DOI:
10.1039/D2RA00705C
(Paper)
RSC Adv., 2022,
12, 11454-11476
Synthesis, gene silencing activity, thermal stability, and serum stability of siRNA containing four (S)-5′-C-aminopropyl-2′-O-methylnucleosides (A, adenosine; U, uridine; G, guanosine; and C, cytidine)†
Received
2nd February 2022
, Accepted 30th March 2022
First published on 12th April 2022
Abstract
Herein, we report the synthesis of (S)-5′-C-aminopropyl-2′-O-methyladenosine and (S)-5′-C-aminopropyl-2′-O-methylguanosine phosphoramidites and the properties of small interfering RNAs (siRNAs) containing four (S)-5′-C-aminopropyl-2′-O-methylnucleosides (A, adenosine; U, uridine; G, guanosine; and C, cytidine). The siRNAs containing (S)-5′-C-aminopropyl-nucleosides at the 3′- and 5′-regions of the passenger strand were well tolerated for RNA interference (RNAi) activity. Conversely, the (S)-5′-C-aminopropyl modification in the central region of the passenger strand decreased the RNAi activity. Furthermore, the siRNAs containing three or four consecutive (S)-5′-C-aminopropyl-2′-O-methylnucleosides at the 3′- and 5′-regions of the passenger strand exhibited RNAi activity similar to that of the corresponding 2′-O-methyl-modified siRNAs. Finally, it was observed that (S)-5′-C-aminopropyl modifications effectively improved the serum stability of the siRNAs, compared with 2′-O-methyl modifications. Therefore, (S)-5′-C-aminopropyl-2′-O-methylnucleosides would be useful for improving the serum stability of therapeutic siRNA molecules without affecting their RNAi activities.
Introduction
Small interfering RNA (siRNA) comprising double-strand RNA (dsRNA) triggers the degradation of complementary mRNA in a sequence-specific manner by mediated RNA interference (RNAi) machinery, which is a biological process in cells.1–3 Accordingly, siRNAs possess potential as clinical tools for unmet medical needs. However, it is known that oligonucleotides such as siRNAs are degraded by nuclease inside and outside cells and are difficult to internalize into the cells, because they have negative charges over all the strands.4,5 Chemical modifications such as 2′-O-methyl-RNA, 2′-fluoro-RNA, and phosphorothioate linkage enhance the cell membrane permeability of siRNAs and the stability of siRNAs toward nuclease.6–11 In particular, chemically modified nucleosides are required for the clinical application of ligand-siRNA conjugates because this siRNA duplex is not protected from nucleolytic degradation.12,13 To date, four siRNA drugs including patisiran, givosiran, lumasiran, and inclisiran, with 2′-O-methyl-, 2′-fluoro-, and phosphorothioate modifications have been approved by the Food and Drug Administration (FDA) and/or European Medicines Agency (EMA).
Recently, Manoharan et al. reported the synthesis and properties of C4′ and C5′-modified nucleosides, such as the 4′-C-methoxy-modified nucleosides, (R)-, or (S)-5′-C-methyl-modified nucleosides.14–19 These nucleosides enhanced the nuclease resistance of oligonucleotides, compared with C2′-modified nucleosides without thermally destabilizing RNA duplexes and inhibiting gene silencing activity. Moreover, (R)- or (S)-5′-C-methylnucleosides improved the stability of oligonucleotides to snake venom phosphodiesterase (SVPD), compared with 4′-C-methoxy-nucleoside. This was due to the 5′-C modifications close to the phosphate linkages, compared with 4′-C modifications.16 Recently, we reported the synthesis of RNA oligomers containing (S)-5′-C-aminopropyl-2′-O-methyluridine (1).20 We observed that analog 1 significantly increased the stability of the RNAs in a buffer containing bovine serum and the binding affinity of the RNAs toward complementary RNA, compared with 4′-C-aminopropyl-2′-O-methyluridine and (R)-5′-C-aminopropyl-2′-O-methyluridine. The results suggested that (S)-5′-C-aminopropyl-2′-O-methyl modifications were useful for oligonucleotide-based therapeutics.
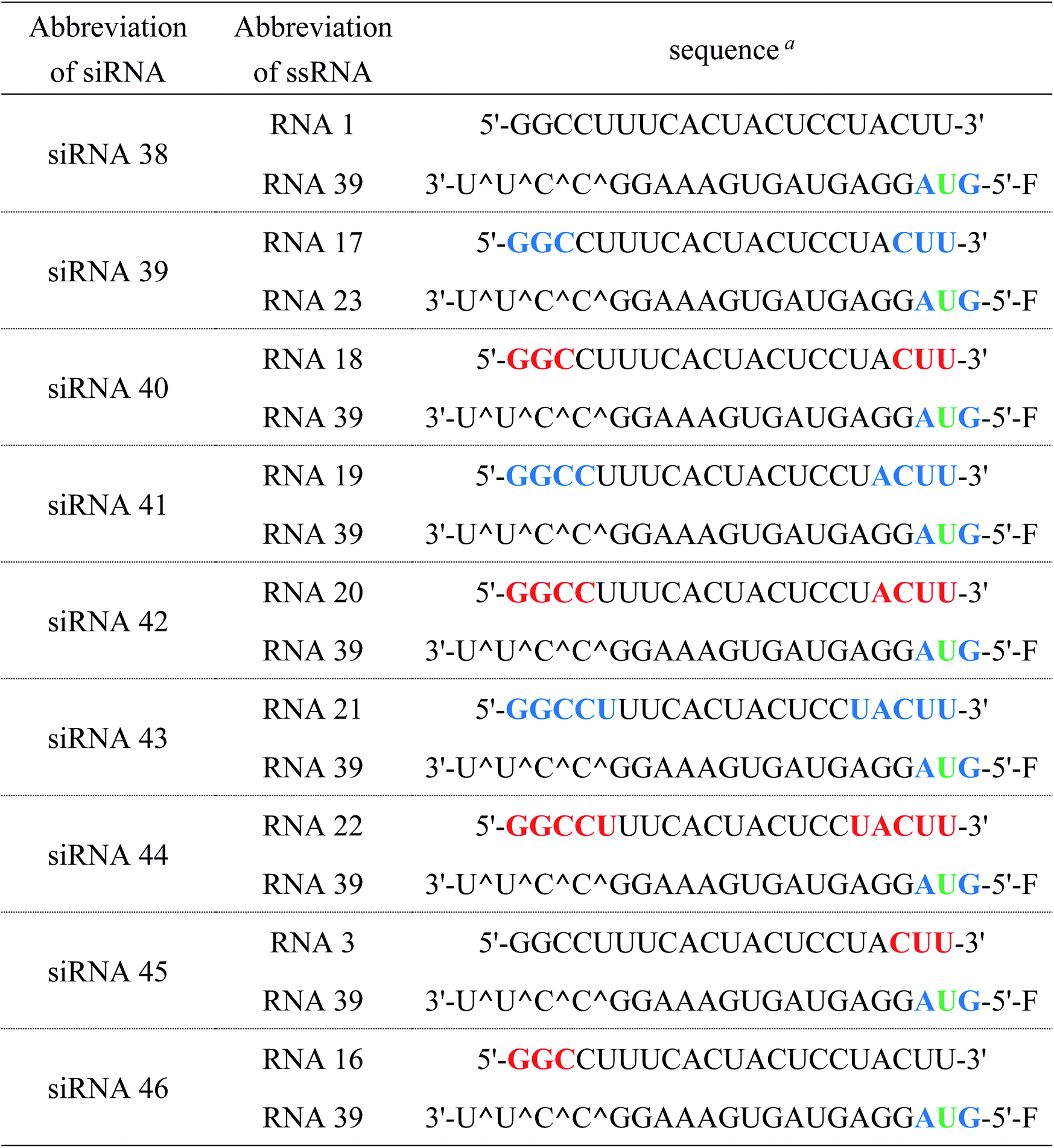
Here, we synthesized the corresponding adenosine and guanosine analogs, (S)-5′-C-aminopropyl-2′-O-methyladenosine (3) and (S)-5′-C-aminopropyl-2′-O-methylguanosine (4). The properties of RNAs and siRNAs containing four (S)-5′-C-aminopropyl-2′-O-methylnucleosides (1, 2, 3, and 4) were investigated in terms of thermal stability, serum stability, and the position-dependent effects of the analogs on the RNAi activity (Fig. 1).
 |
| | Fig. 1 Structures of (S)-5′-C-aminopropyl-2′-O-methyluridine20 (1), (S)-5′-C-aminopropyl-2′-O-methylcytidine21 (2), (S)-5′-C-aminopropyl-2′-O-methyladenosine (3), and (S)-5′-C-aminopropyl-2′-O-methylguanosine (4). | |
Results and discussion
Synthesis of nucleoside analogs
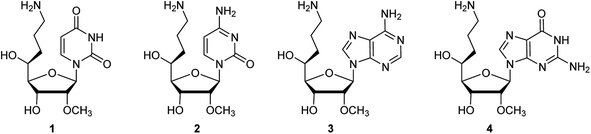
The synthetic route of common synthetic intermediate 12 for the synthesis of the phosphoramidites of (S)-5′-C-aminopropyl-2′-O-methyladenosine (3) and (S)-5′-C-aminopropyl-2′-O-methylguanosine (4) is shown in Scheme 1. We used aldehyde derivative 5, which was prepared according to a previously used procedure,22 as a starting material. The allylation reaction of 5 using allyltrimethylsilane and boron trifluoride diethyl ether (BF3·OEt2)23 afforded 5-C-allyl-ribofuranoside derivatives 6 and 7. The configurations of the 5-carbons in 6 and 7 were determined by nuclear overhauser effect spectroscopy (NOESY).21,24,25 To fix the conformations of the sugar moieties of 6 and 7, the 3- and 5-hydroxy groups of 6 and 7 were protected by cyclic silyl groups. After removing the 3-O-tert-butyldiphenylsilyl (TBDPS) groups of 6 and 7, the resulting 3, 5-dihydroxy derivatives were treated with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2) in pyridine to afford 3,5-O-TIPDS derivatives 34 and 35 in 81% and 79% yields, respectively (Scheme 4). After the NOESY, a strong NOE was observed between the H-5 and H-3 of 35, while no NOE was observed between the H-5 and H-3 of 34. The results of the proton nuclear magnetic resonance (1H NMR) measurement of 34 and 35 showed that the spin–spin coupling constants between H-4 and H-5 of 34 and 35 were 2.0 and 8.0 Hz, respectively. The difference in the spin–spin coupling constants indicated that the conformations of H-4 and H-5 on the C4–C5 bond were gauche-form in 34 and anti-form in 35, respectively. Thus, we concluded that the stereochemistry of C5 in ribofuranoside derivative 6 was an (S)-configuration, whereas that in ribofuranoside derivative 7 was an (R)-configuration. Next, we attempted to protect the 5-hydroxy function of 6 with a benzyl (Bn) group. It was revealed that the 3-O-TBDPS group migrated to the 5-hydroxy group of 6 when 6 was treated with sodium hydride (NaH).26,27 Consequently, the benzylation reaction of 6 using NaH and benzyl bromide (BnBr) afforded the 5-O-TBDPS and 3-O-Bn derivative 8 in 78% yield. The hydroboration and oxidation of the allyl moiety of 8 produced the 5-C-hydroxypropyl-ribofuranose derivative 9 in 91% yield. The tosylation and azidation of the hydroxy function of 9 afforded the azidopropyl derivative 11 in a 50% yield from 9. The deprotection of the isopropylidene moiety of 11 using 50% CF3CO2H in water, followed by the acetylation of 1,2-dihydroxyl groups using acetic anhydride (Ac2O) in pyridine afforded a diastereomeric mixture of 1,2-diacetylated ribofuranoside 12 in 81% yield over two steps.
 |
| | Scheme 1 Synthetic route of the common synthetic intermediate 12 for the synthesis of (S)-5′-C-aminopropyl-2′-O-methyladenosine and (S)-5′-C-aminopropyl-2′-O-methylguanosine phosphoramiditea. a Reagents and conditions: (a) allyltrimethylsilane, BF3·OEt2, CH2Cl2, −40 °C, 30 min, 82% (compounds 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 = 4:1); (b) BnBr, NaH, DMF, room temperature (rt), 16 h, 78%; (c) 9-BBN, THF, 30% H2O2 aq., 3 N NaOH aq., 40 °C, 1 h, 91%; (d) p-TsCl, pyridine, CH2Cl2, rt, 16 h, 95%; (e) NaN3, DMF, 60 °C, 8 h, 90%; and (f) (i) 50% CF3CO2H aq., CH2Cl2, rt, 4.5 h and (ii) Ac2O, pyridine, rt, 24 h, 81% (2 steps). :7 = 4:1); (b) BnBr, NaH, DMF, room temperature (rt), 16 h, 78%; (c) 9-BBN, THF, 30% H2O2 aq., 3 N NaOH aq., 40 °C, 1 h, 91%; (d) p-TsCl, pyridine, CH2Cl2, rt, 16 h, 95%; (e) NaN3, DMF, 60 °C, 8 h, 90%; and (f) (i) 50% CF3CO2H aq., CH2Cl2, rt, 4.5 h and (ii) Ac2O, pyridine, rt, 24 h, 81% (2 steps). | |
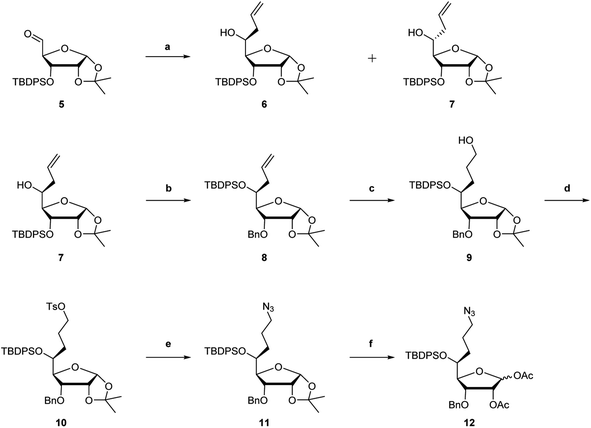
(S)-5′-C-aminopropyl-2′-O-methyladenosine phosphoramidite 22 was synthesized from the common synthetic intermediate 12 (Scheme 2). The stereoselective glycosylation of the 5-C-substituted ribofuranoside derivative 12 with N6-benzoyl adenine using tin(IV) chloride (SnCl4),28,29 followed by deacetylation using potassium carbonate (K2CO3) in CH3OH afforded a 2′-OH derivative 14. The methylation of the 2′-OH moiety of 14 using CH3I and NaH afforded the 2′-O-methyl derivative 15 in 72% yield. After removing the 5′-O-TBDPS and 3′-O-Bn groups in 15, the resulting 5′,3′-dihydroxy nucleoside derivative was treated with TBDPSCl and imidazole in DMF to afford the 3′-O-TBDPS derivative 18 in 76% yield. The tritylation of the 5′-hydroxy function in 19 using dimethoxytrityl chloride (DMTrCl) successfully occurred in the presence of silver nitrate (AgNO3). The reduction of the azide group in 19 by the Staudinger reaction, followed by the protection of the resulting amino function using ethyl trifluoroacetate (CF3CO2Et) afforded the fully protected adenosine derivative 20 in 93% yield. The deprotection of the 3′-O-silyl group in 20 by treatment with n-tetrabutylammonium fluoride (TBAF) afforded 21 in 99% yield. Finally, the 3′-hydroxy function of nucleoside 21 was phosphorylated by treatment with 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (CEP-Cl) to afford (S)-5′-C-aminopropyl-2′-O-methyladenosine phosphoramidite 22 in 80% yield.
