DOI:
10.1039/D2RA00768A
(Paper)
RSC Adv., 2022,
12, 7835-7849
Efficacy of surface-functionalized Mg1−xCoxFe2O4 (0 ≤ x ≤ 1; Δx = 0.1) for hyperthermia and in vivo MR imaging as a contrast agent†
Received
5th February 2022
, Accepted 10th February 2022
First published on 9th March 2022
Abstract
Surface-functionalized Mg1−xCoxFe2O4 (0 ≤ x ≤ 1; Δx = 0.1) can be an exciting candidate as an MRI contrast agent and for thermotherapeutic applications. The figure-of-merit, T2, relaxivity, r2, of MRI and specific loss power, SLP, of hyperthermia depend on the structural and magnetic properties of the nanoparticles. We synthesized cobalt-substituted magnesium ferrite Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles using a chemical co-precipitation method. The lattice parameter and average crystallite size increase with the increase in cobalt content. The force-constant of FTIR of the tetrahedral sites increases, and that of the octahedral sites decreases with an increase in cobalt content. The room temperature Mössbauer spectra of Mg1−xCoxFe2O4 show that the Mössbauer absorption area of the A site decreases, and the Mössbauer absorption area of the B site increases with x. The Mössbauer spectra and M–H hysteresis loops at room temperature confirmed that a transition from fast relaxation (superparamagnetic) to mixed slow/fast (superparamagnetic/ferrimagnetic) relaxation occurs with changing cobalt content. The cobalt ion tends to occupy the octahedral B site, which makes the A–B interaction stronger; therefore, we see the above transition. Cytotoxicity experiments on HeLa cells revealed that both chitosan and chitosan-coated magnesium cobalt ferrite nanoparticles are biocompatible. In the Mg1−xCoxFe2O4 series, both r2 and SLP increase with x because of the increase in magnetization and anisotropy.
Introduction
Multifunctional superparamagnetic spinel ferrite nanoparticles, smaller than many biological objects in size, are suitable for numerous applications such as MRI contrast dyes, tissue repair, immunoassays, detoxification of body fluids, hyperthermia, drug delivery, and cell separation.1–6 To date, these nanoparticles are of great importance for molecular-level detection and therapy. There is ample information on the effects of size, shape, and homogeneity of nanoparticles for colloidal suspensions. Amplifying the magnetic and physical properties relies on the composition of the nanoparticles in the first place. For most practical purposes, the size of the nanoparticles is in the range of <20 nm,3,7 which causes the nanoparticles to have a large surface area, and they are highly surface-active due to their smaller size. This phenomenon provides massive scope for surface modification to achieve targeted delivery and biocompatibility. Coating materials that modify the surfaces of nanoparticles can be organic polymers (dextran, chitosan, polyethylene glycol, polysorbate, polyaniline, etc.), organic surfactants (sodium oleate, dodecylamine, etc.), inorganic materials (gold, silica, carbon, etc.), and bioactive molecules (liposomes, peptides, ligands, etc.).8–11 We investigated samples coated with biocompatible chitosan and PEG to reduce their toxic effects in our previous studies.12–14 Surface modifications by coating and control of the size, shape, pH, surface charge, and hydrodynamic diameter allow an efficient method of controlling the significant parameters necessary for biomedical applications. Particularly for biomagnetic applications, the requirement of higher magnetic moments at smaller particle sizes with uniform physical and chemical properties is crucial.
Magnetic fluid hyperthermia (MFH) has drawn considerable attention in cancer treatment with radio frequency (rf) magnetic fields for localized heating mediated by magnetic nanoparticles. Regarding the magnetic fluid-based localized hyperthermia, the temperature of the tumor rises to 42–46 °C with an application of an rf magnetic field for 30–60 minutes. The heat thus generated causes the damage or weakening of the cancerous tissue. Blood vessels inside the tumor and cancerous tissues are porous due to the disordered formation. It helps to gather nanoparticles inside the malignant tissue, which results in localized heating in the malignant cells. Thus, it causes damage to localized or deeply-seated tumors. Magnetic fluid hyperthermia is advantageous compared to another magnetic thermotherapy such as thermoablation. In the thermoablation process, the temperature of the cancerous cells goes up to 56 °C for a short period. High temperature in thermoablation may cause widespread necrosis, coagulation, and carbonization of the cancerous tissue.15–17
MgFe2O4 nanoparticles are composed of bio-compatible Mg and Fe, which are nontoxic atoms. Inadvertently, magnesium ferrite shows low magnetic moment, zero coercivity, and zero retentivity because nonmagnetic Mg2+ has the tendency to occupy B sites17,18 and therefore, the withdrawal of the magnetic field would demagnetize the nanoparticles faster, which is plausible for biomedical applications. Further, the average daily intake of cobalt is 5 to 60 mg per day19 and for Mg, it is 420 mg per day.20 Therefore, reference daily intake magnesium is about 104 times higher than cobalt. On the other hand, CoFe2O4 nanoparticles have a high magnetic moment and anisotropy, which generates more heat in an alternating magnetic field, which makes them suitable for localized hyperthermia.21–23 The substitution of magnesium by cobalt enhances specific loss power through Néel's and Brownian relaxation, and the hysteresis loss of nanoparticles. We replaced magnesium with cobalt in the entire spec-trum of Mg1−xCoxFe2O4 with Δx = 0.1 to obtain the compositions, which exploits the positive attributes of both MgFe2O4 and CoFe2O4 to deliver higher efficacy and biocompatibility for local magnetic hyperthermia treatment and MRI contrast dye.
Magnetic resonance imaging (MRI) is the most influential diagnostic tool for imaging the brain and central nervous system and the soft tissue of humans, evaluating the cardiac function, and identifying tumors. Various magnetic nanoparticles as MRI contrast agents due to their novel properties, such as small size, large surface area, high magnetic moment, and zero or negligible coercivity, are in use. MRI contrast enhancement occurs due to interaction between the contrast agent and the adjacent water protons influenced by the particle size, magnetic moment, anisotropy constant. Magnetic nanoparticles with high crystallinity are potential candidates as T2 MRI contrast agents.24–30 Superparamagnetic gadolinium-based nanoparticles are in frequent use as contrast agents in hospital MRI, which has the disadvantage of weakening the kidneys. For many applications, especially for the detection of malignant tissues, MRI becomes irrelevant without a contrast agent. In spite of plentiful research on ferrite nanoparticles as MRI contrast agents, finding a suitable composition that is biocompatible still remains an open question. Therefore, another purpose of this study is to observe the ramifications of Mg1−xCoxFe2O4 as an MRI contrast-enhancing dye.
Materials and methods
Sample preparation
We synthesized a series of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) by wet chemical coprecipitation method using NaOH the coprecipitating agent. Analytical grade of Mg(NO3)2·6H2O, CoCl2·6H2O, and FeCl3 were mixed in the required molar ratio and 8 M of NaOH solution was added dropwise under continuous stirring using a magnetic stirrer at a speed of 400 rpm. Extra NaOH was added dropwise to maintain the pH of the solution to a value of 11–13. The mixture was heated to 353 K for one hour to complete the reaction and was retained at room temperature. The excess NaOH was washed out from the mixture by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm ten times. The AgNO3 test confirmed the removal of NaOH. Then, the particles were dried at 343 K for 72 h to complete the ferritization reaction. Finally, we ground the dried nanoparticles in an agate mortar–pestle for further functionalization.
000 rpm ten times. The AgNO3 test confirmed the removal of NaOH. Then, the particles were dried at 343 K for 72 h to complete the ferritization reaction. Finally, we ground the dried nanoparticles in an agate mortar–pestle for further functionalization.
Coating
We prepared 2% chitosan solution by adding 2 g chitosan in 100 mL de-ionized water under continuous stirring at 400 rpm using a magnetic stirrer for 72 hours. A required amount of acetic acid was added to the solution to dissolve chitosan in water. Then, the solution was centrifuged two times at 13000 rpm to remove any undissolved chitosan. We added 20 mg of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) particles to 1 mL of 2% chitosan solution, followed by vortex and sonication several times to get 20 mg mL−1 chitosan-coated Mg–Co nanoparticles as the stock solution. Finally, we prepared other concentrations by diluting the stock solution.
Characterization
We conducted the structural characterization of the as-synthesized Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles by an X-ray diffractometer (XRD), (Model: PW 3040-X'Pert PRO PANalytical, Philips, Netherlands) at 40 kV and 30 mA. The range of 2θ angle was 15–70° using CuKα radiation (λ = 1.54059 A0) with a scan step size of 0.0167°. We carried out transmission electron microscopy studies using a TALOS 200X, Thermofisher, USA, with an operating voltage of 200 kV. For TEM analysis, we dispersed the samples in ethanol and dropcast them on an electron-transparent carbon-coated Cu grid, followed by drying. The bare samples were in the solid state and the coated samples were in the liquid state. Mössbauer spectroscopy was performed using a model: 302 SEE Co., USA, gamma ray resonant spectrometer having a transmission geometry and constant acceleration mode with a transducer velocity of 11 mm s−1. Before starting the measurement, the spectrometer was calibrated using a metallic iron foil as the standard sample, and zero velocity was considered as the centroid of the Mössbauer spectrum. The Mössbauer data were acquired at room temperature, zero magnetic fields, and the acquisition time of 72 hours. The FTIR measurements were carried out by an FTIR spectrometer, model: L1600300 Spectrum PerkinElmer, United Kingdom with the attenuated total reflection (ATR) attachment. The sample in a small amount was placed on the diamond/ZnSe crystal of ATR using a pressure arm, which allows good contact of the sample with the crystal. The FTIR spectra of chitosan, bare, and chitosan-coated magnesium cobalt ferrite nanoparticles were acquired in the range of 350–3000 cm−1. The Raman Spectroscopy measurements were carried out using a CRS+ 500/BX53, MonoVista, S & I Spectroscopy & Imaging, Germany. A diode Laser System of 785 nm was used, which was internally mounted by 100 mW, and the Laser line cleaned up with edge filter width ≤60 cm−1. Raman spectra were acquired for pelletized samples in the range from 200 to 3500 cm−1. The samples' hydrodynamic size and zeta potential were investigated by a dynamic light scattering (DLS) instrument, model: ZEN 3600, Zetasizer, Malvern, U.K. The hydrodynamic size of the chitosan-coated nanoparticles was acquired at 25 °C, 37 °C, and 44 °C. The magnetizations at room temperature of all the samples were measured by the Physical Property Measurement (PPMS) System, model: 10307, Quantum Inc., USA. The M–H hysteresis loops of all the samples were measured from −5 to +5 Tesla in the bare and coated state.
Cytotoxic assay
We carried out the cytotoxic assay on the HeLa cell line by a Bio Safety Cabinet, Model: NU-400 E, Nuaire, U.S.A, CO2 Incubator (Nuaire, U.S.A), Hemocytometer, Optika, Italy, and an inverted light microscope. The media used was DMEM (Dulbecco's Modified Eagles' Medium) containing 1% penicillin–streptomycin (11), 0.2% gentamycin, and 10% fetal bovine serum (FBS). HeLa cells (4 × 105 per 200 μL) were seeded in 96-well plates and incubated for 24 hours in a CO2 incubator containing 37 °C temperature and 5% CO2. Then, they were filtered (using 0.45 μm syringe filter) 50 μL of uncoated and coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles with the concentration of 4 mg mL−1 were added in each well. Duplicate wells were used for each sample. After 48 hours of incubation, insoluble samples were washed out several times with media. The survival of the cells was investigated by cell counting using a hemocytometer and an inverted light microscope using the following equations.
