
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective adsorption of butenes over butanes on isoreticular Ni-IRMOF-74-I and Ni-IRMOF-74-II†
Jay Thakkar,
Winters Kexi Guo,
Michael J. Janik and
Xueyi Zhang*
and
Xueyi Zhang*
Department of Chemical Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: xuz32@psu.edu
First published on 19th July 2022
Abstract
The separation of butanes and butenes using MOF-74 (with two reticular MOFs with different pore sizes, Ni-IRMOF-74-I and Ni-IRMOF-74-II) was evaluated computationally using density functional theory. We identified that C4 alkene versus alkane selectivity stems from π–d chemical interactions, whereas selectivity differences among butenes stem from steric implications.
IRMOF-74-I (or MOF-74) is a highly porous, crystalline metal–organic framework (MOF) molecular sieve with honeycomb topology and one-dimensional pore channels (∼1.1 nm).1 The structure is comprised of divalent metal atoms (e.g. Ni2+, Cu2+, and Co2+), which are coordinated by 2,5-dihyroxyterephthalic acid linkers and homogeneously distributed across the three-dimensional framework (Fig. 1a).1 These metal atoms are accessible via pore channels and are connected to each other by sharing a corner oxygen atom.1 Due to these unique structural features, IRMOF-74-I provides a high concentration of identical and spatially isolated metal atoms, exhibiting long range order. Each metal atom has one axial site occupied by a solvent molecule during synthesis.1 Removal of the axially coordinated solvent molecule by post synthetic vacuum and/or heat treatment reveals coordinatively unsaturated metal atoms that are Lewis acid sites (Fig. 1b). These metal atoms function as binding sites and have thus been utilized for reversible adsorption and separation of hydrocarbon mixtures containing gas molecules such as C2H6, C2H4, C2H2, C3H8, and C3H6.2–9
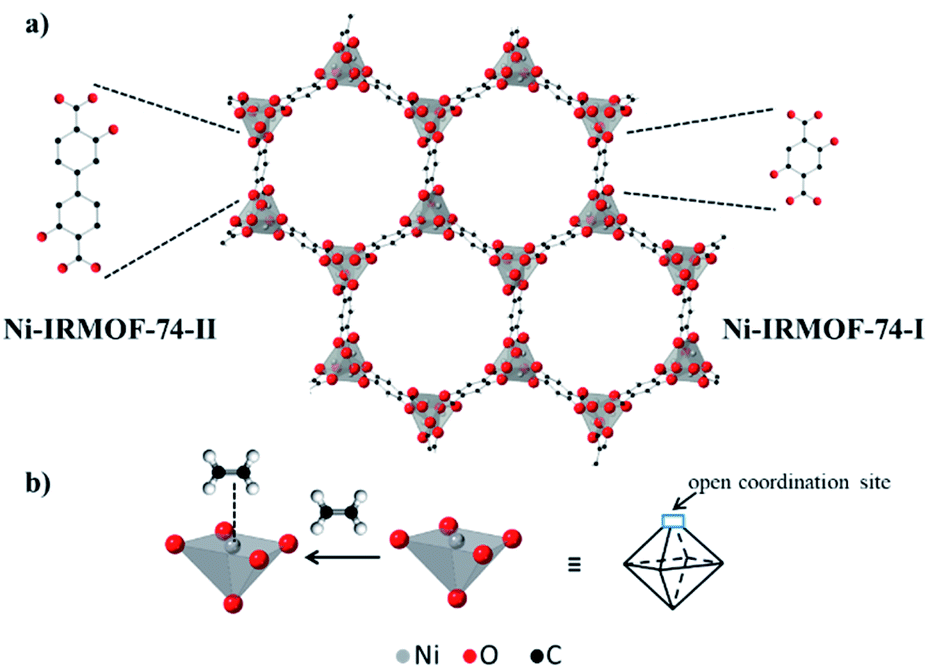 | ||
| Fig. 1 (a) Schematic structure of two isoreticular Ni-MOF-74 structures – smaller pore, Ni-IRMOF-74-I and bigger pore (larger linker), Ni-IRMOF-74-II and (b) open metal site in the structure, and its binding to an ethylene molecule. The framework structure is taken from ref. 1. | ||
Separation of C4 isomers (1-butene, trans-2-butene, cis-2-butene, i-butene, 1,3-butadiene, n-butane, and i-butane) from one another by traditional processes is challenging.10–14 These isomers are important precursors in the synthesis of many commodity chemicals and a high purity isomer is generally desirable for process simplification.10–12 Although IRMOF-74-I and other MOFs with open metal sites have been utilized for the separation of smaller hydrocarbon molecules, separation of C4 isomers is scarce and to the best of our knowledge only a couple of studies focus on utilizing IRMOF-74-I.13,14
Barnett et al. experimentally studied the effect of variation in electronic properties of the metal binding site on the separation selectivity of 1-butene over cis- and trans-2-butene.13 A charge dense cation (e.g. Ni2+) was found to favour 1-butene selectivity whereas a metal cation with comparatively lower charge density (e.g. Mn2+) favoured cis-2-butene selectivity.13 Furthermore, less adsorption selectivity towards trans-2-butene was attributed to the steric hinderance of this isomer to the metal site, whereas the increase in adsorption capacity for 1-butene compared to cis- and trans-2-butene on increasing coverage was ascribed to stabilizing effects from stronger gas–gas interactions.13 Similar results were obtained by Kim et al. in their density functional theory (DFT) work considering the adsorption of C4 alkenes on Fe-IRMOF-74-I.14 The order of DFT-calculated