DOI:
10.1039/D2RA00942K
(Paper)
RSC Adv., 2022,
12, 7229-7236
Alkyl substituent-dependent systematic change in cold crystallization of azo molecules†
Received
13th February 2022
, Accepted 25th February 2022
First published on 2nd March 2022
Abstract
The thermal behavior of alkyl-derivatized 1-(2,4-dimethylphenylazo)-4-naphthol and 1-(2,4-dimethylphenylazo)-2-naphthol (2,4-DM-4-Cn and 2,4-DM-2-Cn, respectively) was investigated. The change in the position of the alkyl substituent led to a variation in the thermal behavior, including the cold crystallization, which is a heat-storing phenomenon. In addition, a comprehensive study of the alkyl chain length revealed that 2,4-DM-4-Cn had better crystallinity and exhibited cold crystallization with short alkyl chains. The π–π, C–H⋯N, and C–H⋯π interactions stabilized the crystal structure of 2,4-DM-4-Cn. On the other hand, the polymorphism of 2,4-DM-2-Cn inhibited the formation of a uniform crystalline phase during cooling, which led to poor crystallinity. The only difference between the compounds, the position of the substituent, resulted in a clear variation in the cold crystallization and heat storage properties.
Introduction
Cold crystallization is a heat storage phenomenon that accompanies supercooling. In general, liquid compounds are transformed to the solid state during cooling. However, if the transition is inhibited, a supercooled state appears.1–4 The amount of heat that the supercooled state stores corresponds to the enthalpy of crystallization (latent heat).5–9 The exothermic crystallization, which accompanies the transition from the supercooled to the solid state, is called cold crystallization. Research on thermal storage is essential in developing a low-carbon and sustainable society. Recently, the cold crystallization of small molecules has attracted attention.10–17 The temperature of cold crystallization for small molecules is generally low, and these molecules can recover waste heat at a low temperature. In the cold crystallization of small molecules, the mobility of the aromatic rings is considered a factor in supercooling.18 In previous studies, the diketopyrrolopyrrole (DPP) pigment and azo dye molecules were found to exhibit cold crystallization.18,19 These molecules have rotatable aromatic rings, and the mobility of the rings leads to polymorphism and/or non-uniform crystallization, resulting in a slow rate of crystallization, supercooling, and cold crystallization. In the study of the azo molecule, the mobility of the aromatic ring was confirmed as polymorphism and disorder in the single-crystal structures.18 Further investigation is necessary to clarify the mechanism of supercooling and cold crystallization of small molecules. In this study, alkyl groups are introduced at different positions on the aromatic ring to investigate the effect of steric hindrance on the aromatic ring.
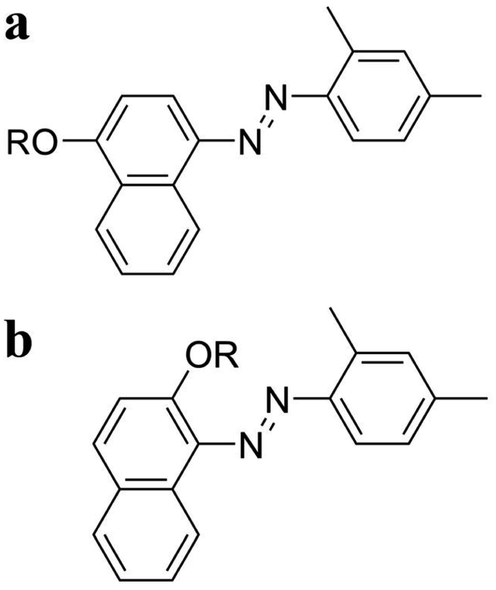
To achieve this objective, alkoxy-derivatized azo molecules are also synthesized (Fig. 1). Fig. 1a shows the alkyl-derivatized 1-(2,4-dimethylphenylazo)-4-naphthol, 2,4-DM-4-Cn, while Fig. 1b shows the alkyl-derivatized 1-(2,4-dimethylphenylazo)-2-naphthol, 2,4-DM-2-Cn. The change in the position of the substituent (4-Cn or 2-Cn) and alkyl chain length (Cn) should result in variations in the thermal behavior and crystallinity of the compounds. The results show that 2,4-DM-4-Cn exhibits a normal solid–liquid transition, and the shorter alkyl derivatives (2,4-DM-4-C1 and 2,4-DM-4-C3) exhibit cold crystallization. On the other hand, the 2,4-DM-2-Cn compounds exhibit poorer crystallinity than 2,4-DM-4-Cn. The short-chain 2,4-DM-2-Cn (n = 2, 5–11) adopts the oily liquid state; once the virgin solid sample is melted, only glass transition is observed. The virgin sample means a non previously thermally-treated sample. The middle-chain derivatives (n = 12–14) exhibit cold crystallization. The long-chain 2,4-DM-2-Cn (n = 15–18, 20) exhibits a normal solid–liquid transition. 2,4-DM-4-Cn and 2,4-DM-2-Cn exhibit systematic changes in the thermal behavior according to the alkyl chain length and substitution position.
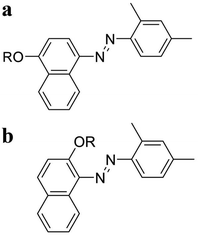 |
| | Fig. 1 Chemical structure of (a) 2,4-DM-4-Cn and (b) 2,4-DM-2-Cn. R is an alkyl chain CnH2n+1. | |
Experimental
Synthesis of 2,4-DM-4-Cn
2,4-Dimethylaniline was dissolved in hydrochloric acid (1/1 v/v distilled H2O). The mixture was then diazotized below −5 °C in a solution of sodium nitrite (0.8 g, 10 mmol, and 30 mL distilled H2O). The diazonium chloride was then combined with an alkaline solution of 1-naphthol (1.7 g, 10 mmol) in distilled H2O (20 mL). The crude product (1-(2,4-dimethylphenylazo)-4-naphthol) was collected after filtration. Potassium carbonate (1.5 g, 11 mmol) was added to a solution of the crude product (0.83 g, 3 mmol) in acetone (100 mL) and stirred for 10 min at room temperature. n-Bromoalkane (10 mmol) was then added to the mixture and stirred for 2 d at 70 °C. The residue was then evaporated. The resultant concentrate was dissolved in CHCl3 (50 mL), and washed with saturated saline. The organic layer was dried over Na2SO4, and evaporated until it was concentrated. Silica gel column chromatography with CHCl3/hexane (1/1.5 v/v) was used to purify the crude product, which afforded 1-(2,4-dimethylphenylazo)-4-alkoxynaphthalene (2,4-DM-4-Cn).
Synthesis of 2,4-DM-2-Cn
Potassium carbonate (1.5 g, 11 mmol) was added to a solution of 1-(2,4-dimethylphenylazo)-2-naphthol (Sudan II, 0.83 g, 3 mmol) in acetone (100 mL), and stirred for 10 min at room temperature. n-Bromoalkane (10 mmol) was added to the mixture and stirred for 2 d at 70 °C. The residue was evaporated to form a concentrate, which was dissolved in CHCl3 (50 mL), and washed with saturated saline. The organic layer was dried over Na2SO4, and evaporated until it became concentrated. Silica gel column chromatography with CHCl3/hexane (1/1.5 v/v) was used to purify the crude product, which afforded 1-(2,4-dimethylphenylazo)-2-alkoxynaphthalene (2,4-DM-2-Cn).
