DOI:
10.1039/D2RA01031C
(Paper)
RSC Adv., 2022,
12, 11933-11941
Controllable in situ growth of novel octahedral TiO2 nanoparticles on nickel/titanium alloy fiber substrate for selective solid-phase microextraction of ultraviolet filters in water samples†
Received
16th February 2022
, Accepted 5th April 2022
First published on 19th April 2022
Abstract
The nature and fabrication of fiber coatings with good adsorption capacity and selectivity play a decisive role in solid-phase microextraction (SPME). In this work, a novel SPME fiber was fabricated through hydrothermal in situ growth of octahedral TiO2 nanoparticles (TiO2NPs) on a superelastic nickel/titanium alloy (NiTi) wire substrate in acid solution. The resulting fiber coatings were characterized by scanning electron microscopy and energy dispersive X-ray spectroscopy. Acid types, acid concentration as well as hydrothermal temperature and time were found to be effective route to manipulate the morphologies and composition of TiO2-based nanoflakes grown on the NiTi fiber substrates. At the concentration of 0.4 mol L−1 HCl as well as hydrothermal temperature of 150 °C and hydrothermal time of 12 h, TiO2NPs were in situ grown on the NiTi wire substrates. The obtained NiTi wire with the TiO2NPs coating (NiTi@TiO2NPs fiber) was employed to investigate the adsorption of some representative aromatic analytes in water samples coupling with high-performance liquid chromatography with UV detection (HPLC/UV). The results clearly demonstrate that the fiber exhibits good extraction selectivity for ultraviolet filters (UVFs). In view of good extraction selectivity for the selected UVFs, the key experimental parameters were optimized. Under the optimum conditions, the calibration curves were linear in the ranges of 0.05–100 μg L−1 with the correlation coefficients greater than 0.998. Limits of detection (LODs) were 0.007 to 0.064 μg L−1. Furthermore, the intra-day and inter-day repeatability of the proposed method with the single fiber varied from 4.3% to 6.1% and from 4.5% to 6.8%, respectively. The fiber-to-fiber reproducibility ranged from 5.8% to 8.2%. The developed SPME-HPLC/UV method was applied to selective preconcentration and sensitive determination of target UVFs from real water samples. Moreover, the fabricated fiber showed precisely controllable growth and 150 extraction and desorption cycles.
1. Introduction
Over the past decades, solid-phase microextraction (SPME) has attracted great interest in the analysis of various analytes in environmental, food, pharmaceutical, biological and clinical samples because it is solvent-free, reusable and cost-effective.1–3 Moreover, this technique integrates sampling, extraction, enrichment, cleanup and sample introduction into a single step. In the conventional fiber configuration, the sensitivity and selectivity of SPME strongly depend on the intrinsic properties of the fiber coating because SPME is based on the establishment of the extraction equilibrium of the analytes between the fiber coating and the sample matrix.4 Currently commercially available SPME fibers are mainly fused-silica fibers coated with organic polymers, such as polydimethylsiloxane (PDMS) for nonpolar analytes and polyacrylate (PA) for polar analytes.5 However, the fused-silica fiber has some potential drawbacks such as mechanical fragility, relatively low working temperature and poor solvent resistance, and must be handled with great care in sampling and injection procedures. In addition, in the case of adsorption, the surface adsorption amount of target analytes is subject to the specific surface area of SPME fibers coatings in practical applications. High specific surface area of the fiber coatings is highly desired to achieve high extraction capacity of SPME fiber coatings. Hence, a variety of novel SPME fibers have been developed to increase the surface area of the fiber coatings, enhance the extraction capability and improve the extraction selectivity at the same time.6 Due to their excellent adsorption performance, different nanomaterials have been used as promising candidates for the development of the SPME fiber coatings.7,8 Meanwhile, metal wires were of strong physical strength, and can be handled with great convenience.9 For these reasons, great efforts have been made to prepare novel metallic SPME fibers with nanomaterial coatings instead of commercial fused-silica fibers with organic polymers coatings. In particular, the in situ growth of nanostructured coatings on the metallic fibers have attracted more attention due to their excellent adsorption properties as well as high chemical, thermal and mechanical stability.9,10
Metal oxides are a class of appealing inorganic functional materials. Their surface properties are highly dependent on their morphologies and sizes.11 Moreover, their characteristic structures allow for hydrogen bonding, electrostatic, hydrophilic/hydrophobic and Lewis acid–base interactions with organic analytes.12,13 In comparison with the commercial polymeric coatings, metal oxide coatings are highly versatile with diverse nanostructures and multiple interactions in extraction process. These interactions along with their nanostructures make them good candidates for use as a class of promising fiber coatings to address the challenge for enhancing extraction capacity and improving extraction selectivity at the same time. Among these metal oxides, as a promising material, titanium dioxide (TiO2) has gained great interest because of its diverse nanostructures, excellent adsorption properties, high chemical and thermal stability, and good compatibility.14–18 Nanostructured TiO2 sorbents as SPME coatings on the metal fiber substrates have been applied to the analysis of various analytes such as, polycyclic aromatic hydrocarbons (PAHs),19,20 ultraviolet filters (UVFs)21 and pesticides.22
Among the metal-based fiber substrates, nearly equiatomic nickel/titanium alloy (NiTi) substrates have some additional interesting characteristics such as shape-memory effect, superelasticity and corrosion resistance.23 In practical application, the NiTi fiber substrates are able to withstand any potential deformation in microextraction and injection procedures. These unique properties have enabled NiTi wires to become one of the most frequently used metallic fiber substrates in SPME applications.24–29 In recent years, the in situ growth of TiO2/NiO composite coatings for SPME applications was reported by electrochemical anodization,30 chemical oxidation31,32 and hydrothermal reaction.33 In some cases, the novel TiO2/NiO nanostructures with different surface morphologies and compositions demonstrate high adsorption capacity and good selectivity toward aromatic analytes.30,32 However, the presence of NiO would lower the acid resistance of the developed NiTi fibers and affect their adsorption performance at the same time.30,32,33 Therefore, the in situ growth of individual stable TiO2 coatings on the NiTi fiber substrate is highly desired. However, the in situ growth of pure TiO2 coatings on the NiTi fiber substrate was rarely reported for the SPME application. Herein, we firstly report controllable in situ growth of novel octahedral TiO2 nanoparticles (TiO2NPs) on the NiTi fiber substrates by direct hydrothermal reaction in strong acid solution for SPME. Surface morphology and elemental composition of oxide coatings were tuned under different acidic conditions. The extraction performance of the obtained TiO2NPs coatings was evaluated using typical chlorophenols (CPs), phthalic acid esters (PAEs), ultraviolet filters (UVFs) and polycyclic aromatic hydrocarbons (PAHs) as model analytes coupled to high-performance liquid chromatography with UV detection (HPLC-UV). In view of its higher extraction efficiency and better extraction selectivity for UVFs, the TiO2NPs coated NiTi (NiTi@TiO2NPs) fiber was employed to optimize the effect of ionic strength, extraction temperature, stirring, extraction and desorption time. Moreover, this fiber was compared with the commercially available polyacrylate (PA) fiber. Finally, the proposed method was applied to selective enrichment and determination of target UVFs in snow water, river water and wastewater samples.