 |
| | Scheme 2 Synthetic route of (S)-5′-C-aminopropyl-2′-O-methyladenosine phosphoramidite 22a. a Reagents and conditions: (a) N6-benzoyl adenine, SnCl4/CH2Cl2, CH3CN, rt, 2 h, 70%; (b) K2CO3, CH3OH, 0 °C, 30 min, 92%; (c) CH3I, NaH, THF, 0 °C, 2.5 h, 72%; (d) TBAF/THF, THF, rt, 16 h, 92%; (e) BCl3/CH2Cl2, CH2Cl2, −78 °C, 2 h, 89%; (f) TBDPSCl, imidazole, DMF, rt, 24 h, 76%; (g) DMTrCl, AgNO3, pyridine, THF, 40 °C, 12 h, 72%; (h) (i) Ph3P, H2O, THF, 45 °C, 12 h and (ii) CF3CO2Et, Et3N, CH2Cl2, rt, 24 h, 93%; (i) TBAF/THF, THF, rt, 24 h, 99%; and (j) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, DIPEA, THF, rt, 1 h, 80%. | |
The synthetic route of (S)-5′-C-aminopropyl-2′-O-methylguanosine phosphoramidite is shown in Scheme 3. Diacetate 12 was glycosylated by treatment with the silylated 2-amino-6-chloropurine and trimethylsilyl triflate (TMSOTf)30 to afford nucleoside derivative 23 in 75% yield. Compound 23 was treated with K2CO3 in CH3OH, followed by the methylation of the resulting 2′-hydroxyl function of 24 by treatment with CH3I and NaH to afford the 2′-O-methylnucleoside derivative 25. The substitution reaction of the chlorine atom at the 6-position of the 2-amino-6-chloropurine moiety of 25 with a hydroxyl group successfully proceeded by treatment with 3-hydroxypropionitrile and NaH to afford the guanosine derivative 26 in 85% yield. Thereafter, the exocyclic amino function of 26 was protected with the isobutyryl (iBu) group to afford the protected guanosine derivative 27 in 77% yield. Similar to the synthesis of the (S)-5′-C-aminopropyl-2′-O-methyladenosine phosphoramidite, 22, 27 was converted into the corresponding phosphoramidite 33.
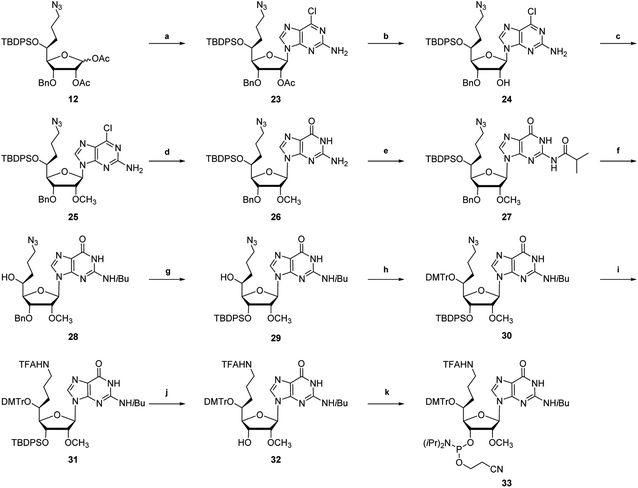 |
| | Scheme 3 Synthetic route of (S)-5′-C-aminopropyl-2′-O-methylguanosine phosphoramidite 33a. a Reagents and conditions: (a) 2-amino-6-chloropurine, N,O-bis(trimethylsilyl)acetamide, TMSOTf, toluene, 80 °C, 15 h, 75%; (b) K2CO3, CH3OH, 0 °C, 30 min, 93%; (c) CH3I, NaH, DMF, 0 °C, 7 h, 69%; (d) 3-hydroxypropionitrile, NaH, THF, 0 °C, 6 h, 85%; (e) isobutyric anhydride, DMAP, DMF, 60 °C, 13 h, 77%; (f) TBAF/THF, THF, rt, 46 h, 83% (g) (i) BCl3/CH2Cl2, CH2Cl2, −78 °C, 4 h; (ii) TBDPSCl, imidazole, DMF, 0 °C, 29 h, 50% (2 steps); (h) DMTrCl, AgNO3, pyridine, THF, 40 °C, 12 h, 92%; (i) (i) Ph3P, H2O, THF, 45 °C, 22 h and (ii) CF3CO2Et, Et3N, CH2Cl2, rt, 19 h, 77% (2 steps); (j) TBAF/THF, THF, rt, 24 h, 70%; and (k) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, DIPEA, THF, rt, 1 h, 73%. | |
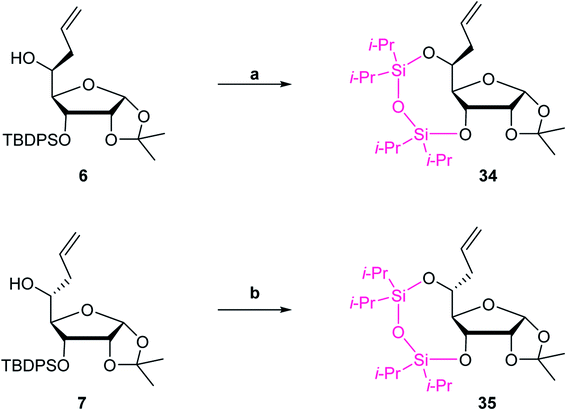 |
| | Scheme 4 Synthetic route of 3-, 5-O-TIPDS derivatives 34 and 35a. a Reagents and conditions: (a) (i) TBAF/THF, THF, rt, 3.5 h and (ii) TIPDSCl2, pyridine, rt, 21 h, 81% (2 steps) and (b) (i) TBAF/THF, THF, rt, 1 h and (ii) TIPDSCl2, pyridine, rt, 21 h, 79% (2 steps). | |
Oligonucleotide synthesis
The nucleoside analogs 3 and 4 were incorporated into oligonucleotides using phosphoramidites 22 and 33 with a DNA/RNA synthesizer. RNA phosphoramidites and 2′-O-methyl-modified phosphoramidites were prepared as 0.10 M solutions in CH3CN. (S)-5′-C-aminopropyl-2′-O-methyl-modified phosphoramidites were prepared as 0.15 M solutions in CH3CN. 5-Benzylthio-1H-tetrazole as 0.25 M solution in CH3CN was used as activator. The coupling time used for all phosphoramidites was 12 min. In this condition, the coupling efficiency of (S)-5′-C-aminopropyl-2′-O-methyl-modified phosphoramidites was nearly equal to that of RNA phosphoramidites. After the synthesis, to prevent the additional reaction of acrylonitrile with the 5′-C-aminopropyl functional groups, the controlled-pore glass (CPG) beads were treated with 10% diethylamine in CH3CN at room temperature for 5 min and with concentrated NH3/40% methylamine (1:1, v/v) solution at 65 °C for 10 min. The 2′-O-TBDMS groups were removed by treatment with Et3N·3HF in dimethyl sulfoxide (DMSO) at 65 °C for 1.5 h. RNAs were purified using 20% denaturing polyacrylamide gel electrophoresis (PAGE). The RNA sequences synthesized in this study are listed in Tables 1–4 and S1–S7.†
Table 1 Sequences of ssRNAs, siRNAs, and Tm values of siRNAs
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2′-O-methylnucleosides, respectively. The Tm values were determined using 3 μM dsRNA in a buffer containing 10 mM sodium phosphate (pH of 7.0) and 100 mM NaCl. All experiments were performed thrice, and data are presented as the mean ± SD. ΔTm represents [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (siRNA2′-O-methyl)]. |
 |
Table 2 Sequences of ssRNAs, siRNAs, and Tm values of siRNAs
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2-O-methylnucleosides, respectively. The Tm values were determined using 3 μM dsRNA in a buffer containing 10 mM sodium phosphate (pH of 7.0) and 100 mM NaCl. All experiments were performed thrice, and data are presented as the mean ± SD. ΔTm represents [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (siRNA2′-O-methyl)]. |
 |
Table 3 Sequences of ssRNAs, siRNAs, and Tm values of siRNAs
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2′-O-methylnucleosides, respectively. The Tm values were determined using 3 μM dsRNA in a buffer containing 10 mM sodium phosphate (pH of 7.0) and 100 mM NaCl. All experiments were performed thrice, and data are presented as the mean ± SD. ΔTm represents [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (siRNA2′-O-methyl)]. |
 |
Table 4 Sequences of ssRNAs, siRNAs, and Tm values of siRNAs
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2-O-methylnucleosides, respectively. The Tm values were determined using 3 μM dsRNA in a buffer containing 10 mM sodium phosphate (pH of 7.0) and 100 mM NaCl. All experiments were performed thrice, and data are presented as the mean ± SD. ΔTm represents [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (siRNA2′-O-methyl)]. |
 |
Thermal stability of siRNA duplexes
We evaluated the effect of (S)-5′-C-aminopropyl modifications on the thermal stability of siRNAs (siRNA 1–15). Temperature-induced melting was investigated by ultraviolet (UV) spectroscopy in 10 mM sodium phosphate buffer (pH of 7.0) containing 100 mM NaCl. The melting temperature (Tm) values are listed in Tables 1–4. As shown in Tables 1–3, the Tm values of the 2′-O-methyl-modified siRNAs (siRNA 2, 4, 6, 8, 10, 12, and 14) were siRNA 2, 77.4 °C; siRNA 4, 77.4 °C; siRNA 6, 77.7 °C; siRNA 8, 77.6 °C; siRNA 10, 77.4 °C; siRNA 12, 77.8 °C; and siRNA 14, 77.9 °C, respectively. Those of the (S)-5′-C-aminopropyl-2′-O-methyl-modified siRNAs (siRNA 3, 5, 7, 9, 11, 13, and 15) were siRNA 3, 75.7 °C; siRNA 5, 73.3 °C; siRNA 7, 75.2 °C; siRNA 9, 73.9 °C; siRNA 11, 74.1 °C; siRNA 13, 75.1 °C; and siRNA 15, 78.0 °C, respectively. Therefore, the ΔTm [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (dsRNA2′-O-methyl)] values of siRNAs 3, 5, 7, 9, 11, 13, and 15 were calculated to be −1.7, −4.1, −2.5, −3.7, −3.3, −2.7, and 0.0 °C, respectively. These results suggested that the (S)-5′-C-aminopropyl-2′-O-methyl modification at the passenger strand decreased the thermal stability of the siRNA duplex compared to the 2′-O-methyl modification, except for the modification of the 5′-end of the passenger strand.
Next, we measured the Tm of the siRNAs containing 2′-O-methylnucleosides or (S)-5′-C-aminopropyl-2′-O-methylnucleosides at the 3′- and 5′-regions of the passenger strand (siRNA 16–21). As shown in Table 4, the Tm values of the 2′-O-methyl-modified siRNAs (siRNA 16, 18, and 20) were siRNA 16, 77.6 °C; siRNA 18, 77.6 °C; siRNA 20, 78.6 °C, respectively. Those of the (S)-5′-C-aminopropyl-2′-O-methyl-modified siRNAs (siRNAs 17, 19, and 21) were siRNA 17, 76.3 °C; siRNA 19, 76.0 °C; and siRNA 21, 72.6 °C, respectively. The ΔTm [Tm (siRNA(S)-5′-C-aminopropyl-2′-O-methyl) − Tm (dsRNA2′-O-methyl)] values of siRNAs 17, 19, and 21 were calculated as −1.3, −1.6 and −6.0 °C, respectively. Previously, we reported that the incorporation of eight (S)-5′-C-aminopropyl-2′-O-methyluridines in the passenger strand of an siRNA resulted in a change in Tm of −0.8 °C/modification, compared with that in the unmodified siRNA.20 Oppositely, the change in the Tm of siRNA 19 containing eight (S)-5′-C-aminopropyl-2′-O-methylnucleosides was −0.18 °C/modification. These results suggested that the thermal stability of siRNAs can be improved by the consecutive introduction of the analogs.