binding strengths was, 1-butene > isobutene > cis-2-butene > trans-2-butene, the weaker binding for cis- and trans-2-butene was due to the increased steric hinderance experienced by these isomers on metal sites.14 These studies, however, did not address the interactions between IRMOF-74 and C4 alkanes. Furthermore, no MOF design attempts were made to vary steric hinderance effects by modulating pore sizes. Additionally, Hubbard U corrections were not considered by Kim et al. while calculating the binding energies or performing energy decomposition analysis.14–18 Hubbard U corrections are shown to improve the description of localized transition metal d states, including improved agreement with experimentally observed high spin states.15,16,18
The work described in this paper builds on the computational results obtained by Barnett et al. and Kim et al. respectively.13,14 We utilize smaller pore-sized Ni-IRMOF-74-I and its isoreticular structure, a larger pore Ni-IRMOF-74-II, to computationally understand the effect of pore size variation on the binding of C4 isomers (1-butene, trans-2-butene, cis-2-butene, i-butene, 1,3-butadiene, n-butane, and i-butane) and their separation.19 The Ni form of IRMOF-74 was chosen due to the high Ni charge density, higher 1-butene adsorption capacity, and its possible role in separating C4 isomers as observed in literature.13,14 DFT methods were used to calculate the binding energies and to decompose these binding energies into constituent components. The data obtained in this paper provides a connection between structure and the adsorption and separation of these C4 isomers on MOF-74.
Periodic DFT calculations of C4 adsorption within the NI-IRMOF-74 structure were performed using the Vienna Ab initio Simulation Package (VASP). Spin polarized electronic structure calculations were performed using the PBE exchange–correlation functional including the vdW-DF3 dispersion corrections, and Hubbard U corrections on Ni d states. Further details of the electronic structure methods and energy decomposition approach are given in the ESI.†
The adsorbed geometries and the computed binding energies for the adsorption of C4 isomers on open Ni metal sites at complete coverage (i.e., 1 mol gas per mol Ni) for both MOFs are presented in Fig. 2 and Table 1. The order of binding energy (and in turn the binding strength and or affinity) of C4 isomers for Ni-IRMOF-74-I is, 1-butene (strongest binding) > cis-2-butene > i-butene > 1,3-butadiene > trans-2-butene > n-butane > i-butane. For the larger pore Ni-IRMOF-74-II, the order changes to, i-butene ≈ 1-butene > 1,3-butadiene > trans-2-butene > cis-2-butene > i-butane ≈ n-butane. These trends in binding energies for the adsorption of C4 isomers are also roughly translated in the metal (Ni) to isomer distances (Fig. 2), where generally the isomer with the stronger interaction is located closer to binding site. Additionally, based on the adsorbed geometries of the C4 molecules, the closest atoms to the Ni site for alkenes are the double bonded carbons, whereas for alkanes, the closest adsorbate atom to Ni is hydrogen. Similar geometrical observations have been reported for smaller hydrocarbon alkane and alkene molecules.20 Binding via the C–C double bond is due to the higher electron density of double bond and the resulting π-complexation with the open metal site, generating stronger interactions with alkenes as is reported in Table 1.20 The weaker interactions of the alkanes stem from the weak electron donation from the σ orbital of the C–H bond to the unsaturated metal site.20 Alkenes, irrespective of the pore size, exhibit stronger binding energies compared to alkanes.