2. Experimental
2.1. Materials and reagents
Commercially available NiTi wires (0.25 mm, OD) were obtained from Alfa Aesar (Ward Hill, MA, USA). A 85 μm PA fiber was purchased from Supelco (Bellefonte, PA, USA). A 0.45 mm microporous membrane of polyvinylidene fluoride was supplied by Xingya Purification Material Factory (Shanghai, China).
Glacial acetic acid and sodium chloride (NaCl) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Nitric acid (HNO3), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O) and disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O) were purchased from Yantai Shuangshuang Chemical Ltd (Shandong, China). Hydrochloric acid (HCl) was purchased from Beijing chemical works (Beijing, China). Hydrofluoric acid (HF) was obtained from Tianjin Damao Chemical Reagent Factory. HPLC-grade methanol was obtained from Yuwang Chemical Company (Shandong, China). Certified individual standards of phenanthrene (Phe), fluoranthene (Flu), pyrene (Pyr), benzo[a]pyrene (B[a]p), and Benzo[ghi]perylene (B[ghi]p) were purchased from Aldrich (St. Louis, MO, USA). 2-Ethylhexyl salicylate (EHS) was purchased from Dr Ehrenstorfer (Augsburg, Ger-many). 4-Mehylbenzyidene camphor (MBC), octocrylene (OC), 2-ethylhexyl 4-(dimethylamino) benzoate (OD-PABA), 2-ethyl-hexyl 4-methoxycinnamate (EHMC), 2-chlorophenol (2-CP), 2,4-dichlorophenol (2,4-DCP), 2,6-dichlorophenol (2,6-DCP), 2,4,6-trichlorophenol (2,4,6-TCP), 2-(2,4-dichlorophenoxy)-5-chlorophenol (Triclosan), dimethyl phthalate (DMP), diethyl phthalate (DEP), di-n-butyl phthalate (DBP), dioctyl phthalate (DOP), and 2-ethylhexyl phthalate (DEHP) were obtained from AccuStandard (New Haven, CT, USA). Unless otherwise stated, all reagents were of analytical grade.
2.2. Apparatus
Surface morphology and elemental composition of the fabricated fibers were characterized by an Ultra Plus scanning electronic microscope (SEM) (Zeiss, Oberkochen, Germany) equipped with an energy dispersive X-ray (EDX) spectrometer. SPME was carried out in a B13-3 thermostat with magnetic stirrer (Shanghai, China). Desorption was carried out in a commercially available SPME-LC interface (Bellefonte, PA, USA). Separation and detection were performed on Waters 600E multi-solvent delivery system (Milford, MA, USA) with a Waters Sunfire C18 column (150 mm × 4.6 mm, 5 μm) and a Waters 2487 dual λ absorbance detector. Data acquisition was performed on a N2000 chromatography workstation (Zhejiang University, China). Ultra-pure water was obtained from an Ulupure water purification system (Chengdu, Sichuan, China).
2.3. Preparation of sample solutions
Standard stock solutions of CPs (2-CP, 2,4-DCP, 2,6-DCP, 2,4,6-TCP and Triclosan), PAEs (DMP, DEP, DBP, DOP and DEHP), UVFs (EHS, MBC, OC, OD-PABA and EHMC), and PAHs (Phe, Flu, Pyr, B[a]p and B[ghi]p) were prepared in HPLC-grade methanol at a concentration of 100 mg L−1, and stored in amber bottles in a refrigerator at 4 °C, shielding from light. Mixtures of standard solutions were prepared by mixing individual standard stock solutions with ultra-pure water to the desired concentration to examine the extraction performance. Real water samples were collected from the local river and the effluents of local wastewater treatment plant in April, 2020. Snow water was collected on campus. All these water samples were filtrated through microporous membrane to remove suspended particulates and stored in brown glass bottles at 4 °C.
2.4. Fabrication of the octahedral TiO2 coatings on the NiTi wires
The NiTi wires (5.5 cm in length) were ultrasonically cleaned with acetone and ultra-pure water for 10 min, respectively. Before the hydrothermal treatment, the NiTi wires were activated in a solution of HF, HNO3 and H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:5, v/v/v) for 2 min. After ultrasonic cleaning in ultra-pure water for 10 min, the pretreated ends were immediately dipped into 20 mL HCl solutions of 0.4 mol L−1 in 50 mL Teflon-lined autoclaves, and kept in an oven at 150 °C for 12 h. Thereafter, the hydrothermally treated NiTi wires were rinsed with ultra-pure water, and dried in air. The fabricated NiTi fibers were conditioned in the SPME-LC interface for 30 min prior to use.
:4:5, v/v/v) for 2 min. After ultrasonic cleaning in ultra-pure water for 10 min, the pretreated ends were immediately dipped into 20 mL HCl solutions of 0.4 mol L−1 in 50 mL Teflon-lined autoclaves, and kept in an oven at 150 °C for 12 h. Thereafter, the hydrothermally treated NiTi wires were rinsed with ultra-pure water, and dried in air. The fabricated NiTi fibers were conditioned in the SPME-LC interface for 30 min prior to use.