RNAi activity
We evaluated the RNAi activity of the 2′-O-methyl-modified, (S)-5′-C-aminopropyl-modified, and unmodified siRNAs by a dual-luciferase reporter assay using HeLa cells, in which the target luciferase genes were constitutively expressed. All the siRNAs targeted the Renilla luciferase genes, while the expression of firefly luciferase genes was used as a control. The HeLa cells were transfected with the siRNAs using RNAiMAX, and the expression of both luciferase genes was analyzed after 24 h of incubation. The relative percentages of Renilla and firefly luciferase activities, compared with the controls containing no siRNAs are shown in Fig. 2–7, Tables 5, 6, and S1–S6.†
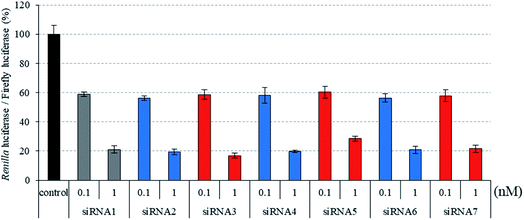 |
| | Fig. 2 RNAi activity of siRNAs modified by three consecutive analogs at the passenger strand. siRNAs were transfected into HeLa cells at concentrations of 0.1 and 1 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using the dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
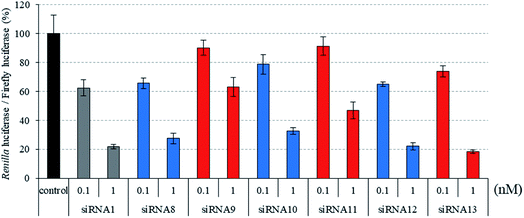 |
| | Fig. 3 RNAi activity of siRNAs modified by three consecutive analogs at the passenger strand. siRNAs were transfected into HeLa cells at concentrations of 0.1 and 1 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using the dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
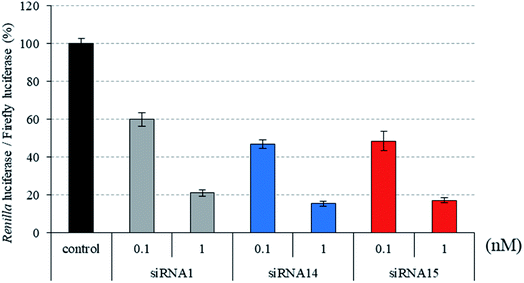 |
| | Fig. 4 RNAi activity of siRNAs modified by three consecutive analogs at the passenger strand. siRNAs were transfected into HeLa cells at concentrations of 0.1 and 1 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using the dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
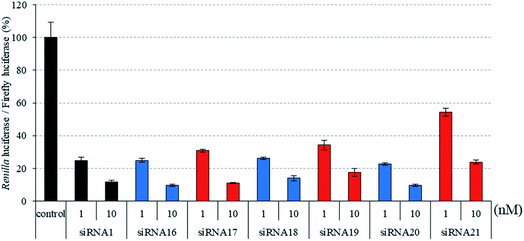 |
| | Fig. 5 RNAi activity of siRNAs modified by three, four, or five consecutive analogs at the 3′- and 5′-regions of the passenger strand. siRNAs were transfected into HeLa cells at concentrations of 1 and 10 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using the dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
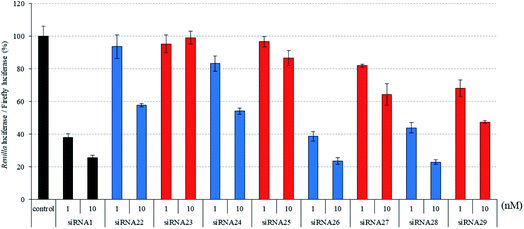 |
| | Fig. 6 RNAi activity of siRNA modified by analogs at the guide strand. siRNAs were transfected into HeLa cells at concentrations of 1 and 10 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using the dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
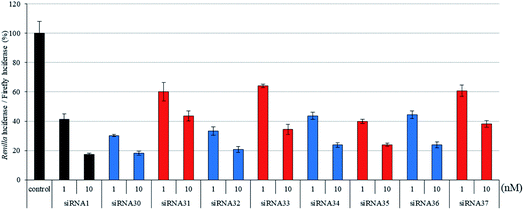 |
| | Fig. 7 RNAi activity of siRNAs modified by analogs at the guide strand. siRNAs were transfected into HeLa cells at concentrations of 1 and 10 nM. After a 24 h incubation, the activities of Renilla and firefly luciferases in the cells were determined using a dual-luciferase reporter assay system. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average of four experiments as the mean ± SD. | |
Table 5 Sequences of ssRNAs and siRNAs used for RNAi activity study
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2-O-methylnucleosides, respectively. |
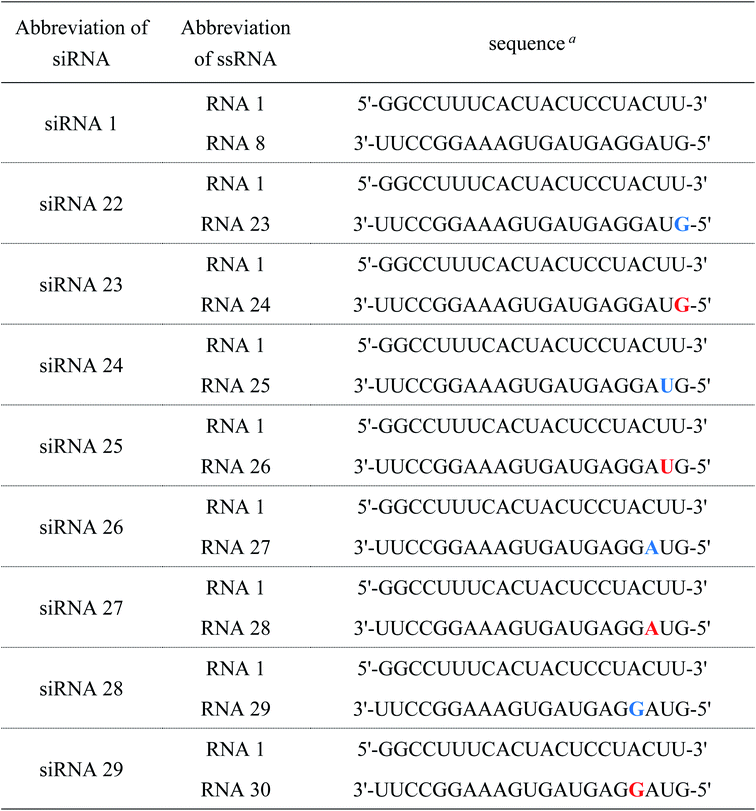 |
Table 6 Sequences of ssRNAs and siRNAs used for RNAi activity study
| Blue and red letters denote 2′-O-methylnucleosides and (S)-5′-C-aminopropyl-2′-O-methylnucleosides, respectively. |
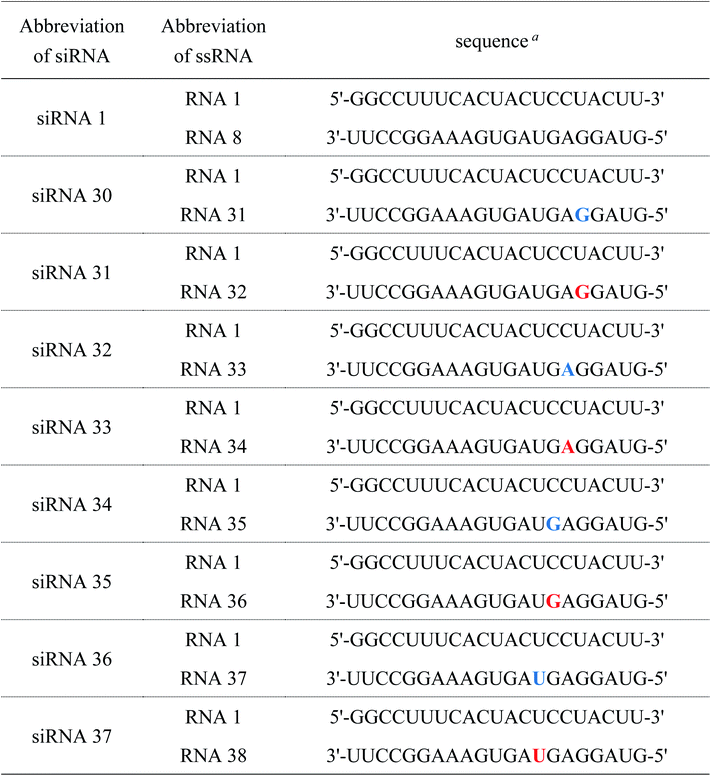 |
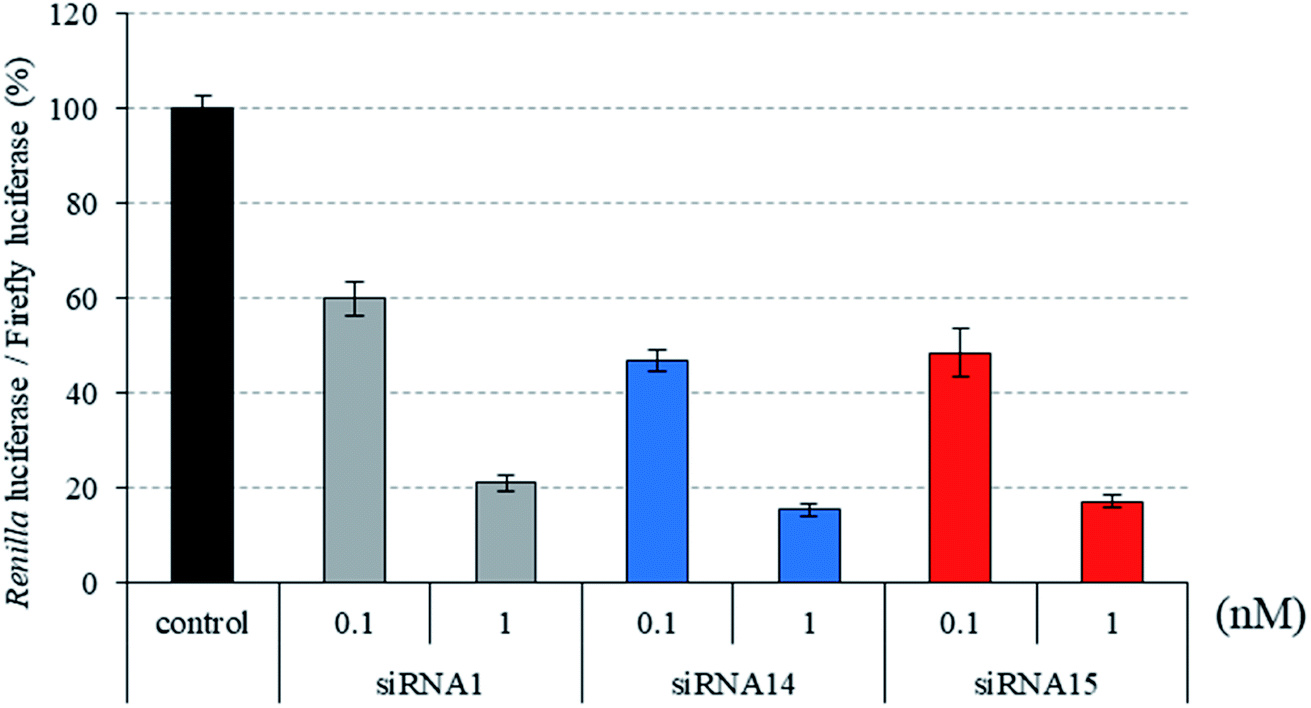
Recently, we reported that siRNAs containing (S)-5′-C-aminopropyl-2′-O-methyluridines at the passenger strand downregulated the expression of the target Renilla luciferase gene with equal or slightly lower activity, compared with the unmodified siRNA. Here, we investigated the position-dependent effects of (S)-5′-C-aminopropyl modifications on the RNAi activity of siRNAs. First, we synthesized modified siRNAs (siRNAs 2–15), which contained three (S)-5′-C-aminopropyl-2′-O-methylnucleosides or three 2′-O-methylnucleosides in succession at every position of the passenger strand. As shown in Fig. 2–4, siRNAs 3, 5, 7, 13, and 15, which contain three consecutive (S)-5′-C-aminopropyl-nucleosides at the 3′- or 5′-regions of the passenger strand, repressed the expression of the Renilla luciferase gene with activities similar to those of the corresponding 2′-O-methyl-modified siRNAs 2, 4, 6, 12, and 14 and the unmodified siRNA 1 at concentrations of 0.1 and 1 nM. Oppositely, the incorporation of three consecutive (S)-5′-C-aminopropyl-nucleosides at the central regions of the passenger strand decreased the silencing activities of siRNAs 9 and 11, compared with the corresponding 2′-O-methyl-modified siRNAs 8 and 10 and the unmodified siRNA 1 at concentrations of 0.1 and 1 nM (Fig. 2). The introduction of (S)-5′-C-aminopropyl-nucleosides at the 3′- or 5′-regions of the passenger strand was well tolerated for eliciting RNAi activity, and the incorporation of (S)-5′-C-aminopropyl-nucleosides at the central regions of the passenger strand decreased the gene silencing activity of the siRNAs. When argonaute-2 and siRNA formed the RNA-induced silencing complex (RISC) in cells, the passenger strand of siRNA was hydrolyzed by the slicer activity of the P-element-induced wimpy testis (PIWI) domain of argonaute-2.31,32 Therefore, it was suggested that siRNAs 9 and 11 were prevented from the hydrolysis of the passenger strand because of the (S)-5′-C-aminopropyl modification near the cleavage site. The results showed that siRNAs containing (S)-5′-C-aminopropyl-nucleosides at the 3′- or 5′-regions of the passenger strand could efficiently suppress the expression of the target gene.