Cell viability was studied manually by a hemocytometer as,
| |
 | (1) |
where, viable cells (per mL) = average viable cell count per square
× dilution factor of 10
4; average viable cells (count per square) = (total number of viable cells in 4 squares)/4; dilution factors = (volume of sample + volume of diluting liquid)/volume of samples.
Hyperthermia studies
We investigated the hyperthermia properties by a hyperthermia setup, model: EASY HEAT 5060LI, Ambrell, U.S.A. The hyperthermia setup consists of a sample coil of 8 turns that has a diameter of 4 cm. During the hyperthermia experiment, the current passing through the rf coil was 283 A, and the frequency was 343 kHz, which generated an rf magnetic field of 26 mT. The volume of the chitosan-coated solution was 600 μL, and the concentration was 0.5, 1, 2, and 4 mg mL−1. An Eppendorf tube containing the solution was placed inside the rf coil for induction heating for different time intervals at a 26 mT magnetic field. We measured the temperature using an optical fiber thermometer. The following equation yielded the specific loss power (SLP).| |
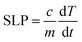 | (2) |
C is the heat capacity of the solution, and m is the relative mass of the magnetic nanoparticles. The slope of the linear range of temperature vs. time curves provided ΔT/Δt. The concentration of the magnetic nanoparticle is small, and therefore we considered the heat capacity of water to be 4.18 J g−1 K−1 for the sample.22,31
Magnetic resonance imaging
We acquired phantom images containing chitosan-coated nanoparticles. The T2 weighted fast spin-echo (FSE) MR images were acquired using the machine of model: MRS7017, MR solution, United Kingdom. During imaging, the repetition time (TR) was 4000 ms, slice thick-ness (THK) was 1.0 mm, and FOV was 40 × 40. We acquired T2 mapping with several echo times (TE) of 7, 14, 21, 28, 35, 42, 49, 56, 63, 70, 77, 84, 91, 98, 105, and 112 ms. Eleven phantoms were prepared by filling small Eppendorf tubes with five different concentrations of 0.04, 0.1, 0.2, 0.3, and 0.4 mM for each cobalt content x of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1). The phantoms of each x with five different concentrations were placed within a mouse body coil at a time. The mouse body coil was situated isocentrally with the static magnet B0 = 7 T within the homogeneous region. The B1 was the perturbation field with the Larmour frequency 2048 Hz generated by the rf coil.
In vivo magnetic resonance imaging
We procured the in vivo T2 weighted FSE MR images using Albino Wister male rats aged 12 to 13 weeks, and body weight 200–250 g. During the T2 W FSE experiment, the rats were made unconscious by administering intramuscular ketamine in a prescribed dose (15 mg kg−1).29 The unconscious rat reclined on a horizontal bed and its head laid inside a head coil generating rf magnetic field B1. The head coil was isocentric with a static magnetic field (7 T). Throughout the experiment, animals were kept warm with a heater, air supplied at a constant rate, and inspected with a cardiac monitor. At first, T2 W FSE images of the rat brain were acquired without a contrast agent with flip angle (FA) 90°, repetition time (TR) 4000 ms, echo time 45 ms, field of view (FOV) 40 × 40 mm, and image matrix 256 × 256. We inoculated 1 mL chitosan-coated Mg1−xCoxFe2O4 nanoparticles of 2 mg mL−1 concentrated through the tail vein without altering the position. We acquired T2 weighted Fast Spin Echo (FSE) images at intervals of 15, 30, 60, 90, and 120 minutes after inoculating the contrast agents Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1).
Animal handling
We handled the animals following The Guide for the Care and Use of Laboratory Animals (1996). We maintained the ARRIVE guidelines for reporting animal research. All necessary efforts were taken to minimize the sufferings of experimental animal according to the requirement of the Ethical Review Committee of Anima Experiments of Atomic Energy Centre Dhaka, which approved the protocol of with the Memo No: AECD/ROD/EC/21/211.
Results and discussion
Structural characterizations
Fig. 1(a and b) presents the X-ray diffraction (XRD) patterns of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) in the as-dried condition, which were acquired for the structural characterization of all compositions. There was no impurity peak in the XRD spectra within the resolution level of the XRD machine. The samples have a spinel structure31–33 with the space group of Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. Fig. 1(a) shows that the patterns of Mg-rich compositions up to x = 0.4 consist of broad diffused peaks. The peaks become narrower with the increase in Co. Fig. 1(a and b) shows that while the peaks become sharper, and their positions shifted toward lower values of 2θ. CoFe2O4 has a higher degree of crystallinity than MgFe2O4 when NaOH was used as the co-precipitating agent. Absolute lattice parameter was calculated using the Nelson–Riley function. We calculated the lattice parameter for each plane using Bragg's law, and absolute lattice parameters were determined using the Nelson–Riley function,
m. Fig. 1(a) shows that the patterns of Mg-rich compositions up to x = 0.4 consist of broad diffused peaks. The peaks become narrower with the increase in Co. Fig. 1(a and b) shows that while the peaks become sharper, and their positions shifted toward lower values of 2θ. CoFe2O4 has a higher degree of crystallinity than MgFe2O4 when NaOH was used as the co-precipitating agent. Absolute lattice parameter was calculated using the Nelson–Riley function. We calculated the lattice parameter for each plane using Bragg's law, and absolute lattice parameters were determined using the Nelson–Riley function, | |
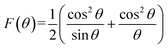 | (3) |
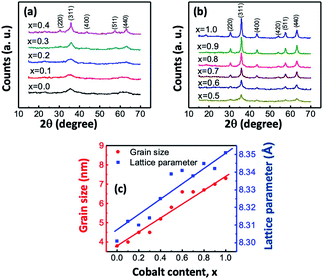 |
| | Fig. 1 (a) and (b) XRD traces of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) in the as-synthesized condition acquired in the range of 15–70° using CuKα radiation with a scan step size of 0.0167°. The peaks are indexed in the figures. (c) Variation of the lattice parameter and grain size with the cobalt content x in the Mg1−xCoxFe2O4 series. | |
In Fig. 1(c), the lattice parameter increases with the increase in the cobalt content x because the ionic radius of the Mg2+ (0.065 nm) ion is smaller than that of the Co2+ (0.072 nm) ion.34,35 Fig. 1(c) shows that the dependence of the lattice parameter on the composition follows a linear relationship, which follows Vegard's law. Abraham et al.36 synthesized spinel Mg1−xCoxFe2O4 (x = 0.0, 0.2, 0.4, 0.6, 0.8, and 1.0) nanocomposites by modified sol–gel combustion method and found that the lattice parameter increases with an increase in the cobalt content x and the value of the lattice constant is 8.303 Å for MgFe2O4, which is close to the values obtained in the current study. Stein et al.37 synthesized CoFe2O4 nanoparticles by chemical coprecipitation method, and its lattice parameter was 8.358 Å, which is close to the lattice parameters of CoFe2O4 of 8.351 Å in this study.
We calculated the grain size of each composition using the Debye–Scherrer's formula:
| |
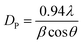 | (4) |
where,
DP is the average crystallite size,
β is the full width at half the maximum of the highest intensity (311) peak,
θ is the Bragg angle, and
λ is the X-ray wavelength. The peak width was estimated using the peak fit option of the X'Pert Highscore Software.
Fig. 1(c) presents the variations in the grain size with
x. The grain sizes of
x = 0 and 1 were 3.8 and 7.3 nm, respectively. The grain size varies almost linearly with
x. Lodhi
et al.32 also obtained that the particle size increases with an increase in the cobalt content,
x of Mg
0.5Co
xZn
0.5−xFe
2O
4 prepared by the micro emulsion method.
Transmission electron microscopy
Fig. 2 shows a representative TEM image of bare and chitosan-coated Mg0.1Co0.9Fe2O4 nanoparticles from the Mg1−xCoxFe2O4 series. The magnetic dipole interactions between ferrite nanoparticles cause the bare par-ticles to cluster.36 The dipole interactions are decreased and the particles are dispersed after coating with chitosan. The histogram of the size distribution follows a log-normal distribution (in the inset of the BF image). The average nanoparticle size in the BF image of Mg0.1Co0.9Fe2O4 was 7.4 nm, which fits reasonably well with the 7.2 nm reported by XRD. The selected area electron diffraction (SAED) patterns of bare and chitosan-coated Mg0.1Co0.9Fe2O4 nanoparticles are shown in Fig. 2. The SAED patterns show that the (311) is the highest intensity plane along with the other planes (220), (400), (420), (511), and (440) indexed in the figure. The d-values of the SAED pattern were determined using the Velox software, and the diffractograms were indexed accordingly. The SAED patterns are compatible with the noncrystalline structure reported in the literature.37–39 The Debye circles are more intense for uncoated than the coated nanoparticles because of the higher degree of dispersion in the coated particles in a region. Fig. 2 also presents the high-resolution transmission electron microscopy (HRTEM) images of bare and chitosan-coated particles. The lattice fringes of the HRTEM image of both bare and chitosan-coated particles in Fig. 2 indicate high crystallinity. The HRTEM image yielded a d value of 252 pm, which corresponds to the (311) plane. In Fig. S-1,† the HRTEM images of all Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) compositions in the as-synthesized condition are shown.
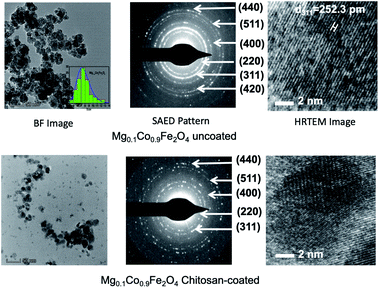 |
| | Fig. 2 TEM images of uncoated and chitosan-coated Mg0.1Co0.9Fe2O4 nanoparticles. In this figure representative TEM images of the Mg1−xCoxFe2O4 series are presented. In the upper panel, the BF image, SAED pattern, and HRTEM images for the uncoated nanoparticles are presented. In the bottom panel, the BF images, SAED patterns, and HRTEM images of chitosan-coated nanoparticles are presented. We see from the BF images in the figure that the uncoated nanoparticles are agglomerated and the coated nanoparticles are dispersed. The SAED patterns show that the lines are more intense for the uncoated samples, as compared to the coated samples since the coated nanoparticles are more dispersed. HRTEM images of the uncoated and coated nanoparticles demonstrate good crystallinity in the as-dried condition. | |
The HRTEM images show that all the nanoparticles are crystalline, with the degree of crystallinity increasing as the cobalt concentration x increases. Lattice fringes are more prominent in the HRTEM image with higher cobalt content. This is because of the higher degree of crystallinity with the increase of Co. The figure also indicates that the particle size increases with the increase of cobalt in Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) in the as-synthesized conditions.