2.5. SPME-HPLC procedure
In a typical SPME procedure, 15 mL of working solution was transferred into a 20 mL sample vial sealed with a Teflon septum and a PEEK cap. NaCl was added to control the ionic strength of working solution or sample solution and phosphate buffer was added to adjust the solution pH. Thereafter, the fabricated fiber was pierced the septum and exposed to the stirred solution at preset temperature for a time interval. After extraction, the fiber was withdrawn and immediately inserted into the SPME-LC interface for static desorption in mobile phase. Subsequently, the mobile phase would pass through the SPME-LC interface for HPLC analysis. The phase ratio of methanol and water was 85/15, 70/30, 75/25 and 87/13 (v/v) for PAHs, CPs, PAEs and UVFs. Corresponding absorption wavelengths were set at 254 nm, 282 nm, 280 nm and 310 nm to detect chromatographic responses. In order to avoid possible carry-over effect, the fabricated fiber was sequentially cleaned in methanol and ultra-pure water for 5 min after extraction, respectively.
3. Results and discussion
3.1. Surface morphology and composition of the oxide coatings
Commercial NiTi wire is straight annealed with surface oxide according to its specifications. As shown in Fig. S1a and b,† the bare NiTi wire has relatively smooth surface with some microcracks at high magnification. A native surface passivation layer is present according to the composition of Ni, Ti and O elements. For this reason, the bare NiTi wire was pretreated in the mixed acids to remove the passivation layer. After acid treatment, the pretreated NiTi wires were immediately dipped into various acid solutions at different concentrations in Teflon-lined autoclaves, and kept in an oven at different temperatures for different time intervals. The in situ growth of the oxide coatings on the NiTi fiber substrates was investigated in detail by hydrothermal reaction in acid solution.
3.1.1. Effect of different acids. Fig. 1 shows the effect of different acids (glacial acetic acid, HNO3 and HCl) on the morphology and composition of surface oxide coatings. The morphologies of the oxide coatings grown in acidic solutions are very different. According to Fig. 1b and d, pure TiO2 coatings were in situ grown on the NiTi fiber substrates. In particular, the well-defined octahedral TiO2NPs coating was formed in HCl solution. During hydrothermal process, the formation of the TiO2NPs coating in HNO3 is due to the Ni dissolution, which occurs simultaneously along with the oxidation of Ti.34 In HCl solution, the formation of TiO2NPs occurs as a consequence of the oxidation and hydrolysis of surface Ti along with the dissolution of Ni.35 It can also be seen from Fig. 1e that compared with those of 50–150 nm obtained in HNO3 solution and those of 150–300 nm obtained in HCl solution, the particle sizes of the oxide composite grown in glacial acetic acid are about 10 nm. Corresponding EDX analysis confirms that the oxide coating is composed of O, Ti and Ni elements (Fig. 1f), indicating that glacial acetic acid is too weak to remove Ni element from the Ni and Ti oxide composite coating. Therefore, the types of acids can affect the morphologies and compositions of the oxide coatings grown on the NiTi fiber substrates. Due to the formation of well-defined octahedral TiO2NPs with high specific surface area, HCl was used as a representative acid to examine several preliminary hydrothermal parameters in the following study.
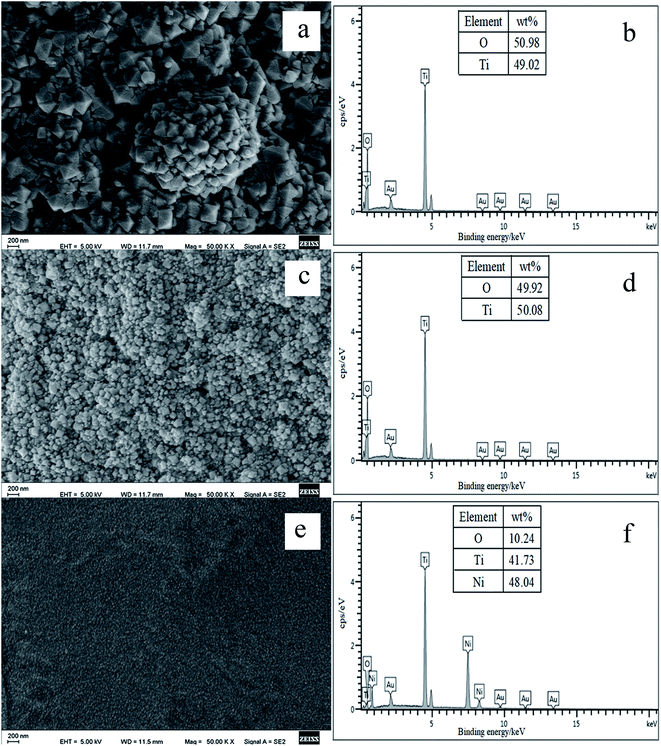 |
| | Fig. 1 Surface SEM images and EDX spectra of the oxide coatings grown on the pretreated NiTi wire by hydrothermal reaction in HCl solution (a and b), HNO3 solution (c and d) and glacial acetic acid (e and f) at 150 °C for 12 h. | |
3.1.2. Effect of HCl concentration. Fig. S2† shows SEM images and EDX spectra of the oxide coatings at HCl concentrations of 0.1 mol L−1 (Fig. S2a and b†), 0.2 mol L−1 (Fig. S2c and d†), 0.3 mol L−1 (Fig. S2e and f†) and 0.4 mol L−1 (Fig. S2g and h†). As the concentration increases, the Ni content rapidly decreased. At the concentration of 0.4 mol L−1 HCl, pure octahedral TiO2 was directly grown on the NiTi fiber substrate (Fig. S2h†), indicating that the Ni element in the surface oxide coating was completely dissolved in the solution of 0.4 mol L−1 HCl. At the same time, the sizes of the oxide nanoparticles remarkably increases. When the concentration was higher than 0.4 mol L−1, the NiTi wire becomes very brittle and was not suitable for SPME. Therefore, 0.4 mol L−1 HCl was employed during hydrothermal process.
3.1.3. Effect of temperature. Effect of temperature on the oxide coatings was further examined in the solution of 0.4 mol L−1 HCl. As shown in Fig. S3a,† the NiTi fiber shows a winkled surface structure at 100 °C. The EDX analysis shown in Fig. S3b† confirms that a thin oxide coating was grown on the NiTi fiber surface due to its less content of O element. At 150 °C and 200 °C, the uniform octahedral TiO2NPs coatings were obtained (Fig. S3c–f†). However, in Fig. S3e,† the TiO2NPs coating was partially detached from the fiber substrate at 200 °C. Therefore, 150 °C was chosen for hydrothermal reaction.
