
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlucose hydrothermal encapsulation of carbonized silicone polyester to prepare anode materials for lithium batteries with improved cycle stability†
Xuan Biea,
Man Xiongb,
Ben Wanga,
Yawei Donga,
Zhongxue Chen a and
Ronghua Huang*a
a and
Ronghua Huang*a
aSchool of Power & Mechanical Engineering, Wuhan University, Wuhan 430072, PR China. E-mail: huangronghua@whu.edu.cn
bSchool of Materials Science and Engineering, Hubei University, Wuhan, 430060, PR China
First published on 24th March 2022
Abstract
A silicon polyester (Si-PET) was synthesized with ethylene glycol and phthalic anhydride, and then it was carbonized and hydrothermally coated with glucose. The formed SiOx with layered graphene as the 3D network had an amorphous carbon layer. The graphene oxide (rGO) after carbothermal reduction was completely retained in SiOx, which improved the conductivity of the SiOx anode material. SiOx were encapsulated with a flexible amorphous carbon layer on the surface, which can not only improve the electrical performance, but also effectively relieve the huge volume changes of the compound. Further, the key point is that, the solid electrolyte interphase (SEI) film was mainly formed on the surface carbon layer. This would keep a stable SEI film during volume pulverization, and result in a good cycle stability. The SiOx/C-rGO material maintained a reversible capacity of 660 mA h g−1 at a current density of 100 mA g−1 for 100 cycles, a reversible capacity of 469.7 mA h g−1 at a current density of 200 mA g−1 for 300 cycles. The Coulomb efficiency was maintained at 98% except for the first cycle. After long cycling, the electrode expansion was 16%, which was much lower than those of silicon based materials. Therefore, this article provides a cheap, simple, and commercially valuable anode material for lithium batteries.
Introduction
With society developing, energy shortage becomes a problem should be solved, and the requirement of energy storage equipment is gradually increasing. As a highly efficient rechargeable secondary battery, lithium ion batteries (LIBs) are widely used in portable equipment, electronic vehicles, medical apparatus and other fields because of their high specific capacity, good safety, long cycle life, and environmental protection.1 With the pursuit of high energy density, stable cycle performance, and excellent safety of LIBs, the optimization of anode materials is a vital part of the current lithium-ion battery research.2,3 The current commercial lithium battery negative electrode is graphite with a specific capacity of 372 mA h g−1. Si-based anode materials are considered a promising choice for lithium ion anode materials because of their ultra-high specific capacity and low delithiation potential. However, they have the following disadvantages, which are caused by huge volumetric changes (∼300%) during charging and discharging, and must be improved: cracks between active material and electrode, unstable solid electrolyte interphase (SEI) layer, low initial coulombic efficiency (ICE), and low conductivity (10−5 to 10−3 S cm−1).4–8As a form of silicon-based anode, SiOx (silicon protoxide, 0 < x < 2) has attracted much attention due to its relatively high specific capacity (∼1600 mA h g−1), low charge–discharge potential and smaller volume expansion. The high-strength Si–O bond and the formation of Li2O and Li4SiO4 effectively reduce volume expansion. Nonetheless, the conductivity and electron migration rate of SiOx anode materials are very low, which would affect its electrical performance.9,10 The main method is to introduce carbon to improve conductivity and cycle stability while alleviating volume changes.11–15 The carbon coating could be achieved by chemical synthesis,16,17 in situ synthesis,18,19 introduction or modification of nanostructures,20 ball milling,21,22 and spraying,23 etc. Jiang et al.20 directly pyrolyzed (methyl methacrylate) (PMMA) polymer on the surface of silicon nanoparticles to construct a dual core–shell structure Si@SiOx@C nanocomposite. The introduction of graphene into SiOx composite materials is due to the unique single-atomic sheet structure of graphene, which exhibits good electrical conductivity, large specific surface area, and special mechanical properties. This allows graphene to be applied to the negative electrode material of lithium batteries to buffer the volume shrinkage in structure, improve the conductivity of the material, provide more tracks for lithium ions, and inlay space for silicon-containing particles.24–27 Bai et al.16 synthesized Si@SiOx/graphene hydrogel composite so that Si@SiOx nanoparticles can be evenly dispersed within the GH structure with a 3D network. Daniel et al.28 mixed polydopamine-coated Si NPs and graphene oxide (GO) through a simple solution mixing. The above methods have been proved to improve the performance of SiOx anode materials, and the pursing of Si-based anode materials about cycle stability is continuing.
In this research, graphene oxide (GO) was evenly dispersed in silicon-containing polyester (Si-PET), and then the resulting material was thermally reduced and carbonized. Finally, using hydrothermal of glucose to form a carbon layer on the surface. The obtained SiOx/C-rGO composite regarded graphene as the 3D network, and Si-PET was reduced to silicon with carbon on it. At the meanwhile, a double-layer structure of SiOx and carbon layers were formed on the surface. The advantages of this method is simple, low in cost, and feasible in practical application. It is worth noting that this SiOx/C-rGO composite has good electrical cycle stability. The excellent electrical performance of it due to the layer structure of graphene, which provides tracks for the insertion and desertion of lithium ions, increasing the conductivity of the material as well. The carbon–silicon structure obtained by reduction provides high capacity, and the flexible carbon layer with a certain mechanical strength effectively relieves volume expansion. The special structure is conducive to forming a stable solid electrolyte interphase (SEI) layer and reducing volume changes of active materials.29–32 Carbon coating can also effectively hinder the formation of silicon crystals and reduce the polarization resistance of the electrode, thereby improving the conductivity and electrical performance of it.33–35 This paper also studied the ratio of glucose hydrothermal coating. When the ratio of glucose to the coated material is 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the performance of the material is the best. At a current density of 200 mA g−1, the specific capacity of it can still be 469.7 mA h g−1 after 300 cycle.
:1, the performance of the material is the best. At a current density of 200 mA g−1, the specific capacity of it can still be 469.7 mA h g−1 after 300 cycle.
Materials
The graphene oxide (GO, >99.9%) used in this article is commercial product. Trimethoxysilane (industrial purity) was purchased in Hubei Wuda Silica New Material Co., Ltd. Ethylene glycol ((CH2OH)2, 99.0% wt), phthalic anhydride (C8H4O3, 99.7% wt), ammonia (NH3·H2O, 25.0–28.07% wt), tin(II) chloride dihydrate (SnCl2·2H2O, 98.0% wt), ethanol absolute (C2H6O, 99.7% wt) and D(+)-glucose anhydrous (C6H12O6) were purchased in Sinopharm Group Chemical Reagent Co., Ltd.Synthesis of SiOx/C-rGO
The fabrication of SiOx/C-rGO with a simple solution blending process is shown in Scheme 1. Ethylene glycol (40 g) and a slight excess of phthalic anhydride (100 g) were esterified at 120 °C, and then trimethoxysilane was introduced to synthesize silicon-containing polyester (Si-PET). 0.08 g of graphene oxide (GO) was added to 4 g of deionized water, and then ultrasonically dispersed for 15 minutes. In order to obtain a uniformly dispersed graphene oxide aqueous solution, a few drops of ammonia were added. The graphene oxide solution was slowly dropped into 20 g of the silicon-containing polyester prepared above, ultrasonicated for 30 minutes, and then tin(II) chloride dihydrate was added as a catalyst, and the obtained mixed solution was exposed to room temperature for curing. Next, the cured material was reduced at 750 °C with a heating rate of 5 °C min−1 under Ar atmosphere for 2 h. The obtained sample was denoted as SiOx/C-rGO-0. The sample of the above pure silicon polyester directly cured without graphene oxide was recorded as SiOx/C. Finally, the carbonized material and D(+)-glucose anhydrous in different proportions were uniformly dispersed in 50 ml of deionized water, and then placed in an autoclave for hydrothermal reaction at 160 °C for 6 hours. The obtained sample was washed with ethanol absolute and deionized water, then dried under vacuum at 80 °C for 24 hours. The ratio of glucose anhydrous to the carbonized sample is 2:1, 3:1, 4:1, 5:1, respectively denoted as SiOx/C-rGO-1, SiOx/C-rGO-2, SiOx/C-rGO-3. SiOx/C-rGO-4.