Identification of 2,4-DM-4-Cn and 2,4-DM-2-Cn
A PerkinElmer 2400II CHN analyzer was used to perform elemental analysis (C, H, N). The nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AVANCE NEO 400 spectrometer. A JASCO FT/IR-4200 spectrometer was used to acquire the Infrared (IR) spectra.
2,4-DM-4-C1. Yield: 0.010 g (1.2%); elemental analysis for C19H18N2O, calc: C, 78.59; H, 6.25; N, 9.65%, found: C, 78.77; H, 6.19; N, 9.62%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 8.99 (d, J = 8.3 Hz, 1Harom), 8.31 (d, J = 8.2 Hz, 1Harom), 7.87 (d, J = 8.4 Hz, 1Harom), 7.72 (d, J = 8.2 Hz, 1Harom), 7.66 (t, J = 7.6 Hz, 1Harom), 7.57 (t, J = 7.6 Hz, 1Harom), 7.16 (s, 1Harom), 7.11 (d, J = 8.6 Hz, 1Harom), 6.91 (d, J = 8.4 Hz, 1Harom), 4.09 (s, 3H, O–CH3), 2.78 (s, 3H, Carom–CH3), 2.40 (s, 3H, Carom–CH3); IR (KBr): ν 1577 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 2840–2952 (C–H), 3011–3076 (C–Harom).
C), 2840–2952 (C–H), 3011–3076 (C–Harom).
2,4-DM-4-C4. Yield: 0.0062 g (0.62%); elemental analysis for C22H24N2O, calc: C, 79.48; H, 7.28; N, 8.43%, found: C, 79.36; H, 6.98; N, 8.36%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 9.00 (d, J = 8.4 Hz, 1Harom), 8.34 (d, J = 8.3 Hz, 1Harom), 7.86 (d, J = 8.4 Hz, 1Harom), 7.72 (d, J = 8.2 Hz, 1Harom), 7.66 (t, J = 7.6 Hz, 1Harom), 7.56 (t, J = 7.6 Hz, 1Harom), 7.16 (s, 1Harom), 7.11 (d, J = 8.2 Hz, 1Harom), 6.90 (d, J = 8.5 Hz, 1Harom), 4.23 (t, J = 6.3 Hz, 2H, O–CH2–C), 2.74 (s, 3H, Carom–CH3), 2.40 (s, 3H, Carom–CH3), 1.97 (quin, J = 7.5 Hz, 2H, O–C–CH2–C), 1.65 (sext, J = 6.9 Hz, 2H, C–CH2–C), 1.05 (t, J = 7.4 Hz, 3H, C–CH3); IR (KBr): ν 1582 cm−1 (CC), 2873–2960 (C–H), 3020–3076 (C–Harom).
2,4-DM-2-C5. Yield: 0.28 g (27%); elemental analysis for C23H26N2O, calc: C, 79.73; H, 7.56; N, 8.09%, found: C, 79.85; H, 7.66; N, 8.19%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 8.36 (d, J = 8.4 Hz, 1Harom), 7.82 (d, J = 9.1 Hz, 1Harom), 7.81 (d, J = 8.5 Hz, 1Harom), 7.68 (d, J = 8.2 Hz, 1Harom), 7.50 (t, J = 8.2 Hz, 1Harom), 7.40 (t, J = 7.6 Hz, 1Harom), 7.38 (d, J = 9.1 Hz, 1Harom), 7.19 (s, 1Harom), 7.12 (d, J = 8.0 Hz, 1Harom), 4.16 (t, J = 6.6 Hz, 2H, O–CH2–C), 2.72 (s, 3H, Carom–CH3), 2.41 (s, 3H, Carom–CH3), 1.80 (quin, J = 7.0 Hz, 2H, O–C–CH2–C), 1.44–1.31 (m, 4H, C–CH2–C), 0.87 (t, J = 7.1 Hz, 3H, C–CH3); IR (KBr): ν 1592 cm−1 (CC), 2860–2947 (C–H), 3011–3043 (C–Harom).
2,4-DM-2-C13. Yield: 0.20 g (15%); elemental analysis for C31H40N2O, calc: C, 81.17; H, 9.23; N, 6.11%, found: C, 81.14; H, 9.37; N, 6.10%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 8.40 (d, J = 8.4 Hz, 1Harom), 7.82 (d, J = 9.0 Hz, 1Harom), 7.81 (d, J = 8.0 Hz, 1Harom), 7.68 (d, J = 8.2 Hz, 1Harom), 7.50 (t, J = 7.7 Hz, 1Harom), 7.40 (t, J = 7.5 Hz, 1Harom), 7.38 (d, J = 9.1 Hz, 1Harom), 7.19 (s, 1Harom), 7.12 (d, J = 8.4 Hz, 1Harom), 4.15 (t, J = 6.5 Hz, 2H, O–CH2–C), 2.72 (s, 3H, Carom–CH3), 2.41 (s, 3H, Carom–CH3), 1.78 (quin, J = 7.1 Hz, 2H, O–C–CH2–C), 1.46–1.22 (m, 20H, C–CH2–C), 0.88 (t, J = 6.8 Hz, 3H, C–CH3); IR (KBr): ν 1595 cm−1 (CC), 2852–2951 (C–H), 3025–3050 (C–Harom).
2,4-DM-2-C15. Yield: 0.24 g (17%); elemental analysis for C33H44N2O, calc: C, 81.43; H, 9.53; N, 5.76%, found: C, 81.73; H, 9.57; N, 5.66%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 8.40 (d, J = 8.4 Hz, 1Harom), 7.82 (d, J = 9.0 Hz, 1Harom), 7.80 (d, J = 7.6 Hz, 1Harom), 7.68 (d, J = 8.2 Hz, 1Harom), 7.49 (t, J = 7.7 Hz, 1Harom), 7.40 (t, J = 7.3 Hz, 1Harom), 7.38 (d, J = 9.1 Hz, 1Harom), 7.19 (s, 1Harom), 7.11 (d, J = 8.4 Hz, 1Harom), 4.15 (t, J = 6.5 Hz, 2H, O–CH2–C), 2.72 (s, 3H, Carom–CH3), 2.41 (s, 3H, Carom–CH3), 1.78 (quin, J = 7.0 Hz, 2H, O–C–CH2–C), 1.49–1.22 (m, 24H, C–CH2–C), 0.88 (t, J = 6.8 Hz, 3H, C–CH3); IR (KBr): ν 1595 cm−1 (CC), 2851–2951 (C–H), 3019–3052 (C–Harom).