Fig. 3 displays the STEM image of chitosan-coated Mg0.1Co0.9Fe2O4 nanoparticles using the HAADF detector, as well as electron diffraction spectroscopy (EDS) spectra and mapping of the constituent elements Fe, Mg, Co, and O. The mapping of the chitosan constituent atoms, such as carbon (C) and nitrogen (N), is also shown. The STEM-EDS mapping in the figure indicates that the ferrite nanoparticles are chemically homogeneous. The chitosan coating on ferrite nanoparticles is remarkable, as seen by the uniform distribution of C and N atoms. Chitosan is only applied to the nanoparticles as a coating, and the thickness of the coating is very thin. From the STEM EDS map ping, we found the atomic percent of Mg, Co, Fe, and O to be 4.11% (±0.86), 8.6%5 (±1.31), 12.86% (±1.94), and 74.39% (±5.36), and the mass percent of Mg, Co, Fe, and O to be 3.96% (±0.81), 20.25% (±) 2.87, 28.52 (±4.03), and 47.27% (±2.31), respectively. From the EDS spectrum of chitosan-coated Mg0.1Co0.9Fe2O4 particles, we see the peaks of C (0.28 keV), N (0.39 keV), O (0.53 keV), Fe-L (0.71 keV), Cu-L (0.79 keV), Co-L (0.93 keV), Mg (1.27 keV), Fe-ka (6.42 keV), Co-ka (6.96 keV), Co-kb (7.68 keV), Cu-ka (8.07 keV), and Cu-kb (8.91 keV). Because of the carbon-coated copper grid, the C and Cu peaks in the EDS spectrum have a high intensity. Also, there are no impurity peaks.
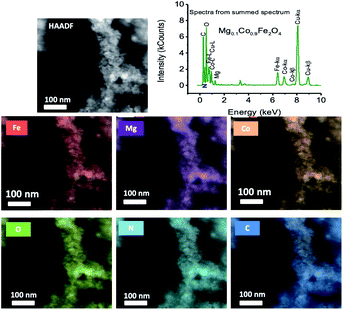 |
| | Fig. 3 Scanning transmission electron microscopy (STEM) image of chitosan-coated Mg0.1Co0.9Fe2O4 nanoparticles using the HAADF detector, along with the electron diffraction spectroscopy (EDS) mapping of the constituent elements Fe, Mg, Co and O. The mapping of the constituent atoms of chitosan such as carbon (C) and nitrogen (N) atoms are also presented. In the figure, the EDS spectrum of chitosan-coated Mg0.1Co0.9Fe2O4 particles is given. | |
Mössbauer spectroscopy
Fig. 4 presents the Mössbauer spectra at room temperature of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) in the dried condition. We used the WMOSS 4R program to analyze the spectrum. Table 1 presents the hyperfine parameters of isomer shift, quadruple splitting, and hyperfine magnetic field with x. We obtained the hyperfine parameters by model fitting the experimental data. The goodness of fit χ2 varies between 0.641 to 3.112, which is within acceptable limits.38,39 Manova et al.40 showed that the Fe3+ sub-spectrum with a larger isomer shift represents octahedral B-sites while the Fe3+ sub-spectrum with a lower isomer shift represents tetrahedral A-sites. The covalent bond of the A-sites is more potent than that of the B-sites because of the larger internuclear separation of ferric and oxygen ions at the B-site than the A-site, and the orbital overlap is smaller at the B-site than the A-site.41 However, the site occupancy cannot be determined accurately with zero field measurements for this series of spinel because of the short-range order of the ferric ions surrounded by the nonmagnetic ions. The area of doublets decreases, and that of the sextets increases. Lin et al.39 observed a decrease in the hyperfine field on both the A- and B-sites of CoAlxFe2−xO4 ferrite nanoparticles with an increase in the nonmagnetic Al3+ content. The spectra with lower cobalt content (0 ≤ x ≤ 0.3) were superparamagnetic with two distinct doublets. In these samples, nonmagnetic Mg2+ ions surround the Fe3+ ions, which are magnetically short-range ordered, which lowers the magnetic moment of these samples, and we observed fast relaxation.39 For spectra with higher cobalt content (0.4 ≤ x ≤ 1.0), model fitting was satisfactory considering one sextet and one doublet subspectra, which shows mixed relaxation. Quadruple splitting is higher for the doublet subspectra than for the sextets.42 For cobalt ferrite, the area of the subspectra representing slow relaxation of the ferrimagnetic contribution was the highest. The isomer shifts are in the range of 0.250–0.338, which matches well with the literature.43 The values of isomer shift reveal that iron exists only in the trivalent state (Fe3+), which belong to the range of 0.1–0.5 and there is no Fe2+ having isomer shift >0.5. The fact that the areas of the subspectra representing the sextet decrease with the decrease in cobalt (Table 1) because of the ferrimagnetic to superparamagnetic transition, which corroborates with the results obtained from the physical property measurement system (PPMS) presented later.40,41,44–46
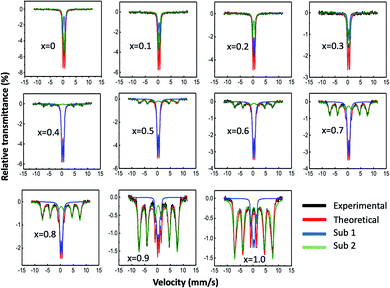 |
| | Fig. 4 Mössbauer spectra at room temperature of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) in the as-dried condition. The spectra were acquired at room temperature without any applied field. In the figure, the black lines represent the experimental data, and the red lines represent the model fitting. The blue and green lines represent the contributions of subspectra 1 and 2, respectively, determined by model fitting using the WMOSS 4R software. | |
Table 1 Hyperfine parameters derived from the Mössbauer spectroscopy acquired at room temperature and zero-field conditions
| x |
Position of Fe3+ |
IS (mm s−1) |
QS (mm s−1) |
Hyperfine magnetic field (kG) |
Area |
| 0 |
Doublet |
0.327 |
0.939 |
— |
0.395 |
| Doublet |
0.338 |
0.555 |
— |
0.597 |
| 0.1 |
Doublet |
0.337 |
0.531 |
— |
0.670 |
| Doublet |
0.338 |
0.903 |
— |
0.431 |
| 0.2 |
Doublet |
0.335 |
0.506 |
— |
0.473 |
| Doublet |
0.334 |
0.864 |
— |
0.480 |
| 0.3 |
Doublet |
0.323 |
0.532 |
— |
0.348 |
| Doublet |
0.320 |
0.871 |
— |
0.596 |
| 0.4 |
Doublet |
0.332 |
0.631 |
— |
0.651 |
| Sextet |
0.327 |
0.022 |
449 |
0.405 |
| 0.5 |
Doublet |
0.325 |
0.663 |
— |
0.862 |
| Sextet |
0.250 |
0.169 |
457 |
0.202 |
| 0.6 |
Doublet |
0.329 |
0.650 |
— |
0.652 |
| Sextet |
0.303 |
0.060 |
453 |
0.430 |
| 0.7 |
Doublet |
0.326 |
0.656 |
— |
0.453 |
| Sextet |
0.300 |
0.014 |
456 |
0.688 |
| 0.8 |
Doublet |
0.325 |
0.669 |
— |
0.418 |
| Sextet |
0.293 |
0.047 |
494 |
0.730 |
| 0.9 |
Doublet |
0.276 |
0.674 |
— |
0.207 |
| Sextet |
0.295 |
0.270 |
512 |
0.895 |
| 1.0 |
Doublet |
0.304 |
0.220 |
— |
0.055 |
| Sextet |
0.299 |
0.299 |
518 |
0.997 |
Fourier-transform infrared spectroscopy
Fig. 5(a) shows the representative FTIR spectrum of chitosan, bare, and chitosan-coated Co0.2Mg0.8Fe2O4 nanoparticles in the range of 350–3000 cm−1. A small amount of sample was placed on the diamond/ZnSe crystal of ATR using a pressure arm, which allows good contact of the sample with the crystal. The bare and chitosan samples were in the powder form and the coated samples were in the liquid form. The ESI, Fig. 2† represents the FTIR spectrum of all compositions of the series Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1).
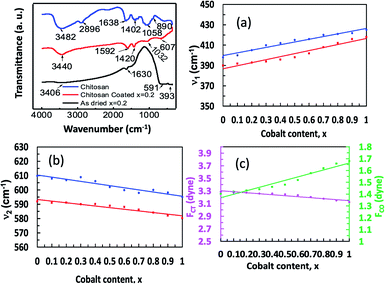 |
| | Fig. 5 FTIR spectrum of chitosan, bare nanoparticles and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles in the range of 350–3000 cm−1. The samples were placed on the diamond/ZnSe crystal of ATR using a pressure arm, which allows good contact between the sample and the crystal. The bare and chitosan samples were in the powder form and the coated samples were in the liquid form. (a) A representative FTIR spectrum of chitosan, bare and chitosan-coated Co0.2Mg0.8Fe2O4 nanoparticles in the range of 350–3000 cm−1. (b) Variations of lower frequency absorption band (ν1), (c) higher frequency absorption band (ν2) of the bare (red) and coated (blue) nanoparticles. (d) Variations of force constants FCT (magenta) and FCO (green) of tetrahedral and octahedral sites, respectively, with x. | |
We see in Fig. 5(a) and the ESI, Fig. 2† that the peaks of pure chitosan are at 890, 1402, 1638, 2896, and 3482 cm−1 which are the characteristic absorption bands of chitosan. The absorption peaks at 3482 cm−1 and 2896 cm−1 were present due to the stretching vibration of O–H and –CH2 pyranose rings, respectively. The presence of absorption peaks at 1638 cm−1 was due to the N–H bending vibration of the amino (–NH2) group. An absorption peak at 1402 cm−1 represents the C–H bending vibration of the alkali group. The absorption peaks at 1058 and 890 cm−1 represent the antisymmetric stretching vibration of the C–O–C bridges of the glucopyranose ring in the chitosan matrix.47,48 Two peaks of the bare nanoparticles at the lower frequency region are due to the characteristic absorption bands of the cubic spinel structure.32–35 A lower frequency absorption band (ν1) is observed in the range of 390 to 418 cm−1, and a higher frequency absorption band (ν2) is observed in the range of 580 to 592 cm−1. The lower frequency band (ν1) represents the stretching vibration of the metal-oxide bond at the octahedral site, while the higher frequency band (ν2) represents the stretching vibration of the metal-oxide bond at the tetrahedral site.
Fig. 5(b) and (c) show the variations of ν1 and ν2 of bare and coated nanoparticles with the cobalt content x. It is interesting to note that the variations of ν1 and ν2 with x are linear for both bare and coated nanoparticles. The standard mode of vibration of the tetrahedral site (ν2) is higher than that at the octahedral site (ν1) because of the shorter bond length of the tetrahedral site than the octahedral site.35,36 With the increase in x, the occupation of Co2+ ions at the octahedral sites increases, thus reducing the bond length at this site. The preference of Mg2+ ions is for the tetrahedral sites. Therefore, with the increase in x, the bond length at the octahedral sites reduces with a corresponding increase in the bond length at the tetrahedral sites for which ν1 increases and ν2 decreases with cobalt content x. For the coated Mg1−xCoxFe2O4 nanoparticles, however, the ν1 and ν2 show a shift of the absorption band toward the higher wave number compared to the as-dried nanoparticles due to the bonding of ferrite with chitosan.49 The force constant tells us the stiffness of the Fe3+ ions to the vibrational displacement considering other nuclei at their equilibrium position. Force constants are inversely related to the bond length of Fe3+ and the nearest neighbor ions. We obtained the force constants (FC) for the tetrahedral A site (FCT) and the octahedral B site (FCO) by the following relation.