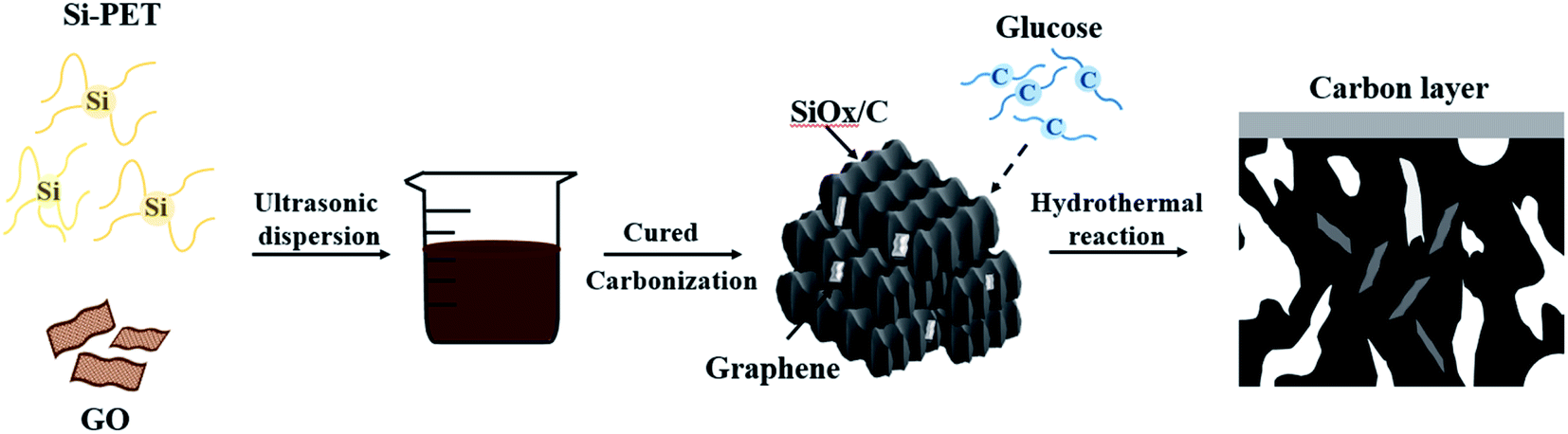 | ||
| Scheme 1 Schematic illustration of the synthesize process of the SiOx/C-rGO. | ||
Structural characterization
X-ray diffraction (XRD, XPert Pro, Netherlands) was carried out to characterize the crystal structure by using a Cu Kα (λ = 1.54178 Å) in the 2θ ranging of 5–80°. Fourier transform infrared spectroscope (FTIR, Thermo 5700, USA) was collected from 400 to 4000 cm−1. The morphologies and energy-dispersive X-ray spectroscopy of the samples were observed by scanning electron microscope (SEM, Tescan – MIRA3, Czech Republic) and high resolution transmission electron microscopy (HR-TEM, JEOL JEM F200, Japan). Raman spectra were recorded using a Scientific LabRam HR Evolution with an argon ion laser operating at 514 nm for excitation. The Brunauer–Emmett–Teller (BET, AUTOSORB IQ, Quantachrome, USA) was used to measure the specific surface area of samples. The pore size distribution was confirmed by Barrett–Joyner–Halenda (BET, AUTOSORB IQ, Quantachrome, USA) at liquid nitrogen temperature. Thermogravimetric analysis (TGA) was tested using a Henven HTG-1 analyzer at a heating rate of 10 °C min−1 from room temperature to 750 °C in air. X-ray photoelectron spectroscopy (XPS) was conducted with Thermo Scientific ESCALAB 250Xi using Al Kα (1486.6 eV) excitation.Electrochemical measurement
The working electrode was fabricated via pasting slurries of the active material, acetylene black (super P) and polyvinylidene fluoride (PVDF) at a weight ratio of 8:1:1 on the thin copper foil. After vacuum drying, the electrode was stamped into a disk with a diameter of 12 mm. Finally, using the lithium electrode as counter electrode and Celgard 2300 as separator in a glove box filled with argon to form the coin cell CR-2032. The electrolyte consisted of 1 mol l−1 LiPF6 in a mixed solvent with ethylene carbonate (EC)/ethyl methyl carbonate (EMC) = 1:1 by volume. Charge/discharge measurement were performed by LAND (CT2001A) battery-test system in the voltage range of 0.01–2.0 V. Cyclic voltammetry curves (CV) were measured at a scanning rate of 0.1 mV s−1 within the potential range of 0.01–2.0 V vs. Li/Li+ using an electrochemistry working station (Squidstat Plus). The electrochemical impedance spectroscopy (EIS) test was carried out in the frequency range from 100 kHz to 10 mHz on an electrochemical workstation (Squidstat Plus).
Results and discussion
XRD patterns of SiOx/C-rGO-0 and SiOx/C-rGO-2 are compared in Fig. 1a. The similar XRD spectra of the two samples have same crystal structure. The SiOx/C-rGO material in this study is composed of carbon and amorphous silicon oxide (SiOx). The broad peak in the 20°–25° region corresponds to SiOx. The main peak of carbon overlaps with the SiOx peak, and the small peak at 43° represents amorphous carbon (JCPDS No. 26-1081).36,37 Fourier transform infrared (FT-IR) spectra of three samples of SiOx/C, SiOx/C-rGO-0 and SiOx/C-rGO-2, as shown in Fig. 1b, have similar absorption peaks. The FT-IR spectra indicate the several bonding in the above composites mentioned. The strong and shape peaks at 3430 cm−1 are assigned to the Si–OH or C–OH. There is no significant difference in these peaks of the three samples in intensity, indicating that the addition of graphene oxide (GO) and hydrothermal treatment will not significantly affect the interaction between molecular. At the meanwhile, the SiOx/C has a small peak at 2930 cm−1, which is attributed to the C–H bond.38 Compared with SiOx/C, the peak intensity of SiOx/C-rGO-0 is obviously smaller, and the peak of SiOx/C-rGO-2 is almost absent. It illustrates that the addition of GO and hydrothermal reaction contribute to carbonization completely. The absorption peaks at 1060 cm−1, 792 cm−1 and 449 cm−1 are ascribed to three different structural vibrations: asymmetric stretching vibration (asymmetric Si–O–Si bond stretching) and symmetric stretching vibration (SiO4 tetrahedral ring), bending vibration (Si–O–Si bond angle deformation), the above three peaks are all represented to SiOx.39 It is worth mentioning that the peak intensity at 792 cm−1 is obviously smaller accompanied by the addition of GO and hydrothermal treatment, which proves that carbon coating indeed formed around silicon. The peak near 1602 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, indicating that glucose has been carbonized during the hydrothermal treatment.40
C, indicating that glucose has been carbonized during the hydrothermal treatment.40
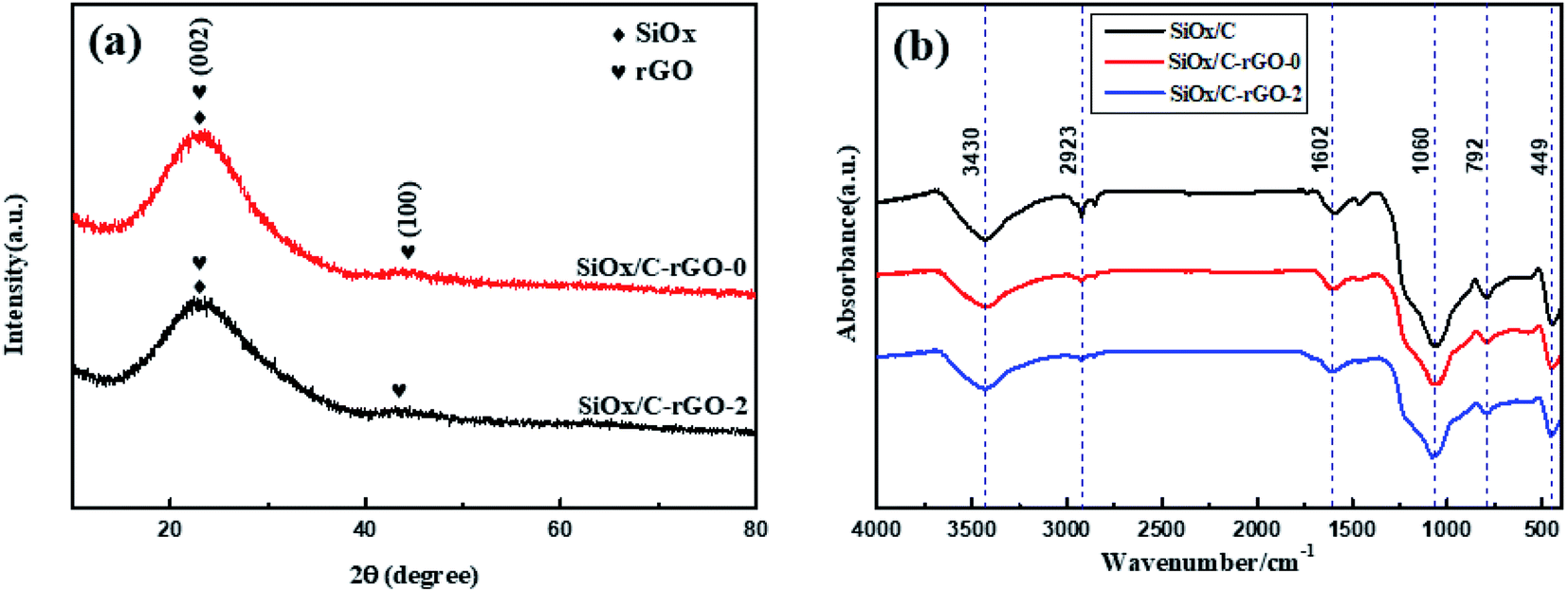 | ||