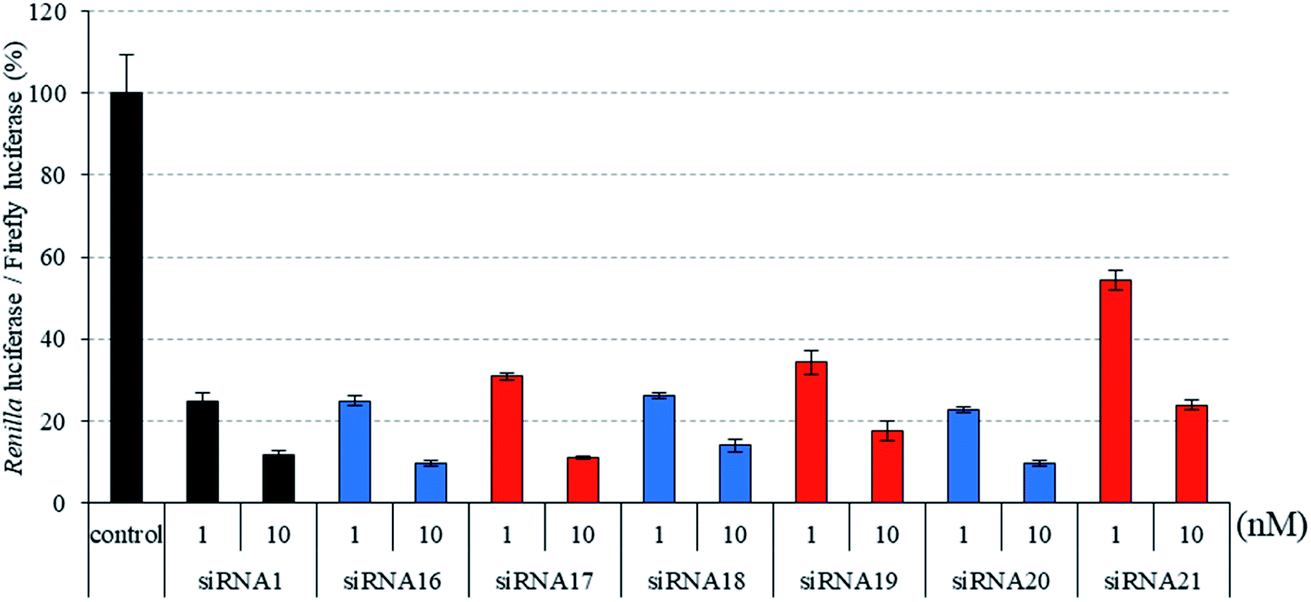
Next, we assessed the silencing activity of siRNAs (siRNA 16–21) containing three, four, or five consecutive analogs at the 3′- and 5′-regions of the passenger strand. As shown in Fig. 5, the RNAi activities of siRNAs 17 and 19, which contained three and four consecutive analogs at the 3′- and 5′-regions, respectively, were equal to or slightly lower than those of the corresponding 2′-O-methyl-modified siRNAs 16 and 18, whereas that of siRNA 21 containing five consecutive analogs was lower than that of the corresponding 2′-O-methyl-modified siRNA 20. These results suggested that the simultaneous incorporation of (S)-5′-C-aminopropyl-nucleosides at the 3′- and 5′-regions of the passenger strand was tolerable for eliciting RNAi activity. However, it was observed that increasing the number of contiguous analogs in the 3′- and 5′-regions of the passenger strand tended to further reduce RNAi activity.
To investigate the position-dependent effects of (S)-5′-C-aminopropyl modifications in the seed region of the siRNA, we synthesized siRNAs containing one (S)-5′-C-aminopropyl-nucleoside at positions 1–8 from the 5′-end of the guide strand and evaluated their silencing activity. It was found that the thermal stability of these siRNA duplexes containing one (S)-5′-C-aminopropyl-nucleoside at the guide strand were similar to those of the corresponding 2′-O-methyl-modified siRNA duplexes (Fig. S5–S7 and Tables S7–S9†). As shown in Fig. 6, incorporation of (S)-5′-C-aminopropyl-nucleoside at positions 1 or 2 was detrimental to the RNAi activity of the siRNAs. Pradeepkumar and Harikrishna reported that the 5′-phosphates of positions 1 and 2 in the guide strand of siRNA interacted with many amino acid residues, such as T526, Y529, Q545, Q548, K550, N551, K566, K570, and R814.33 Thus, it was considered that the (S)-5′-C-aminopropyl modification incorporated at the 5′-end of the guide strand disturbed the important interaction between the guide strand and argonaute-2 protein to form RISC. As shown in Fig. 6 and 7, incorporating the (S)-5′-C-aminopropyl-nucleoside at positions 3, 4, 5, 6, or 8 tended to decrease the RNAi activity of the siRNAs, compared with that of the 2′-O-methyl modification. However, the (S)-5′-C-aminopropyl-modification at position 7 from the 5′-end of the guide strand was well tolerated for RNAi activity. Therefore, to understand why the (S)-5′-C-aminopropyl modification at position 7 of the guide strand was well tolerated for the RNAi activity of siRNA, we performed a modeling study on the interaction between argonaute-2 and the (S)-5′-C-aminopropyl modification in siRNA. As shown in Fig. S8,† a pocket on the protein surface that can accommodate the aminopropyl side-chain existed near position 7 from the 5′-end of the guide strand. Thus, it was considered that the RNAi activity of siRNA 35 containing the analog at position 7 was retained by accommodating the aminopropyl side chain in the pocket on the argonaute-2 protein.
Serum stability
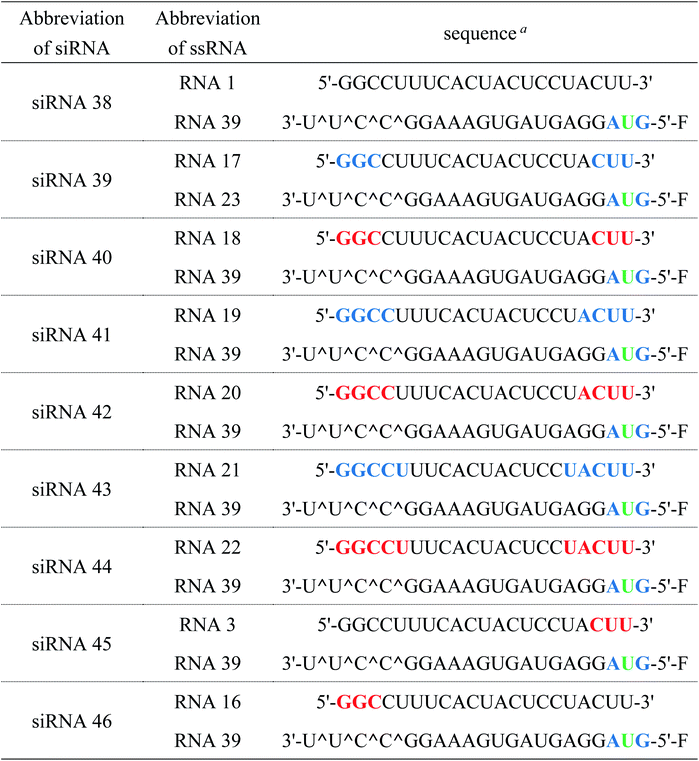
Chemical modifications such as 2′-O-methyl-RNA and 2′-fluoro-RNA are essential for siRNA-based therapeutics because unmodified siRNAs are rapidly degraded by nucleases in serum. Thus, to investigate the effects of (S)-5′-C-aminopropyl modifications at the 3′- and 5′-regions of the passenger strand on the serum stability of the siRNAs, we assessed the stability of the modified siRNAs in a buffer containing bovine serum. Chemical modifications at the guide strand of siRNA are important for achieving serum stability. Considering that the introduction of the (S)-5′-C-aminopropyl modification at the guide strand decreased the RNAi activity of siRNAs by disturbing the interaction with the argonaute-2 protein, we used siRNAs comprising the guide strand containing phosphorothioate, 2′-O-methyl, and 2′-fluoro modifications in this study. The fluorescein-labeled siRNAs 38–44 (Table 7) were incubated in a buffer containing 50% bovine serum, and the reaction mixtures at various incubation times (0, 15, and 30 min and 1, 3, 6, 12, and 24 h) were analyzed by PAGE.
As shown in Fig. 8, when the (S)-5′-C-aminopropyl-modified siRNAs 40, 42, and 44 were used, the bands corresponding to the full-length of the siRNAs remained intact even after 24 h of incubation. With the unmodified siRNA 38 or the 2′-O-methyl-modified siRNAs 39, 41, and 43, it was observed that the bands corresponding to the full-length of the siRNAs gradually degraded and became faint. Thus, it was observed that the (S)-5′-C-aminopropyl modifications enhanced the serum stability of the siRNA, compared with the 2′-O-methyl modifications.
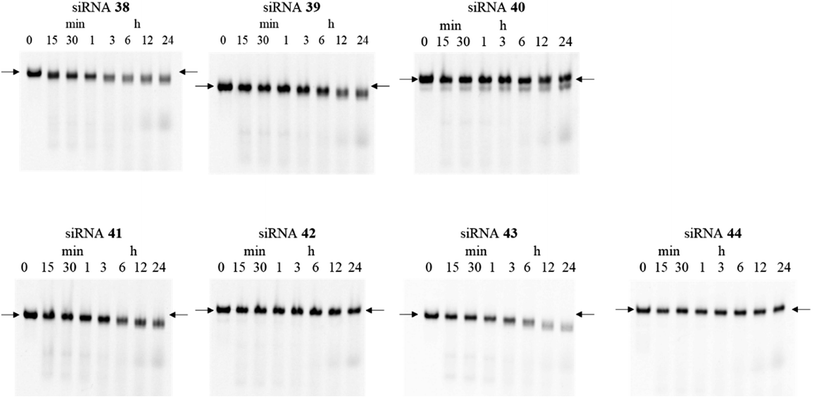 |
| | Fig. 8 Polyacrylamide gel electrophoresis (PAGE) analysis donated stability of siRNA (4.5 μM) in 50% bovine serum. Fluorescein-labeled siRNAs 38–44 (600 pmol) were incubated in a buffer containing 50% bovine serum. Subsequently, the reaction mixtures at various incubation times (0, 15, and 30 min and 1, 3, 6, 12, and 24 h) were analyzed by nondenaturing PAGE. Arrows refer to the full-length band of the siRNAs. | |
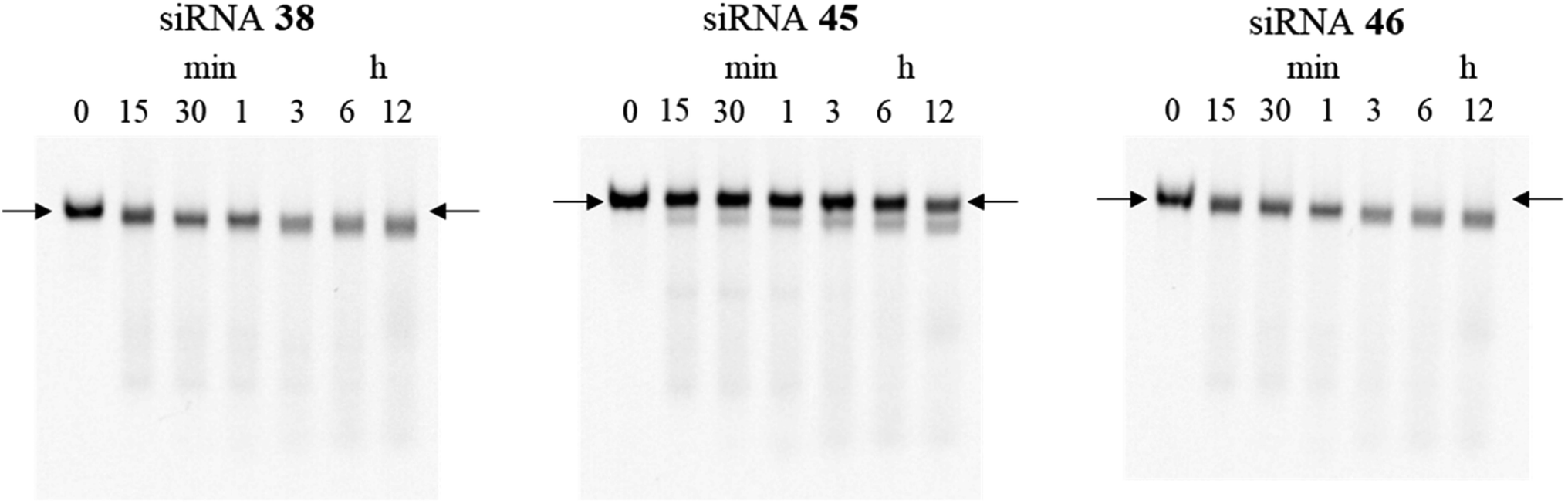
Furthermore, to investigate the effect of the site-specific incorporation of the (S)-5′-C-aminopropyl modifications on serum stability, we evaluated the stability of siRNAs 45 and 46 containing three consecutive (S)-5′-C-aminopropyl-nucleosides at the 3′- or 5′-regions of the passenger strand in 50% bovine serum. As shown in Fig. 9, when siRNA 45 with three consecutive (S)-5′-C-aminopropyl-nucleosides at the 3′-region was used, the band corresponding to the full-length siRNA remained intact even after 12 h of incubation, whereas with siRNA 46 with three consecutive (S)-5′-C-aminopropyl-nucleosides at the 5′-region, the band corresponding to the full-length siRNA gradually degraded and became faint. These results indicated that incorporating the (S)-5′-C-aminopropyl modification at the 3′-region of siRNA effectively improved the resistance toward the nuclease in serum, compared with that of the analog at the 5′-region of siRNA. Previously, Gait et al. reported that siRNA degradation in serum occurred similar to RNase A-like mechanism, cleaving at UpA sequences close to the end of each strand in the siRNA.34 siRNA 38 used in this study contains the UpA sequence at the 3′-end region of the passenger strand. The siRNAs were considered to be degraded at the UpA site in bovine serum. siRNAs 42 and 44 contained the (S)-5′-C-aminopropyl-uridine analog instead of the U of the UpA site; therefore, they were hardly degraded. Thus, the incorporation of the (S)-5′-C-aminopropyl modification at the most vulnerable degradation site of siRNA such as the UpA sequence near the 3′-end region of the strands would be useful for improving the biological stability of siRNAs in mammalian serum.