In the above equation, c is the speed of light ∼2.99 × 1010 cm s−1, ν is the vibrational frequency of the ions at the A- and B-sites, m is the reduced mass for the Fe2+ and O2− ions, which is 2.061 × 10−23 g. Fig. 5(d) shows that the value of FCT is lower than FCO because of the higher orbital overlap at the tetrahedral site. However, FCT decreases and FCO increases with increasing cobalt content x, which indicates that the orbital overlap at the A-site decreases and that at the B-site increases with x, i.e., Co concentration46 because of the preferance of Co at the B-site.
Raman spectroscopy
We used Raman spectroscopy to further investigate the structure of the nanoparticles by identifying several vibrational modes.50–53 Fig. 6(a) shows the Raman spectra of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles in the range of 190–1000 cm−1 at ambient temperature. We saw earlier that Mg1−xCoxFe2O4 ferrites have a partially inverse spinel structure, which is associated with the space group (Fdm).51,54–57 The vibrational modes associated with this space group are A1g (R), Eg (R), F1g, 3F2g (R), 2A2u, 2Eu, 4F1u (IR), and 2 F2u. Notation R implies Raman active vibrational modes, IR symbolizes infrared-active vibrational modes, and the rest are silent modes. Therefore, five Raman active modes A1g, Eg, F2g(3), F2g(2), and F2g(1) are expected to be observed in the Raman spectra. Fig. 6(b) presents the discernible Raman modes of the studied samples. The A1g modes are associated with symmetric stretching of MeO4 (Me represents Co and/or Mg) and stretching of FeO4 at the A-site. The Eg modes represent bending of oxygen with respect to Fe at the B-site. The F2g(3) modes represent the antisymmetric bending of oxygen with respect to Fe, the F2g(2) modes represent the asymmetric stretching of Fe and O, and the F2g(1) modes represent the complete translational motion of Fe and O.55,58 The Raman peaks in the region of 660–720 cm−1 represent the vibrational modes of the A-site, and the Raman peaks in the region of 460–660 cm−1 represent the vibrational modes of the B-site of the ferrites. Table 2 presents the wavenumbers of five Raman active modes A1g, Eg, F2g(1), F2g(2), and F2g(3) of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) assigned to the Raman spectra. The wavenumber of the A1g vibrational modes lowers with an increase in the cobalt content x because the atomic mass of Co (58.9332 amu) ions is higher than that of Mg (24.3050 amu) ions. Mund et al.52 also found an increase in the vibrational mode (blue shift) when Mg is increased in magnesium cobalt ferrite prepared by the sol–gel auto-combustion method because of the lower atomic mass of Mg than Co.
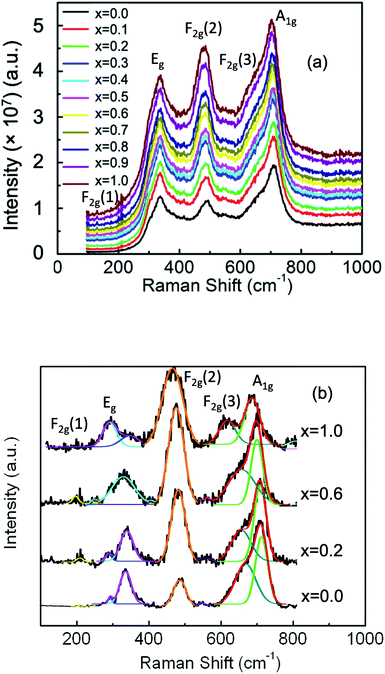 |
| | Fig. 6 (a) Room temperature Raman spectra of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles in the range of 190–1000 cm−1 using the pelletized solid samples. Five Raman active modes A1g, Eg, F2g(1), F2g(2), and F2g(3) are assigned in the Raman spectra. (b) Representative best fitting of the Raman spectra using the Gaussian function after background subtraction and deconvolution. | |
Table 2 Wavenumbers of the five Raman active modes A1g, Eg, F2g(1), F2g(2), and F2g(3) of Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) assigned to the Raman spectra
| x |
Main Raman mode peak energy (cm−1) |
| F2g(1) |
Eg |
F2g(2) |
F2g(3) |
A1g Me–O |
A1g Fe–O |
| 0.0 |
211 |
292 |
487 |
549 |
667 |
712 |
| 0.1 |
205 |
333 |
480 |
548 |
667 |
707 |
| 0.2 |
208 |
335 |
478 |
553 |
668 |
706 |
| 0.3 |
214 |
338 |
478 |
557 |
658 |
704 |
| 0.4 |
211 |
345 |
479 |
620 |
652 |
701 |
| 0.5 |
223 |
330 |
474 |
555 |
661 |
697 |
| 0.6 |
200 |
329 |
474 |
555 |
660 |
697 |
| 0.7 |
— |
292 |
478 |
566 |
655 |
695 |
| 0.8 |
— |
294 |
478 |
577 |
653 |
696 |
| 0.9 |
— |
320 |
471 |
— |
619 |
691 |
| 1.0 |
— |
300 |
472 |
— |
619 |
690 |
Fig. 7 presents the variations in the area integrals with the cobalt concentration x, which were obtained by the Gaussian fitting of A1g, Eg, F2g(3), F2g(2), and F2g(1) peaks assigned to the Raman spectra. After background subtractions and deconvolution, the Gaussian function produced the best fit between the experimental and theoretical data. Fig. 7 shows that for x = 0, the area integral for A1g, i.e., the peaks representing tetrahedral sites for both Me–O and Fe–O, is the highest, indicating that the cation occupancy at the A-site is the highest (where Me represents Mg and Co and Fe for iron). The area integral of A1g of both Me–O and Fe–O is depicted in Fig. 7, which shows that when x increases, the occupancy of Me and Fe ions decreases. The integral area of Eg, F2g(2) and F2g(3), on the other hand, increases with x, indicating that the occupancy of Me and Fe at the B-site increases with x, resulting in a higher degree of inverse spinel structure. We know that cobalt prefers to occupy the B-sites, while Mg prefers to occupy the A-sites. As the cobalt concentration x increases, the area integral of the peaks of A1g decreases, resulting in a rise of the lower modes, namely, Eg and F2g.
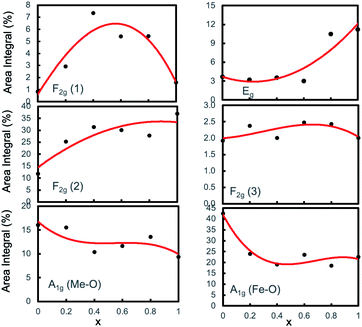 |
| | Fig. 7 Variation of area integral with Co concentration x of the A1g, Eg, F2g(1), F2g(2), and F2g(3) peaks assigned to the Raman spectra of the Mg1−xCoxFe2O4 nanoparticles obtained by Gaussian fitting and deconvolution. | |
Hydrodynamic diameter and zeta potential
We measured the hydrodynamic size distribution of chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles at 25 °C temperature for the concentration of 2 and 4 mg mL−1. Fig. 8(a) and S-3† show some representative data of the intensity distribution of the hydrodynamic sizes for x = 0.2, 0.5, 0.8, and 1.0. Dynamic light scattering (DLS) measures Brownian motion, which is the random movement of particles due to the interactions with the solvent molecules surrounding them, correlating to the particle size in the solution.59 It is noticeable from Fig. 8(a) that the curves are symmetric. The hydrodynamic size of the particles from the translational diffusion coefficient uses the Stokes–Einstein equation59| |
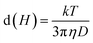 | (6) |
where d(H) is the hydrodynamic diameter, D is the translational diffusion coefficient, k is the Boltzmann's constant, T is the absolute temperature, and η is the viscosity of the solution. The hydrodynamic diameters of bare nanoparticles at 2 mg mL−1 were in the range from 2000 to 8000 nm. In the as-dried condition, agglomeration causes the formation of large particles in water, which results in sedimentation.59 The TEM images presented in Fig. 2 confirmed the agglomeration of the uncoated samples, whereas the chitosan-coated particles are highly dispersed. Fig. 8(b) presents the concentration dependence of the hydro-dynamic diameters of the chitosan-coated nanoparticles of Mg1−xCoxFe2O4 for different values of x. The hydrodynamic diameters of the nanoparticles in Fig. 8(b) increase with an increase in the concentration of the solution in water. Fig. 8(c) and S-3† present the temperature dependence of hydrodynamic diameters of coated nanoparticles for 2 and 4 mg mL−1, respectively. The hydrodynamic diameter decreases with an increase in the temperature for both the concentrations. The corresponding polydispersity indexes were in the range from 0.11 to 0.29 only, which are suitable for biomedical applications.
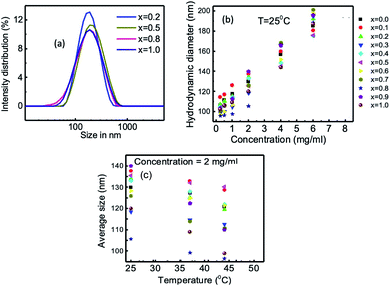 |
| | Fig. 8 Hydrodynamic size distribution of chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles measured at 25 °C for the concentrations of 2 and 4 mg mL−1. (a) Representative intensity distribution of hydrodynamic sizes of x = 0.2, 0.5, 0.8, and 1.0. (b) Variation of hydrodynamic size with concentration of the solution of chitosan-coated Mg1−xCoxFe2O4 nanoparticles. (c) Variation of average hydrodynamic size with temperature at the concentration of 2 mg mL−1 chitosan-coated Mg1−xCoxFe2O4 nanoparticles. | |
Fig. 9 and S-4† present the variation in the zeta potential of chitosan-coated Mg1−xCox Fe2 O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles with the pH of the solution. The zeta potential of the chitosan-coated Mg1−xCoxFe2O4 nanoparticles was greater than 30 mV at pH 2.5 to 5, which means that the nanoparticles are stable in this region. The isoelectric zeta potentials of the solution were from pH 9 to 10.5. The un-coated Mg1−xCoxFe2O4 particles with concentration 4 mg mL−1, which were from −2 mV to 6.45 mV, and their pH was in the range from 6.25 to 6.70, which also means that the particles are unstable in water. In Fig. 9, the zeta potential of the chitosan-coated Mg1−xCoxFe2O4 particles of concentration 4 mg mL−1 was in the range from 30 to 50 mV, and their pH was from 3.45 to 4.55. Zahraei et al.60 found the highest value of the zeta potential of chitosan-coated manganese zinc ferrite at pH 3. Arakha et al.61 measured the zeta potential of chitosan-coated iron oxide nanoparticles, resulting in a stable suspension, and that value is 36.3 mV. The zeta potentials of the coated particles of our samples were higher than 30 mV, which means that in this pH region, the solution is stable. Even after one year, no precipitation or agglomeration was noticeable in the vial of the solution, indicating that the samples were well coated and dispersed. Therefore, the electrostatic stability occurs in the region of pH 2.5 to 5.5. The isoelectric zeta potentials of the samples were in the pH range from 7 to 8.5. Zahraei et al.60 found that the isoelectric point of chitosan-coated manganese zinc ferrite is 8.5, which is supported by our data. For a higher pH, the zeta potential decreases slowly, and particles may settle down due to flocculation, aggregation, and coagulation. Zeta potential is related to the electrophoretic mobility by Henry equation,62 which is,
| |
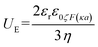 | (7) |
where
UE is electrophoretic mobility,
εr dielectric constant,
ε0 is the permittivity of vacuum,
ζ is zeta potential,
F (
κα) is Henry's function, and
η is the viscosity of the solution at the experimental temperature. Thus, zeta potential depends on the concentration of the particles, the viscosity, and the pH of the solution.