| Fig. 1 (a) Powder XRD patterns of SiOx/C-rGO-0 and SiOx/C-rGO-2 (b) FT-IR spectra of SiOx/C, SiOx/C-rGO-0 and SiOx/C-rGO-2. | ||
The Raman spectrum of the SiOx/C-rGO-2 sample is shown in Fig. 2a. Although there is no obvious Si band, the D band and G band that typically characterize the graphene structure appear in it. The D band near 1330 cm−1 attribute to the structural defects on the graphitic carbon plane, and the G band near 1590 cm−1 associate with a strong tangential mode of the graphene structure. The ID/IG = 0.84, which can indicate the degree of graphitization of the sample. From the Raman spectrum, it is obvious that there is amorphous carbon in the composite. The X-ray photoelectron spectroscopy (XPS) of SiOx/C-rGO-2 and SiOx/C-rGO-0 are shown in Fig. 2b. The three dominant elements C (C 1s), O (O 1s) and Si (Si 2p and Si 2s) can be seen. It is easy to conclude from the figure that the three main elements of the both composites are quite different. The XPS high-resolution spectrum comparison of the C 1s spectrum of SiOx/C-rGO-2 and SiOx/C-rGO-0 are shown in Fig. 2c and S1.† After the hydrothermal reaction, a significant peak appeared near 286 eV. The strong peak at 284.7 eV corresponds to the C–C bond in graphitic carbon, 285.9 eV and 290.7 eV correspond to the C–O bond and CO bond, respectively.41 The peak area of the C–C bond is the largest, which indicates that the functional groups containing carbon–oxygen bonds in the complex have been reduced. Glucose hydrothermal coating, on the one hand, fully carbonized on the surface to increase the carbon content, on the other hand, the introduction of carbon-containing groups made up for the insufficient conductivity of the SiOx anode material. The O 1s spectrum (Fig. 2d and S2†) prove that glucose is fully reduced in the hydrothermal process to form an effective carbon layer, leading to a significant reduction in surface oxygen. The strong peak at 532.3 eV corresponds to the multiplicity of adsorbed H2O molecule at or near the surface, and 530.8 eV correspond to the metal–oxygen bond.42 Fig. 2e and f illustrate respectively the Gaussian fitting of the Si 2p spectrum of the SiOx/C-rGO-2 and SiOx/C-rGO-0. The peak of Si 2p can be split into four peaks, which correspond to different spins of Si element in p orbital under different oxidation states. Peaks at 103.8 eV, 102.8 eV, 102.1 eV, and 101.3 eV are respectively referred to Si4+, Si3+, Si2+, Si+,43 and the absence of Si0 in the composite is also consistent with the results of Raman spectrum. The proportions of Si in different valence states of SiOx/C-rGO-2 and SiOx/C-rGO-0 are compared in Table 1 according to the area of the fitted peaks. After hydrothermal reduction, the proportion of high-valence silicon is significantly lower, which demonstrates that a carbon layer has formed on the surface to cover the silicon oxide compound and part of the silicon is reduced to lower valence state. It is concluded that SiOx/C-rGO-2 is composed of silicon-oxygen (SiOx) and amorphous carbon, and the silicon element is further reduced to a mixed valence state after hydrothermal reaction.
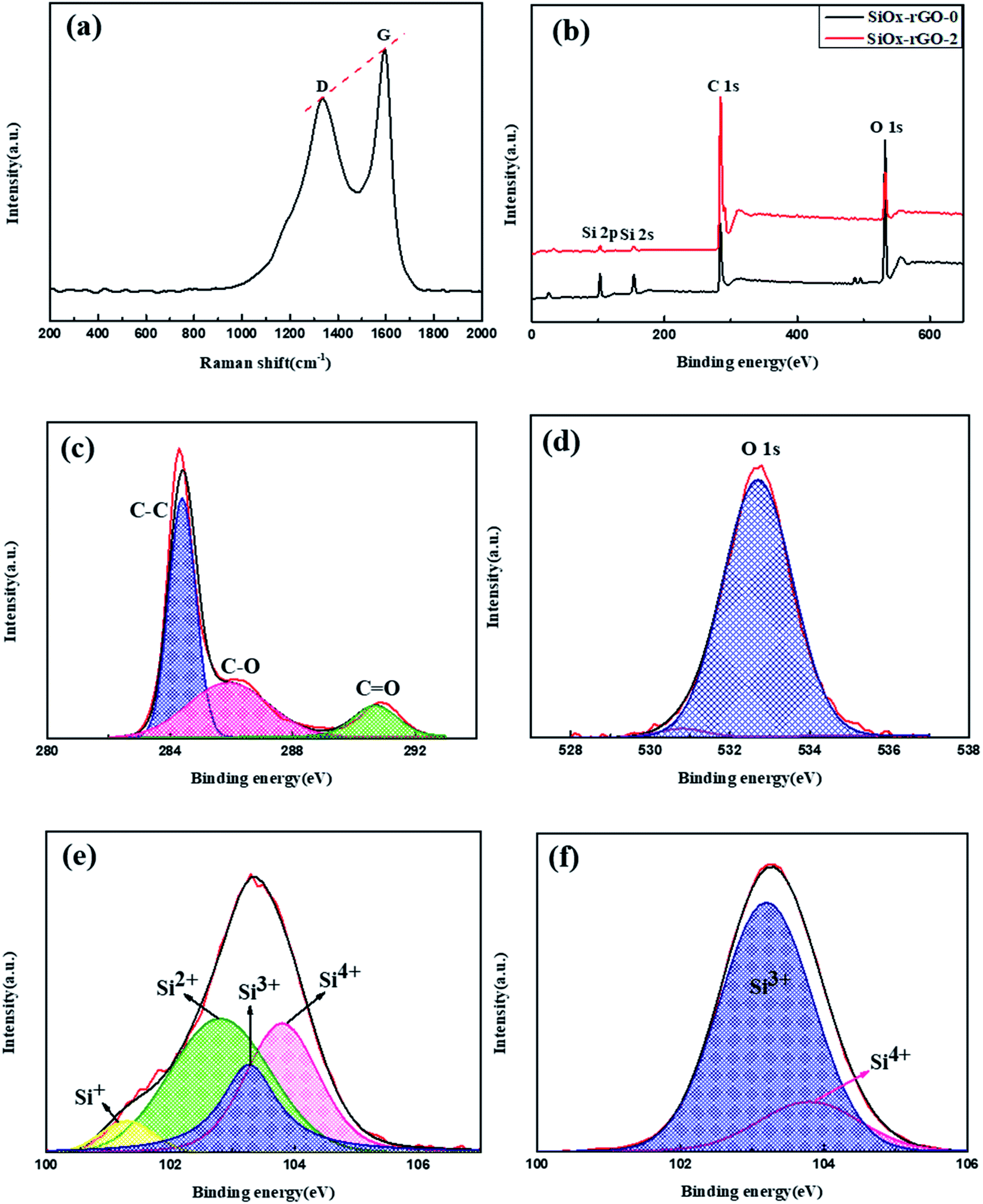 | ||
| Fig. 2 (a) Raman spectra of SiOx/C-rGO-2 with an excitation laser wavelength of 514.5 nm. (b) XPS spectra of SiOx/C-rGO-0 composite and SiOx/C-rGO-2 composite. High-resolution XPS spectra for the SiOx/C-rGO-2 composite of C 1s (c), O 1s (d) and Si 2p (e). (f) High-resolution XPS spectra of Si 2p for the SiOx/C-rGO-0. | ||
| Sample | Si4+ (%) | Si3+ (%) | Si2+ (%) | Si+ (%) |
|---|---|---|---|---|
| SiOx/C-rGO-2 | 31.39 | 20.19 | 42.88 | 5.54 |
| SiOx/C-rGO-0 | 17.63 | 82.37 | 0 | 0 |
Scanning electron microscope (SEM) observes the microscopic morphology of the sample as shown in Fig. 3. The structure of SiOx/C without graphene (Fig. 3a) is flat and smooth, which is a typical SiOx morphology.44 The SiOx/C-rGO-0 with graphene oxide (rGO) added is shown in Fig. 3b and c, which are macroporous, and the graphene fold-like structure can be clearly observed at higher magnification. The necessity of introducing graphene has been verified, because the generation of holes is conducive to the lithiation and delithiation, so that it has better electrical performance.45 The hydrothermally treated SiOx/C-rGO-2 is shown in Fig. 3d and e. The graphene fold-like structure and pores still exist, but the pore size is smaller than before. The agglomerate structure that did not exist before can be clearly seen in the depression of the fracture (Fig. 3f), which is presumed to be an irregular carbon layer formed by hydrothermal reduction, and this inference was further verified.