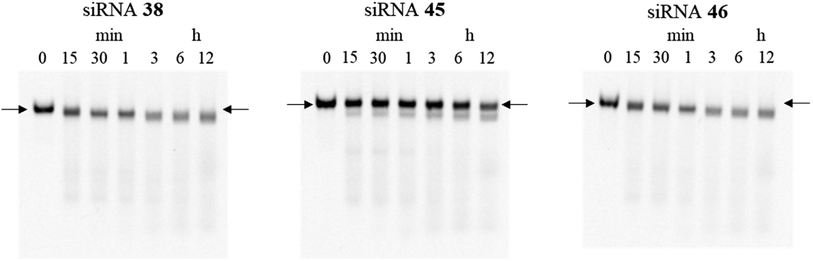 |
| | Fig. 9 Polyacrylamide gel electrophoresis (PAGE) analysis donated stability of siRNA (4.5 μM) in 50% bovine serum. Fluorescein-labeled siRNAs 38, 45, and 46 (600 pmol) were incubated in a buffer containing 50% bovine serum. Subsequently, the reaction mixtures at various incubation times (0, 15, and 30 min and 1, 3, 6, and 12 h) were analyzed by nondenaturing PAGE. Arrows refer to the full-length band of the siRNAs. | |
Table 7 Sequences of ssRNAs and siRNAs for serum stability test
| F, blue letters, green letters, red letters, and ^ denote fluorescein, 2′-O-methylnucleosides, 2′-fluoro-nucleosides, (S)-5′-C-aminopropyl-2′-O-methylnucleosides, and phosphorothioate linkages, respectively. |
 |
Quantitative reverse-transcriptional PCR (RT-qPCR) analysis
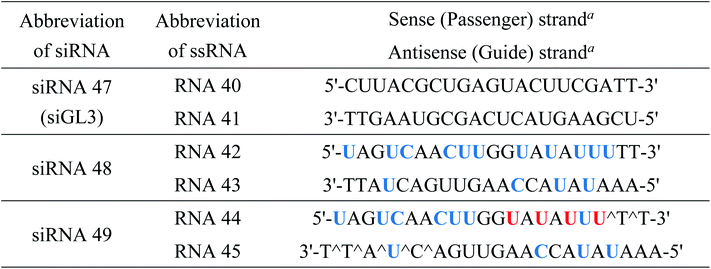
We also examined the effect of the (S)-5′-C-aminopropyl modifications on RNAi activity in another siRNA targeting human KNTC2 gene, which is considered to be a therapeutic target for several cancers.35,36 We synthesized a modified KNTC2-siRNA (siRNA 49) which contained four (S)-5′-C-aminopropyl methyluridines at the 3′-regions of the passenger strand (Table 8) and investigated its RNAi activity on the KNTC2 mRNA level in human colon cancer HCT116 cells (Fig. 10). A quantitative reverse-transcriptional PCR (RT-qPCR) analysis revealed that the RNAi activity of the (S)-5′-C-aminopropyl modified siRNA (siRNA 49) was nearly equal to that of the corresponding 2′-O-methyl-modified KNTC2-siRNA (siRNA 48). This result supports our mention that the simultaneous incorporation of (S)-5′-C-aminopropyl-nucleosides at the 3′-regions of the passenger strand was tolerable for eliciting RNAi activity.
Table 8 Sequences of ssRNAs and siRNAs for RT-qPCR analysis
| Blue letters, red letters, and ^ denote 2′-O-methylnucleosides, (S)-5′-C-aminopropyl-2′-O-methylnucleosides, and phosphorothioate linkages, respectively. |
 |
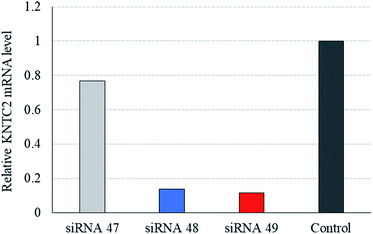 |
| | Fig. 10 RNAi activity of KNTC2 siRNA modified by (S)-5′-C-aminopropyl-2′-O-methylnucleosides. siRNAs targeting KNTC2 gene (siRNA 48, siRNA 49) or firefly luciferase gene (siRNA 47) were transfected to HCT116 cells at a concentration of 10 nM. After a 24 hours incubation, cells were replated in fresh cell culture media and were further incubated for another 24 hours. Then, total RNA of the cells was extracted and KNTC2 mRNA levels were determined by RT-qPCR. Data were normalized to the level of ACTB mRNA and are presented as relative mRNA levels compared to the un-transfected control group (control). Data represent the mean of two independent experiments. | |
Conclusion
Here, we successfully synthesized (S)-5′-C-aminopropyl-2′-O-methyladenosine phosphoramidite (22) and (S)-5′-C-aminopropyl-2′-O-methylguanosine phosphoramidite (33). It was observed that the introduction of (S)-5′-C-aminopropyl-nucleosides at the 3′- and/or 5′-regions of the passenger strand of siRNA was tolerable for the RNAi activity of the siRNAs, whereas the incorporation of analogs at the central region of the passenger strand of siRNA was detrimental to the RNAi activity. Furthermore, the (S)-5′-C-aminopropyl modifications at the passenger strand thermally destabilized siRNA duplexes, compared with the corresponding 2′-O-methyl modifications, except for the consecutive incorporation of the (S)-5′-C-aminopropyl modifications at the 5′-region of the passenger strand. Finally, it was revealed that the incorporation of the (S)-5′-C-aminopropyl modifications at the vulnerable degradation sites of siRNA effectively enhanced the serum stability of siRNA. Therefore, incorporating the (S)-5′-C-aminopropyl-2′-O-methylnucleosides into appropriate sites of siRNAs would be useful for improving the serum stability of therapeutic siRNA molecules without affecting their RNAi activities.
Experimental section
General remark
All chemicals and dry solvents (THF, DMF, CH2Cl2, CH3CN, toluene and pyridine) were obtained from commercial sources and used without any further purification. Thin layer chromatography (TLC) was performed on silica gel plates precoated with fluorescent indicator with visualization by UV light or by dipping into a solution of 5% (v/v) concentrated H2SO4 in mixture of p-anisaldehyde and methanol and then heating. Silica gel (63–210 mesh) was used for column chromatography. 1H NMR (400 or 600 MHz), 13C {1H}NMR (101 or 151 MHz), 31P NMR (162 MHz) were recorded on 400 or 600 MHz NMR equipment. CDCl3 or DMSO-d6 was used as a solvent for obtaining NMR spectra. Chemical shifts (δ) are given in parts per million (ppm) from CDCl3 (7.26 ppm), DMSO-d6 (2.50 ppm) for 1H NMR spectra and from CDCl3 (77.2 ppm) DMSO-d6 (39.5 ppm) for 13C {1H}NMR spectra. The abbreviations s, d, t, q, and m signify singlet, doublet, triplet, quadruplet, and multiplet, respectively. High resolution mass spectra (HRMS) were obtained in positive ion electrospray ionization (ESI-TOF) mode.
Experimental procedures
(S)-5-C-Allyl-1,2-O-isopropylidene-3-O-[(1,1-dimethylethyl)diphenylsilyl]-α-D-ribose (6) and (S)-5-C-allyl-1,2-O-isopropylidene-3-O-[(1,1-dimethylethyl)diphenylsilyl]-α-D-ribose (7). To a solution of 5 in CH2Cl2 (50 mL) cooled to −40 °C was added BF3·OEt2 (1.27 mL, 10.12 mmol), allyltrimethylsirane (1.28 mL, 8.09 mmol) under argon atmosphere. After 30 min at −40 °C, the reaction mixture was quenched with saturated NaHCO3 aqueous. The organic layer was washed with saturated NaHCO3 aqueous and brine, dried (Na2SO4). After concentration, the residue was purified by a silica gel column chromatography (hexane:EtOAc = 5:1) to afford product as a colorless oil (1.94 g, 4.14 mmol, 82%, S:R = 4:1). Compound 6: 1H NMR (400 MHz, CDCl3): δ 7.78–7.76 (m, 2H), 7.71–7.69 (m, 2H), 7.47–7.35 (m, 6H), 5.89–5.78 (m, 1H), 5.53 (d, J = 3.6 Hz, 1H), 5.14–5.11 (m, 2H), 4.08 (dd, J = 8.6, 4.0 Hz, 1H), 3.99 (dd, J = 8.8, 1.2 Hz, 1H), 3.94 (t, J = 4.0 Hz, 1H), 3.69–3.64 (m, 1H), 2.41–2.32 (m, 2H), 1.56 (s, 3H), 1.50 (d, J = 8.4 Hz, 1H), 1.23 (s, 3H), 1.10 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 136.2, 136.0, 134.8, 133.7, 133.5, 130.1, 130.0, 127.9, 127.6, 117.8, 112.7, 104.0, 81.4, 79.2, 72.7, 68.3, 39.6, 27.1, 27.0, 26.5, 19.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C27H36NaO5Si 491.2230, found 491.2229.Compound 7: 1H NMR (400 MHz, CDCl3): δ 7.79–7.71 (m, 4H), 7.47–7.34 (m, 6H), 5.88–5.77 (m, 1H), 5.47 (d, J = 3.6 Hz, 1H), 5.11–5.07 (m, 2H), 4.10–4.09 (m, 2H), 3.89–3.84 (m, 1H), 3.79–3.77 (m, 1H), 2.31–2.27 (m, 2H), 1.98 (d, J = 3.6 Hz, 1H), 1.54 (s, 3H), 1.14 (s, 3H), 1.10 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 136.4, 136.1, 135.0, 133.7, 133.4, 130.1, 130.0, 127.9, 127.5, 117.8, 112.5, 103.4, 81.4, 79.1, 73.5, 70.6, 36.8, 27.1, 26.9, 26.3, 19.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C27H36NaO5Si 491.2230, found 491.2223.
(S)-5-C-Allyl-3-O-benzyl-5-O-[(1,1-dimethylethyl)diphenylsilyl]-1,2-O-isopropylidene-α-D-ribose (8). To a solution of 6 (1.00 g, 2.13 mmol) in DMF (10 mL) was added NaH (0.17 g, 4.25 mmol) under argon atmosphere. After 30 min, to a reaction mixture was added BnBr (0.50 mL, 4.25 mmol) at 0 °C, stirred for 16 h at room temperature. After the reaction mixture was quenched with CH3OH, the mixture was washed with saturated NaHCO3 aqueous and brine, dried (Na2SO4). After concentration, the residue was purified by a silica gel column chromatography (hexane:EtOAc = 15:1) to afford desired product 8 as a colorless oil (0.92 g, 1.65 mmol, 78%). 1H NMR (600 MHz, CDCl3): δ 7.72–7.69 (m, 4H), 7.43–7.28 (m, 11H), 5.80 (d, J = 3.6 Hz, 1H), 5.52–5.45 (m, 1H), 4.87–4.82 (m, 2H), 4.65 (d, J = 11.4 Hz, 1H), 4.56 (t, J = 3.0 Hz, 1H), 4.38 (d, J = 12.0 Hz, 1H), 4.10 (dd, J = 9.0, 1.8 Hz, 1H), 4.02 (dd, J = 8.4, 4.2 Hz, 1H), 3.92 (ddd, J = 9.6, 4.8, 1.8 Hz, 1H), 1.53 (s, 3H), 1.37 (s, 3H), 1.00 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 138.0, 136.1, 136.0, 134.4, 134.1, 133.9, 129.8, 129.7, 128.5, 128.1, 127.9, 127.7, 127.6, 117.8, 113.2, 104.5, 80.2, 78.2, 78.1, 72.1, 71.9, 38.7, 27.2, 27.1, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C34H42NaO5Si 581.2699, found 581.2677.
3-O-Benzyl-(S)-5-C-hydroxypropyl-5-O-[(1,1-dimethylethyl)diphenylsilyl]-1,2-O-isopropylidene-α-D-ribose (9). Under argon atmosphere, 9-borabicyclo[3.3.1]nonane (9-BBN, 0.5 M in THF, 9.50 mL) was added dropwise to a solution of compound 8 (0.92 g, 1.65 mmol) in THF (16 mL) and stirred for 13 h at room temperature. Water was added to the reaction mixture until evolution of gas ceased. 3 N NaOH solution (3.6 mL) was added, and then, slowly 30% aqueous hydrogen peroxide solution (1.87 mL) was added while keeping the temperature between 40 °C. The mixture was stirred and extracted with water and ethyl acetate. The organic layer was washed with neutral phosphate buffer solution and brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 9 as a white solid (0.86 g, 1.50 mmol, 91%). 1H NMR (600 MHz, CDCl3): δ 7.72–7.69 (m, 4H), 7.43–7.29 (m, 11H), 5.77 (d, J = 3.6 Hz, 1H), 4.68 (d, J = 10.8 Hz, 1H), 4.57 (t, J = 3.6 Hz, 1H), 4.38 (d, J = 12.0 Hz, 1H), 4.11 (dd, J = 8.4, 2.4 Hz, 1H), 3.96 (dd, J = 8.7, 4.8 Hz, 1H), 3.93–3.90 (m, 1H), 3.30–3.27 (m, 2H), 1.70–1.63 (m, 2H), 1.56 (s, 3H), 1.44–1.39 (m, 2H), 1.37 (s, 3H), 1.02 (s, 9H), 0.98 (bs, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 137.9, 136.1, 134.4, 134.0, 129.8, 129.7, 128.5, 128.1, 128.0, 127.7, 127.6, 113.0, 104.4, 80.7, 78.2, 78.0, 72.1, 72.1, 62.7, 30.0, 28.5, 27.2, 27.2, 27.0, 19.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C34H44NaO6Si 599.2805, found 599.2783.