2,4-DM-2-C17. Yield: 0.24 g (15%); elemental analysis for C35H48N2O, calc: C, 81.66; H, 9.79; N, 5.44%, found: C, 81.50; H, 9.74; N, 5.40%. 1H NMR (400 MHz, CDCl3, Me4Si): δ 8.40 (d, J = 8.4 Hz, 1Harom), 7.82 (d, J = 9.0 Hz, 1Harom), 7.80 (d, J = 7.9 Hz, 1Harom), 7.68 (d, J = 8.1 Hz, 1Harom), 7.49 (t, J = 7.7 Hz, 1Harom), 7.40 (t, J = 7.5 Hz, 1Harom), 7.38 (d, J = 9.1 Hz, 1Harom), 7.19 (s, 1Harom), 7.11 (d, J = 8.2 Hz, 1Harom), 4.15 (t, J = 6.5 Hz, 2H, O–CH2–C), 2.72 (s, 3H, Carom–CH3), 2.41 (s, 3H, Carom–CH3), 1.78 (quin, J = 7.0 Hz, 2H, O–C–CH2–C), 1.44–1.22 (m, 28H, C–CH2–C), 0.88 (t, J = 6.8 Hz, 3H, C–CH3); IR (KBr): ν 1595 cm−1 (CC), 2850–2952 (C–H), 3014–3052 (C–Harom).Identification of the other 2,4-DM-4-Cn and 2,4-DM-2-Cn molecules is described in the ESI.†
Analyses of thermal behavior
A NETZSCH DSC 3500 Sirius calorimeter was used to perform differential scanning calorimetry (DSC). The powdered crystals of 2,4-DM-4-Cn or 2,4-DM-2-Cn were enclosed in an aluminum pan, and the DSC scans were then performed under dry nitrogen gas. The heating and cooling rates were both set to 10 °C min−1. A Nikon ECLIPSE LV100POL microscope was used to record the optical microscopic data. A heat stage (Linkam, THMS600) was used to control the temperature of the sample. The 2,4-DM-4-Cn or 2,4-DM-2-Cn crystals, which were inserted between cover glasses, were heated and cooled, and microscopic images of the thermal behavior were collected.
Single crystal X-ray crystallography
Single crystals of 2,4-DM-4-C8 compounds suitable for X-ray crystallography were obtained by slow evaporation of the CHCl3–CH3OH mixed solution. A single crystal was then mounted on a glass capillary, transferred to a Bruker AXS SMART diffractometer equipped with a CCD area detector and Mo-Kα (λ = 0.71073 Å) radiation, and centered in the beam at 173 K. SHELX,20,21 which involved the direct method and expanded Fourier techniques, was used to solve and refine the structures. All the non-hydrogen and hydrogen atoms were refined anisotropically and isotropically, respectively. The crystal data are presented in Table S1.†
Theoretical study
The model diagram of molecular rotation around azo group was created with a Materials Studio 2020 software22 (Biovia, San Diego, CA, US). To optimize the molecular structure, the generalized gradient approximation (GGA) proposed by Perdew et al.23 was used to perform density functional theory (DFT) calculations. The rotation model around azo group was created by using Conformers module in Materials Studio.
Results and discussion
Influence of the alkyl chain length on the thermal behavior of 2,4-DM-4-Cn and 2,4-DM-2-Cn
A comprehensive study of the alkyl chain lengths of 2,4-DM-4-Cn and 2,4-DM-2-Cn was conducted. The thermal behavior of various alkyl derivatives of 2,4-DM-4-Cn (n = 1–18, 20) and 2,4-DM-2-Cn (n = 2, 3, 5–18, 20) was analyzed and the results are listed in Table 1. The compounds of 2,4-DM-4-Cn with shorter alkyl chains (C1 and C3) exhibited cold crystallization. In contrast, cold crystallization was observed in the longer chains (C12, C13, and C14) of 2,4-DM-2-Cn. Most of the 2,4-DM-4-Cn compounds exhibited a normal solid–liquid transition; the compounds melted during heating and crystallized during cooling. The shorter-chain derivatives of 2,4-DM-2-Cn (n = 2, 3, 5–11) were oily; the virgin solid sample melted during heating and only the glass transition was observed in subsequent cooling–heating. The oily state of 2,4-DM-2-Cn suggests that this compound was more difficult to crystallize than 2,4-DM-4-Cn. The longer-chain derivatives of 2,4-DM-2-Cn (n = 15–18, 20) exhibited a solid–liquid transition. The strong van der Waals interactions between the long alkyl chains increased the crystallinity,24–28 and induced crystallization of the compound during cooling. The change in the position of the alkyl substituent in 2,4-DM-4-Cn versus 2,4-DM-2-Cn resulted in variations in the thermal behavior, such as cold crystallization.
Table 1 List of thermal behavior depending on the alkyl chain length
| Alkyl chain length |
Thermal behavior for 2,4-DM-4-Cn |
