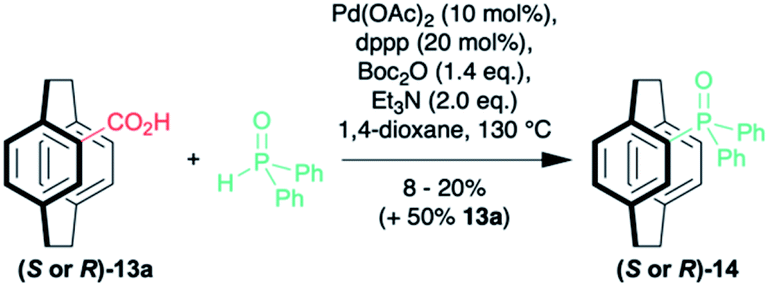 | ||
| Scheme 3 Decarboxylative phosphorylation. | ||
[2.2]Paracyclophane offers a useful planar chiral skeleton but its widespread adoption has been frustrated by a lack of simple methods to resolve the planar chirality. In this paper, we have shown how oxazolines can be coupled to bromo-derivatives to give separable diastereomers. The methodology permits a range of disubstituted [2.2]paracyclophanes to be rapidly prepared in enantiomerically pure form. We report the first example of a decarboxylative coupling on [2.2]paracyclophane. Optimisation of the decarboxylative coupling offers an exciting new route to enantiomerically enriched planar chiral molecules that are challenging to synthesise by conventional methods. We intend to extend the utility of this reaction to other derivatives, and will publish these studies in due course. Simpler, more rapid access to enantiomerically pure [2.2]paracyclophane derivatives will facilitate their wider use in catalysts, bioactive compounds, and materials.
Author contributions
ST and MNM contributed equally to this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Massey University for support.References
- M. N. Mungalpara, J. Wang, M. P. Coles, P. G. Plieger and G. J. Rowlands, Tetrahedron, 2018, 74, 5519–5527 CrossRef CAS.
- C. Zippel, Z. Hassan, M. Nieger and S. Bräse, Adv. Synth. Catal., 2020, 362, 3431–3436 CrossRef CAS.
- J. Paradies, Synthesis, 2011, 3749–3766 CrossRef CAS.
- S. Felder, S. Wu, J. Brom, L. Micouin and E. Benedetti, Chirality, 2021, 33, 506–527 CrossRef CAS PubMed.
- M. Skultety, H. Hubner, S. Lober and P. Gmeiner, J. Med. Chem., 2010, 53, 7219–7228 CrossRef CAS PubMed.
- R. Rajan, D. Schepmann, R. Steigerwald, J. A. Schreiber, E. El-Awaad, J. Jose, G. Seebohm and B. Wünsch, ChemMedChem, 2021, 16(20), 3201–3209 CrossRef CAS PubMed.
- K. J. Weiland, A. Gallego and M. Mayor, Eur. J. Org. Chem., 2019, 2019, 3073–3085 CrossRef CAS.
- Y. Morisaki and Y. Chujo, Bull. Chem. Soc. Jpn., 2019, 92, 265–274 CrossRef CAS.
- O. R. P. David, Tetrahedron, 2012, 68, 8977–8993 CrossRef CAS.
- Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC.
- G. J. Rowlands, Org. Biomol. Chem., 2008, 6, 1527–1534 RSC.
- Z. Hassan, E. Spuling, D. M. Knoll and S. Bräse, Angew. Chem., Int. Ed., 2020, 59, 2156–2170 CrossRef CAS PubMed.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284–PR437 CrossRef CAS PubMed.
- G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
- S. A. Babu, K. K. Krishnan, S. M. Ujwaldev and G. Anilkumar, Asian J. Org. Chem., 2018, 7, 1033–1053 CrossRef CAS.
- D. K. Whelligan and C. Bolm, J. Org. Chem., 2006, 71, 4609–4618 CrossRef CAS PubMed.