 | ||
| Fig. 3 SEM images of composites: (a) SiOx/C ×1.00k, (b) SiOx/C-rGO-0 ×1.00k, (c) SiOx/C-rGO-0 ×2.00k and 10.00k, (d) SiOx/C-rGO-2 ×5.00k, (e) SiOx/C-rGO-2 ×20.00k, (f) SiOx/C-rGO-2 ×100.00k. | ||
The specific surface area and pore size distribution of these samples were evaluated by nitrogen adsorption–desorption measurements in Fig. 4a–c. The isotherms of these three materials show distinct hysteresis loops, which are type IV plots, showing the existence of mesoporous structures in these materials.46,47 Based on the multi-point BET method, the specific surface area of SiOx/C, SiOx/C-rGO-0 and SiOx/C-rGO-2 are 6.065 m2 g−1, 8.165 m2 g−1 and 12.725 m2 g−1, respectively. With the addition of graphene and the formation of carbon layer on the surface, the specific surface area increased significantly. The specific surface area of the sample is improved by the hydrothermal method, which is beneficial to provide lithium ion migration channels, reduce power assistance, release volume expansion, and have better electrical properties.
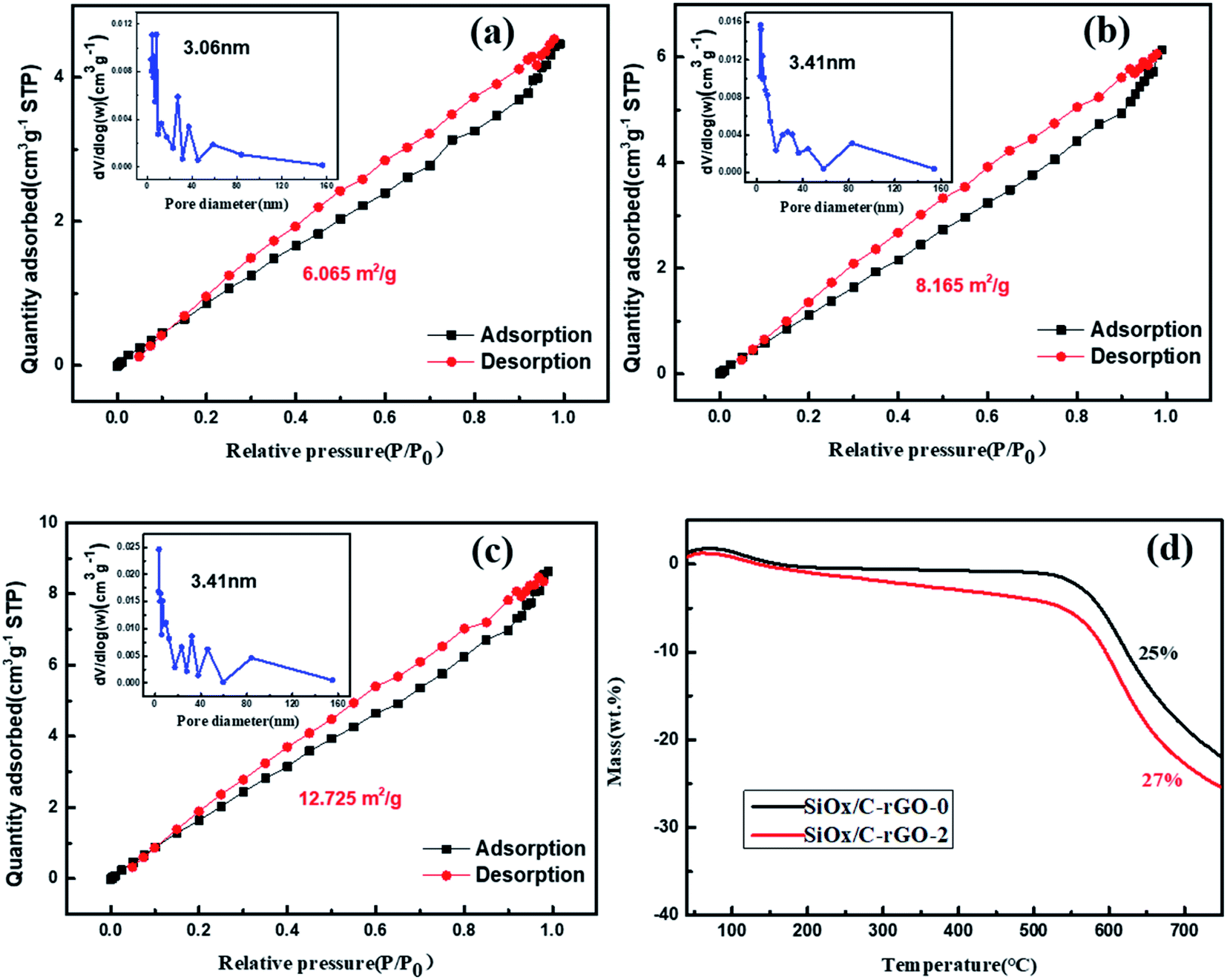 | ||
| Fig. 4 N2 adsorption/desorption isotherms of SiOx/C (a), SiOx/C-rGO-0 (b) and SiOx/C-rGO-2 (c), inset shows the pore size distribution. (d) Thermogravimetry analysis of SiOx/C-rGO-0 and SiOx/C-rGO-2 in air. | ||
Thermogravimetric analysis (TGA) test is carried out under air atmosphere, and the carbon is completely oxidized by heating to 750 °C. The weight increase of silicon into silicon dioxide at a temperature (<800 °C) is negligible. Therefore, the remaining weight after heating to >700 °C can reflect the carbon content of the composite. Fig. 4d is the comparison between SiOx/C-rGO-0 and SiOx/C-rGO-1. The weight loss increased from 25 wt% to 27 wt% after hydrothermal treatment, demonstrating that glucose by hydrothermal method does introduce more carbon elements. The thermal weight loss data of different ratios of glucose added are shown in Table 2. With the added amount of glucose increasing, the weight loss percentage of the sample becomes larger and the carbon content increases. The difference between the values is not great because the relative atomic mass of carbon is relatively small compared to that of silicon oxide.
| Sample | SiOx/C-rGO-1 | SiOx/C-rGO-2 | SiOx/C-rGO-3 | SiOx/C-rGO-4 |
|---|---|---|---|---|
| Ratio of glucose | 2:1 |
3:1 |
4:1 |
5:1 |
| Weight (wt%) | 27.75 | 29.56 | 30.38 | 32.17 |
The structure and morphology of the sample were characterized by field emission scanning electron microscope (FESEM), transmission electron microscope (TEM) and high resolution transmission electron microscope (HRTEM). From the TEM images of SiOx/C-rGO-2 (Fig. 5a), at low magnification, it is obvious that the graphene sheets are distributed in SiOx, which is consistent with the previously envisaged structure. Fig. 5c can more clearly see that the carbon layer is coated on the SiOx surface, and amorphous carbon formed on the surface of the sample, which further verifies the results of the previous XRD and Raman spectroscopy. The lattice lines in Fig. 5b are clearly marked, and the d-spacing measured 0.33 nm corresponds to the C nanocrystals, indicating that in addition to the presence of amorphous carbon in the sample, the carbonized graphene crystal structure is still intact.48 The d-spacing measured 0.27 nm corresponds to SiC structure,49 which is in agreement with the XRD analysis. The disordered domain in the Fig. 5c is an amorphous SiOx (0 < x < 2) microstructure. The actual morphology and structure of the sample are consistent with the above analysis of XRD and XPS, which proves that there is no silicon crystal in the sample. The STEM images (Fig. 5d–g) show the existence of the three elements, including C, O and Si in this sample. The silicon is obviously reduced near the edge of the surface, while the carbon has no obvious decrease, indicating that the hydrothermal reaction happened indeed in the SiOx to form carbon layer on the surface. What's more, the interface between SiOx particle and graphene is very tight without interstice from HR-TEM image. EDS line scan curve (Fig. 5h) also indicates the existence of C, Si and O. This is consistent with above speculations about its structural performance for electrical cycling stability.