Thermal behavior for 2,4-DM-2-Cn |
| 1 |
Cold crystallization |
— |
| 2 |
No cold crystallization |
Oily liquid state |
| 3 |
Cold crystallization |
Oily liquid state |
| 4 |
No cold crystallization |
— |
| 5 |
No cold crystallization |
Oily liquid state |
| 6 |
No cold crystallization |
Oily liquid state |
| 7 |
No cold crystallization |
Oily liquid state |
| 8 |
No cold crystallization |
Oily liquid state |
| 9 |
No cold crystallization |
Oily liquid state |
| 10 |
No cold crystallization |
Oily liquid state |
| 11 |
No cold crystallization |
Oily liquid state |
| 12 |
No cold crystallization |
Cold crystallization |
| 13 |
No cold crystallization |
Cold crystallization |
| 14 |
No cold crystallization |
Cold crystallization |
| 15 |
No cold crystallization |
No cold crystallization |
| 16 |
No cold crystallization |
No cold crystallization |
| 17 |
No cold crystallization |
No cold crystallization (unique) |
| 18 |
No cold crystallization |
No cold crystallization |
| 20 |
No cold crystallization |
No cold crystallization |
Thermal behavior of 2,4-DM-4-Cn
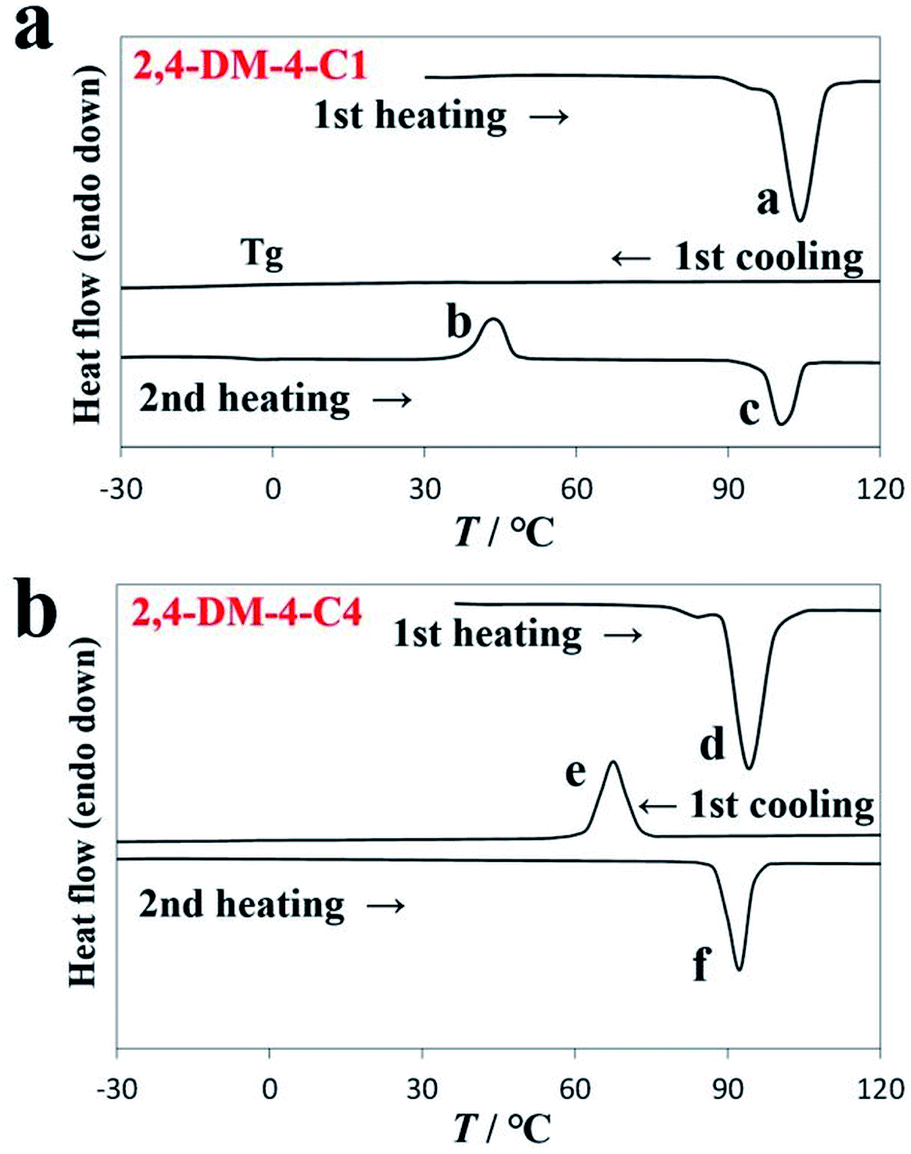
As aforementioned, the alkyl chain length of 2,4-DM-4-Cn influenced the occurrence of either cold crystallization or normal solid–liquid transition. Fig. 2 shows the DSC diagrams of 2,4-DM-4-C1 (cold crystallization, Fig. 2a) and 2,4-DM-4-C4 (normal solid–liquid transition, Fig. 2b). Fig. S1† shows the thermal behavior of the other 2,4-DM-4-Cn compounds. The virgin solid sample of 2,4-DM-4-C1 was heated at a rate of 10 °C min−1, and melted at 100.3 °C, which resulted in an enthalpy change (ΔH) of 27.0 kJ mol−1 (peak ‘a’). There were no peaks in the first cooling curve, which suggests that 2,4-DM-4-C1 adopted the supercooled state. The glass transition (Tg) was observed at −8.1 °C as a baseline shift in the DSC curve. When the compound was re-heated (second heating), a glass transition was observed at −17.8 °C. An exothermic peak ‘b’ (42.2 °C, 18.4 kJ mol−1) was subsequently observed. The exothermic peak during heating occurred because of cold crystallization. Finally, the cold-crystallized 2,4-DM-4-C1 melted to generate peak ‘c’ (103.8 °C, 23.3 kJ mol−1). The thermal behavior during further heating-cooling was the same as the first cooling and second heating. The virgin solid sample of 2,4-DM-4-C4 melted at peak ‘d’ (Fig. 2b, 89.4 °C, 29.0 kJ mol−1) in the first heating. The liquid 2,4-DM-4-C1 was supercooled during cooling; in contrast, the liquid 2,4-DM-4-C4 transformed into a solid with a clear exothermic peak ‘e’ (72.3 °C, 28.0 kJ mol−1). The corresponding melting peak ‘f’ (88.3 °C, 30.1 kJ mol−1) was observed in the second heating. The thermal behavior of 2,4-DM-4-C4 was reproducible during further heating-cooling.
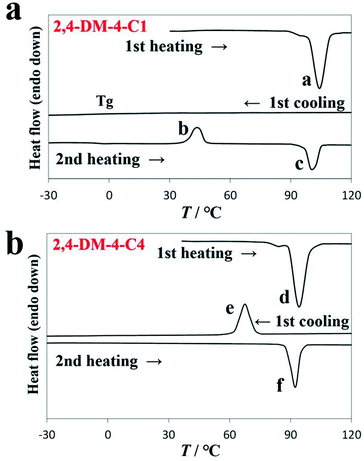 |
| | Fig. 2 DSC diagrams of (a) 2,4-DM-4-C1 and (b) 2,4-DM-4-C4. | |
Polarized optical microscopy was used to analyze the thermal behavior of 2,4-DM-4-C1 and 2,4-DM-4-C4. Fig. 3 shows the crossed Nicol images of 2,4-DM-4-C1 and -C4. Fig. 3a shows the virgin orange powder crystals of 2,4-DM-4-C1 obtained by solvent (CHCl3) evaporation with a rotary evaporator. A temperature control stage was used to heat and cool the sample. The crystals were melted at approximately 110 °C. The crossed Nicol image (Fig. 3b) is dark, suggesting that the compounds transformed into an isotropic liquid state. The liquid sample was cooled to −40 °C and the microscopic image was kept dark. The compounds did not crystallize during cooling and were supercooled instead. After the compounds were reheated, they became cold-crystallized. The cold-crystallized sample is shown in Fig. 3c. Fig. 3d–f show the crossed Nicol images of 2,4-DM-4-C4. The virgin orange sample is shown in Fig. 3d. The sample melted at 99 °C during heating, and the image of the liquid state is shown in Fig. 3e. The 2,4-DM-4-C4 compounds crystallized at 60 °C after they were cooled. The crystallized sample is shown in Fig. 3f. Microscopic images confirmed the cold crystallization and normal solid–liquid transition of 2,4-DM-4-C4.