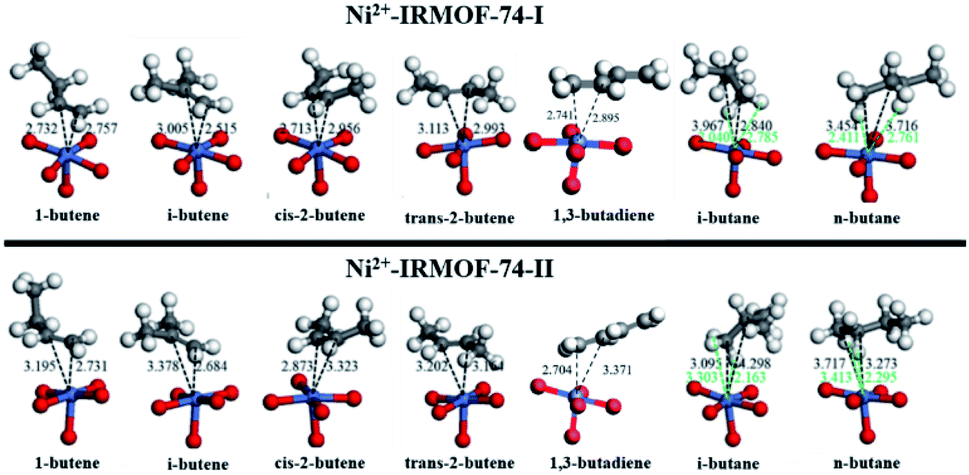 | ||
| Fig. 2 Adsorbed geometries of C4 isomers on Ni-IRMOF-74-I (top) and Ni-IRMOF-74-II (bottom). Black dotted lines represent Ni–C distances, while green dotted lines represent Ni–H distances. Distances are labelled in Angstrom. | ||
| C4 isomer | Binding energy (kJ mol−1) | |
|---|---|---|
| Ni-IRMOF-74-I | Ni-IRMOF-74-II | |
| 1-Butene | −79.53 | −66.45 |
| i-Butene | −76.72 | −66.77 |
| cis-2-Butene | −78.10 | −53.73 |
| trans-2-Butene | −67.99 | −60.61 |
| 1,3-Butadiene | −74.13 | −64.65 |
| i-Butane | −51.60 | −48.41 |
| n-Butane | −56.08 | −48.14 |
In order to further investigate, analyse and compare the binding energies of each individual C4 isomer, energy decomposition analysis similar to that in Kim et al., was performed and is depicted in Fig. 3.14 Binding and consequently the adsorption capacity of a specific molecule is a combination of the stabilizing molecule–molecule interactions, dispersion interactions and the binding affinity towards the Lewis acid metal sites. To facilitate these attractive interactions, destabilizing MOF framework and C4 molecule geometry distortions occur, as well as steric repulsions induced during adsorption. The adsorption of C4 isomers on Ni sites generate significant MOF framework distortions, with alkenes causing positive distortion energies of greater than 30 kJ mol−1 whereas alkanes cause significantly less MOF distortion. Non-dispersion interactions among C4 adsorbates in the adsorbed structure are attractive in the Ni-IRMOF-74-I small pore size structure, contributing −10 to −15 kJ mol−1 to the binding energy. The dispersion stabilization, including both gas–gas interactions and gas–MOF interactions, contributes significantly to the overall binding energy, however, are comparable for both alkanes and alkenes and irrespective of the MOF pore size. Dispersion interactions, therefore, do not contribute significantly to dictating the selectivity for adsorption among the various C4 species. For example, in Ni-IRMOF-74-I, the dispersions interactions are −53.06 and −47.39 kJ mol−1 for 1-butene and n-butane, respectively. However, the large difference in framework distortion (49.18 and 8.23 kJ mol−1 for 1-butene and n-butane) and the bond and steric interactions (−62.46 kJ mol−1 and −0.46 kJ mol−1 for 1-butene and n-butane) mainly dictate the difference in binding energy between the alkenes and alkanes. The combination of bond and steric interactions is near zero for the alkanes, while contributing significantly to the binding of alkenes due to π–Ni complexation. Alkane binding is due only to C4–C4 interactions and dispersion terms. Comparing to Kim et al.,14 the bond and steric interactions between C4 and Ni-IRMOF-74-I are lower than those for C4 and Fe-MOF-74, which contributed to the main difference in binding energies between these two MOFs.