3-O-Benzyl-(S)-5-O-[(1,1-dimethylethyl) diphenylsilyl]-1,2-O-isopropylidene-5-C-p-toluenesulfonyloxypropyl-α-D-ribose (10). Under argon atmosphere, p-TsCl (5.98 g, 31.36 mmol) and pyridine (5.2 mL, 62.71 mmol) were added to a solution of compound 9 (5.17 g, 8.96 mmol) in CH2Cl2 (52 mL) at 0 °C. The mixture was stirred for 16 h at room temperature. The mixture was extracted with CHCl3 and saturated NaHCO3 aqueous solution; organic layer was washed with brine. The organic layer was dried over N2SO4, filtered, and concentrated. The crude material was purified by column chromatography (17% ethyl acetate in hexane) to afford desired product 10 as a colorless oil (6.19 g, 8.47 mmol, 95%). 1H NMR (600 MHz, CDCl3): δ 7.67–7.65 (m, 6H), 7.42–7.39 (m, 2H), 7.34–7.26 (m, 11H), 5.71 (d, J = 3.0 Hz, 1H), 4.64 (d, J = 10.8 Hz, 1H), 4.52 (t, J = 4.2 Hz, 1H), 4.33 (d, J = 10.8 Hz, 1H), 4.00 (dd, J = 8.4, 2.4 Hz, 1H), 3.84–3.81 (m, 2H), 3.73–3.65 (m, 2H), 2.43 (s, 3H), 1.58–1.55 (m, 1H), 1.53 (s, 3H), 1.53–1.48 (m, 2H), 1.43–1.37 (m, 1H), 1.35 (s, 3H), 1.00 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 144.6, 137.7, 136.1, 136.0, 134.1, 133.8, 133.2, 129.9, 129.8, 129.8, 128.5, 128.2, 128.0, 128.0, 127.7, 113.0, 104.3, 80.8, 78.1, 77.8, 72.0, 71.9, 70.5, 29.8, 27.1, 26.9, 24.6, 21.7, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C41H50NaO8SSi 753.2893, found 753.2886.
(S)-5-C-azidopropyl-3-O-benzyl-5-O-[(1,1-dimethylethyl)diphenylsilyl]-1,2-O-isopropylidene-α-D-ribose (11). Under argon atmosphere, NaN3 (4.63 g, 71.15 mmol) was added to a solution of compound 10 (6.19 g, 8.47 mmol) in DMF (62 mL). The mixture was stirred for 8 h at 60 °C. The mixture was extracted with ethyl acetate and brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (9% ethyl acetate in hexane) to afford desired product 11 as a colorless oil (4.57 g, 7.59 mmol, 81%) 1H NMR (600 MHz, CDCl3): δ 7.71–7.68 (m, 4H), 7.43–7.40 (m, 2H), 7.36–7.30 (m, 9H), 5.76 (d, J = 3.6 Hz, 1H), 4.68 (d, J = 10.8, 1H), 4.56 (t, J = 4.2 Hz, 1H), 4.37 (d, J = 10.8, 1H), 4.07 (dd, J = 8.1, 2.4 Hz, 1H), 3.92 (dd, J = 8.7, 4.2 Hz, 1H), 3.89–3.87 (m, 1H), 2.92–2.86 (m, 2H), 1.65–1.63 (m, 1H), 1.56 (s, 3H), 1.45–1.40 (m, 3H), 1.36 (s, 3H), 1.02 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 137.8, 136.1, 136.1, 134.3, 133.9, 129.8, 129.8, 128.5, 128.1, 128.0, 127.7, 127.7, 113.0, 104.4, 80.8, 78.2, 77.9, 72.1, 72.0, 51.3, 31.0, 27.2, 27.0, 24.7, 19.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C34H43N3NaO5Si 624.2870, found 624.2843.
(S)-5-C-Azidopropyl-3-O-benzyl-1,2-O-diacetyl-5-O-[(1,1-dimethylethyl)diphenylsilyl]-β-D-ribose (12). To a solution of 11 in CH2Cl2 (20 mL) was added 50% CF3CO2H aqueous solution (120 mL) and stirred for 4.5 h at room temperature. The mixture was washed with saturated NaHCO3 aqueous and brine, dried (Na2SO4). After concentration, the residue was dissolved in pyridine (130 mL). Acetic anhydride (32.6 mL, 350.8 mmol) was added to the mixture under argon atmosphere. After the reaction mixture was stirred for 20 h at room temperature, the mixture was washed with saturated NaHCO3 aqueous and brine, dried (Na2SO4). After concentration, the residue was purified by a silica gel column chromatography (hexane:EtOAc = 4:1) to afford desired product 12 as a colorless oil (11.89 g, 18.41 mmol, 81%). β-anomer: 1H NMR (600 MHz, CDCl3): δ 7.70–7.68 (m, 4H), 7.45–7.29 (m, 10H), 6.16 (s, 1H), 5.35 (d, J = 4.2 Hz, 1H), 4.57 (d, J = 10.8 Hz, 1H), 4.43 (dd, J = 8.4, 4.2 Hz, 1H), 4.33 (d, J = 10.8 Hz, 1H), 4.09 (dd, J = 8.4, 3.6 Hz, 1H), 3.86–3.84 (m, 1H), 2.89–2.75 (m, 2H), 2.13 (s, 3H), 1.97 (s, 3H), 1.67–1.60 (m, 1H), 1.47–1.40 (m, 1H), 1.36–1.25 (m, 2H), 1.06 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3) δ 170.0, 169.4, 137.3, 136.0, 136.0, 134.3, 133.5, 130.0, 129.9, 128.6, 128.2, 128.1, 127.8, 127.7, 98.2, 83.2, 76.7, 74.0, 73.4, 72.2, 51.3, 30.6, 27.1, 24.4, 21.3, 20.9, 19.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C35H43N3NaO7Si 688.2768, found 688.2769.
2′-O-Acetyl-(S)-5′-C-azidopropyl-6-N-benzoyl-3′-O-benzyl-5′-O-[(1,1-dimethylethyl)diphenylsilyl]adenosine (13). Under argon atmosphere, 1 M SnCl4 in CH2Cl2 (14.69 mL, 14.69 mmol) was added to a solution of 12 (6.32 g, 9.79 mmol) and N6-benzoyl adenine (2.81 g, 11.75 mmol) in CH3CN (63 mL) at −20 °C. After the reaction mixture was stirred for 2 h at room temperature, the mixture was quenched with saturated NaHCO3 aqueous. The mixture was extracted with saturated NaHCO3 aqueous and brine, dried (Na2SO4). After concentration, the residue was purified by a silica gel column chromatography (hexane:EtOAc = 1:1) to afford desired product 13 as a white form (5.65 g, 6.85 mmol, 70%). 1H NMR (600 MHz, CDCl3): δ 9.07 (s, 1H), 8.77 (s, 1H), 8.22 (s, 1H), 8.04 (d, J = 7.2 Hz, 2H), 7.62–7.58 (m, 5H), 7.53 (t, J = 7.2 Hz, 2H), 7.41–7.25 (m, 11H), 6.15 (d, J = 4.8 Hz, 1H), 5.82 (t, J = 4.2 Hz, 1H), 4.63 (t, J = 5.4 Hz, 1H), 4.60 (d, J = 11.4 Hz, 1H), 4.38 (d, J = 11.4 Hz, 1H), 4.13 (dd, J = 6.3, 3.6 Hz, 1H), 3.82–3.79 (m, 1H), 2.94–2.83 (m, 2H), 2.12 (s, 3H), 1.75–1.70 (m, 1H), 1.44–1.28 (m, 3H), 1.05 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 170.0, 164.7, 153.0, 151.6, 149.7, 141.7, 137.2, 136.0, 135.9, 133.8, 133.5, 133.1, 132.9, 130.0, 129.9, 129.0, 128.7, 128.3, 128.3, 128.1, 128.0, 127.8, 127.7, 123.5, 86.6, 84.2, 76.5, 74.5, 73.6, 72.8, 51.2, 30.6, 27.2, 24.6, 20.8, 19.5; HRMS (ESI-TOF) m/z: [M + K]+ calcd for C45H48KN8O6Si 863.3103, found 863.3113.
(S)-5′-C-Azidopropyl-6-N-benzoyl-3′-O-benzyl-5′-O-[(1,1-dimethylethyl)diphenylsilyl]adenosine (14). K2CO3 (1.89 g, 13.70 mmol) was added to a solution of compound 13 (5.65 g, 6.85 mmol) in methanol (56 mL), and the mixture was stirred for 30 min at 0 °C. The mixture was extracted with ethyl acetate and water; the organic layer was washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 14 as a white solid (4.96 g, 6.33 mmol, 92%). 1H NMR (400 MHz, CDCl3): δ 8.97 (s, 1H), 8.76 (s, 1H), 8.15 (s, 1H), 8.05–8.03 (m, 2H), 7.66–7.53 (m, 7H), 7.44–7.28 (m, 11H), 5.98 (d, J = 5.2 Hz, 1H), 4.70 (dd, J = 11.4, 6.0 Hz, 1H), 4.65 (d, J = 11.2 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 4.36 (t, J = 4.8 Hz, 1H), 4.11 (t, J = 3.6 Hz, 1H), 3.77–3.73 (m, 1H), 3.45 (d, J = 6.0 Hz, 1H), 2.99–2.83 (m, 2H), 1.74–1.68 (m, 1H), 1.45–1.38 (m, 2H), 1.30–1.26 (m, 1H), 1.02 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.7, 152.8, 151.6, 149.7, 141.7, 136.9, 135.9, 135.9, 133.8, 133.5, 133.0, 132.9, 130.1, 130.0, 129.0, 128.9, 128.7, 128.5, 128.0, 127.8, 127.7, 123.4, 88.8, 84.3, 78.0, 74.3, 73.4, 73.1, 51.2, 30.5, 27.1, 24.6, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C43H46N8NaO5Si 805.3258, found 805.3243.
(S)-5′-C-Azidopropyl-6-N-benzoyl-3′-O-benzyl-5′-O-[(1,1-dimethylethyl)diphenylsilyl]-2-O-methyladenosine (15). NaH (0.63 g, 15.66 mmol) was added to a solution of compound 14 (4.09 g, 5.22 mmol) in THF (41 mL) at 0 °C. Then, CH3I (1.6 mL, 26.10 mmol) was added in dropwise and stirred 2.5 hours at 0 °C. Saturated NaHCO3 aqueous solution was added, then the mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 15 as a white solid (2.99 g, 3.75 mmol, 72%). 1H NMR (600 MHz, CDCl3): δ 9.11 (s, 1H), 8.79 (s, 1H), 8.33 (s, 1H),8.04–8.03 (m, 2H), 7.65–7.60 (m, 5H), 7.54–7.52 (m, 2H), 7.43–7.30 (m, 11H), 6.18 (d, J = 4.2 Hz, 1H), 4.61 (d, J = 11.4 Hz, 1H), 4.48 (d, J = 11.4 Hz, 1H), 4.42 (t, J = 4.8 Hz, 1H), 4.36 (t, J = 5.4 Hz, 1H), 4.20 (dd, J = 5.7, 2.4 Hz, 1H), 3.85–3.82 (m, 1H), 3.48 (s, 3H), 2.93–2.85 (m, 2H), 1.83–1.75 (m, 1H), 1.48–1.39 (m, 2H), 1.30–1.23 (m, 1H), 1.06 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.7, 152.9, 151.5, 149.6, 141.7, 137.4, 136.0, 135.9, 133.9, 133.5, 132.9, 130.1, 130.0, 129.0, 128.6, 128.3, 128.0, 127.9, 127.8, 123.6, 86.8, 83.7, 82.4, 76.2, 72.9, 72.7, 58.7, 51.2, 30.8, 27.3, 27.2, 24.6, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for 819.3415, found 819.3417.
(S)-5′-C-Azidopropyl-6-N-benzoyl-3′-O-benzyl-2′-O-methyladenosine (16). n-Tetrabuthylammonium fluoride (TBAF, 1.35 mL of a 1 M solution in THF) was added to a solution of compound 15 (0.72 g, 0.90 mmol) in THF (7.2 mL) at room temperature under argon atmosphere. After being stirred at room temperature for 16 h, the mixture was concentrated. The residue was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 16 as a white solid (0.46 g, 0.83 mmol, 92%). 1H NMR (600 MHz, CDCl3): δ 9.19 (s, 1H), 8.74 (s, 1H), 8.04 (s, 1H), 8.02–8.00 (m, 2H), 7.61–7.59 (m, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.14–7.32 (m, 5H), 5.99 (d, J = 7.2 Hz, 1H), 5.78 (d, J = 12.0 Hz, 1H), 4.74 (d, J = 12.0 Hz, 1H), 4.71 (d, J = 11.4 Hz, 1H), 4.68 (dd, J = 7.8, 4.8 Hz, 1H), 4.32 (d, J = 4.8 Hz, 1H), 4.27 (s, 1H), 3.61–3.57 (m, 1H), 3.29 (s, 3H), 3.27–3.26 (m, 2H), 1.79–1.75 (m, 1H), 1,66–1.62 (m, 2H), 1.58–1.53 (m, 1H); 13C {1H} (151 MHz, CDCl3): δ 164.7, 152.0, 150.5, 143.4, 137.4, 133.5, 133.1, 129.0, 128.8, 128.4, 128.3, 128.0, 124.9, 89.8, 87.7, 81.7, 77.6, 72.7, 72.1, 58.6, 51.4, 31.1, 25.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C28H30N8NaO5 581.2237, found 581.2238.
(S)-5′-C-Azidopropyl-6-N-benzoyl-2′-O-methyladenosine (17). Under argon atmosphere, BCl3 (4.96 mL of a 1 M solution in CH2Cl2) was added to a solution of compound 16 (0.46 g, 0.83 mmol) in CH2Cl2 (6.9 mL) at −78 °C. After the mixture was stirred at −78 °C for 3 h, the reaction mixture was quenched with 50% Et3N in EtOH. After concentration, the residue was purified by column chromatography (5% methanol in CHCl3) to afford desired product 17 as a white solid (0.35 g, 0.74 mmol, 89%). 1H NMR (400 MHz, CDCl3): δ 9.04 (s, 1H), 8.80 (s, 1H), 8.07 (s, 1H), 8.04–8.02 (m, 2H), 7.66–7.62 (m, H), 7.55 (t, J = 7.6 Hz, 2H), 5.93 (d, J = 7.6 Hz, 1H), 5.78 (d, J = 12.0 Hz, 1H), 4.74 (dd, J = 7.2, 4.4 Hz, 1H), 4.58 (d, J = 4.4 Hz, 1H), 4.28 (s, 1H), 3.80–3.74 (m, 1H), 3.37 (s, 3H), 3.31 (t, J = 6.4 Hz, 2H), 2.72 (d, J = 1.2 Hz, 1H), 1.85–1.80 (m, 1H), 1.72–1.60 (m, 3H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.8, 152.2, 150.7, 150.6, 143.3, 133.5, 133.2, 129.1, 128.1, 124.8, 89.5, 89.4, 82.3, 71.9, 71.3, 59.0, 51.4, 31.0, 25.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C21H24N8NaO5 491.1767, found 491.1781.