 |
| | Fig. 3 Crossed Nicol images of 2,4-DM-4-C1: (a) virgin sample at room temperature, (b) 160 °C during the 1st heating, (c) 60 °C during the 2nd heating. Crossed Nicol images of 2,4-DM-4-C4: (d) virgin sample at room temperature, (e) 160 °C during the 1st heating, (f) 60 °C during the 1st cooling. | |
Thermal properties of 2,4-DM-2-Cn
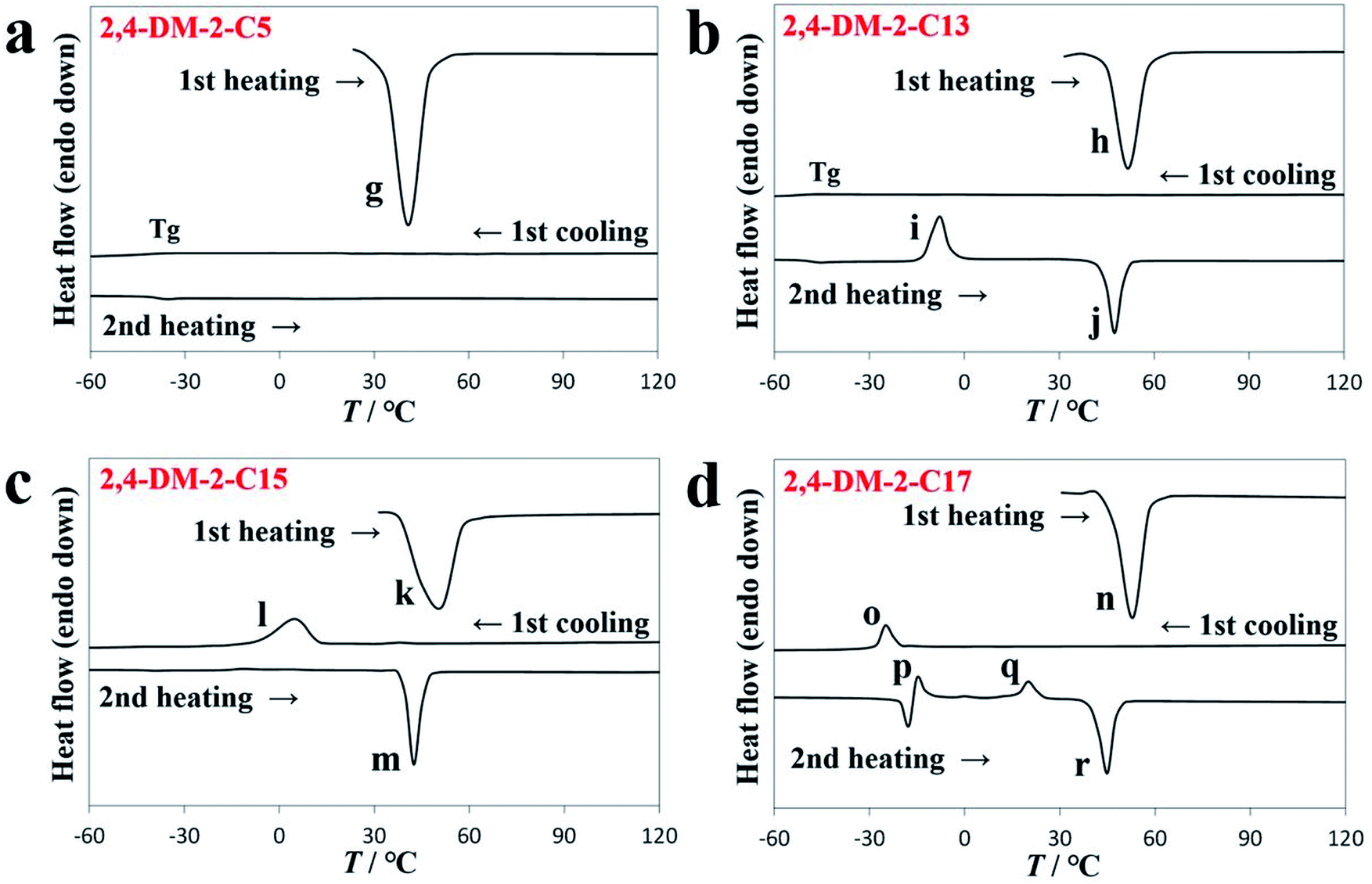
Table 1 shows the three thermally induced states of 2,4-DM-2-Cn: oily liquid, cold crystallization, and solid–liquid transition states. The DSC diagram in Fig. 4a shows that 2,4-DM-2-C5 existed as a viscous oily liquid. The cold crystallization of 2,4-DM-2-C13 is shown in Fig. 4b. The DSC curve in Fig. 4c shows that the compounds of 2,4-DM-2-C15 exhibit a normal solid–liquid transition. The multiple exothermic peaks are observed in the DSC diagram of 2,4-DM-2-C17, which indicate that 2,4-DM-2-C17 exhibited a unique solid–liquid transition (Fig. 4d). The thermal behavior of 2,4-DM-2-Cn is shown in Fig. S2.† The virgin sample of 2,4-DM-2-C5 was obtained as orange needle crystals, and the sample melted during the first heating cycle (peak ‘g’, 36.1 °C, 30.6 kJ mol−1). During subsequent cooling–heating, these transition peaks were not observed, and only the glass transition state was observed at −31.6 °C (during cooling) and −39.3 °C (during heating). The DSC results suggest that the crystallinity of 2,4-DM-2-C5 was low. In the oily liquid state, 2,4-DM-2-C5 was highly viscous. The 2,4-DM-2-C13 compounds melted at 46.9 °C (peak ‘h’, 52.0 kJ mol−1) during the first heating, and were supercooled after the first cooling. A Tg was observed at −49.6 °C, and the corresponding Tg appeared at −48.5 °C in the second heating. The cold crystallization peak ‘i’ was observed at −12.3 °C with an ΔH of 36.2 kJ mol−1. Finally, the cold-crystallized sample was melted at 44.0 °C (peak ‘j’, 50.6 kJ mol−1). The virgin 2-C15 sample was melted at 43.2 °C (peak ‘k’, 50.0 kJ mol−1) during the first heating. The clear crystallization peak ‘l’ (11.5 °C, 35.2 kJ mol−1) was observed in the first cooling, and the melting peak ‘m’ (39.2 °C, 48.7 kJ mol−1) appeared in the second heating. As mentioned, 2,4-DM-2-C15 exhibited a normal solid–liquid transition. The thermal behavior of the derivatives with long alkyl chains (2,4-DM-2-C16, -C18, and -C20) was similar to that of 2,4-DM-2-C15. In contrast, 2,4-DM-2-C17 exhibited a unique solid–liquid transition. During the first heating, the powdered crystals of 2,4-DM-2-C17 melted at peak ‘n’ (46.2 °C, 65.6 kJ mol−1). When the 2,4-DM-2-C17 compounds were first cooled (first cooling), they exhibited a clear exothermic peak ‘o’ (−22.1 °C, 15.5 kJ mol−1), which suggests that the samples crystallized. When the 2,4-DM-2-C17 compounds were subsequently heated (second heating), two exothermic peaks (p and q) were observed. The thermal behavior of peaks ‘p’ and ‘o’ were likely similar (endotherm: −19.7 °C, 13.1 kJ mol−1; exotherm: −16.7 °C, 10.4 kJ mol−1) because both peaks appeared at almost the same temperature. The 2,4-DM-2-C17 compounds partially crystallized during cooling, and the remaining supercooled compounds possibly cold-crystallized subsequent to heating. The second exothermic peak, ‘q’ (17.0 °C, 16.0 kJ mol−1), was associated with a solid–solid transition. Long-alkyl-chain derivatives often exhibit polymorphism,29–34 therefore, a kinetically-formed metastable structure crystallizes first. The structure subsequently forms a thermodynamically stable structure after heating. Peak ‘r’ (41.2 °C, 56.0 kJ mol−1) was associated with the final melting of the 2,4-DM-2-C17 compounds. To obtain additional insight into the thermal behavior of 2,4-DM-2-C17, DSC experiments at a slow cooling rate (1 °C min−1) and powder X-ray diffraction (XRD) analyses were conducted. The DSC diagram is shown in Fig. S3.† A normal solid–liquid transition was observed with slow cooling; only one crystallization peak during cooling and only one melting peak during heating were observed. When the compounds were first heated, they melted at 46.3 °C (65.2 kJ mol−1). The compounds were then cooled at a rate of 1 °C min−1, which resulted in a crystallization peak (20.7 °C, 51.8 kJ mol−1). In the subsequent heating step, a melting peak (41 °C, 61.7 kJ mol−1) was observed. The temperature of the melting peak was almost the same as that of peak ‘r’. These results suggest that crystallization at a slow cooling rate led to thermodynamically stable crystals during cooling. Fig. S4† shows the powder XRD patterns of 2,4-DM-2-C17. The sample was heated to 100 °C, melted, and then cooled to −60 °C (under peak ‘o’) at a rate of 10 °C min−1. The sample was transferred into a powder XRD instrument, and the XRD pattern was determined at room temperature (blue line in Fig. S4†). The sample was heated and kept at 30 °C (between the temperature of peak ‘q’ and the melting peak ‘r’) for 24 h, and the XRD pattern of the sample was acquired (red line in Fig. S4†). Both peaks had distinct patterns, where peak ‘q’ was associated with a solid–solid transition. The sample was reheated, melted, and cooled to −60 °C at a rate of 1 °C min−1. The gray line in Fig. S4† shows the XRD pattern of the crystallized sample. The pattern partially corresponded to the blue line and included other peaks. The XRD patterns in Fig. S4† suggest that the polymorphism of the 2,4-DM-2-C17 compounds (different crystalline phases) depends on the conditions.