- S. Kitagaki, S. Murata, K. Asaoka, K. Sugisaka, C. Mukai, N. Takenaga and K. Yoshida, Chem. Pharm. Bull., 2018, 66, 1006–1014 CrossRef CAS PubMed.
- B. Hong, Y. Ma, L. Zhao, W. Duan, F. He and C. Song, Tetrahedron: Asymmetry, 2011, 22, 1055–1062 CrossRef CAS.
- X.-L. Hou, X.-W. Wu, L.-X. Dai, B.-X. Cao and J. Sun, Chem. Commun., 2000, 1195–1196 RSC.
- D. M. Knoll, Y. Hu, Z. Hassan, M. Nieger and S. Bräse, Molecules, 2019, 24, 4122 CrossRef PubMed.
- C. Bolm, K. Wenz and G. Raabe, J. Organomet. Chem., 2002, 662, 23–33 CrossRef CAS.
- C. Braun, M. Nieger, W. R. Thiel and S. Bräse, Chem.–Eur. J., 2017, 23, 15474–15483 CrossRef CAS PubMed.
- A. Marchand, A. Maxwell, B. Mootoo, A. Pelter and A. Reid, Tetrahedron, 2000, 56, 7331–7338 CrossRef CAS.
- J. R. Fulton, J. E. Glover, L. Kamara and G. J. Rowlands, Chem. Commun., 2011, 47, 433–435 RSC.
- J. E. Glover, P. G. Plieger and G. J. Rowlands, Aust. J. Chem., 2014, 67, 374–380 CrossRef CAS.
- L. Ackermann, S. Barfüsser, C. Kornhaass and A. R. Kapdi, Org. Lett., 2011, 13, 3082–3085 CrossRef CAS PubMed.
- D. Ghorai, V. Müller, H. Keil, D. Stalke, G. Zanoni, B. A. Tkachenko, P. R. Schreiner and L. Ackermann, Adv. Synth. Catal., 2017, 359, 3137–3141 CrossRef CAS.
- T. Xi, Y. Mei and Z. Lu, Org. Lett., 2015, 17, 5939–5941 CrossRef CAS PubMed.
- S. Kitagaki, K. Sugisaka and C. Mukai, Org. Biomol. Chem., 2015, 13, 4833–4836 RSC.
- X. C. Wang, Z. Chen, W. Z. Duan, C. Song and Y. D. Ma, Tetrahedron: Asymmetry, 2017, 28, 783–790 CrossRef CAS.
- D. M. Knoll, H. Simek, Z. Hassan and S. Brase, Eur. J. Org. Chem., 2019, 2019, 6198–6202 CrossRef CAS.
- M. N. Mungalpara, P. G. Plieger and G. J. Rowlands, Adv. Synth. Catal., 2021, 363, 1069–1080 CrossRef CAS.
- K. P. Jayasundera, D. N. M. Kusmus, L. Deuilhe, L. Etheridge, Z. Farrow, D. J. Lun, G. Kaur and G. J. Rowlands, Org. Biomol. Chem., 2016, 14, 10848–10860 RSC.
- H. J. Reich and D. J. Cram, J. Am. Chem. Soc., 1969, 91, 3527–3533 CrossRef CAS.
- J.-S. Zhang, T. Chen and L.-B. Han, Eur. J. Org. Chem., 2020, 2020, 1148–1153 CrossRef CAS.
- K. Xu, L. Liu, Z. Li, T. Huang, K. Xiang and T. Chen, J. Org. Chem., 2020, 85, 14653–14663 CrossRef CAS PubMed.
- R. Yu, X. Chen, S. F. Martin and Z. Wang, Org. Lett., 2017, 19, 1808–1811 CrossRef CAS PubMed.
- R. Parmar, M. P. Coles, P. B. Hitchcock and G. J. Rowlands, Synthesis, 2010, 4177–4187 CAS.
- P. B. Hitchcock, G. J. Rowlands and R. Parmar, Chem. Commun., 2005, 4219–4221 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra01075e |
| This journal is © The Royal Society of Chemistry 2022 |