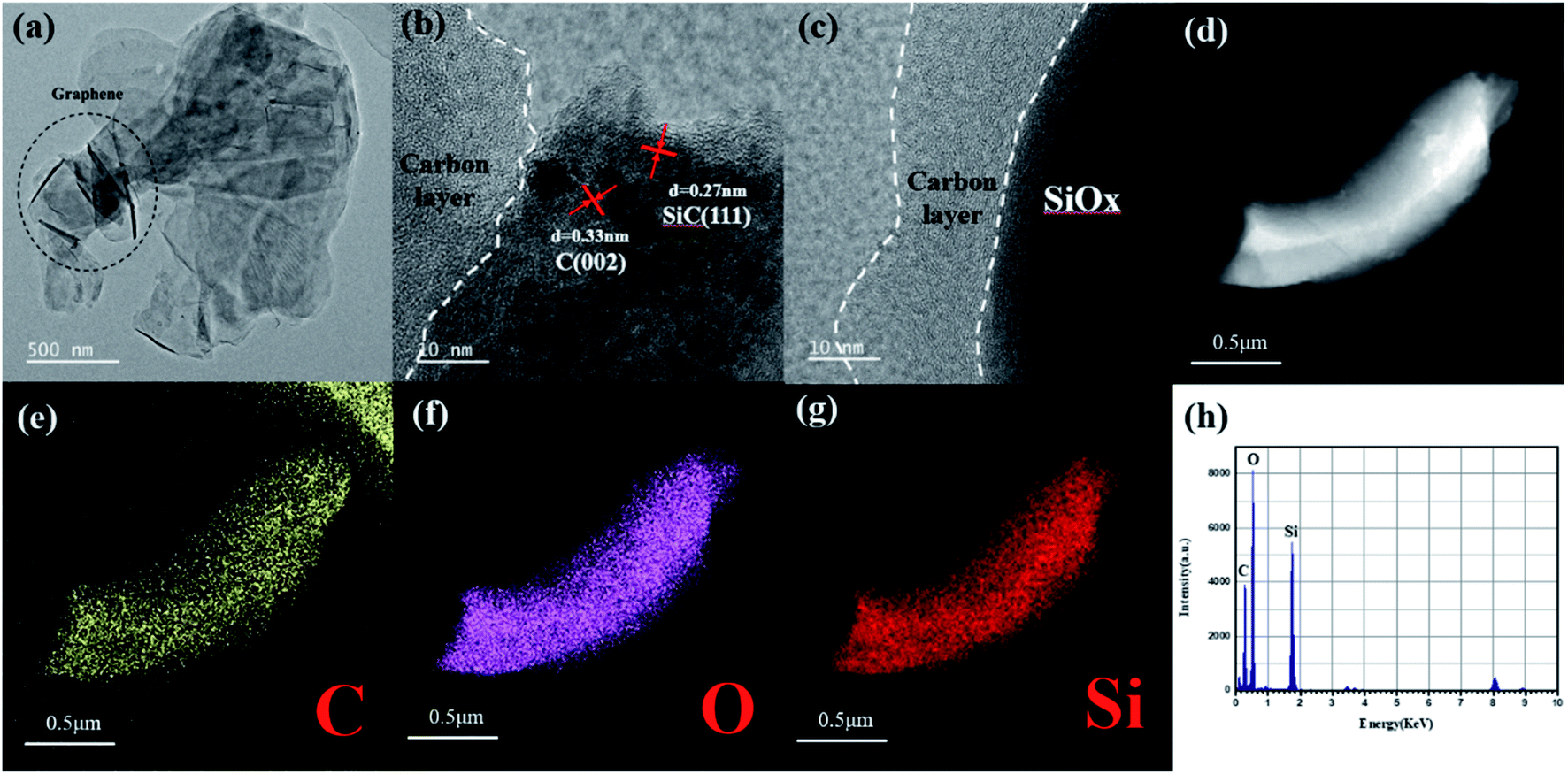 | ||
| Fig. 5 (a) TEM and (c) and (d) HRTEM images of SiOx/C-rGO-2. (d)–(g) Elements mapping and EDS line scan curve (h) of SiOx/C-rGO-2. | ||
Electrochemical performance
SiOx/C-rGO-0, SiOx/C-rGO-1, SiOx/C-rGO-2, SiOx/C-rGO-3 and SiOx/C-rGO-4 were tested for cycle stability. As shown in Fig. 6a, the charge–discharge experiment was carried out in the voltage range of 0.01–2 V and the current density of 100 mA g−1. The cycling stability of SiOx/C-rGO-2 is the best. After 100 cycles, the capacity remains at 568 mA h g−1, showing good cycling stability. In contrast, the cycle stability of the other four samples is not much different. However, SiOx/C-rGO-4 and SiOx/C-rGO-3 have obvious activation stages, and the activation stage is longer with the increase of glucose. Because the proportion of glucose is too much, which leads to a significant increase of carbon content in the composites. It is also reflected in the weight loss of TGA in oxygen atmosphere in Table 2. Too much carbon coating on the surface, on the one hand, reduces the theoretical capacity of the material, and at the same time increases the resistance in the process of lithium ion deintercalation. On the other hand, there will be an activation stage in the first few cycles of charging and discharging, which will significantly affect the electrical capacity of the material. It can be illustrated that the more glucose is added, the better the electrical performance, but when over a certain ratio, the worse the electrical performance. Therefore, the ratio of glucose to polyester in 3:1 is undoubtedly the most suitable, and the cycle performance of SiOx/C-rGO-2 is consistent with the structural advantages mentioned above. The first-cycle charge–discharge volt–ampere curve of the above sample is shown in Fig. S3,† showing the typical behavior of Si: the discharge curve is a plateau below 0.25 V, and the gentle slope of the volt–ampere curve between 0.3 and 0.6 V corresponds to the lithium insertion of Si and delithiation with LixSi phase respectively. The charge and discharge platform of SiOx/C-rGO-2 in the first cycle is significantly better than other samples, and has a higher first-cycle capacity.