3.1.4. Effect of hydrothermal reaction time. Fig. S4† shows the effect of hydrothermal reaction time on the morphology and composition of the oxide coatings. From Fig. S4a and b,† the interconnected Ni and Ti oxide nanoparticles with smaller sizes were grown on the NiTi fiber substrate within 3 h. When the longer time was employed, the pure octahedral TiO2NPs coatings were grown. Compared with the TiO2NPs coating of 6 h (Fig. S4c†), the well-defined TiO2NPs coating with larger sizes was grown on the NiTi fiber substrate within 12 h (Fig. S4e†). As a result, 12 h was employed for the hydrothermal reaction.
3.2. Extraction capability and selectivity
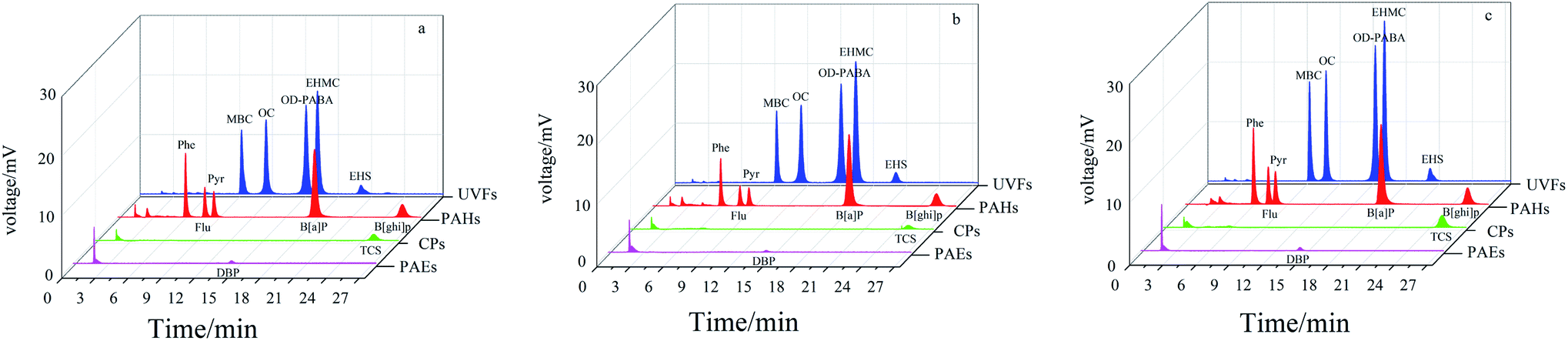
Metal oxides have high affinity toward aromatic compounds because of multiple interactions such as van der Waals forces, hydrogen bonding, Lewis acid-base, and hydrophilic/hydrophobic interaction interactions, which can induce adsorption selectivity toward aromatic analytes.36,37 For this purpose, four groups of typical aromatic compounds (CPs, PAEs, UVFs and PAHs) with different molecular structures were employed as model analytes to study the extraction performance of the pure TiO2 coatings. Fig. 2 shows the extraction capability and selectivity of the NiTi@TiO2NPs fiber obtained in HNO3 solution, and obtained in HCl solution within 6 h and 12 h. All of the fibers showed high extraction capability for the studied UVFs and PAHs, and poor/negligible extraction capability for PAEs and CPs. Notably the highest adsorption efficiency was achieved for SPME with the NiTi@TiO2NPs fiber obtained in HCl solution within 12 h. In view of its highest extraction capability for UVFs, the NiTi@TiO2NPs fiber within 12 h was further exploited as a potential fiber for the extraction of UVFs in the following study.
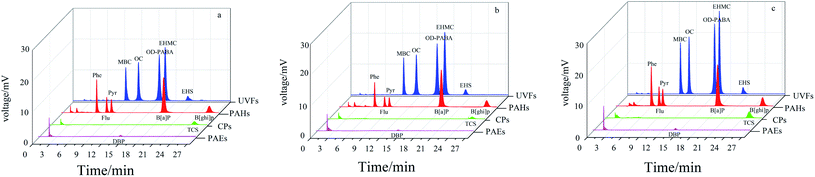 |
| | Fig. 2 Typical chromatograms of SPME-HPLC with the NiTi@TiO2NPs fiber obtained in HNO3 solution (a), the NiTi@TiO2NPs fibers obtained in HCl solution within 6 h (b) and 12 h (c) for CPs, PAEs, UVFs and PAHs. | |
3.3. Comparison of the NiTi@TiO2NPs fiber with commercial PA fiber
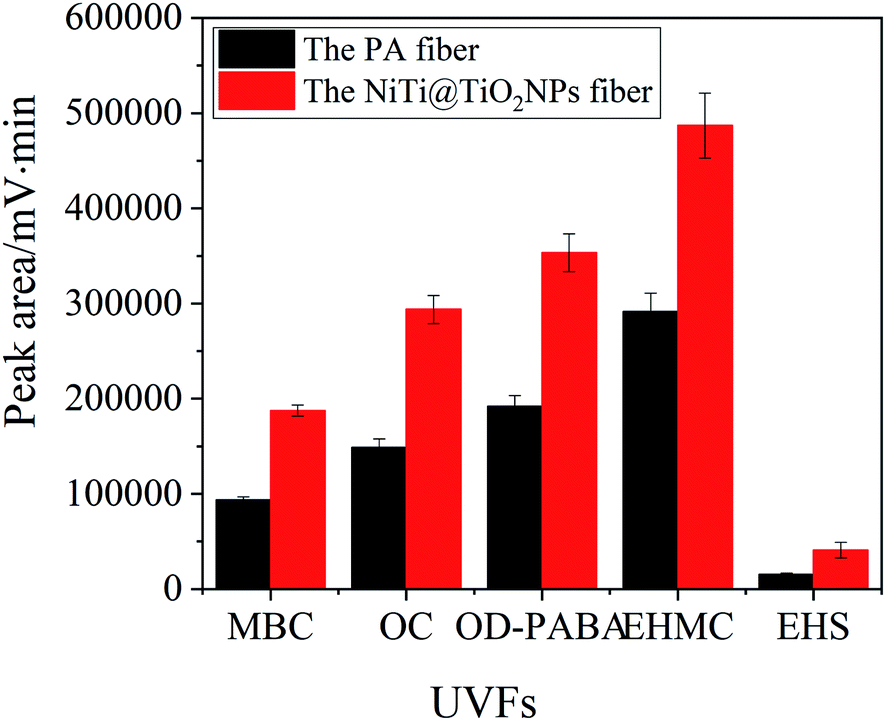
Fig. 3 exhibites the extraction capability of the NiTi@TiO2NPs fiber and the commercial PA fiber for UVFs. The NiTi@TiO2NPs fiber exhibited high extraction efficiency for the studied UVFs than the PA fiber. The peak areas of MBC, OC, OD-PABA, EHMC and EHS obtained by the TiO2NPs coating are 1.7–2.6 times higher than those obtained by the PA coating. In view of its excellent extraction capability, the NiTi@TiO2NPs fiber was further employed to examine the feasibility of the SPME procedure in combination with HPLC-UV.