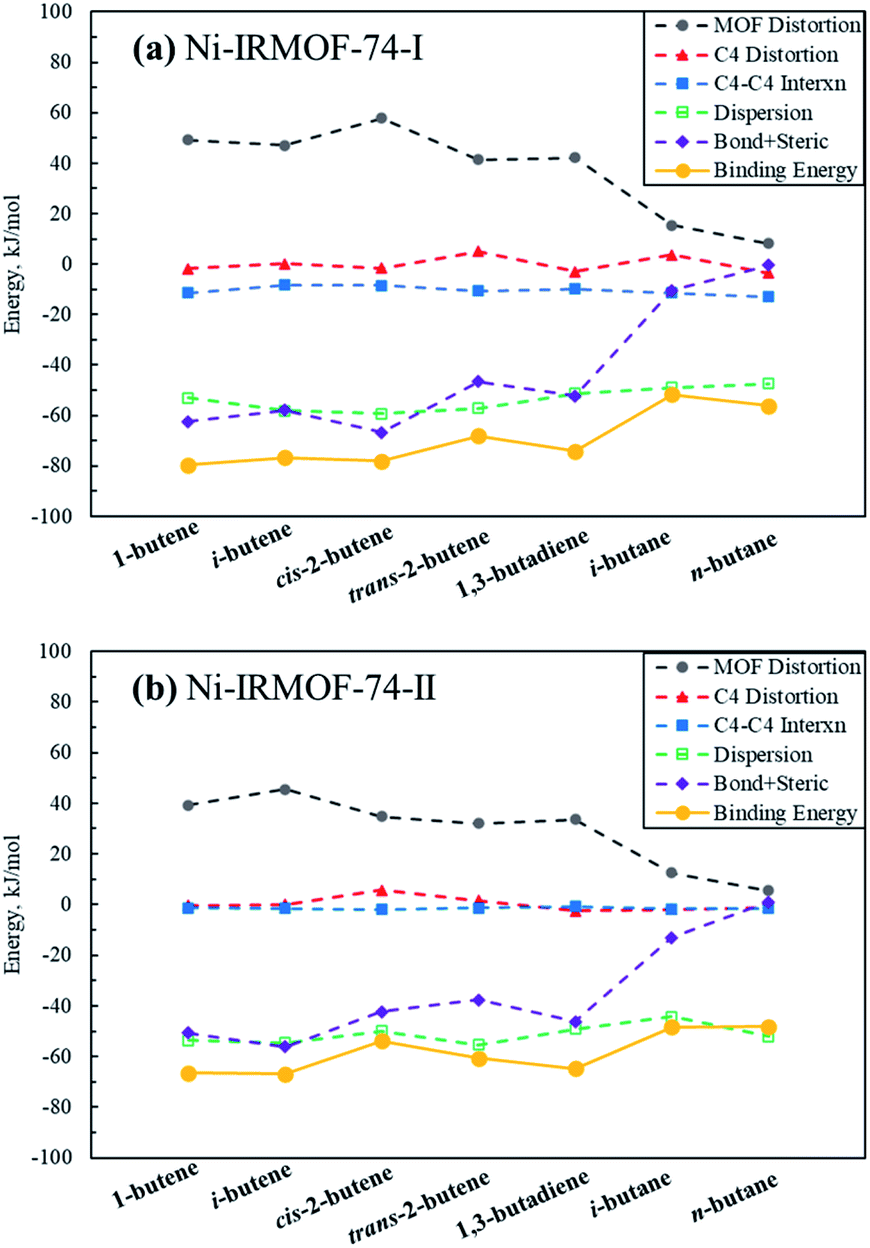 | ||
| Fig. 3 Decomposition of C4 binding energies to the Ni-IRMOF-74 structures. The individual contributing terms to the binding energies are defined in eqn (2)–(6) in the ESI,† and sum to the total binding energy (also included in the figure). | ||
Fig. 3a and b show the binding energy decomposition differences between the different linker sizes. The reduced Lewis acidity of the Ni metal sites in Ni-IRMOF-74-II, due to the addition of an extra benzene ring in the linker, imparts weaker interactions with the adsorbing C4 alkenes, thus weaker binding energies compared to Ni-IRMOF-74-I. This weaker binding is reflected in longer Ni–C distances for adsorbed alkenes in the larger pore MOF. This weaker interaction leads to less framework distortion energy in Ni-IRMOF-74-II as well as weaker bond + steric interactions. The greater available volume about the Ni sites in the larger pore MOF do not directly alter the binding interaction, as the C4 distortion energies are small in both MOFs and do not impact binding energy trends, and the C4-MOF dispersion interactions are equivalent between the two MOF structures. Instead, the electronic differences associated with the reduced Lewis acidity of the Ni-IRMOF-74-II sites mainly dictate the weaker binding in the larger pore MOF. Additionally, C4–C4 gas molecule interactions contribute somewhat to promoting adsorption in the small pore structure, whereas the greater distance between Ni binding sites in the Ni-IRMOF-74-II structure makes C4–C4 interactions negligible. The distances of closest Ni binding sites are 6.835 Å and 8.139 Å for Ni-IRMOF-74-I while the distances increase to 6.848 Å and 11.118 Å for Ni-IRMOF-74-II.