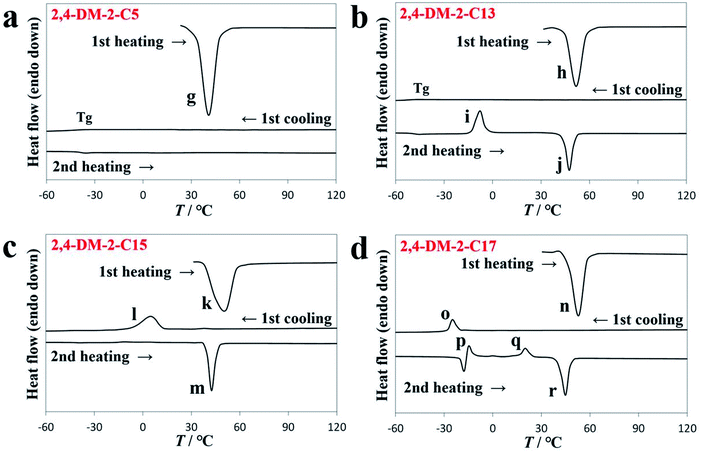 |
| | Fig. 4 DSC diagrams of (a) 2,4-DM-2-C5, (b) 2,4-DM-2-C13, (c) 2,4-DM-2-C15, and (d) 2,4-DM-2-C17. | |
The polarized microscopic images of 2,4-DM-2-Cn confirmed the thermal behavior of its compounds. Fig. 5a shows that after solvent extraction, the virgin sample of 2,4-DM-2-C5 comprised orange crystals. During heating, the crystals melted at 50 °C, and the crossed Nicol image became dark (Fig. 5b). The image, which remained dark after cooling–heating, indicates that the 2,4-DM-2-C5 compounds adopted only the liquid and glass states after melting. Fig. 5c shows the virgin orange crystals of 2,4-DM-2-C13 and the crystals after melting at 52 °C. An image of the isotropic liquid state (dark image) is shown in Fig. 5d. Crystallization was not observed during cooling, and the compounds exhibited supercooling instead. Cold crystallization was observed after subsequent heating, where the cold-crystallized crystals are shown in Fig. 5e. Fig. 5f and g show the virgin orange crystals of 2,4-DM-2-C15 and the liquid state after melting (50 °C), respectively. Fig. 5h shows that the compounds of 2,4-DM-2-C15 crystallized into orange crystals during cooling, which resulted in a normal solid–liquid transition. The virgin sample of 2,4-DM-2-C17 comprised orange crystals (Fig. 5i), which melted at 55 °C (Fig. 5j). During subsequent cooling, small crystals were observed in the polarized optical microscope (POM) image as bright and dark spots (Fig. 5k). The dark spots disappeared during subsequent heating, and the bright spots and darker regions remained (Fig. 5l). Consistent with the XRD patterns, the POM images suggest that 2,4-DM-2-C17 exhibits polymorphism.
 |
| | Fig. 5 Crossed Nicol images of 2,4-DM-2-C5: (a) virgin sample at room temperature, (b) 160 °C during the 1st heating. Crossed Nicol images of 2,4-DM-2-C13: (c) virgin sample at room temperature, (d) − 40 °C during the 1st cooling, (e) 15 °C during the 2nd heating. Crossed Nicol images of 2,4-DM-2-C15: (f) virgin sample at room temperature, (g) 160 °C during the 1st heating, (h) −15 °C during the 1st cooling. Crossed Nicol images of 2,4-DM-2-C17: (i) virgin sample at room temperature, (j) 160 °C during the 1st heating, (k) −40 °C during the 1st cooling, (l) 30 °C during the 1st heating. | |
Single crystal structure of 2,4-DM-4-C8
After solvent evaporation, a single crystal of 2,4-DM-4-C8 was obtained, and was subjected to X-ray diffraction. The crystal structure of 2,4-DM-4-C8 is shown in Fig. 6. The details of the crystal structure are shown in Table S1† and crystallographic information file (CIF). Fig. 6a shows the molecular structure of 2,4-DM-4-C8, which is almost flat. The angle between the phenyl and naphthalene plane is 6.43°. The intermolecular interactions between the flat molecules are shown in Fig. 6b. The figure shows that π–π interactions35 (distance between the centroids is 3.72 Å) resulted in the stacking of neighboring phenyl groups. Additionally, the methyl group at the 4-position and azo groups formed C–H⋯N interactions36–39 (C⋯N: 3.64 Å, H⋯N: 2.71 Å, and C–H⋯N: 159.0°). The dimer structure shown in Fig. 6b is formed by these intermolecular interactions. The methyl groups at the 2-position were arranged to avoid steric hindrance. Fig. 6c shows the molecular packing in a single crystal of 2,4-DM-4-C8. The dimer did not have a 1-D columnar structure, and π–π stacking between the dimers was not observed. The phenyl and naphthalene planes overlapped with the alkyl chains. A few C–H⋯π interactions40,41 were observed, namely the C–H (alkyl)⋯π (centroid of phenyl ring), C⋯centroid (3.64 Å), H⋯centroid (2.79 Å), C–H⋯centroid (144.8°), C–H (alkyl)⋯π (centroid of naphthalene ring), C⋯centroid (3.79 Å), H⋯centroid (2.97 Å), and C–H⋯centroid (140.5°) interactions. These π–π, C–H⋯N, and C–H⋯π interactions in the single crystal were likely the major stabilizing factors. In addition, the disorder and pedal motion42 around azo group were not observed, suggesting that the uniformity and good crystallinity of the crystal. The methyl and alkyl groups of 2,4-DM-4-C8 stabilized its crystal structure and increased its crystallinity.