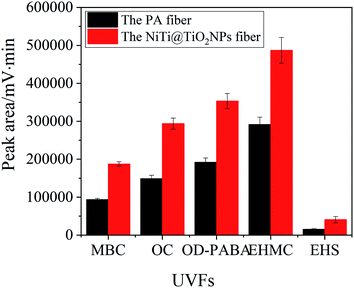 |
| | Fig. 3 The extraction capability of the PA fiber and the NiTi@TiO2NPs fiber for UVFs. | |
3.4. Optimization of SPME conditions for UVFs
In SPME, adsorption is subject to the thermodynamic and kinetic factors. For this purpose, the effects of ionic strength, stirring rate, extraction temperature, pH, as well as extraction and desorption time on the extraction efficiency of the selected UVFs from water were further optimized.
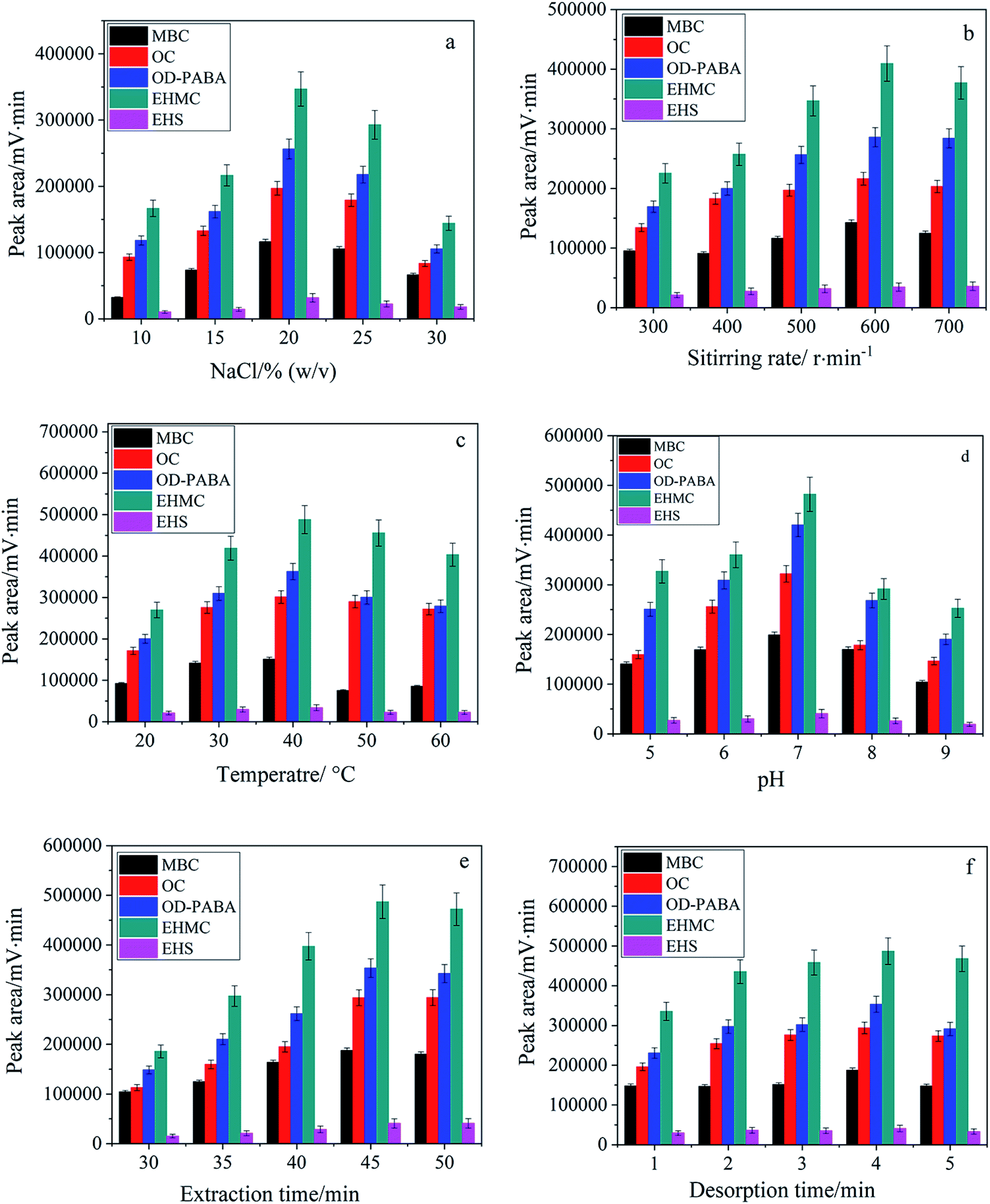
3.4.1. Effect of ionic strength. Generally addition of inert salt can decrease the solubility of analytes in aqueous phase due to salting-out effect and enhance their partitioning onto the fiber for SPME. Nevertheless more salt addition also leads to increase viscosity of the aqueous phase which negatively affects the kinetics of the adsorption process. Therefore, addition of salt is favorable from thermodynamic point of view but unfavorable from kinetic point of view.38 In this study, NaCl of 10–30%(w/v) was investigated. As shown in Fig. 4a, the extraction efficiency was gradually increased with the increasing content of NaCl from 10% to 20%, and thereafter decreased at higher content of NaCl. In the following experiments, NaCl of 20% was selected to maintain the ionic strength of aqueous solution.