(S)-5′-C-Azidopropyl-6-N-benzoyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-O-methyladenosine (18). Under argon atmosphere, imidazole (1.16 g, 17.08 mmol), TBDPSCl (1.48 mL, 5.69 mmol) was added to a solution of 17 (2.05 g, 4.38 mmol) in DMF (20 mL) at 0 °C, and the mixture was stirred for 24 h at 0 °C. The mixture was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 18 as a white solid (2.36 g, 3.34 mmol, 76%). 1H NMR (600 MHz, CDCl3): δ 9.14 (s, 1H), 8.70 (s, 1H), 8.13 (s, 1H), 8.02 (d, J = 7.2 Hz, 2H), 7.75–7.70 (m, 4H), 7.62–7.60 (m, 1H), 7.53–7.40 (m, 8H), 6.06 (d, J = 6.6 Hz, 1H), 5.64 (d, J = 11.4 Hz, 1H), 4.56–4.54 (m, 2H), 4.00 (d, J = 1.2 Hz, 1H), 3.15 (s, 3H), 3.15–3.12 (m, 2H), 2.95–2.92 (m, 1H), 1.54–1.50 (m, 1H), 1.47–1.42 (m, 2H), 1.31–1.24 (m, 1H), 1.16 (s, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.6, 152.0, 150.6, 150.5, 143.5, 136.1, 136.0, 133.6, 133.5, 133.1, 133.0, 130.3, 130.2, 129.0, 128.1, 128.0, 128.0, 124.9, 90.6, 89.7, 82.1, 73.0, 71.0, 58.9, 51.2, 30.9, 27.1, 25.3, 19.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C37H42N8NaO5Si 729.2945, found 729.2958.
(S)-5′-C-Azidopropyl-6-N-benzoyl-5′-O-(4,4′-dimethoxytrityl)-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-O-methyladenosine (19). Under argon atmosphere, DMTrCl (0.50 g, 1.46 mmol), AgNO3 (0.25 g, 1.46 mmol) were added to a solution of 18 (0.52 g, 0.73 mmol) in THF/pyridine (v:v = 3:1, 16 mL) at room temperature, and the mixture was stirred for 12 h at 40 °C. The mixture was filtered through Celite, and the filtrate was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 19 as a yellow solid (0.53 g, 0.53 mmol, 72%). 1H NMR (600 MHz, CDCl3): δ 9.05 (s, 1H), 8.80 (s, 1H), 8.40 (s, 1H), 8.06–8.05 (m, 2H), 7.63–7.60 (m, 3H), 7.55–7.49 (m, 4H), 7.44–7.37 (m, 4H), 7.33 (t, J = 7.8 Hz, 2H), 7.26–7.23 (m, 7H), 7.18–7.16 (m, 2H), 6.70–6.66 (m, 4H), 6.22 (d, J = 7.2 Hz, 1H), 4.86 (dd, J = 7.8, 4.8 Hz, 1H), 4.51 (dd, J = 4.8, 1.2 Hz, 1H), 3.96 (dd, J = 2.7, 1.2 Hz, 1H), 3.77 (s, 3H), 3.76 (s, 3H), 3.13 (s, 3H), 3.01–2.99 (m, 1H), 2.78–2.73 (m, 1H), 2.67–2.62 (m, 1H), 1.40–1.34 (m, 1H), 1.08 (s, 9H), 1.06–1.02 (m, 1H), 0.98–0.91 (m, 1H), 0.56–0.49 (m, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.7, 158.7, 158.6, 152.9, 152.4, 149.6, 146.2, 142.7, 136.6, 136.2, 136.1, 135.8, 133.9, 133.8, 133.2, 132.9, 130.6, 130.5, 130.1, 129.9, 129.0, 128.4, 128.0, 127.9, 127.7, 126.9, 123.9, 113.1, 113.0, 87.2, 86.7, 85.7, 82.4, 74.1, 72.1, 58.6, 55.4, 51.1, 27.8, 27.1, 19.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C58H60N8NaO7Si 1031.4252, found 1031.4272.
6-N-Benzoyl-(S)-5′-O-(4,4′-dimethoxytrityl)-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-O-methyl-5′-C-trifluoroacetylaminopropyladenosine (20). Ph3P (1.46 g, 5.55 mmol) and H2O (1.6 mL, 88.8 mmol) were added to a solution of compound 19 (2.24 g, 2.22 mmol) in THF (44 mL). After being stirred at 40 °C for 12 h, the mixture was concentrated in vacuo. The residue was dissolved in CH2Cl2 (20 mL). Et3N (0.46 mL, 3.33 mmol) and CF3CO2Et (0.79 mL, 6.66 mmol) were added to the mixture. After being stirred at room temperature for 24 h, the mixture was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 20 as a yellow solid (2.22 g, 2.06 mmol, 93%). 1H NMR (600 MHz, CDCl3): δ 9.04 (s, 1H), 8.80 (s, 1H), 8.38 (s, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.63–7.58 (m, 3H), 7.54 (t, J = 7.8 Hz, 2H), 7.49 (d, J = 6.6 Hz, 2H), 7.45–7.42 (m, 1H), 7.39–7.37 (m, 2H), 7.36–7.32 (m, 3H), 7.26–7.23 (m, 6H), 7.17–7.16 (m, 3H), 6.69–6.65 (m, 4H), 6.20 (d, J = 7.2 Hz, 1H), 5.94 (bs, 1H), 4.92 (dd, J = 7.8, 4.8 Hz, 1H), 4.50 (dd, J = 4.8, 1.2 Hz, 1H), 3.92 (d, J = 1.2 Hz, 1H), 3.77 (s, 3H), 3.76 (s, 3H), 3.15 (s, 3H), 2.99–2.97 (m, 1H), 2.80–2.77 (m, 2H), 1.32–1.24 (m, 1H), 1.08 (s, 9H), 1.02–0.97 (m, 1H), 0.83–0.78 (m, 1H), 0.54–0.50 (m, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.7, 158.7, 158.6, 152.9, 152.4, 149.7, 146.1, 142.2, 136.6, 136.1, 135.9, 134.0, 133.8, 133.0, 133.0, 130.5, 130.4, 130.0, 130.0, 129.1, 128.3, 128.0, 127.9, 127.8, 127.0, 123.9, 113.1, 113.0, 87.2, 86.6, 85.7, 82.1, 73.9, 72.1, 58.6, 55.4, 39.7, 27.6, 27.0, 19.5; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C60H61F3N6NaO8Si 1101.4156, found 1101.4170.
6-N-Benzoyl-(S)-5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-5′-C-trifluoroacetylaminopropyladenosine (21). TBAF (1.31 mL of a 1 M solution in THF) was added to a solution of compound 20 (0.94 g, 0.87 mmol) in THF (9.4 mL) at room temperature under argon atmosphere. After being stirred at room temperature for 24 h, the mixture was concentrated. The residue was purified by column chromatography (66% ethyl acetate in hexane) to afford desired product 21 as a white solid (0.73 g, 0.87 mmol, 99%). 1H NMR (600 MHz, CDCl3): δ 9.09 (s, 1H), 8.77 (s, 1H), 8.20 (s, 1H), 8.04 (d, J = 7.2 Hz, 2H), 7.61 (t, J = 7.2 Hz, 1H), 7.53 (t, J = 7.8 Hz, 2H), 7.47 (d, J = 6.6 Hz, 2H), 7.36 (dd, J = 9.0, 3.0 Hz, 4H), 7.22 (t, J = 7.2 Hz, 2H), 7.19–7.16 (m, 1H), 6.78–6.74 (m, 4H), 6.43 (bs, 1H), 6.07 (d, J = 4.2 Hz, 1H), 4.61 (dd, J = 5.7, 4.2 Hz, 1H), 4.38 (d, J = 4.8 Hz, 1H), 4.12 (t, J = 4.8 Hz, 1H), 3.77 (s, 3H), 3.76 (s, 3H), 3.55 (dd, J = 11.1, 4.8 Hz, 1H), 3.53 (s, 3H), 3.90–2.97 (m, 2H), 2.84 (d, J = 4.8 Hz, 1H), 1.45–1.41 (m, 1H), 1.36–1.34 (m, 2H), 1.29–1.25 (m, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 164.8, 158.8, 158.7, 157.6, 157.3, 157.1, 156.8, 152.8, 151.6, 149.8, 146.1, 142.0, 136.6, 136.5, 133.7, 133.0, 130.1, 129.1, 128.5, 128.0, 127.8, 127.1, 123.9, 118.8, 116.8, 114.9, 113.2, 113.1, 113.0, 87.1, 87.0, 86.7, 85.5, 82.7, 73.5, 69.8, 59.1, 55.3, 40.0, 28.1, 24.2; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C44H43F3N6NaO8 863.2992, found 863.2966.
6-N-Benzoyl-3′-O-[2-cyanoethoxy(diisopropylamino) phosphino]-(S)-5′-O-(4,4′-dimethoxytrityl)-2′-O-methyl-5′-C-trifluoroacetylaminopropyladenosine (22). Under argon atmosphere, DIPEA (1.31 mL, 7.5 mmol), 2-cyanoethyl-N,N-diisopropylchlorophosphoroamidite (0.67 mL, 3.0 mmol) were added to a solution of 21 (1.26 g, 1.50 mmol) in THF (13 mL) at room temperature, and the mixture was stirred for 1 h. The mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 22 as a white solid (1.25 g, 1.20 mmol, 80%) 31P NMR (151 MHz, CDCl3): δ 151.7, 150.2; HRMS (ESI-TOF) m/z: [M + K]+ calcd for C53H60F3KN8O9P 1079.3810, found 1079.3836.
2′-O-Acetyl-(S)-5′-C-azidopropyl-3′-O-benzyl-6-chloro-5′-O-[(1,1-dimethylethyl)diphenylsilyl] guanosine (23). N,O-Bis(trimethylsilyl)acetamide (2.0 mL, 8.16 mmol) was added to a solution of 12 (1.75 g, 2.72 mmol) and 2-amino-6-chloropurine (0.51 g, 2.99 mmol) in toluene (15 mL). The mixture was stirred for 1 h at 80 °C. The solution was cooled to 0 °C, and TMSOTf (1.0 mL, 5.44 mmol) was added in dropwise; the mixture was warmed to 80 °C and stirred for 15 h. The mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 23 as a white solid (1.54 g, 2.04 mmol, 75%). 1H NMR (600 MHz, CDCl3): δ 7.96 (s, 1H), 7.63–7.59 (m, 4H), 7.42–7.29 (m, 10H), 7.26–7.25 (m, 1H), 5.97 (d, J = 4.8 Hz, 1H), 5.73 (t, J = 4.8 Hz, 1H), 5.14 (s, 2H), 4.59 (d, J = 10.8 Hz, 1H), 4.53 (t, J = 5.4 Hz, 1H), 4.37 (d, J = 11.4 Hz, 1H), 4.10 (dd, J = 5.4, 3.6 Hz, 1H), 3.78–3.75 (m, 1H), 2.95–2.91 (m, 1H), 2.86–2.81 (m, 1H), 2.12 (s, 3H), 1,72–1.69 (m, 1H), 1.41–1.38 (m, 2H), 1.30–1.26 (m, 1H), 1.05 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 170.0, 159.1, 153.4, 151.7, 140.8, 137.2, 136.0, 135.9, 133.5, 133.1, 130.1, 129.9, 128.7, 128.4, 128.3, 127.8, 127.7, 125.9, 86.0, 84.3, 76.4, 74.2, 73.5, 72.8, 51.1, 30.4, 27.2, 24.6, 20.8, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C38H43ClN8NaO5Si 777.2712, found 777.2700.
(S)-5′-C-Azidopropyl-3′-O-benzyl-6-chloro-5′-O-[(1,1-dimethylethyl)diphenylsilyl] guanosine (24). K2CO3 (2.77 g, 20.18 mmol) was added to a solution of 23 (8.00 g, 10.59 mmol) in methanol (80 mL), and the mixture was stirred for 30 min at 0 °C. The mixture was extracted with ethyl acetate and water; the organic layer was washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 24 as a white solid (7.03 g, 9.86 mmol, 93%). 1H NMR (600 MHz, CDCl3): δ 7.95 (s, 1H), 7.61–7.59 (m, 4H), 7.44–7.28 (m, 11H), 5.80 (d, J = 5.4 Hz, 1H), 5.03 (s, 2H), 4.64–4.60 (m, 2H), 4.54 (d, J = 11.4 Hz, 1H), 4.28 (dd, J = 5.4, 4.2 Hz, 1H), 4.07 (t, J = 4.2 Hz, 1H), 3.74–3.71 (m, 1H), 3.22 (d, J = 7.2 Hz, 1H), 2.98–2.94 (m, 1H), 2.86–2.82 (m, 1H), 1.75–1.70 (m, 1H), 1.42–1.34 (m, 2H), 1.28–1.24 (m, 1H), 1.04 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 159.1, 153.5, 151.6, 140.7, 136.7, 135.9, 135.9, 133.5, 132.8, 130.2, 130.1, 128.9, 128.7, 128.4, 127.9, 127.8, 125.8, 88.2, 84.1, 77.8, 74.1, 73.4, 73.1, 51.1, 30.4, 27.3, 24.6, 19.6; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C36H41ClN8NaO4Si 735.2606, found 735.2599.