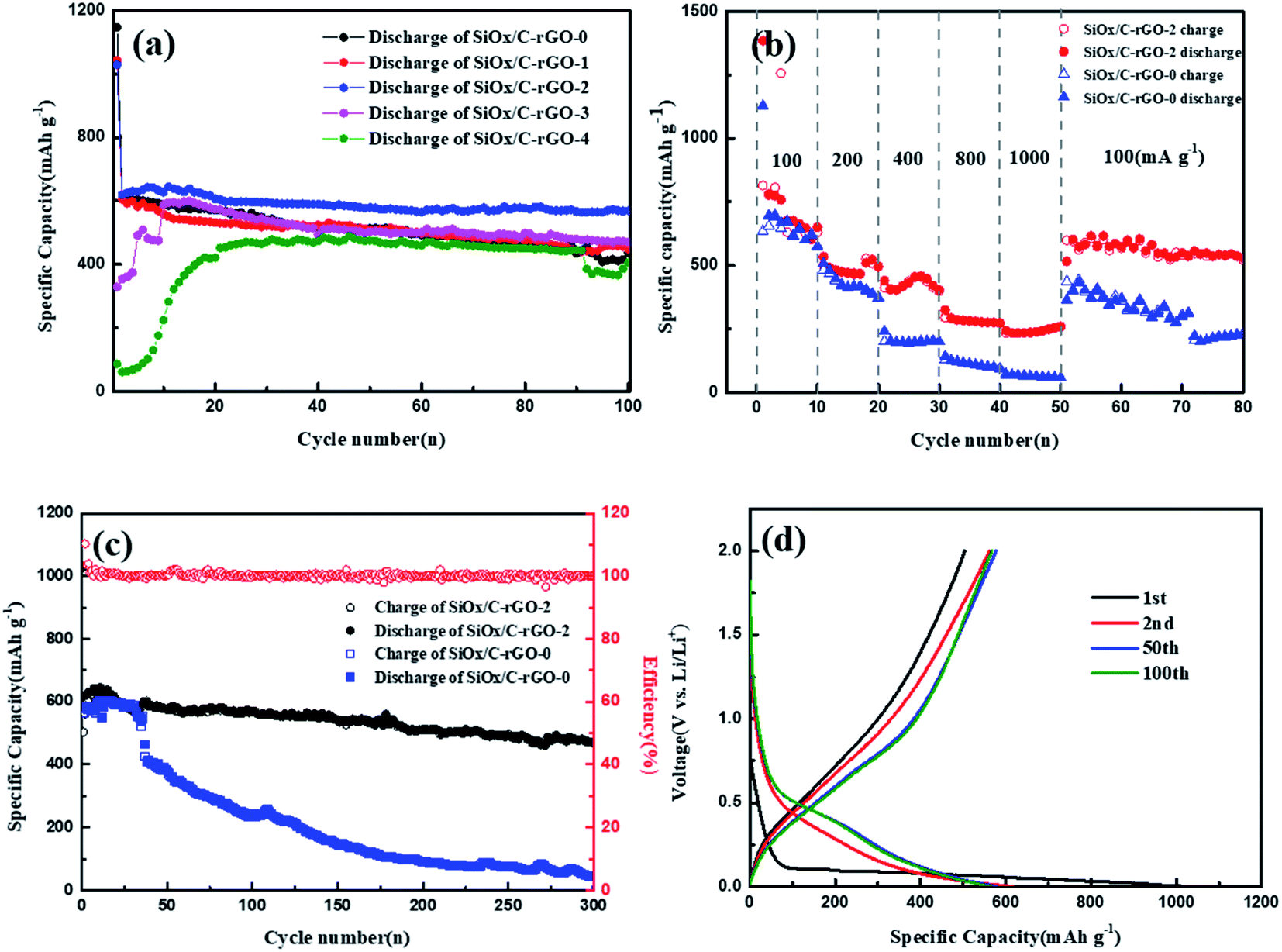 | ||
| Fig. 6 (a) Cycling stability comparison of SiOx/C-rGO-0, SiOx/C-rGO-1, SiOx/C-rGO-2, SiOx/C-rGO-3 and SiOx/C-rGO-4 at 100 mA g−1. (b) Rate capability of SiOx/C-rGO-2 and SiOx/C-rGO-0 at various current densities. (c) The long-term cycling stability of SiOx/C-rGO-2 and SiOx/C-rGO-0 at 200 mA g−1. (d) Discharge–charge curves of SiOx/C-rGO-2 cycled between 0.01 and 2.0 V under a current density of 200 mA g−1 for the 1st, 2nd, 50th and 100th cycle. | ||
The rate capability of SiOx/C-rGO-0 and SiOx/C-rGO-2 was performed at different current densities from 100 to 1000 mA g−1, as shown in Fig. 6b. Compared with SiOx/C-rGO-0, SiOx/C-rGO-2 obviously has excellent rate stability and capacity recovery. The capacities are 676.9 mA h g−1, 471.9 mA h g−1, 431.8 mA h g−1, 281.6 mA h g−1, 234.4 mA h g−1 at current densities of 100, 200, 400, 800, and 1000 mA g−1 respectively. When the current density returned from 1000 mA g−1 to 100 mA g−1, the capacity rose again to 616.5 mA h g−1, indicating that the electrode material structure was not damaged under high current density test. The excellent rate stability is due to the flexible carbon layer formed on the surface by the hydrothermal reaction of glucose, which not only improves the conductivity but also acts as a buffer for the shrinkage and expansion of the silicon-containing electrode material. The activation process is also evident from the rate capability of SiOx/C-rGO-3 and SiOx/C-rGO-4 (Fig. S5†), which are the same as the performances in Fig. 6a. Moreover, as shown in Fig. 6c, the long-cycle stability of SiOx/C-rGO-2 is very significant. At a current density of 200 mA g−1, the reversible capacity of 469.7 mA h g−1 is maintained after 300 cycles, and the capacity retention rate is 77.5%; at a current density of 100 mA g−1, it still maintains 660 mA g−1 after 100 cycles, the capacity retention rate is 84.6% (Fig. S5†). However, the SiOx/C-rGO-0 has only 43.8 mA h g−1 after 300 cycles at 200 mA g−1, which is more than ten times lower than the former. This once again proves the improvement of the cycle stability after glucose hydrothermal coating. Fig. 6d shows the charge–discharge curves of SiOx/C-rGO-2 from 0.01 to 2.0 V at 200 mA g−1 in the 1st, 2nd, 50th, and 100th. The discharge curve between the first cycle and the second cycle has a certain difference due to the irreversible capacity loss of the silicon-containing material, and the low coulomb conversion rate in the first cycle is due to the generation of SEI film, inert lithium oxide and Li4SiO4.22 The Coulomb efficiency of SiOx/C-rGO-2 was 60.1% in the first cycle, quickly reached 90.7% in the second cycle, and then maintained at around 98% in the following cycles.
The cyclic voltammetry (CV) curve of SiOx/C-rGO-2 is shown in Fig. 7a. The cathodic peak at around 0.74 V (vs. Li/Li+) of first cycle is due to the decomposition of the electrolyte and the formation of the SEI film. During the formation of the SEI film, silicon dioxide is reduced to Si, accompanied by the formation of Li2O and Li4SiO4.9,11,50 The cathodic peak at 0.01 V corresponds to the alloying of Si and Li and the reversible reaction between Li and carbon (including carbon coating and rGO), and the obvious peak change in the second cycle is attributed to the further formation of the SEI film.51,52 During the anodic polarization of the first cycle, the insignificant anodic peak at 0.35 V is de-lithiation of LixSi phase.53,54 Fig. S6† shows Nyquist plots for the six samples (SiOx/C, SiOx/C-rGO-0, SiOx/C-rGO-1, SiOx/C-rGO-2, SiOx/C-rGO-3, SiOx/C-rGO-4). For a clearer comparison, the Nyquist plots of SiOx/C-rGO-0 and SiOx/C-rGO-2 are shown in Fig. 7b. The Nyquist plots of the two samples both present a semicircle in the high frequency region and a straight line in the low frequency region. The former corresponds to the parallel combination of capacity and charge transfer resistance (Rct) and/or resistance from the SEI layer (RSEI), while the latter reflects Li+ diffusion resistance.55 The charge transfer resistance (Rct) of SiOx/C-rGO-0 is much larger than that of SiOx/C-rGO-2. The reduced resistance of glucose hydrothermally is due to the formation of a stable carbon coated structure, while the introduction of carbon improves the conductivity of the material, which is consistent with our previous speculation and subsequent characterization of the composite. The SiOx/C-rGO-2 Nyquist diagram after three cycles of charging and discharging is shown in Fig. S7.† The three curves are composed of two semi-circular arcs in the high frequency range and a straight line in the low frequency range. Obviously, it can be seen that the impedance becomes significantly smaller after the first cycle, and the material properties tend to be stable.