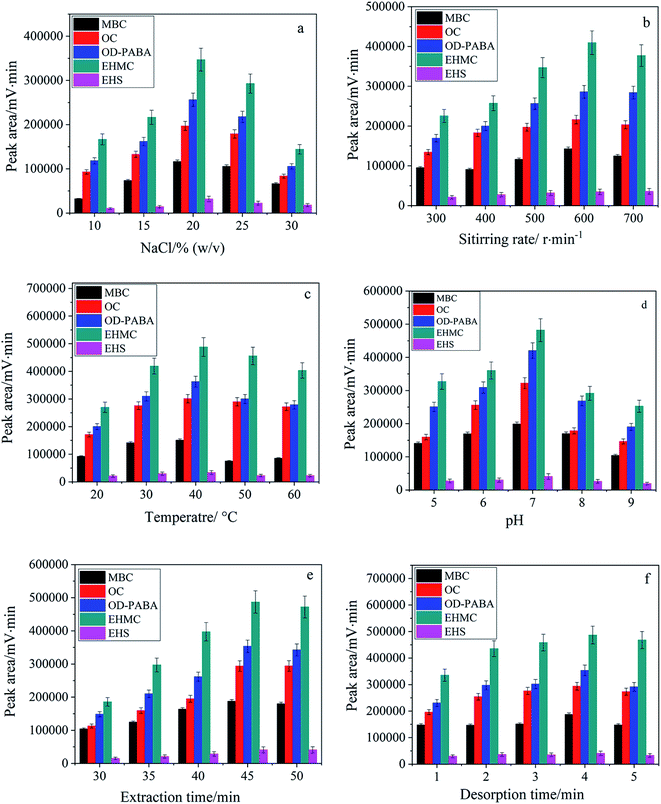 |
| | Fig. 4 Dependence of extraction efficiency on ionic strength (a), stirring rate (b), extraction temperature (c), pH (d) as well as extraction time (e) and desorption time (f). | |
3.4.2. Effect of stirring rate and extraction temperature. In fiber SPME, stirring is an essential process to improve mass transfer and reduce the equilibration time. Therefore, stirring rate was studied over the range of 300–700 rpm. As shown in Fig. 4b, the highest extraction efficiency was obtained at the stirring rate of 600 rpm. Furthermore, the elevated temperature is kinetically favorable for improving the diffusion of the analyte molecules but thermodynamically unfavorable for adsorption process due to its exothermic nature. Moreover, the increased water solubility and volatilization of the analytes result in a less amount of the analytes extracted. Therefore, temperature is required to be examined in SPME.38 As shown in Fig. 4c, the best extraction efficiency was achieved at 40 °C.
3.4.3. Effect of pH. According to the molecular structures of UVFs and TiO2NPs, the solution pH will affect the existing forms of UVFs in aqueous solution and the surface charge of TiO2NPs at the same time.39 Therefore, the effect of solution pH on the extraction efficiency of five UVFs was investigated within the pH range varying from 5 to 9. As shown in Fig. 4d, pH strongly affected the extraction efficiency of target species and presented the highest extraction efficiency for UVFs at pH 7. Therefore, the microextraction was performed at pH 7 in subsequent study.
3.4.4. Effect of extraction and desorption time. SPME is an equilibrium-based extraction process, and thereby the optimal extraction time should be the time taken for the system to reach equilibrium. The influence of the extraction time was studied by varying the extraction time from 30 to 50 min. In Fig. 4e, the extraction equilibrium was reached within 45 min. Also, the effect of desorption time was examined within 1–5 min (Fig. 4f). Desorption of the extracted analytes from the fiber coating is faster when desorption was carried out in the mobile phase. Sufficient solvent desorption of UVFs in the mobile phase was accomplished within 4 min.
3.5. Analytical performance
To validate the analytical performance of the proposed method, linear ranges, limits of detection (LODs) and limits of quantification (LOQs) were evaluated under the optimum conditions. In Table 1, LODs were in the range of 0.007 to 0.064 μg L−1 (S/N = 3) along with LOQs between 0.023 and 0.215 μg L−1 (S/N = 10). Good linear responses were obtained in the concentration range of 0.05 μg L−1 to 100 μg L−1 with correlation coefficients (r) > 0.998. To examine the repeatability of the proposed method with the single fiber, five replicate analyses of a standard sample were performed at the concentration levels of 50 μg L−1. Relative standard deviations (RSDs) of the intra-day and inter-day analyses varied from 4.3% to 6.1% and from 4.5% to 6.8%, respectively. RSDs for the fiber-to-fiber reproducibility of the proposed method ranged from 5.8% to 8.2% with respect to five fibers fabricated in different batches, demonstrating the precisely controllable fabrication of the NiTi@TiO2NPs fiber. Furthermore, no significant decrease in peak areas of the tested UVFs was observed even after 150 cycles of extraction and desorption. The recoveries from 91.8% to 95.3% were achieved for spiking water at the level of 50 μg L−1, indicating that the NiTi@TiO2NPs fiber presents high recycling stability.
Table 1 Analytical parameters of the proposed method with the NiTi@TiO2NPs fiber (n = 5)
| Analytes |
Linear ranges (μg L−1) |
r |
Recovery (%) |
RSDs for single fiber repeatability |
RSDs for fiber-to-fiber reproducibility (%) |
LODs (μg L−1) |
LOQs (μg L−1) |
| Intra-day (%) |
Inter-day (%) |
| MBC |
0.1–100 |
0.9991 |
99.1 |
4.3 |
4.5 |
5.8 |
0.019 |
0.063 |
| OC |
0.1–100 |
0.9987 |
103 |
5.7 |
6.0 |
7.5 |
0.021 |
0.071 |
| OD-PABA |
0.1–100 |
0.9981 |
96.4 |
6.1 |
6.3 |
8.0 |
0.019 |
0.065 |
| EHMC |
0.05–100 |
0.9989 |
101 |
4.5 |
5.9 |
6.9 |
0.007 |
0.023 |
| EHS |
0.5–100 |
0.9994 |
105 |
5.4 |
6.8 |
8.2 |
0.064 |
0.215 |
3.6. Real water analysis
Organic UVFs are synthetic compounds with single or multiple aromatic structures, sometimes conjugated with carbonyl and/or hydroxyl groups. They are frequently used as ingredients in the formulation of sunscreens, cosmetics and personal care products to protect human skin from direct exposure to the deleterious wavelengths of sunlight. UVFs enter the aquatic environment directly as a result of water recreational activities or indirectly through wastewater resulting from the use of personal care products, washing clothes and industrial discharges. Currently extensive use of UVFs would lead to environmental pollution as well as accumulative negative effect on human health.40 Therefore, UVFs have been recently considered as emerging contaminants, and more attention should be paid to their monitoring in environmental water. However, it is difficult to determine UVFs in real water samples due to their low concentration and the co-existing interferences. As a result, an appropriate sample pretreatment and enrichment procedure is necessary prior to analysis.21,27 In order to examine its applicability, the proposed method was applied to the selective enrichment and the determination of target UVFs in snow water, river water and wastewater samples. In Table S1,† the relative recoveries of 76.2%–110% were achieved for snow water and river water spiked at 5.0 μg L−1 and 10.0 μg L−1. However, the relative recoveries were between 61.2% and 83.6% for wastewater samples. The representative chromatograms of UVFs obtained from snow water sample were shown in Fig. S5.† These data indicate that acceptable recoveries were obtained for target UVFs in real water samples.