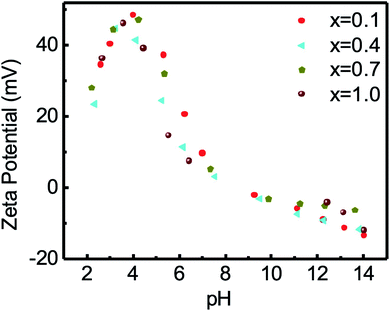 |
| | Fig. 9 The variation of zeta potential of chitosan-coated samples with the pH of the solution. The zeta potential of the chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) particles was greater than 30 at pH 2.5 to 5, which means that the particles are stable in that region. The isoelectric zeta potentials of the solution were at pH 9 to 10.5. In this figure, the representative data for x = 0.1, 0.4, 0.7, and 1.0 are shown. | |
Magnetic measurements
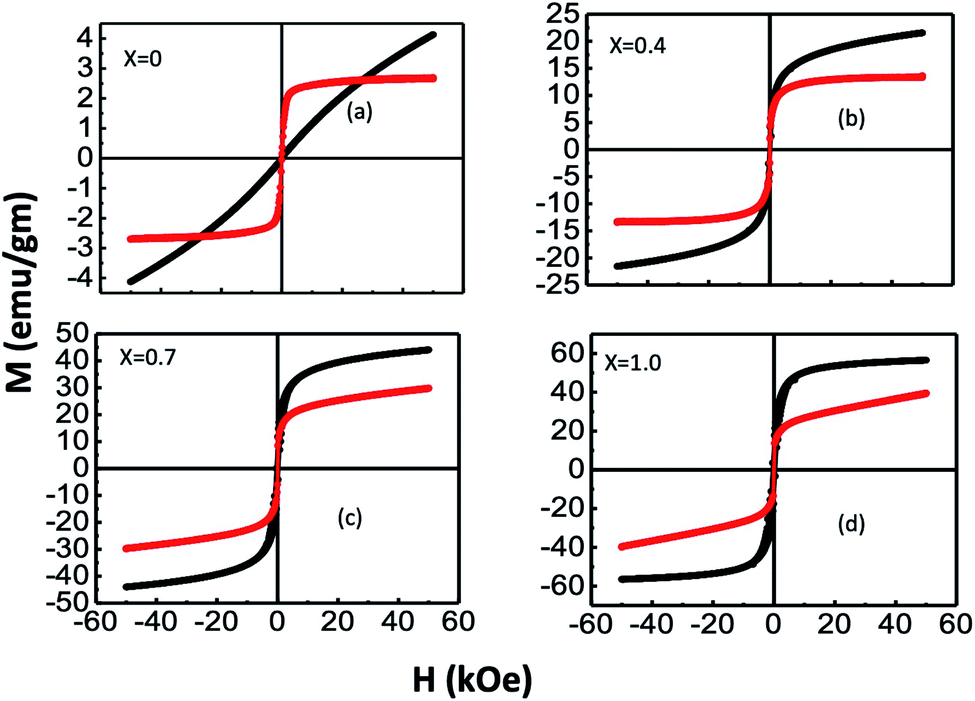
We measured the variations in the magnetization (M) with an applied field (H) for bare and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles with a maximum magnetic field of 5 Tesla. Fig. 10 presents the representative M–H curves of x = 0, 0.4, 0.7, and 1.0, and Fig. S-5† presents the M–H curves for other values of x. It is interesting to note that a large difference in the shape of the M–H curves occurred with composition and chitosan-coating. The surface functionalization of the coated samples causes changes in the shape of M– the H curves of bare and coated samples. For instance, pure MgFe2O4, i.e., x = 0, bare nanoparticles are superparamagnetic. With the increase x, there is a transition from superparamagnetic to a mixed superparamagnetic/ferrimagnetic relaxation as evidenced further by the Mössbauer spectroscopy in Fig. 4. Wide variations in the shape of the hysteresis loop between bare and coated particles for x = 0 might be due to the clustering effect in the coated in particles that facilitate exchange interaction and thereby causes ferrimagnetic relaxations with a negligible coercivity and remanance.63 Because of the enormous surface-to-volume ratio, the magnetic properties of nanoparticles are greatly influenced by their surface atoms. A–B coupling via superexchange interaction mediated by oxygen is disrupted at the surface due to insufficient atom coordination. As a result, surface anisotropy is induced by the distribution of the exchange field and crystal field perturbations.40 The stress anisotropy at the nanoparticles' surface is reduced by chitosan coating, which reduces the total effective anisotropy. As a result, coated particles respond to applied fields faster than bare particles, despite the fact that bare particles have higher magnetization than coated particles. Magnetocrystalline anisotropy increases as x increases, raising the effective anisotropy,40 and thereby, magnetic hardness increases with x.
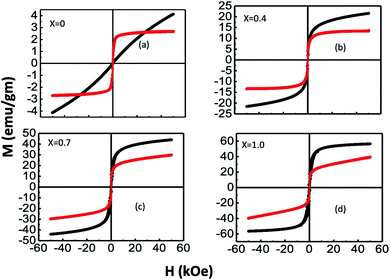 |
| | Fig. 10 Representative M–H curves of (a) x = 0, (b) x = 0.4, (c) x = 0.7, and (d) x = 1.0. The M–H loops for bare and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles were measured on the powder samples in the bare and liquid samples in coated nanoparticles with a maximum magnetic field of 5 T. Extensive variation in the M–H curves of bare and coated samples demonstrates that the nanoparticles of all compositions have undergone surface functionalization. | |
Using the law of approach to saturation, we estimated the saturation magnetization Ms for each composition.64 Fig. 10(a) presents the variation of Ms with x for bare and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles. Ms increases with x because of the higher magnetic moment of cobalt (3.88 μB) than magnesium (0 μB).18,65 The Ms of the as-dried Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles are 6.9, 11.4, 14.67, 22.26, 25.21, 32.29, 43.44, 49.12, 55.54, 56.54, and 59.29 emu g−1, respectively. Fig. 11(b–d) shows an increase in the coercive field, remanent magnetization, and anisotropy constant with cobalt supported by Mund et al.18. When the Mg2+ is higher, i.e., at lower x, the magnetic moment is diluted. With the increase in x, the magnetic moment increases and the magnetocrystalline anisotropy of Co2+ raises the effective anisotropy. Thus, we see an increase in the coercivity and remanent ratio in the bare state. Pervaiz et al.66 synthesized CoFe2O4 by sol–gel auto combustion technique. They found the saturation magnetic moment and anisotropy constant to be 63 emu g−1 and 0.35 × 106 erg cm−3, respectively, which are close to the values we reported in the current study.
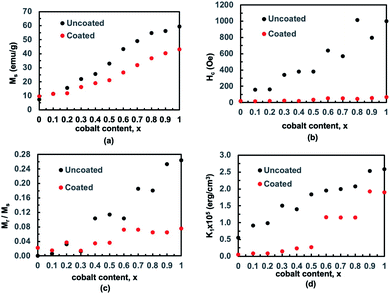 |
| | Fig. 11 We determined the saturation magnetization for each composition using the law of approach to saturation. (a) The variation of saturation magnetization with x for bare and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles. The variations in (b) coercive field, (c) remanent magnetization, and (d) anisotropy constant with x. | |
Fig. 11(a) shows that the magnetization of coated particles is lower than that of the uncoated particles. This is because of the chitosan coating, which is a magnetically dead layer on the surface of the nanoparticles, which inhibits inter-particle exchange interactions of magnetic moments. The chitosan coating also lessens the surface anisotropy and effective anisotropy. Therefore, the values of coercivity and remanent magnetization of the coated particles are lower than that of the uncoated particles.67–69 The cubic anisotropy constant, K1, was determined by the relation70
| |
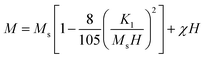 | (8) |
where
K1 is the cubic anisotropy constant,
Ms is the saturation magnetization by the law of approach to saturation, which characterizes the dependence of magnetization
M on the applied magnetic field for
H ≫
Hc, and
χ is the high-field susceptibility. The cubic anisotropy constant
K1 increases with
x but decreases with the coating. At lower
x, the surface anisotropy effect contributes to the cubic anisotropy. At higher
x, both surface and magnetocrystalline anisotropy contribute to the cubic anisotropy. The coating reduces the surface and stress anisotropy but is unable to lessen the magnetocrystalline anisotropy. Therefore, in the complete range of chitosan-coated Mg
1−xCo
xFe
2O
4, cubic anisotropy decreases with the coating but increases with
x,
i.e., the cobalt content.
Cytotoxicity
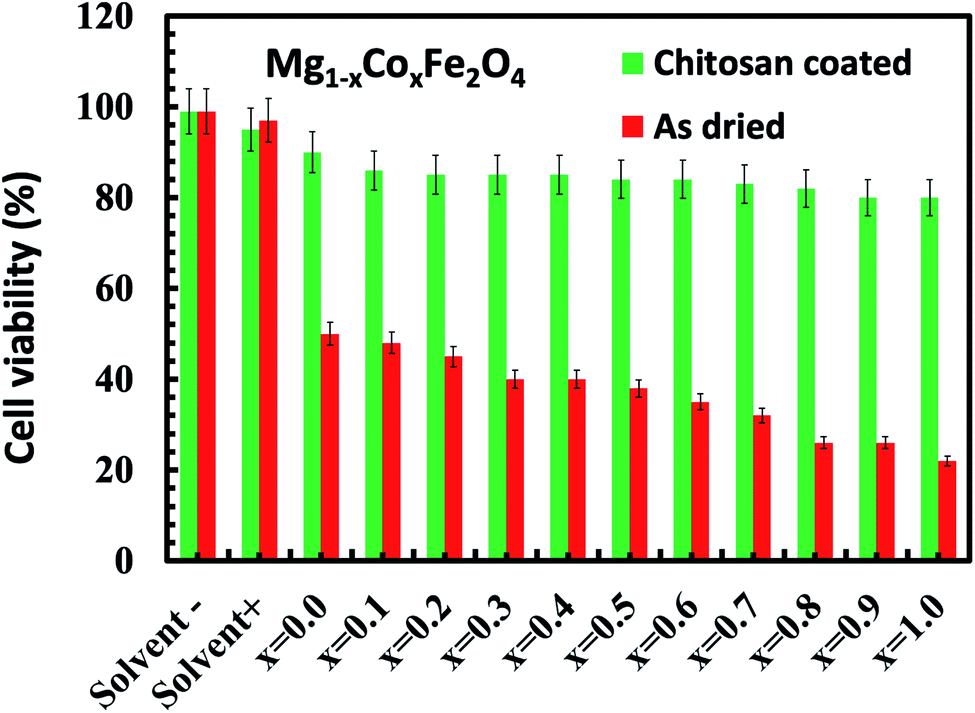
We studied the cytotoxic effect of bare and chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles using the HeLa cell line. We studied the toxicity effect on the HeLa cell line for the future in vivo MRI and hyperthermia studies by inoculating the tumor using HeLa cell lines. For in vivo tumor studies, we need to exclude any cytotoxic effect of the nanoparticles on the HeLa cell line. Fig. 12 shows that the uncoated Mg1−xCoxFe2O4 nanoparticles in the bare conditions are not viable. While we see a remarkable difference for chitosan-coated Mg1−xCoxFe2O4 nanoparticles, both chitosan and chitosan-coated Mg1−xCoxFe2O4 nanoparticles were biocompatible because their cell viability was more than 80%. For the as-synthesized Mg1−xCoxFe2O4, the toxicity increases with an increase in the cobalt content x for the entire range of composition. The cell viability slightly decreases with an increase in the cobalt content x.71–73
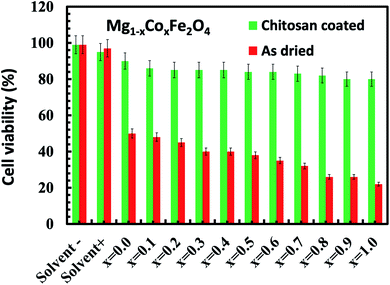 |
| | Fig. 12 Cytotoxicity assay of HeLa cells incubated for 48 hours in the DMEM media with the as-dried and chitosan coated as-dried Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) sample. The figure shows that the uncoated Mg1−xCoxFe2O4 nanoparticles in the bare conditions are not viable, while we see a remarkable difference for the chitosan-coated Mg1−xCoxFe2O4 nanoparticles, which are fully viable. | |
Hyperthermia
Fig. 13(a) presents the heating profiles of chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1), which shows the rise in the temperature with time for different values of x and reaches a plateau temperature. The temperature rises with time because of the Néel and Brownian relaxations and hysteresis loss. The Néel relaxation time is,| |
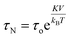 | (9) |
where K is the anisotropy constant, and V is the volume of the magnetic particles, kB is the Boltzmann constant, and T is the temperature. In Fig. 13(a), the plateau temperature rises with Co because of the increase in the anisotropy constant and particle size, which generates more heat through Néel's relaxation.