Comparing the factors that dictate the relative binding of butene isomers is not straightforward, as the binding energies are comparable, and the decomposition does not provide reliable nor straightforward explanations for the differences. There is some difference in the binding energies of 1-butene and i-butene from cis-2- and trans-2-butene in Ni-IRMOF-74-II, suggesting that the separation of these isomers might be possible using the larger pore Ni-IRMOF-74-II.
Conclusions
This study computationally investigated the adsorption of C4 alkanes and alkenes on IRMOF-74-I and its isoreticular IRMOF-74-II with larger pores. This study set up computational models for hydrocarbon adsorption on IRMOF-74 series, which also detailed different types of interactions contributing to the adsorption. We applied periodic DFT to reliably represent the extended MOF structure, and provided a detailed binding energy decomposition analysis to explain the mechanistic source of binding energy differences between alkenes and alkanes and with the variation in MOF pore size. The results indicate that C4 isomers can be separated in both IRMOF-74. C4–C4 interactions in IRMOF-74-I are stronger. Although the adsorption strength of C4 hydrocarbons on IRMOF-74-II is weaker, there is still obvious difference in adsorption energies, especially among alkenes.Author contributions
Jay Thakkar: writing – original draft, methodology, investigation; Winters Kexi Guo: investigation; Michael J. Janik: writing – review & editing, supervision, resources; Xueyi Zhang: writing – review & editing, supervision, conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Penn State Department of Chemical Engineering. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575.Notes and references
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellavag, Chem. Commun., 2006, 959 RSC.
- J. Li, R. J. Kuppler and H. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606 CrossRef CAS PubMed.
- Z. Bao, S. Alnemrat, L. Yu, I. Vasiliev, Q. Ren, X. Lu and S. Deng, Langmuir, 2011, 27, 13554 CrossRef CAS PubMed.
- Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107 RSC.
- Z. Bao, G. Chang, H. Xing, R. Krishna, Q. Ren and B. Chen, Energy Environ. Sci., 2016, 9, 3612 RSC.
- H. Liu, Y. He, J. Jiao, D. Bai, D. L. Chen, R. Krishna and B. Chen, Chem.–Eur. J., 2016, 22, 14988 CrossRef CAS PubMed.
- D. Bai, Y. Wang, M. He, X. Gao and Y. He, Inorg. Chem. Front., 2018, 5, 2227 RSC.
- J. Jiao, H. Liu, D. Bai and Y. He, Inorg. Chem., 2016, 55, 3974 CrossRef CAS PubMed.
- M. Gehre, Z. Guo, G. Rothenberg and S. Tanase, ChemSusChem, 2017, 10, 3947 CrossRef CAS PubMed.
- P.-A. Breuil, L. Magna and H. Olivier-Bourbigou, Catal. Lett., 2015, 145, 173 CrossRef CAS.
- A. Forestiere, H. Olivier-Bourbigou and L. Saussine, Oil Gas Sci. Technol., 2009, 64, 649 CrossRef CAS.
- B. R. Barnett, S. T. Parker, M. V. Paley, M. I. Gonzalez, N. M. Biggins, J. Oktawiec and J. R. Long, J. Am. Chem. Soc., 2019, 141, 18325 CrossRef CAS PubMed.
- H. Kim and Y. Jung, J. Phys. Chem. Lett., 2014, 5, 440 CrossRef CAS PubMed.
- L. Wang, T. Maxisch and C. Gerbrand, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- G. W. Mann, K. Lee, M. Cococcioni, B. Smit and J. B. Neaton, J. Chem. Phys., 2016, 144, 174104 CrossRef PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- K. Lee, J. D. Howe, L. Lin, B. Smit and J. B. Neaton, Chem. Mater., 2015, 27, 668 CrossRef CAS.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed.
- P. Verma, X. Xu and D. G. Truhlar, J. Phys. Chem. C, 2013, 117, 12648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00817c |
| This journal is © The Royal Society of Chemistry 2022 |