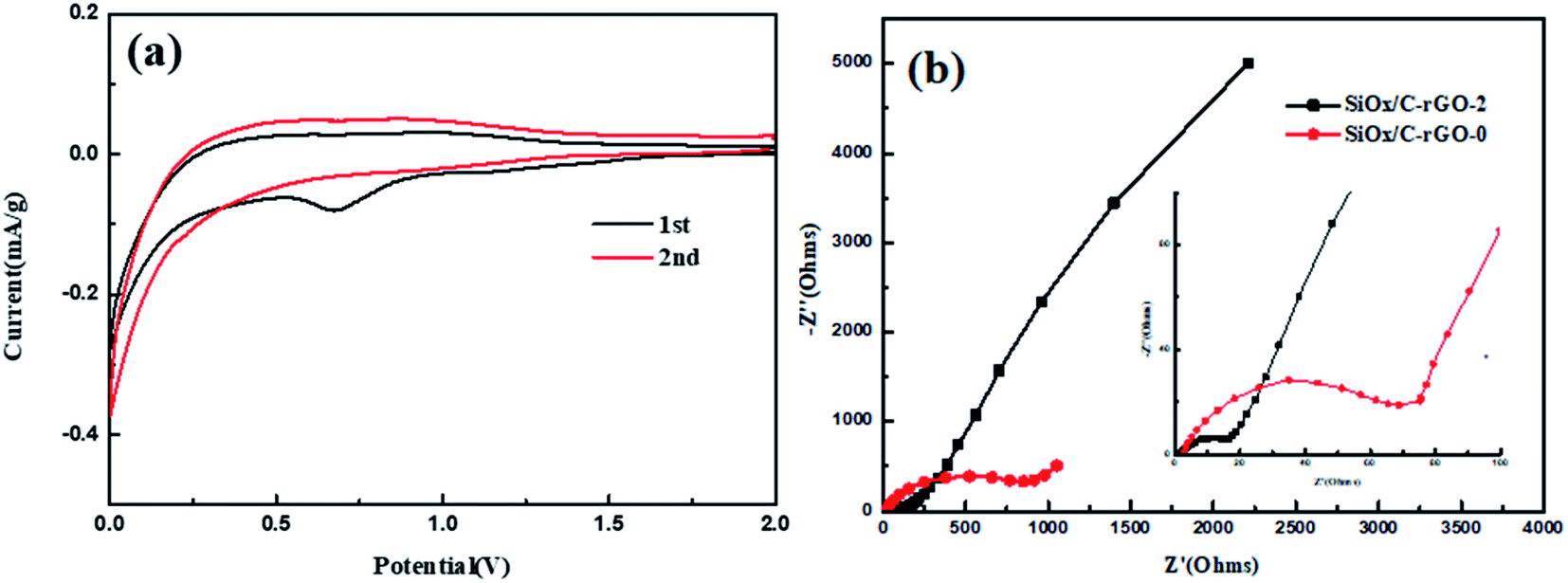 | ||
| Fig. 7 (a) Cyclic voltammetry curves of the SiOx/C-rGO-2 measured in the voltage range of 0.01–2.0 V with a scan rate of 0.1 mV s−1, (b) Nyquist plots of the SiOx/C-rGO-0 and SiOx/C-rGO-2. | ||
The formation of SEI film has always been proved to be a very important factor affecting the electrochemical stability of materials. Since the specific SEI film composition is still controversial, this article will not describe it in detail. However, it is not difficult to draw a conclusion from the chemical composition of the two SEI films, and we can distinguish them through element characterization. Fig. 8a–e and f–j are the high-magnification scanning electron micrographs and the mappings of SiOx/C-rGO-0 and SiOx/C-rGO-2 after 100 cycles at 100 mA g−1. The SEI film is a stable solidified layer that is insoluble in organic solvents.56 In order to further study the principle of the significant increase in the stability of SiOx/C-rGO-2 after the hydrothermal coating of glucose, the electrodes after a long cycle were soaked in an organic solvent to remove the residual electrolyte on the surface, and then they were ultrasonically dispersed in ethanol solution to test its in situ elemental composition. Although after a long cycle, the material structure has changed a little. However, the information about the SEI film can still find from its elemental composition. The phosphorus and fluorine of SiOx/C-rGO-0 are much higher than that of SiOx/C-rGO-2. On the basis that the remaining LiPF6 had been removed, the P and F detected were all derived from the already formed SEI film. The decrease of P and F after glucose hydrothermal coating is because the carbon layer formed on the original SiOx/C-rGO-0 in this study, the SEI film mainly formed on the carbon surface. However, the carbon layer formed by hydrothermal might fall off the surface during ultrasonic dispersion, and the SEI film on the surface would also falls off. Therefore, it is not easy for us to find an area with a special structure where the carbon layer is ultrasonically removed, and there is a significant reduction in elements. It can be known that after hydrothermal coating, the formation of the SEI film on the carbon surface is more conducive to the cycle stability of the material than the formation on the silicon surface. In addition, we also investigated the dimensional stability of the SiOx/C-rGO composite electrode after cycling.31 We compared the cross-sectional FE-SEM images of the SiOx/C-rGO-2 composite electrode before and after 300 cycles (Fig. 8k and l). Before cycling, the electrode thickness of the SiOx/C-rGO-2 composite was measured to be 10.52 μm (Fig. 8k), which increased to 12.22 μm after 300 cycles (Fig. 8l), and the electrode expansion was 16%, which is much lower than those of silicon based materials. The composite electrode has good dimensional stability in terms of structure. After long cycling, the structural properties of the electrodes are still stable, so the electrical cycling stability is remarkable.
 | ||
| Fig. 8 (a) SEM image and (b)–(e) the corresponding elemental mappings of C, Si, P and F of SiOx/C-rGO-0 after 100 cycles at 100 mA h g−1, (f) STEM image and (g)–(j) the corresponding elemental mappings of C, Si, P and F of SiOx/C-rGO-2 after 100 cycles at 100 mA h g−1. Cross-sectional FE-SEM images of the SiOx/C-rGO-2 composite electrode (k) before and (l) after 300 cycles at room temperature. | ||
Conclusions
This research innovatively obtained a SiOx/C-rGO composite material that can maintain stability in long cycles by hydrothermally coating silicon-containing polyester with glucose. The holes formed in the material due to the introduction of graphene are conducive to the deintercalation of lithium ions. At the same time, the flexible carbon layer on the surface not only buffers the volume shrinkage and expansion during the charge and discharge of the silicon oxide compound, but also increases the electrical conductivity of it. Its resistance to electron migration becomes smaller. The structural advantage of the stability of the composite is that the SEI film mainly formed on the carbon surface, avoiding the problem of low coulomb conversion efficiency of the Si-containing anode materials, but it also utilizes the superior theoretical specific capacity of the SiOx. Therefore, the SiOx/C-rGO material still maintains a reversible capacity of 469.7 mA h g−1 at a current density of 200 mA g−1 for 300 cycles, and still maintains a reversible capacity of 660 mA h g−1 at a current density of 100 mA g−1 for 100 cycles. The Coulomb efficiency maintained at 98% except for the first cycle, and it has good cycle stability. This study provides a method for preparing lithium-ion anode materials with simple preparation, low cost, high industrial feasibility, and excellent cycle stability, and finds a new reference for optimizing the performance of existing commercial lithium-ion batteries.Conflicts of interest
There are no conflicts to declare.References
- C. M. Park, et al., Li-alloy based anode materials for Li secondary batteries, Chem. Soc. Rev., 2010, 39(8), 3115–3141 Search PubMed.
- J. M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414(6861), 359–367 Search PubMed.
- X. Cheng, et al., Insight into the Synergistic Effect of N, S Co-Doping for Carbon Coating Layer on Niobium Oxide Anodes with Ultra-Long Life, Adv. Funct. Mater., 2021, 31(19), 1–9 Search PubMed.
- J. Li, C. Daniel and D. Wood, Materials processing for lithium-ion batteries, J. Power Sources, 2011, 196(5), 2452–2460 Search PubMed.
- X. Shen, et al., Research progress on silicon/carbon composite anode materials for lithium-ion battery, J. Energy Chem., 2018, 27(4), 1067–1090 Search PubMed.
- Y. Zhang, et al., Recent progress and perspectives on silicon anode: synthesis and prelithiation for LIBs energy storage, J. Energy Chem., 2022, 64, 615–650 Search PubMed.
- T. F. Yi, et al., Structural and thermodynamic stability of Li4Ti5O12 anode material for lithium-ion battery, J. Power Sources, 2013, 222, 448–454 Search PubMed.
- T. F. Yi, et al., Facile synthesis of polypyrrole-modified Li5Cr7Ti6O25 with improved rate performance as negative electrode material for Li-ion batteries, Composites, Part B, 2019, 167, 566–572 Search PubMed.
- F. Dai, et al., Amorphous Si/SiOx/SiO2 nanocomposites via facile scalable synthesis as anode materials for Li-ion batteries with long cycling life, RSC Adv., 2012, 2(33), 12710 Search PubMed.
- W. Wu, et al., A low-cost and advanced SiOx-C composite with hierarchical structure as an anode material for lithium-ion batteries, Phys. Chem. Chem. Phys., 2015, 17(20), 13451–13456 Search PubMed.
- Z. Favors, et al., Stable cycling of SiO(2) nanotubes as high-performance anodes for lithium-ion batteries, Sci. Rep., 2014, 4, 4605 Search PubMed.
- C. Guo, et al., A three dimensional SiOx/C@RGO nanocomposite as a high energy anode material for lithium-ion batteries, J. Mater. Chem. A, 2014, 2(10), 3521–3527 Search PubMed.
- J. Im, et al., Two-dimensional, P-doped Si/SiOx alternating veneer-like microparticles for high-capacity lithium-ion battery composite, Chem. Eng. J., 2020, 402, 126292 Search PubMed.
- G. Li, et al., An integral interface with dynamically stable evolution on micron-sized SiOx particle anode, Nano Energy, 2020, 74, 104890 Search PubMed.
- D. Shao, et al., Nanostructured silicon/porous carbon spherical composite as a high capacity anode for Li-ion batteries, J. Mater. Chem. A, 2013, 1(47), 15068 Search PubMed.
- X. Bai, et al., Si@SiOx/graphene hydrogel composite anode for lithium-ion battery, J. Power Sources, 2016, 306, 42–48 Search PubMed.