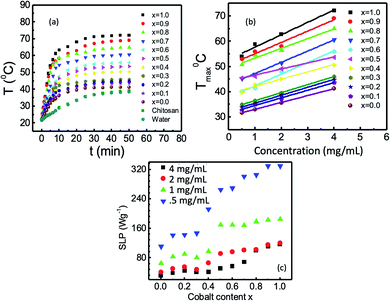 |
| | Fig. 13 (a) Rise of temperature as a function of time of chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) with 4 mg mL−1 concentration in a rf field of 26 mT; (b) variation of maximum temperature with the concentration of the coated nanoparticles for different values of x, and (c) variation of specific loss power (SLP) with cobalt content x of chitosan-coated nanoparticles at concentrations of 0.5, 1, 2, and 4 mg mL−1. | |
Brownian relaxation is associated with the particle size and viscosity of the solutions of nanoparticles. Brownian relaxation also contributes to the rise in the temperature with time in Fig. 13(a). An increase in the concentration of the colloidal suspension increases the viscosity. With the increase in the Co content, the size of the nanoparticles increases. Therefore, Brownian relaxation also contributes to the rise in the plateau temperature with x and magnetic nanoparticles' concentration.22,74,75 The Brownian relaxation time is,
| |
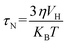 | (10) |
where
η is the dynamic viscosity of the carrier liquid,
VH is the hydrodynamic volume of the particles,
kB is the Boltzmann constant, and
T is the temperature. The effective relaxation time is
| |
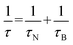 | (11) |
Fig. 13(b) shows an increase in the maximum temperature (at plateau region) Tmax with the solution concentration for 0 ≤ x ≤ 1 with Δx = 0.1. For the applied field of 26 mT and concentration of 4 mg mL−1, the maximum temperature varies between 41 to 72 °C. Fig. 13(b) also demonstrates that for the AC magnetic field of 26 mT, the concentration needed to reach the hyperthermia temperature (42–46 °C) is 8 mg mL−1 for chitosan-coated MgFe2O4. With the increase in the Co content, i.e., for x = 0.1, 0.2, and 0.3, the concentration of 4 mg mL−1, for x = 0.4 and 0.5, a concentration 1 mg mL−1, for x = 0.6 and 0.7, a concentration of 0.5 mg mL−1, and for x = 0.8, 0.9, and 1, a concentration of 0.25 mg mL−1 are needed. Fig. 13(c) presents the variations in the specific loss power (SLP) at different compositions and concentrations. The SLP increases with an increase in the cobalt content x and decreases with the increase in the nanoparticles' concentration.22,74,75 MgFe2O4 does not show hyperthermia properties up to 4 mg mL−1 concentration. However, the minimal substitution of magnesium with cobalt makes the studied samples suitable for hyperthermia at 4 mg mL−1 concentration.
Magnetic resonance imaging (MRI)
We acquired the T2 map of phantoms containing chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles with different concentrations. The parameters for the imaging were 7 Tesla static magnetic field with a flip angle (FA) of 90°, repetition time (TR) of 4000 ms, and echo times (TE) were 7 ms, 14 ms, 21 ms, 28 ms, 35 ms, 42 ms, 49 ms, 56 ms, 63 ms, 70 ms, 77 ms, 84 ms, 91 ms, 98 ms, 105 ms, and 112 ms. The field of view (FOV) was 40 × 40 mm2, and the image matrix was 128 × 128. The transverse relaxation time T2 is the time of relaxation of net magnetization when it changes by applying a radio-frequency (RF) pulse. Fig. 14(a) presents the T2 map of phantoms containing chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles of different concentrations. The darkening effect of each voxel increases with an increase in the concentration for every x because of the shorter T2 r relaxation time.28 We found, from the magnetic characterization by Mösssbauer and PPMS studies, a transition from the superparamagnetic to the ferromagnetic state with increasing x. The ferrite nanoparticles accelerate transverse or spin–spin relaxation, which expedites the dephasing of magnetic moments as a consequence of magnetic field gradients generated by the ultrafine magnetic nanoparticles by the subsequent onset of an RF pulse. The spin–spin relaxation is28 | |
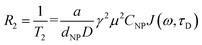 | (12) |
where a is a constant, dNP is the nanoparticle diameter, D is the diffusion coefficient, μ is the magnetic moment of the nanoparticles, γ is the gyromagnetic ratio of the water proton, CNP is the concentration of the nanoparticles, and J is the spectral density function.
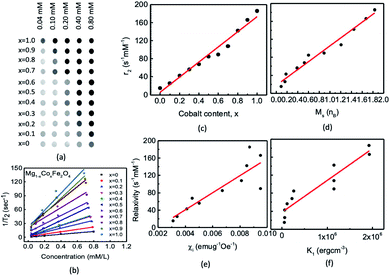 |
| | Fig. 14 We acquired phantom images for the chitosan-coated Mg1−xCoxFe2O4 (0 ≤ x ≤ 1 with Δx = 0.1) nanoparticles. The T2 weighted fast spin-echo (FSE) magnetic resonance images were acquired using the machine of model Bo = 7 T; the repetition time (TR) was 4000 ms and the slice thickness (THK) was 1.0 mm, and the FOV was 40 × 40. We acquired the T2 mapping at different echo times (TE) of 7 ms, 14 ms, 21 ms, 28 ms, 35 ms, 42 ms, 49 ms, 56 ms, 63 ms, 70 ms, 77 ms, 84 ms, 91 ms, 98 ms, 105 ms, and 112 ms. (a) Slices of different cobalt content x with concentrations of 0.04, 0.10, 0.20, 0.40, and 0.80 mM in each phantom. (b) Concentration dependence of relaxation with linear fitting, the slope of which provides the values of relaxivity for different values of x; (c) variation of relaxivity with cobalt content x, (d) with saturation magnetization, (e) with susceptibility, and (f) with magnetocrystalline anisotropy. | |
We measured the intensity drop I(t) with echo time TE. The first-order exponential fitting provides spin–spin relaxation 1/T2 by the relation
| |
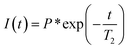 | (13) |
where
t is the decay time,
P* is the initial intensity, and
I(
t) is the peak intensity.
75 From the values of
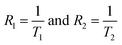
relaxation rate with contrast agent of concentration C found from the exponential fitting of
eqn (13). The relaxivities of nuclear spins of magnetic nanoparticles in an aqueous solution can be calculated using
| |
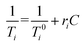 | (14) |
where
i is 1 or 2,
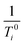
is the relaxation rate of the control, and
ri is the relaxivity of nuclear spins of the nanoparticles (
eqn (14)).
Fig. 14(b) shows the variations of the relaxation rate with the concentrations. We found a linear relationship between 1/
T2 with concentrations of nanoparticles, and the slope gives the values of relaxivities for each
x.
Fig. 14(c) shows the variation of
r2 relaxivities with
x because of the change in the magnetic properties with the composition. The obtained values of
r2 relaxivities of chitosan-coated Mg
1−xCo
xFe
2O
4 ferrite nanoparticles vary between 15.2 to 185.5 mM
−1 s
−1 with
x, which shows almost a linear relationship.
Fig. 14(d) shows the variations of the relaxivities with the saturation magnetic moment that shows a linear variation. However the variations of the relaxivity with the initial susceptibility and anisotropy in
Fig. 14(e) and (f) are scattered but there is an increasing trend of relaxivity with the susceptibility and anisotropy. Kim
et al.30 reported similar results for MnFe
2O
4, CoFe
2O
4, and Fe
3O
4 synthesized by a diol reduction of organic metals. Wang
et al.26 reported the
r2 relaxivities of commercially approved MRI phase-contrast agent Ferumoxtran-10 (AMI-227; Combidex, AMAG Pharma; Sinerem, Guerbet), Ferucarbotran (Resovist, Bayer Healthcare), and Ferumoxides (Feridex IV, Berlex Laboratories; and Endorem, Guerbet) as 60, 151, and 98.3, respectively. The
r2 relaxivities are comparable to the relaxivities of chitosan-coated Mg
1−xCo
xFe
2O
4 ferrite nanoparticles. Hence, Mg
1−xCo
xFe
2O
4 ferrite nanoparticles are potential candidates for the MRI contrast agent.
We acquired the T2 weighted in vivo MRI imaging of the rat brain using the Fast Spin Echo (FSE) pulse sequence. The purpose was to study the efficacy of chitosan-coated Mg1−xCoxFe2O4 nanoparticles. Fig. 15(a–c) shows some representative T2 FSE MR images of rat brains before and after administering the chitosan-coated Mg1−xCoxFe2O4 nanoparticles as contrast agents for x = 0.2, 0.5, and 0.8. We marked the region of interest ROI 1 and 2 to observe the (%) intensity drop before and after administering the contrast agents with x. For instance, at x = 0 and ROI 1, the signal intensity was 17818 counts for rats before inoculation. The signal dropped to the values of 17820, 16578, 10398, 11605, and 13247 counts after 15, 30, 60, 90, and 120 minutes of inoculation of contrast agent. Fig. 15(d) shows the data of (%) intensity fall in the brain (ROI 1) and muscle (ROI 2), which shows the average intensity fall is significant after 60 minutes of inoculation of the contrast agent for the entire range of composition Mg1−xCoxFe2O4 (with Δx = 0.1). Intensity loss of the T2 image of rat brain after injecting chitosan-coated Mg1−xCoxFe2O4 nanoparticles varied between 29.2 to 41.6% in Fig. 15(d). Hong et al.27 reported a 31.7% intensity loss in the T2 image of a rabbit liver after injecting the aqueous solution of chitosan-coated Fe3O4, which supported our result.