3.7. Comparison of the proposed method with the reported methods
Comparison of the proposed method with some other methods, such as stir bar sorptive extraction41 and SPME,21,27,42–44 for the determination of UVFs from environmental waters in terms of some analytical parameters is listed in Table 2. Extraction time of the proposed method with the NiTi@TiO2NPs fiber is shorter than that of the reported works.27,41,43 The obtained LODs are lower than those in previous reports21,27,42–44 and comparable to those in previous reports.41 RSDs are also comparable to those in the previously reported methods. The proposed method is comparable to and even better than the other reported methods for the detection of UVFs.
Table 2 Comparison of the proposed method with some reported methods for the enrichment and determination of UVFs in water
| Methods |
Extraction time (min) |
Linear ranges (μg L−1) |
LODs (μg L−1) |
RSDs (%) |
Recovery (%) |
Ref. |
| TiO2NSs, TiO2 nanosheets. PIL, polymeric ionic liquids. SBSE, stir-bar sorptive extraction, MS, mass spectrometry. FID, flame ionization detection. C12, dodecyl. |
| TiO2NSs-SPME-HPLC-UVa |
30 |
0.1–400 |
0.026–0.089 |
4.5–9.6 |
88.8–107 |
21 |
| PIL-SPME-HPLC-UVb |
60 |
0.5–200 |
0.10–5.00 |
1.8–11.6 |
54.5–120 |
27 |
| PDMS-SBSE-HPLC-MSc |
180 |
0.025–1 |
0.0025–0.01 |
<26 |
25–89 |
41 |
| PDMS-SPME-GC-FIDd |
45 |
10–500 |
0.26–0.51b |
<8 |
85–97 |
42 |
| PA-SPME-GC-FID |
45 |
10–500 |
0.35–0.74b |
<8 |
82–99 |
42 |
| C12-SPME-HPLC-UVe |
60 |
5–200 |
0.69–1.37 |
3.5–19.7 |
82–93 |
43 |
| PA-SPME-GC-MS |
45 |
0.5–25 |
0.17–0.29 |
0.7–4.3 |
80–83 |
44 |
| TiO2-SPME-HPLC-UV |
45 |
0.05–100 |
0.007–0.064 |
4.5–9.1 |
61.2–110 |
This method |
4. Conclusion
In the present work, the NiTi wire was used as a Ti source and a fiber substrate for the in situ growth of octahedral TiO2NPs on the NiTi fiber substrates via hydrothermal reaction in acid solutions. The strong acid-based hydrothermal preparation was beneficial to the elimination of Ni element in the oxide coating. Pure octahedral TiO2NPs coating can be obtained in the solution of 0.4 mol L−1 HCl at 150 °C for more than 6 h, which was favorable for the SPME of UVFs and PAHs with a large delocalized π system. The types of acids, acid concentration and hydrothermal reaction time greatly affected the surface morphologies, dimensions and composition of the TiO2-based coatings, and thereby affected their extraction capability. To examine the potential applicability of the NiTi@TiO2NPs fiber, the effects of pH and ionic strength of aqueous solution, stirring rate, temperature as well as adsorption and desorption time on the adsorption efficiency of the selected PAHs were further optimized. At pH 7.0, maximum adsorption efficiency was achieved under the optimized experimental conditions including NaCl of 20%(w/v), stirring rate of 600 rpm, solution temperature of 40 °C, adsorption time of 45 min, and desorption time of 4 min. Under the obtained conditions, linear chromatographic responses were achieved over the concentration ranges of 0.05–100 μg L−1 with the correlation coefficients ranging from 0.9981 to 0.9994. Moreover, the NiTi@TiO2NPs fiber presented high mechanical stability, superior solvent resistance and highly efficient extraction performance for UVFs compared to the commercial PA fiber. This new strategy provides a facile route for controllable in situ growth of robust and highly efficient TiO2 nanostructures on the superelastic NiTi fiber substrates and expand the potential application of TiO2 coatings in SPME.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Scientific Research Fund of Mianyang normal university (QD2021A36).
References
- M. Llompart, M. Celeiro, C. García-Jares and T. Dagnac, Trends Anal. Chem., 2019, 112, 1–12 CrossRef CAS
 .
. - C. H. Xu, G. S. Chen, Z. H. Xiong, Y. X. Fan, X. C. Wang and Y. Liu, Trends Anal. Chem., 2016, 80, 12–29 CrossRef CAS .
- N. Reyes-Garcés and E. Gionfriddo, Trends Anal. Chem., 2019, 113, 172–181 CrossRef .
- V. Jalilia, A. Barkhordarib and A. Ghiasvand, Microchem. J., 2020, 152, 104319 CrossRef .
- M. d. F. Alpendurada, J. Chromatogr. A, 2000, 889, 3–14 CrossRef CAS PubMed .
- J. Zheng, J. L. Huang, Q. Yang, C. Y. Ni, X. T. Xie, Y. R. Shi, J. F. Sun, F. Zhu and G. F. Ouyang, Trends Anal. Chem., 2018, 108, 135–153 CrossRef CAS .
- M. Lashgari and Y. Yamini, Talanta, 2019, 191, 283–306 CrossRef CAS PubMed .
- V. Jalili, A. Barkhordari and A. Ghiasvand, Microchem. J., 2020, 153, 104386 CrossRef CAS .
- J. J. Feng, H. D. Qiu, X. Liu and S. X. Jiang, Trends Anal. Chem., 2013, 46, 44–58 CrossRef CAS .