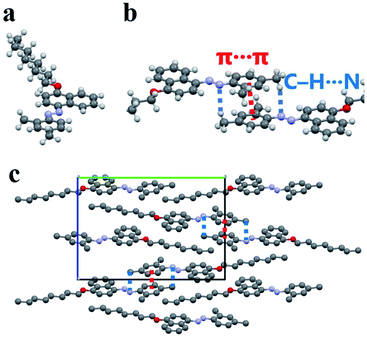 |
| | Fig. 6 Structure of a single 2,4-DM-4-C8 crystal. (a) Almost flat molecular structure of 2,4-DM-4-C8, (b) π–π and C–H⋯N intermolecular interactions, (c) molecular packing viewed along a-axis. | |
Effect of alkyl chain length and substituent position
In previous studies, the mobility of aromatic rings caused cold crystallization. Therefore, the effect of the mobility of the aromatic rings around the azo group was investigated. To estimate the effect of the degree of steric hindrance of the alkyl chain on the rotation around the azo group, diagrams of the molecular structure were created to show the steric features (Fig. S5†). Materials Studio 2020 software22 (Biovia, San Diego, CA, US) was used to calculate the molecular structure. To optimize the molecular structure, the generalized gradient approximation (GGA) proposed by Perdew et al.23 was used to perform density functional theory (DFT) calculations. The Conformers module in Materials Studio was used to create the rotation model of the azo groups. Fig. S5a and S5b† show the rotation models of 2,4-DM-4-Cn and 2,4-DM-2-Cn, respectively. In the Figures, the yellow molecule is rotated by 90°, while the red molecule is rotated by 180°. The protrusion of the alkyl chain from the azo molecular framework was greater in 2,4-DM-2-Cn than in 2,4-DM-4-Cn. This observation suggests that the alkyl chain of 2,4-DM-2-Cn inhibited crystallization, which accounts for the oily liquid state of 2,4-DM-2-Cn. The long alkyl chains in 2,4-DM-2-Cn also generated strong van der Waals interactions, which increased the crystallinity of 2,4-DM-2-Cn and explain its observed cold crystallization and solid–liquid transition. Powder XRD analyses revealed that 2,4-DM-2-Cn exhibited polymorphism. The multiple types of crystals in the POM image also suggest that 2,4-DM-2-Cn exhibits polymorphism, which likely contributes to inhibiting uniform crystallization during cooling. For example, polymorphism led to the poor crystallinity of 2,4-DM-2-Cn. In contrast, the steric hindrance of the alkyl chain in the rotation of 2,4-DM-4-Cn was smaller, which resulted in higher crystallinity when compared to 2,4-DM-2-Cn. Polymorphism was not observed in the POM images of 2,4-DM-4-Cn, which suggests that the crystalline phase of the compounds was uniform. In addition, single crystal X-ray analysis of 2,4-DM-4-C showed that π–π, C–H⋯N, and C–H⋯π interactions were present. Therefore, the methyl and alkyl groups of 2,4-DM-4-Cn contributed to its structural stability, rather than steric hindrance. Consequently, 2,4-DM-4-Cn showed higher crystallinity than 2,4-DM-2-Cn. 2,4-DM-4-Cn exhibited a normal solid–liquid transition, while the shorter alkyl derivatives (2,4-DM-4-C1 and 2,4-DM-4-C3) exhibited cold crystallization.
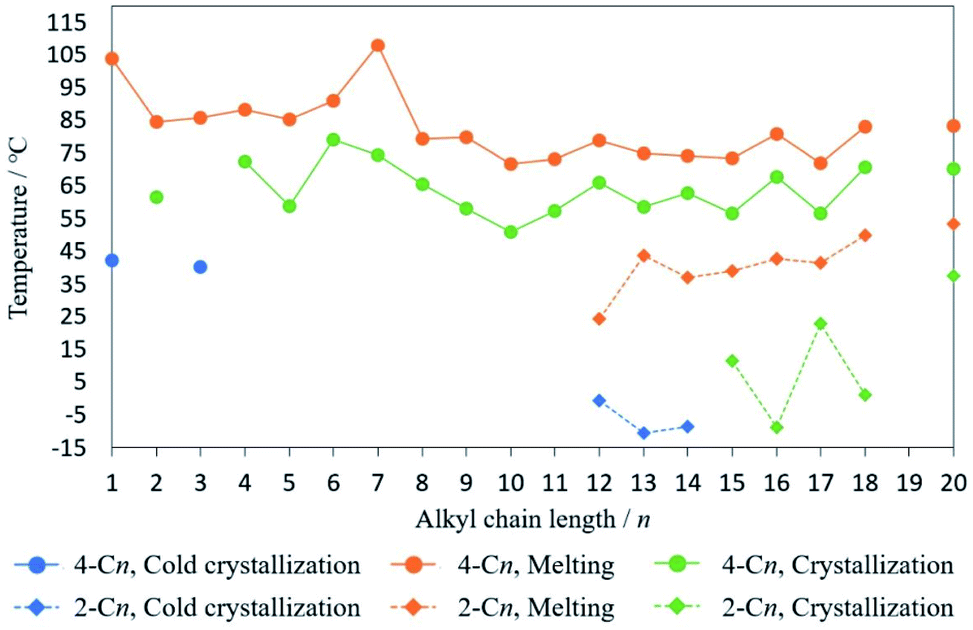
To investigate the effect of alkyl substituents, the phase transition temperature of 2,4-DM-4-Cn and 2,4-DM-2-Cn is summarized in Fig. 7. Additionally, the melting enthalpies in the DSC measurements are summarized in Fig. S6.† The odd-even effect (the zigzag line)24 can be seen in Fig. 7 and S6,† which is evidence that the alkyl chains control the crystal structure and crystallization behavior. The crystallization temperature of 2,4-DM-2-Cn is lower than that of 2,4-DM-4-Cn, indicating that the temperature at which molecular motion freezes is lower, and the alkyl group of 2,4-DM-2-Cn is an obstacle to crystallization. The enthalpies of melting in the middle-chain derivatives are also lower for 2,4-DM-2-Cn, which indicates a smaller energy difference between the liquid and the crystal, i.e., less stability of the crystal structure. This can also be confirmed from the lower melting points of 2,4-DM-2-Cn. These results prove that 2,4-DM-4-Cn has better crystallinity in the short-chain and meddle-chain derivatives. In the long-chain derivatives, 2,4-DM-4-Cn and 2,4-DM-2-Cn with the same chain length show almost the same melting enthalpy, leading to the similar normal solid–liquid transition. In summary, the only difference between these compounds, the position of the alkyl group (4- or 2-Cn), resulted in a clear variation in the thermal behavior.
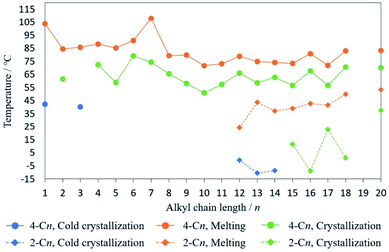 |
| | Fig. 7 Phase transition temperature of 2,4-DM-4-Cn and 2,4-DM-2-Cn: (blue circles) temperature of cold crystallization of 2,4-DM-4-Cn; (orange circles) melting temperature of 2,4-DM-4-Cn; (green circles) crystallization temperature of 2,4-DM-4-Cn; (blue squares) temperature of cold crystallization of 2,4-DM-2-Cn; (orange squares) melting temperature of 2,4-DM-2-Cn; (green squares) crystallization temperature of 2,4-DM-2-Cn. | |
Conclusions
The investigations revealed that the variation in the thermal behavior depends on the position and chain length of the alkyl substituent. 2,4-DM-4-Cn exhibited good crystallinity and a normal solid–liquid transition, while 2,4-DM-4-Cn with shorter alkyl chains exhibited cold crystallization. The π–π, C–H⋯N, and C–H⋯π interactions stabilized the crystal structure of 2,4-DM-4-Cn, which resulted in good crystallinity. In contrast, 2,4-DM-2-Cn exhibited poor crystallinity, which accounted for its glassy and oily liquid states. Additionally, 2,4-DM-2-Cn with longer alkyl chains exhibited cold crystallization. Powder XRD and POM experiments revealed that 2,4-DM-2-Cn exhibited polymorphism. The protrusion of the alkyl chain in the azo molecular framework of 2,4-DM-2-Cn is a possible steric barrier to its crystallization. The polymorphism and steric barrier in 2,4-DM-2-Cn prevent uniform crystallization, accounting for its poor crystallinity. The only difference between the investigated compounds, the position of the alkyl substituent, led to a clear variation in thermal behavior, such as cold crystallization.
Conflicts of interest
There are no conflicts to declare.