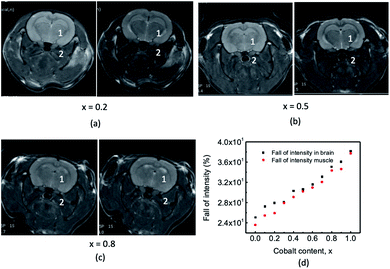 |
| | Fig. 15 The T2 weighted MRI imaging of rat brain using the Fast Spin Echo (FSE) pulse sequence. Some representative T2 FSE MR images of rat brains before and after administering the chitosan-coated Mg1−xCoxFe2O4 nanoparticles as contrast agents for x = 0.2 (a), x = 0.5 (b), and x = 0.8 (c). We marked the regions of interest (ROI) 1 and 2 to observe the (%) intensity fall before and after administering the contrast agents for all the values of x. (d) Fall of intensity at ROI 1 in the brain and ROI 2 in the muscle before and after administering the contrast agents. | |
Conclusion
From the FTIR, Raman, and Mössbauer spectra, we observe an increase in cobalt on the tetrahedral site with the increase in x. Therefore, the addition of cobalt condenses covalent bonds at the tetrahedral site and expands the covalent bonds at the octahedral site. The magnetization increases with x in the bare condition because of the increase in the A–B interactions and higher magnetic moment of Co. The peak shifts of the absorption bands representing the A- and B-sites of FTIR indicate a good coating with optimum hydrodynamic diameter and zeta potential. The Tmax and SLP of hyperthermia increase with x due to a higher magnetic moment and anisotropy. The values of T2 relaxivity, r2, increase with x because of the increase in the magnetic moment, susceptibility, and anisotropy. The in vivo studies of rat brain show 29.2 to 41.6% intensity drop after 60 minutes of inoculation of the contrast agent. The present study validates that the chitosan-coated Mg1−xCoxFe2O4 ferrite nanoparticles of all compositions are potential candidates for hyperthermia therapy and MRI contrast. However, the addition of cobalt reduces the concentration of the dose required for hyperthermia therapy and MR imaging.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to offer special thanks to the Annual Development Project (ADP) of the Planning Commission, Government of Bangladesh (the project ID 5003, Nano Project), the Semiconductor Technology Research Centre, University of Dhaka, Ministry of Science and Technology, Bangladesh, and Materials Science Division, Atomic Energy Centre, Dhaka, Bangladesh Atomic Energy Commission for the support to perform this research. The authors are also highly grateful to the International Science Program, Uppsala University, Sweden, for their great support and encouragement.
References
- A. H. Habib, C. L. Ondeck, P. Chaudhary, M. R. Bock- staller and M. E. McHenry, J. Appl. Phys., 2008, 103, 07A307 CrossRef.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- J. Chomoucka, J. Drbohlavova, D. Huska, V. Adam, R. Kizek and J. Hubalek, Pharmacol. Res., 2010, 62, 144–149 CrossRef CAS PubMed.
- A. Kumar, P. K. Jena, S. Behera, R. F. Lockey, S. Mohapatra and S. Mohapatra, Nanomedicine, 2010, 6, 64–69 CrossRef CAS PubMed.
- S. M. Hoque, M. K. Islam, A. Hoq, M. M. Haque, S. Maritim, D. Coman and F. Hayder, Frontiers in Nanotechnology, 2021, 3, 1–13 CrossRef.
- V. K. Sharma, J. Filip, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 8410–8423 RSC.
- L. A. Frank, G. R. Onzi, A. S. Morawski, A. R. Pohlmann, S. S. Guterres and R. V. Contri, React. Funct. Polym., 2020, 147, 104459 CrossRef CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- N. I. Taib, V. Agarwal, N. M. Smith, R. C. Woodward, T. G. S. Pierre and K. S. Iyer, Mater. Chem. Front., 2017, 1, 2335–2340 RSC.
- J. Simon, L. K. Müller, M. Kokkinopoulou, I. Lieber- wirth, S. Morsbach, K. Landfester and V. Mailänder, Nanoscale, 2018, 10, 10731–10739 RSC.
- S. M. Hoque, M. K. Islam, A. Hoq, M. M. Haque, S. Mar- itim, D. Coman and F. Hyder, Frontiers in Nanotechnology, 2021, 3, 1–13 CrossRef.
- S. M. Hoque, M. S. Hossain, S. Choudhury, S. Akhter and F. Hyder, Mater. Lett., 2016, 162, 60–63 CrossRef CAS PubMed.
- M. S. Shakil, M. A. Hasan, M. F. Uddin, A. Islam, A. Nahar, H. Das, M. N. I. Khan, B. P. Dey, B. Rokeya and S. M. Hoque, ACS Appl. Bio Mater., 2020, 3, 7952–7964 CrossRef CAS PubMed.
- J. T. Tang, X. W. Zeng, Q. S. Xia, X. X. Wang and D. L. Zhao, J. Alloys Compd., 2009, 477, 739–743 CrossRef.
- F. Sonvico, S. Mornet, S. Vasseur, C. Dubernet, D. Jaillard, J. Degrouard, J. Hoebeke, E. Duguet, P. Colombo and P. Couvreur, Bioconjugate Chem., 2005, 16, 1181–1188 CrossRef CAS PubMed.
- L. Khanna and S. K. Tripathi, Res. J. Recent Sci., 2017, 6, 1–9 CrossRef CAS.
- H. S. Mund and B. L. Ahuja, Mater. Res. Bull., 2017, 85, 228–233 CrossRef CAS.
- F. Cámara-Martos and R. Moreno-Rojas, Encyclopedia of Food and Health, 2016, pp. 172–178 Search PubMed.
- US Food and Drug Administration Regulation (FDA), Food Labeling – Revision of the Nutrition and Supplement Facts Labels (US Food and Drug Administration Regulation) (FDA) (2018 Edition), 2018, pp. 903–904, The Law Library, https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-11867.pdf Search PubMed.
- S. Amiri and H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 1–8 CrossRef CAS PubMed.
- S. M. Hoque, Y. Huang, E. Cocco, S. Maritim, A. D. Santin, E. M. Shapiro, D. Coman and F. Hyder, Contrast Media Mol. Imaging, 2016, 11, 514–526 CrossRef CAS PubMed.
- T. Yadavalli, H. Jain and G. Chandrasekharan, AIP Adv., 2016, 6, 55904 CrossRef.
- A. B. Norman, S. R. Thomas, R. G. Pratt, R. C. Samaratunga and P. R. Sanberg, Exp. Neurol., 1990, 109, 164–170 CrossRef CAS PubMed.
- P. A. Valdés-Hernández, A. Sumiyoshi, H. Nonaka, R. Haga, E. Aubert-Vásquez, T. Ogawa, Y. Iturria- Medina, J. J. Riera and R. Kawashima, Front. Neuroinf., 2011, 5, 1–19 Search PubMed.
- Y.-X. J. Wang, Quant. Imag. Med. Surg., 2011, 1, 35–40 Search PubMed.
- S. Hong, Y. Chang and I. Rhee, J. Korean Phys. Soc., 2010, 56, 868–873 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- L. L. Muldoon, S. Gahramanov, X. Li, D. J. Mar-shall, D. F. Kraemer and E. A. Neuwelt, Neuro-Oncology, 2011, 13(3), 51–60 CrossRef PubMed.
- D. H. Kim, H. Zeng, H. Zeng, T. C. Ng and C. S. Brazela, J. Magn. Magn. Mater., 2009, 321, 3899–3904 CrossRef CAS.
- D. Varshney, K. Verma and A. Kumar, J. Mol. Struct., 2011, 1006, 447–452 CrossRef CAS.
- M. Y. Lodhi, K. Mahmood, A. Mahmood, H. Malik, M. F. Warsi, I. Shakir, M. Asghar and M. A. Khan, Curr. Appl. Phys., 2014, 14, 716–720 CrossRef.
- A. C. Druc, A. I. Borhan, A. Diaconu, A. R. Iordan, G. G. Nedelcu, L. Leontie and M. N. Palamaru, Ceram. Int., 2014, 40, 13573–13578 CrossRef CAS.
- M. E. Maguire and J. A. Cowan, BioMetals, 2002, 15, 203–210 CrossRef CAS PubMed.
- M. Barsoum and M. W. Barsoum, Fundamentals of Ceramics, Taylor & Francis Group, New York, 2003, vol. 3, pp. 80–84 Search PubMed.
- A. G. Abraham, A. Manikandan, E. Manikandanb, S. Vadivel, S. K. Jaganathan, A. Baykal and P. SriRen- ganathan, J. Magn. Magn. Mater., 2018, 452, 380–388 CrossRef CAS.
- C. R. Stein, M. T. S. Bezerra, G. H. A. Holanda, J. André- Filho and P. C. Morais, AIP Adv., 2018, 8, 056303 CrossRef.
- J. A. R. Guivar, A. Bustamante, J. Flores, M. M. Santillan, A. M. Osorio, A. I. Martínez, L. D. L. S. Valladares and C. H. W. Barnes, Hyperfine Interact., 2014, 224, 89–97 CrossRef.
- Q. Lin, Y. He, J. Xu, J. Lin, Z. Guo and F. Yang, Nanomaterials, 2018, 8, 750 CrossRef PubMed.
- E. Manova, B. Kunev, D. Paneva, I. Mitov, L. Petrov, C. Estournès, C. D'Orléan, J.-L. Rehspringer and M. Kurmoo, Chem. Mater., 2004, 16, 5689–5696 CrossRef CAS.
- Y. He, X. Yang, J. Lin, Q. Lin and J. Dong, J. Nanomater., 2015, 2015, 854840 Search PubMed.
- S. Kumar, A. M. M. Farea, K. M. Batoo, C. G. Lee, B. H. Koo, A. Yousef and A. Y Alimuddin, Phys. B, 2008, 403, 3604–3607 CrossRef CAS.
- A. Ghasemi, A. Paesano, C. F. C. Machado, S. E. Shirsath, X. Liu and A. Morisako, J. Appl. Phys., 2014, 115, 17A522 CrossRef.
- M. Gupta and B. S. Randhawa, Mater. Chem. Phys., 2011, 130, 513–518 CrossRef CAS.
- G. A. Sawatzky, F. V. D. Woude and A. H. Morrish, Phys. Rev., 1969, 183, 383–386 CrossRef CAS.
- T. Zeeshan, S. Anjum, H. Iqbal and R. Zia, Mater. Sci., 2018, 36, 255–263 CAS.
- B. Babić-Stojić, V. Jokanović, D. Milivojević, Z. Jagličić, D. Makovec, N. Jović and M. Marinović-Cincović, J. Nanomater., 2013, 2013, 1–9 CrossRef.
- R. Vemuria, G. Raju, M. G. Kiran, M. S. N. A. Prasad, E. Rajesh, G. P. Kumar and N. Murali, Results Phys., 2019, 12, 947–952 CrossRef.
- I. L. Ardelean, D. Ficai, A. Ficai, G. Nechifor, D. Dragu and C. Bleotu, UPB Scientific Bulletin, Series B: Chemistry and Materials Science, 2018, 80, 33–46 CAS.
- J. Kreisel, G. Lucazeau and H. Vincent, J. Solid State Chem., 1998, 137, 127–137 CrossRef CAS.