- M. O. Aziz-Zanjani and A. Mehdinia, Anal. Chim. Acta, 2013, 781, 1–13 CrossRef CAS PubMed .
- M. Khajeh, S. Laurent and K. Dastafkan, Chem. Rev., 2013, 113, 7728–7768 CrossRef CAS PubMed .
- F. Pena-Pereira, R. M. B. O. Duarte and A. C. Duarte, Trends Anal. Chem., 2012, 40, 90–105 CrossRef CAS .
- J. Xu, P. Wu, E. C. Ye, B. F. Yuan and Y. Q. Feng, Trends Anal. Chem., 2016, 80, 41–56 CrossRef CAS .
- M. T. García-Valverde, R. Lucena, S. Cardenas and M. Valcárcel, Trends Anal. Chem., 2014, 62, 37–45 CrossRef .
- H. H. El-Maghrabi, A. Barhoum, A. A. Nada, Y. M. Moustafa, S. M. Seliman, A. M. Youssef and M. Bechelany, J. Photochem. Photobiol., A, 2018, 351, 261–270 CrossRef CAS .
- H. Moustafa, D. Darwish, A. Youssef, S. Reda and A. El-Wakil, Egypt. J. Chem., 2018, 61, 23–32 Search PubMed .
- M. H. A. Rehim, A. M. Youssef and A. Ghanem, Polym. Bull., 2015, 72, 2353–2362 CrossRef .
- A. M. Youssef, RSC Adv., 2014, 4, 6811–6820 RSC .
- H. M. Liu, D. A. Wang, L. Ji, J. B. Li, S. J. Liu, X. Liu and S. X. Jiang, J. Chromatogr. A, 2010, 1217, 1898–1903 CrossRef CAS PubMed .
- Y. Tian, J. J. Feng, Y. N. Bu, X. Q. Wang, C. N. Luo and M. Sun, Anal. Bioanal. Chem., 2017, 409, 4071–4078 CrossRef CAS PubMed .
- Y. Li, M. Zhang, Y. X. Yang, X. M. Wang and X. Z. Du, J. Chromatogr. A, 2014, 1358, 60–67 CrossRef CAS PubMed .
- S. Q. Liu, L. J. Xie, J. Zheng, R. F. Jiang, F. Zhu, T. G. Luan and G. F. Ouyang, Anal. Chim. Acta, 2015, 878, 109–117 CrossRef CAS PubMed .
- S. Shabalovskaya, J. Anderegg and J. Van Humbeeck, Acta Biomater., 2008, 4, 447–467 CrossRef CAS PubMed .
- L. Setkova, S. Risticevic, C. M. Linton, G. F. Ouyang, L. M. Bragg and J. Pawliszyn, Anal. Chim. Acta, 2007, 581, 221–231 CrossRef CAS PubMed .
- M. Azenha, M. Ornelas and A. Fernando Silva, J. Chromatogr. A, 2009, 1216, 2302–2306 CrossRef CAS PubMed .
- A. N. Dias, V. Simão, J. Merib and E. Carasek, Anal. Chim. Acta, 2013, 772, 33–39 CrossRef CAS PubMed .
- J. An and J. L. Anderson, Talanta, 2018, 182, 74–82 CrossRef CAS PubMed .
- M. J. Trujillo-Rodríguez, H. Nan and J. L. Anderson, J. Chromatogr. A, 2018, 1540, 11–20 CrossRef PubMed .
- C. C. An, Y. Liu, F. F. Wang, R. Zhang, J. L. Du and X. Z. Du, Microchem. J., 2020, 152, 104318 CrossRef CAS .
- Q. Zhen, M. Zhang, W. L. Song, H. J. Wang, X. M. Wang and X. Z. Du, J. Sep. Sci., 2016, 39, 3761–3768 CrossRef CAS PubMed .
- G. S. Nardini, J. O. Merib, A. N. Dias, J. N. B. Dutra, C. D. S. Silveira, D. Budziak, E. Martendal and E. Carasek, Microchem. J., 2013, 109, 128–133 CrossRef CAS .
- M. Zhang, Q. Zhen, H. J. Wang, M. Guo, S. S. Zhou, X. M. Wang and X. Z. Du, Talanta, 2016, 158, 214–221 CrossRef CAS PubMed .
- H. J. Wang, W. L. Song, M. Zhang, Q. Zhen, M. Guo, Y. D. Zhang and X. Z. Du, J. Chromatogr. A, 2016, 1468, 33–41 CrossRef CAS PubMed .
- D. Vojtěch, M. Voděrová, J. Fojt, P. Novák and T. Kubásek, Appl. Surf. Sci., 2010, 257, 1573–1582 CrossRef .
- L. M. Pérez, L. Gracia-Villa, J. A. Puértolas, M. Arruebo, S. Irusta and J. Santamaría, J. Biomed. Mater. Res., B, 2009, 91, 337–347 CrossRef PubMed .
- Na. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyacı, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef PubMed .
- P. J. Li, B. Hu and X. Y. Li, J. Chromatogr. A, 2012, 1247, 49–56 CrossRef CAS PubMed .
- A. Spietelun, A. Kloskowski, W. Chrzanowski and J. Namieśnik, Chem. Rev., 2013, 113, 1667–1685 CrossRef CAS PubMed .
- G. Sposito, Environ. Sci. Technol., 1998, 32, 2815–2819 CrossRef CAS .
- S. Ramos, V. Homem, A. Alves and L. Santos, Sci. Total Environ., 2015, 526, 278–311 CrossRef CAS PubMed .
- M. Pedrouzo, F. Borrull, R. M. Marcé and E. Pocurull, Anal. Bioanal. Chem., 2010, 397, 2833–2839 CrossRef CAS PubMed .
- D. A. Lambropoulou, D. L. Giokas, V. A. Sakkas, T. A. Albanis and M. I. Karayannis, J. Chromatogr. A, 2002, 967, 243–253 CrossRef CAS PubMed .
- J. Li, L. Y. Ma, M. Q. Tang and L. Xu, J. Chromatogr. A, 2013, 1298, 1–8 CrossRef CAS PubMed .
- O. Yilmazcan, C. Kanakaki, B. Izgi and E. Rosenberg, J. Sep. Sci., 2015, 38, 2286–2297 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b