Author contributions
A. Honda: Conceptualization, Investigation, Visualization, Writing – original draft –, Writing – review & editing. Y. Hibi: Conceptualization, Investigation, Validation, Writing – original draft. K. Matsumoto: Conceptualization, Investigation. M. Kawai: Formal analysis, Investigation. K. Miyamura: Supervision, Writing – review & editing.
Acknowledgements
This work was supported by JSPS KAKENHI (Grant Number JP21K14725) and the Grant for Young or Female Researchers at the Tokyo University of Science.
Notes and references
- M. D. Ediger, C. A. Angell and S. R. Nagel, J. Phys. Chem., 1996, 100, 13200–13212 CrossRef CAS.
- C. A. Angell, K. L. Ngai, G. B. McKenna, P. F. McMillan and S. W. Martin, J. Appl. Phys., 2000, 88, 3113–3157 CrossRef CAS.
- M. D. Ediger, Annu. Rev. Phys. Chem., 2000, 51, 99–128 CrossRef CAS PubMed.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS PubMed.
- S. Puupponen, V. Mikkola, T. A. Nissila and A. Seppälä, Appl. Energy, 2016, 172, 96–106 CrossRef CAS.
- G. Englmair, Y. Jiang, M. Dannemand, C. Moser, H. Schranzhofer, S. Furbo and J. Fan, Energ. Build., 2018, 180, 159–171 CrossRef.
- C. Rathgeber, S. Hiebler, R. Bayón, L. F. Cabeza, G. Zsembinszki, G. Englmair, M. Dannemand, G. Diarce, O. Fellmann, R. Ravotti, D. Groulx, A. C. Kheirabadi, S. Gschwander, S. Höhlein, A. K. Haagen, N. Beaupere and L. Zalewski, Appl. Sci., 2020, 10, 7968 CrossRef CAS.
- H. Zhou, L. Lv, Y. Zhang, M. Ji and K. Cen, Sol. Energy Mater. Sol. Cells, 2021, 230, 111244 CrossRef CAS.
- G. Wang, C. Xu, W. Kong, G. Englmair, J. Fan, G. Wei and S. Furbo, J. Energy Storage, 2021, 40, 102780 CrossRef.
- G. Szklarz, K. Adrjanowicz, J. K. Kowalczuk, K. Jurkiewicz and M. Paluch, Phys. Chem. Chem. Phys., 2017, 19, 9879–9888 RSC.
- R. Chang, Q. Fu, Y. Li, M. Wang, W. Du, C. Chang and A. Zeng, CrystEngComm, 2017, 19, 335–345 RSC.
- K. Iwase, Y. Toyama, I. Yoshikawa, Y. Yamamura, K. Saito and H. Houjou, Bull. Chem. Soc. Jpn., 2018, 91, 669–677 CrossRef CAS.
- A. A. Boopathi, S. Sampath and T. Narasimhaswamy, New J. Chem., 2019, 43, 9500–9506 RSC.
- M. T. Viciosa, J. J. M. Ramos and H. P. Diogo, Int. J. Pharm., 2020, 584, 119410 CrossRef CAS PubMed.
- Ł. Kolek, M. M. Arodź, K. Adrjanowicz, T. Rozwadowski, K. Dychtoń, M. Drajewicz and P. Kula, J. Mol. Liq., 2020, 297, 111913 CrossRef.
- S. E. Lapuk, T. A. Mukhametzyanov, C. Schick and A. V. Gerasimov, Int. J. Pharm., 2020, 599, 120427 CrossRef PubMed.
- A. Dołęga, E. J. Gałązka, A. Deptuch, S. Baran and P. M. Zieliński, Thermochim. Acta, 2022, 707, 179100 CrossRef.
- A. Honda, S. Kakihara, M. Kawai, T. Takahashi and K. Miyamura, Cryst. Growth Des., 2021, 21, 6223–6229 CrossRef CAS.
- A. Honda, Y. Takahashi, Y. Tamaki and K. Miyamura, Chem. Lett., 2016, 45, 211–213 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- BIOVIA Materials Studio 2020, Biovia, San Diego, CA, US Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Boese, H.-C. Weiss and D. Bläser, Angew. Chem., Int. Ed., 1999, 38, 988–992 CrossRef CAS PubMed.
- K. Kim, K. E. Plass and A. J. Matzger, Langmuir, 2005, 21, 647–655 CrossRef CAS PubMed.
- Q. Chen, H.-J. Yan, C.-J. Yan, G.-B. Pan, L.-J. Wan, G.-Y. Wen and D.-Q. Zhang, Surf. Sci., 2008, 602, 1256–1266 CrossRef CAS.
- E. C. Constable, G. Zhang, C. E. Housecroft and J. A. Zampese, Inorg. Chem. Commun., 2012, 15, 113–116 CrossRef CAS.
- N. Komiya, T. Hori, M. Naito and T. Naota, Eur. J. Inorg. Chem., 2014, 1, 156–163 CrossRef.
- R. W. Crowe and C. P. Smyth, J. Am. Chem. Soc., 1950, 72, 1098–1106 CrossRef CAS.
- H. Kuttenreich, H.-J. Hinz, M. I. Marcsek, R. Koynova, B. Tenchov and P. Laggner, Chem. Phys. Lipids, 1988, 47, 245–260 CrossRef CAS.
- J. Swadesh and H. Fischer, Mol. Cryst. Liq. Cryst., 1996, 275, 175–181 CrossRef CAS.
- Y. Wang, J. Li, T. Hu, M. Dong, J. Wu, X. Miao and W. Deng, J. Phys. Chem. C, 2019, 123, 27643–27650 CrossRef CAS.
- C.-Z. Kuo, L.-Y. Hsu, Y.-S. Chen, K. Goto, S. Maity, Y.-H. Liu, S.-M. Peng, K. V. Kong, T. Shinmyozu and J.-S. Yang, Chem. - Eur. J., 2020, 26, 11511–11521 CrossRef CAS PubMed.
- A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, J. Phys. Chem. C, 2021, 125, 21595–21606 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 21, 3885–3896 RSC.
- R. Taylor and O. Kennard, J. Am. Chem. Soc., 1982, 104, 5063–5070 CrossRef CAS.
- Z. B. Yellin and L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 159–165 CrossRef.
- V. R. Thalladi, A. Gehrke and R. Boese, New J. Chem., 2000, 24, 463–470 RSC.
- A. L. Webber, J. R. Yates, M. Zilka, S. Sturniolo, A.-C. Uldry, E. K. Corlett, C. J. Pickard, M. P. Torralba, M. A. Garcia, D. S. Maria, R. M. Claramunt and S. P. Brown, J. Phys. Chem. A, 2020, 124, 560–572 CrossRef CAS PubMed.
- H. Suezawa, T. Yoshida, M. Hirota, H. Takahashi, Y. Umezawa, K. Honda, S. Tsuboyama and M. Nishio, J. Chem. Soc., Perkin Trans. 2, 2001, 11, 2053–2058 RSC.
- D. Sredojević, G. A. Bogdanović, Z. D. Tomić and S. D. Zarić, CrystEngComm, 2007, 9, 793–798 RSC.
- J. Harada and K. Ogawa, Chem. Soc. Rev., 2009, 38, 2244–2252 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2130396. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00942k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yukie Hibi,
Kazuma Matsumoto,
Masato Kawai and
Kazuo Miyamura*
*,
Yukie Hibi,
Kazuma Matsumoto,
Masato Kawai and
Kazuo Miyamura*