- P. Galinetto, B. Albini, M. Bini and M. C. Mozzati, https://www.intechopen.com/chapters/58926, 2018.
- H. S. Mund and B. L. Ahuja, Mater. Res. Bull., 2017, 85, 228–233 CrossRef CAS.
- M. A. G. Soler, E. C. D. Lima, S. W. da Silva, T. F. O. Melo, A. C. M. Pimenta, J. P. Sinnecker, R. B. Azevedo, V. K. Garg, A. C. Oliveira, M. A. Novak and P. C. Morais, Langmuir, 2007, 23, 9611–9617 CrossRef CAS PubMed.
- K. V. Chandekar, Adv. Mater. Lett., 2017, 8, 435–443 CrossRef CAS.
- M. A. G. Solera, T. F. O. Melo, S. W. Silva, E. C. D. Lima, A. C. M. Pimenta, V. K. Garg, A. C. Oliveira and P. C. Morais, J. Magn. Magn. Mater., 2004, 2357–2358 CrossRef.
- X. Wu, Z. Ding, N. Song, L. Li and W. Wang, Ceram. Int., 2015, 1, 10 Search PubMed.
- C. O. Ehi-Eromosele, B. I. Ita and E. E. J. Iweala, J. Sol-Gel Sci. Technol., 2015, 76, 298–308 CrossRef CAS.
- S. Thota, S. C. Kashyap, S. K. Sharma and V. R. Reddy, J. Phys. Chem. Solids, 2016, 91, 136–144 CrossRef CAS.
- E. H. M. Sakho, E. Allahyari, O. S. Oluwafemi, S. Thomas and N. Kalarikka, Thermal and Rheological Measurement Techniques for Nanomaterials Characterization, Elsevier, 2017, pp. 2–5 Search PubMed.
- M. Zahraei, A. Monshi, D. Shahbazi-Gahrouei, M. Amirnasr, B. Behdadfar and M. Rostami, J. Nanostruct., 2015, 5, 77–86 CrossRef.
- M. Arakha, S. Pal, D. Samantarrai, T. K. Panigrahi, B. C. Mallick, K. Pramanik, B. Mallick and S. Jha, Sci. Rep., 2015, 5, 14813 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- Z. Alborzi, A. Hassanzadeh and M. M. Golzan, Int. J. Nanosci. Nanotechnol., 2012, 8, 93–98 Search PubMed.
- A. Franco and M. S. Silva, J. Appl. Phys., 2011, 109, 07B505 CrossRef.
- M. A. ur Rehman, M. A. Malik, K. Khan and A. Maq- sood, J. Nano Res., 2011, 14, 1–9 Search PubMed.
- E. Pervaiz and I. H. Gul, Int. J. Curr. Eng. Technol., 2012, 2, 377–387 Search PubMed.
- B. Issa, I. Obaidat, B. Albiss and Y. Haik, Int. J. Mol. Sci., 2013, 14, 21266–21305 CrossRef CAS PubMed.
- S. M. Hoque, C. Srivastava, N. Venkatesha and K. Chattopadhyay, IEEE Trans. NanoBioscience, 2013, 12, 298–303 Search PubMed.
- E. Girgis, M. M. S. Wahsh, A. G. M. Othman, L. Bandhu and K. V. Rao, Nanoscale Res. Lett., 2011, 6, 460 CrossRef PubMed.
- A. Franco and F. C. e Silva, Appl. Phys. Lett., 2010, 96, 172505 CrossRef.
- E. U. Okon, G. Hammed, P. A. E. Wafa, O. Abraham, N. Case and E. Henry, Int. J. Innovation Appl. Stud., 2014, 5, 192–199 Search PubMed.
- A. K. Veni and S. Mohandass, International Journal of Pharmacognosy and Phytochemical Research, 2014, 6, 320–323 Search PubMed.
- P. W. Prasetyaningrum, A. Bahtiar and H. Hayun, Sci. Pharm., 2018, 86, 25 CrossRef PubMed.
- J. H. Lee, J. Jang, J. Choi, S. H. Moon, S. Noh, J. Kim, J. G. Kim, I. S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS PubMed.
- H. M. Joshi, Y. P. Lin, M. Aslam, P. V. Prasad, E. A. Schultz-Sikma, R. Edelman, T. Meade and V. P. Dravid, J. Phys. Chem. C, 2009, 113, 17761–17767 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00768a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
M. Razibul Hasana,
M. Mahbubabl Haquea,
Rimi Rashida,
Ishtiaque M. Syedc and
S. Manjura Hoque
abc,
M. Razibul Hasana,
M. Mahbubabl Haquea,
Rimi Rashida,
Ishtiaque M. Syedc and
S. Manjura Hoque
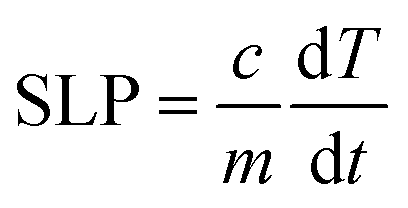









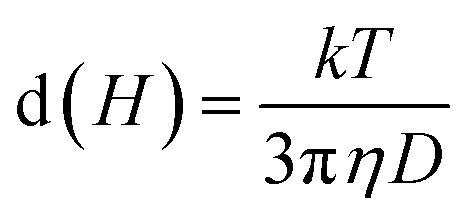




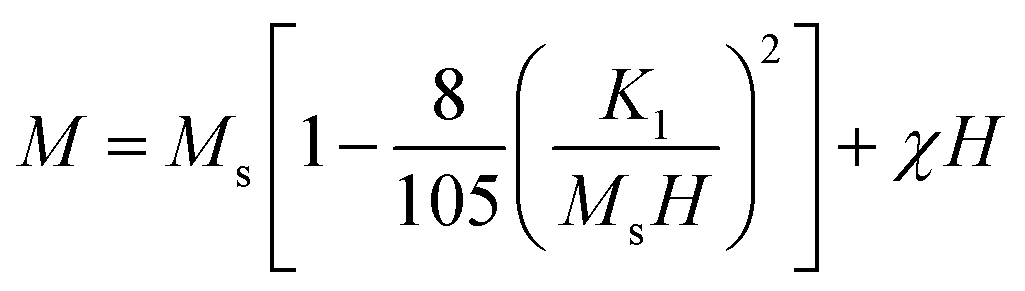


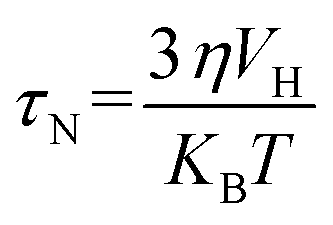



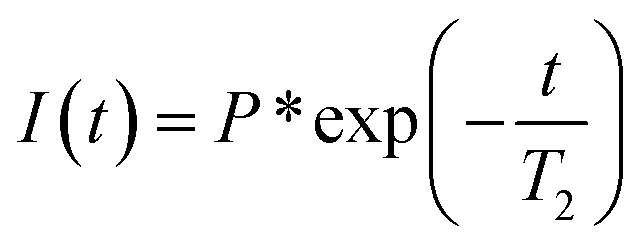
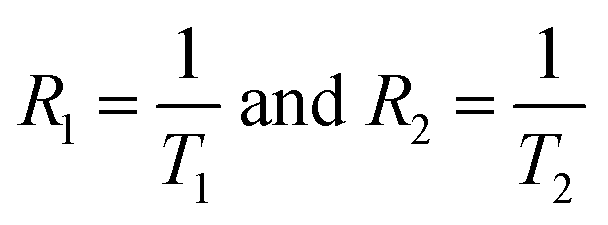 relaxation rate with contrast agent of concentration C found from the exponential fitting of eqn (13). The relaxivities of nuclear spins of magnetic nanoparticles in an aqueous solution can be calculated using
relaxation rate with contrast agent of concentration C found from the exponential fitting of eqn (13). The relaxivities of nuclear spins of magnetic nanoparticles in an aqueous solution can be calculated using 
 is the relaxation rate of the control, and ri is the relaxivity of nuclear spins of the nanoparticles (eqn (14)). Fig. 14(b) shows the variations of the relaxation rate with the concentrations. We found a linear relationship between 1/T2 with concentrations of nanoparticles, and the slope gives the values of relaxivities for each x. Fig. 14(c) shows the variation of r2 relaxivities with x because of the change in the magnetic properties with the composition. The obtained values of r2 relaxivities of chitosan-coated Mg1−xCoxFe2O4 ferrite nanoparticles vary between 15.2 to 185.5 mM−1 s−1 with x, which shows almost a linear relationship. Fig. 14(d) shows the variations of the relaxivities with the saturation magnetic moment that shows a linear variation. However the variations of the relaxivity with the initial susceptibility and anisotropy in Fig. 14(e) and (f) are scattered but there is an increasing trend of relaxivity with the susceptibility and anisotropy. Kim et al.30 reported similar results for MnFe2O4, CoFe2O4, and Fe3O4 synthesized by a diol reduction of organic metals. Wang et al.26 reported the r2 relaxivities of commercially approved MRI phase-contrast agent Ferumoxtran-10 (AMI-227; Combidex, AMAG Pharma; Sinerem, Guerbet), Ferucarbotran (Resovist, Bayer Healthcare), and Ferumoxides (Feridex IV, Berlex Laboratories; and Endorem, Guerbet) as 60, 151, and 98.3, respectively. The r2 relaxivities are comparable to the relaxivities of chitosan-coated Mg1−xCoxFe2O4 ferrite nanoparticles. Hence, Mg1−xCoxFe2O4 ferrite nanoparticles are potential candidates for the MRI contrast agent.
is the relaxation rate of the control, and ri is the relaxivity of nuclear spins of the nanoparticles (eqn (14)). Fig. 14(b) shows the variations of the relaxation rate with the concentrations. We found a linear relationship between 1/T2 with concentrations of nanoparticles, and the slope gives the values of relaxivities for each x. Fig. 14(c) shows the variation of r2 relaxivities with x because of the change in the magnetic properties with the composition. The obtained values of r2 relaxivities of chitosan-coated Mg1−xCoxFe2O4 ferrite nanoparticles vary between 15.2 to 185.5 mM−1 s−1 with x, which shows almost a linear relationship. Fig. 14(d) shows the variations of the relaxivities with the saturation magnetic moment that shows a linear variation. However the variations of the relaxivity with the initial susceptibility and anisotropy in Fig. 14(e) and (f) are scattered but there is an increasing trend of relaxivity with the susceptibility and anisotropy. Kim et al.30 reported similar results for MnFe2O4, CoFe2O4, and Fe3O4 synthesized by a diol reduction of organic metals. Wang et al.26 reported the r2 relaxivities of commercially approved MRI phase-contrast agent Ferumoxtran-10 (AMI-227; Combidex, AMAG Pharma; Sinerem, Guerbet), Ferucarbotran (Resovist, Bayer Healthcare), and Ferumoxides (Feridex IV, Berlex Laboratories; and Endorem, Guerbet) as 60, 151, and 98.3, respectively. The r2 relaxivities are comparable to the relaxivities of chitosan-coated Mg1−xCoxFe2O4 ferrite nanoparticles. Hence, Mg1−xCoxFe2O4 ferrite nanoparticles are potential candidates for the MRI contrast agent.