- Y. Liu, et al., Synthesis of SiOx/C composite with dual interface as Li-ion battery anode material, J. Alloys Compd., 2019, 802, 704–711 Search PubMed.
- X. Bai, et al., In situ synthesis of carbon fiber-supported SiOx as anode materials for lithium ion batteries, RSC Adv., 2016, 6(39), 32798–32803 Search PubMed.
- H. Shi, et al., In situ fabrication of dual coating structured SiO/1D-C/a-C composite as high-performance lithium ion battery anode by fluidized bed chemical vapor deposition, Carbon, 2020, 168, 113–124 Search PubMed.
- B. Jiang, et al., Dual Core-Shell Structured Si@SiOx@C Nanocomposite Synthesized via a One-Step Pyrolysis Method as a Highly Stable Anode Material for Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8(46), 31611–31616 Search PubMed.
- C.-H. Doh, et al., A new SiO/C anode composition for lithium-ion battery, J. Power Sources, 2008, 179(1), 367–370 Search PubMed.
- D. Wang, et al., High performance amorphous-Si@SiOx/C composite anode materials for Li-ion batteries derived from ball-milling and in situ carbonization, J. Power Sources, 2014, 256, 190–199 Search PubMed.
- S. J. Lee, et al., Delicate Structural Control of Si-SiOx-C Composite via High-Speed Spray Pyrolysis for Li-Ion Battery Anodes, Nano Lett., 2017, 17(3), 1870–1876 Search PubMed.
- K. S. Novoselov, et al., Electric field effect in atomically thin carbon films, Science, 2004, 306(5696), 666–669 Search PubMed.
- W. R. Yang, et al., Carbon Nanomaterials in Biosensors: Should You Use Nanotubes Or Graphene?, Angew. Chem., Int. Ed., 2010, 49(12), 2114–2138 Search PubMed.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, An overview of graphene in energy production and storage applications, J. Power Sources, 2011, 196(11), 4873–4885 Search PubMed.
- G. Dai and L. Mishnaevsky, Graphene reinforced nanocomposites: 3D simulation of damage and fracture, Comput. Mater. Sci., 2014, 95, 684–692 Search PubMed.
- D. A. Agyeman, et al., Carbon-Coated Si Nanoparticles Anchored between Reduced Graphene Oxides as an Extremely Reversible Anode Material for High Energy-Density Li-Ion Battery, Adv. Energy Mater., 2016, 6(20), 1600904 Search PubMed.
- C. Stetson, et al., Microscopic Observation of Solid Electrolyte Interphase Bilayer Inversion on Silicon Oxide, ACS Energy Lett., 2020, 5(12), 3657–3662 Search PubMed.
- W. Huang, et al., Dynamic Structure and Chemistry of the Silicon Solid-Electrolyte Interphase Visualized by Cryogenic Electron Microscopy, Matter, 2019, 1(5), 1232–1245 Search PubMed.
- J. Chen, et al., Electrolyte design for LiF-rich solid-electrolyte interfaces to enable high-performance microsized alloy anodes for batteries, Nat. Energy, 2020, 5(5), 386–397 Search PubMed.
- J. Tan, et al., A Growing Appreciation for the Role of LiF in the Solid Electrolyte Interphase, Adv. Energy Mater., 2021, 11(16), 2100046 Search PubMed.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Generation and Evolution of the Solid Electrolyte Interphase of Lithium-Ion Batteries, Joule, 2019, 3(10), 2322–2333 Search PubMed.
- W. Song, et al., Electronic structure influences on the formation of the solid electrolyte interphase, Energy Environ. Sci., 2020, 13(12), 4977–4989 Search PubMed.
- K. Xu, A. von Cresce and U. Lee, Differentiating Contributions to “Ion Transfer” Barrier from Interphasial Resistance and Li+ Desolvation at Electrolyte/Graphite Interface, Langmuir, 2010, 26(13), 11538–11543 Search PubMed.
- W. Ren, et al., Carbon-coated porous silicon composites as high performance Li-ion battery anode materials: can the production process be cheaper and greener?, J. Mater. Chem. A, 2016, 4(2), 552–560 Search PubMed.
- G. Zhu, et al., Engineering the Distribution of Carbon in Silicon Oxide Nanospheres at the Atomic Level for Highly Stable Anodes, Angew. Chem., Int. Ed. Engl., 2019, 58(20), 6669–6673 Search PubMed.
- H. Ren, et al., Competitive adsorption of dopamine and rhodamine 6G on the surface of graphene oxide, ACS Appl. Mater. Interfaces, 2014, 6(4), 2459–2470 Search PubMed.
- P. Lv, et al., Highly efficient and scalable synthesis of SiOx/C composite with core-shell nanostructure as high-performance anode material for lithium ion batteries, Electrochim. Acta, 2015, 152, 345–351 Search PubMed.
- Z. Zhang, et al., Amorphous silicon-carbon nanospheres synthesized by chemical vapor deposition using cheap methyltrichlorosilane as improved anode materials for Li-ion batteries, Nanoscale, 2013, 5(12), 5384–5389 Search PubMed.
- Y. Xu, et al., Functionalized graphene hydrogel-based high-performance supercapacitors, Adv. Mater., 2013, 25(40), 5779–5784 Search PubMed.
- T. F. Yi, et al., Porous spherical NiO@NiMoO4@PPy nanoarchitectures as advanced electrochemical pseudocapacitor materials, Sci. Bull., 2020, 65(7), 546–556 Search PubMed.
- B.-C. Yu, et al., Reaction mechanism and enhancement of cyclability of SiO anodes by surface etching with NaOH for Li-ion batteries, J. Mater. Chem. A, 2013, 1(15), 4820 Search PubMed.
- Z. Zhang, et al., Scalable synthesis of interconnected porous silicon/carbon composites by the Rochow reaction as high-performance anodes of lithium ion batteries, Angew. Chem., Int. Ed. Engl., 2014, 53(20), 5165–5169 Search PubMed.
- Z. Zhang, et al., Low-Cost Synthesis of Porous Silicon via Ferrite-Assisted Chemical Etching and Their Application as Si-Based Anodes for Li-Ion Batteries, Adv. Electron. Mater., 2015, 1(4), 1400059 Search PubMed.
- J. N. Wang, et al., Suppressing the Shuttle Effect and Dendrite Growth in Lithium-Sulfur Batteries, ACS Nano, 2020, 14(8), 9819–9831 Search PubMed.
- L. Zhu, et al., Smart formaldehyde detection enabled by metal organic framework-derived doped electrospun hollow nanofibers, Sens. Actuators, B, 2021, 326, 128819 Search PubMed.
- J. Woo and S.-H. Baek, A comparative investigation of different chemical treatments on SiO anode materials for lithium-ion batteries: towards long-term stability, RSC Adv., 2017, 7(8), 4501–4509 Search PubMed.
- M. B. Ma, et al., Self-assembled homogeneous SiOC@C/graphene with three-dimensional lamellar structure enabling improved capacity and rate performances for lithium ion storage, Carbon, 2022, 186, 273–281 Search PubMed.
- M. Li, et al., Nanosilica/carbon composite spheres as anodes in Li-ion batteries with excellent cycle stability, J. Mater. Chem. A, 2015, 3(4), 1476–1482 Search PubMed.
- C. Sun, et al., Graphene oxide-immobilized NH(2)-terminated silicon nanoparticles by cross-linked interactions for highly stable silicon negative electrodes, ACS Appl. Mater. Interfaces, 2014, 6(14), 11277–11285 Search PubMed.
- F. Li, et al., Flexible free-standing graphene foam supported silicon films as high capacity anodes for lithium ion batteries, Mater. Lett., 2014, 128, 132–135 Search PubMed.
- Y. Yu, et al., Self-adaptive Si/reduced graphene oxide scrolls for high-performance Li-ion battery anodes, Carbon, 2017, 120, 397–404 Search PubMed.
- C.-K. Back, T.-J. Kim and N.-S. Choi, Activated natural porous silicate for a highly promising SiOx nanostructure finely impregnated with carbon nanofibers as a high performance anode material for lithium-ion batteries, J. Mater. Chem. A, 2014, 2(33), 13648 Search PubMed.
- J. Guo, et al., Cyclability study of silicon–carbon composite anodes for lithium-ion batteries using electrochemical impedance spectroscopy, Electrochim. Acta, 2011, 56(11), 3981–3987 Search PubMed.
- M. Winter, B. Barnett and K. Xu, Before Li Ion Batteries, Chem. Rev., 2018, 118(23), 11433–11456 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00960a |
| This journal is © The Royal Society of Chemistry 2022 |