(S)-5′-C-Azidopropyl-3′-O-benzyl-6-chloro-5′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-O-methyl-guanosine (25). CH3I (1.84 mL, 29.58 mmol) and NaH (0.43 g, 10.85 mmol) were added to a solution of 24 (7.03 g, 9.86 mmol) and molecular sieve 3 Å (7.0 g) in DMF (70 mL) at 0 °C under argon atmosphere. After this solution was stirred 7 hours at 0 °C, the reaction was quenched with saturated NaHCO3 aqueous solution. The mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 25 as a white solid (4.95 g, 6.81 mmol, 69%). 1H NMR (600 MHz, CDCl3): δ 8.08 (s, 1H), 7.64–7.61 (m, 4H), 7.37–7.31 (m, 11H), 6.00 (d, J = 3.6 Hz, 1H), 5.11 (s, 2H), 4.64 (d, J = 12.0 Hz, 1H), 4.48 (d, J = 11.4 Hz, 1H), 4.28 (d, J = 4.2 Hz, 1H), 4.15–4.13 (m, 1H), 3.79–3.76 (m, 1H), 3.43 (s, 3H), 2.95–2.90 (m, 1H), 2.85–2.80 (m, 1H), 1.80–1.74 (m, 1H), 1.42–1.35 (m, 2H), 1.28–1.20 (m, 1H), 1.06 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 159.1, 153.5, 151.6, 140.6, 137.5, 136.0, 135.9, 133.5, 132.8, 130.2, 130.1, 128.7, 128.3, 127.9, 127.8, 125.9, 86.0, 83.8, 82.6, 76.2, 72.9, 72.9, 58.7, 51.2, 30.6, 27.3, 24.6, 19.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C37H43ClN8NaO4Si 749.2763, found 749.2741.
(S)-5′-C-Azidopropyl-3′-O-benzyl-5′-O-[(1,1-dimethylethyl)diphenylsilyl]-2′-O-methyl-guanosine (26). NaH (0.40 g, 10.00 mmol) was added to a solution of 3-hydroxypropionitrile (0.68 mL, 10.00 mmol) in THF (25 mL) at 0 °C under argon atmosphere. After this solution was stirred 10 min at 0 °C, compound 25 (3.64 g, 5.00 mmol) in THF (25 mL) was added to the reaction mixture at 0 °C; the mixture was stirred for 6 h at 0 °C. The reaction was quenched with saturated NH4Cl aqueous solution and extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (5% methanol in CHCl3) to afford desired product 26 as a white solid (3.02 g, 4.26 mmol, 85%). 1H NMR (600 MHz, CDCl3): δ 12.09 (s, 1H), 7.88 (s, 1H), 7.69–7.62 (m, 4H), 7.44–7.40 (m, 2H), 7.37–7.29 (m, 9H), 6.10 (s, 2H), 5.97 (d, J = 4.2 Hz, 1H), 4.60 (d, J = 11.4 Hz, 1H), 4.44 (d, J = 12.0 Hz, 1H), 4.30 (t, J = 5.4 Hz, 1H), 4.17–4.14 (m, 2H), 3.84–3.81 (m, 1H), 3.47 (s, 3H), 2.94–2.90 (m, 1H), 2.87–2.81 (m, 1H), 1.83–1.77 (m, 1H), 1.43–1.39 (m, 2H), 1.28–1.22 (m, 1H), 1.07 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 159.3, 153.9, 151.4, 137.5, 136.0, 136.0, 135.5, 133.6, 132.9, 130.2, 130.1, 128.6, 128.3, 128.2, 127.9, 127.9, 117.7, 86.1, 83.2, 82.7, 76.1, 72.7, 58.6, 51.2, 30.8, 27.3, 24.6, 19.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C37H44N8NaO5Si 731.3102, found 731.3082.
(S)-5′-C-Azidopropyl-3′-O-benzyl-5′-O-[(1,1-dimethylethyl)diphenylsilyl]-2-N-isobutyryl-2′-O-methyl-guanosine (27). Isobutylic anhydride (2.84 g, 17.04 mmol) was added to a solution of 26 (3.02 g, 4.26 mmol) and DMAP (0.21 g, 1.70 mmol) in DMF (25 mL) at room temperature under argon atmosphere; the mixture was warmed to 60 °C and stirred for 13 h. The reaction was quenched with methanol and extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (33% ethyl acetate in hexane) to afford desired product 27 as a white solid (2.57 g, 3.30 mmol, 77%). 1H NMR (600 MHz, CDCl3): δ 12.08 (s, 1H), 8.61 (s, 1H), 8.03 (s, 1H), 7.66–7.62 (m, 4H), 7,46–7,42 (m, 2H), 7.38–7.28 (m, 9H), 5.96 (d, J = 5.4 Hz, 1H), 4.61 (d, J = 11.4 Hz, 1H), 4.46 (d, J = 11.4 Hz, 1H), 4.25 (t, J = 4.8 Hz, 1H), 4.15 (t, J = 4.8 Hz, 1H), 4.12 (dd, J = 4.8, 2.4 Hz, 1H), 3.78–3.75 (m, 1H), 3.37 (s, 3H), 2.92–2.88 (m, 1H), 2.82–2.78 (m, 1H), 2.63–2.56 (m, 1H), 1.81–1.73 (m, 1H), 1.41–1.32 (m, 2H), 1.24 (d, J = 7.2 Hz, 6H), 1.20–1.16 (m, 1H), 1.06 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 178.6, 155.7, 148.2, 17.6, 137.4, 137.0, 136.0, 135.9, 133.4, 132.7, 130.3, 130.1, 128.7, 128.3, 128.0, 127.9, 121.8, 85.7, 83.9, 83.3, 76.1, 72.9, 72.8, 58.7, 51.1, 36.6, 30.6, 27.3, 24.6, 19.6, 19.1, 19.1; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C41H50N8NaO6Si 801.3520, found 801.3532.
(S)-5′-C-Azidopropyl-3′-O-benzyl-2-N-isobutyryl-2′-O-methyl-guanosine (28). n-Tetrabuthylammonium fluoride (TBAF, 2.56 mL of a 1 M solution in THF) was added to a solution of compound 27 (1.31 g, 1.71 mmol) in THF (13 mL) at room temperature under argon atmosphere. After being stirred at room temperature for 46 hours, the mixture was concentrated. The residue was purified by column chromatography (66% ethyl acetate in hexane) to afford desired product 28 as a white solid (0.77 g, 1.42 mmol, 83%). 1H NMR (600 MHz, CDCl3): δ 12.03 (s, 1H), 8.21 (s, 1H), 7.73 (s, 1H), 7.39–7.33 (m, 5H), 5.86 (d, J = 6.6 Hz, 1H), 5.21 (d, J = 10.8 Hz, 1H), 4.75 (d, J = 12.6 Hz, 1H), 4.67 (d, J = 11.4 Hz, 1H), 4.35 (dd, J = 7.5, 4.8 Hz, 1H), 4.22 (dd, J = 5.1, 1.2 Hz, 1H), 4.19 (s, 1H), 3.62–3.57 (m, 1H), 3.34–3.27 (m, 5H), 2.66–2.62 (m, 1H), 1.74–1.63 (m, 3H), 1.58–1.54 (m, 1H), 1.28 (dd, J = 6.9, 3.0 Hz, 6H); 13C {1H}NMR (151 MHz, CDCl3): δ 178.7, 155.3, 147.6, 147.0, 139.1, 137.4, 128.7, 128.4, 128.3, 122.7, 88.7, 86.7, 82.3, 72.8, 71.8, 58.6, 51.2, 36.6, 31.4, 25.5, 19.0, 19.0; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C25H32N8NaO6 563.2343, found 563.2340.
(S)-5′-C-Azidopropyl-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2-N-isobutyryl-2′-O-methyl-guanosine (29). Under argon atmosphere, BCl3 (8.30 mL of a 1 M solution in CH2Cl2) was added to a solution of compound 28 (0.75 g, 1.38 mmol) in CH2Cl2 (11 mL) at −78 °C. After the mixture was stirred at −78 °C for 4 h, the mixture was warmed to −50 °C and stirred for 4 hours. The reaction mixture was quenched with 7 M solution of aqueous NH3 in methanol. After concentration, the residue was purified by column chromatography (10% methanol in CHCl3) to afford a white solid compound. This compound was in DMF (5 mL), and imidazole (0.27 g, 4.02 mmol), TBDPSCl (0.35 mL, 1.34 mmol) was added at 0 °C under argon atmosphere and the mixture was stirred for 29 hours. The mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (66% ethyl acetate in hexane) to afford desired product 29 as a white solid (0.48 g, 0.69 mmol, 50% in 2 steps). 1H NMR (600 MHz, CDCl3): δ 11.97 (s, 1H), 7.98 (s, 1H), 7.76 (s, 1H), 7.74–7.69 (m, 4H), 7.50–7.40 (m, 6H), 5.90 (d, J = 7.2 Hz, 1H), 5.18 (d, J = 10.2 Hz, 1H), 4.43 (d, J = 5.4 Hz, 1H), 4.18 (dd, J = 8.4, 4.8 Hz, 1H), 3.94 (s, 1H), 3.20–3.16 (m, 5H), 2.93–2.88 (m, 1H), 2.61–2.55 (m, 1H), 1.51–1.40 (m, 4H), 1.25 (dd, J = 7.5, 4.2 Hz, 6H), 1.14 (s, 9H); 13C {1H}NMR (151 MHz, CDCl3): δ 178.4, 155.1, 147.5, 146.7, 139.3, 136.1, 136.0, 133.6, 133.0, 130.3, 130.2, 128.1, 127.9, 123.1, 89.7, 88.9, 82.5, 72.9, 71.2, 58.9, 51.1, 36.6, 31.3, 27.1, 25.3, 19.5, 19.0, 18.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C34H44N8NaO6Si 711.3051, found 711.3072.
(S)-5′-C-Azidopropyl-5′-O-(4,4′-dimethoxytrityl)-3′-O-[(1,1-dimethylethyl) diphenylsilyl]-2-N-isobutyryl-2′-O-methyl-guanosine (30). Under argon atmosphere, DMTrCl (0.89 g, 2.62 mmol), AgNO3 (0.45 g, 2.62 mmol) were added to a solution of 29 (0.90 g, 1.31 mmol) in THF/pyridine (v:v = 3:1, 30 mL) at room temperature, and the mixture was stirred for 12 h at 40 °C. The mixture was filtered through Celite, and the filtrate was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 30 as a yellow solid (1.19 g, 1.20 mmol, 92%). 1H NMR (600 MHz, CDCl3): δ 11.92 (s, 1H), 7.89 (s, 1H), 7.60–7.58 (m, 2H), 7.52–7.50 (m, 2H), 7.47–7.46 (m, 2H), 7.41–7.27 (m, 9H), 7.23–7.18 (m, 5H), 6.69 (dd, J = 10.8, 9.0 Hz, 4H), 5.83 (d, J = 7.8 Hz, 1H), 5.24 (dd, J = 7.8, 4.8 Hz, 1H), 4.59 (d, J = 5.4 Hz, 1H), 3.90 (s, 1H), 3.78 (s, 3H), 3.75 (s, 3H), 3.22 (s, 3H), 2.81–2.77 (m, 1H), 2.74 (dt, J = 9.0, 2.4 Hz, 1H), 2.66–2.62 (m, 1H), 1.50–1.45 (m, 1H), 1.24–1.20 (m, 1H), 1.08 (s, 9H), 0.91–0.85 (m, 1H), 0.66 (d, J = 6.6 Hz, 3H), 0.55 (d, J = 7.2 Hz, 3H), 0.44–0.36 (m, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 178.0, 158.8, 158.8, 155.6, 148.4, 147.1, 146.9, 140.3, 136.7, 136.0, 135.8, 133.9, 133.0, 130.2, 130.0, 130.0, 129.9, 127.9, 127.9, 127.8, 127.7, 127.1, 123.2, 113.2, 86.7, 86.4, 86.3, 80.5, 74.7, 71.6, 58.8, 55.4, 55.3, 507, 36.1, 27.0, 24.8, 19.4, 18.5, 17.9, 17.8; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C55H62N8NaO8Si 1013.4363, found 1013.4358.
(S)-5′-O-(4,4′-dimethoxytrityl)-3′-O-[(1,1-dimethylethyl)diphenylsilyl]-2-N-isobutyryl-2′-O-methyl-5′-C-trifluoroacetylaminopropyl-guanosine (31). Ph3P (0.79 g, 3.00 mmol) and H2O (0.87 mL, 48.00 mmol) were added to a solution of compound 30 (1.19 g, 1.20 mmol) in THF (24 mL). After being stirred at 40 °C for 22 h, the mixture was concentrated in vacuo. The residue was dissolved in CH2Cl2 (10 mL). Et3N (0.25 mL, 1.80 mmol) and CF3CO2Et (0.45 mL, 3.60 mmol) were added to the mixture. After being stirred at room temperature for 19 h, the mixture was extracted with ethyl acetate and water. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (50% ethyl acetate in hexane) to afford desired product 31 as a yellow solid (0.98 g, 0.92 mmol, 77% in 2 steps). 1H NMR (600 MHz, CDCl3): δ 11.93 (s, 1H), 7.85 (s, 1H), 7.59–7.57 (m, 2H), 7.51–7.49 (m, 2H), 7.47–7.45 (m, 2H), 7.41–7.27 (m, 9H), 7.21–7.17 (m, 5H), 6.69 (dd, J = 10.8, 9.0 Hz, 4H), 6.37 (t, J = 5.4 Hz, 1H), 5.81 (d, J = 9.0 Hz, 1H), 5.25 (dd, J = 8.1, 5.4 Hz, 1H), 4.61 (d, J = 5.4 Hz, 1H), 3.89 (s, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.24 (s, 3H), 2.86–2.80 (m, 2H), 2.72–2.70 (m, 1H), 1.45–1.38 (m, 1H), 1.27–1.21 (m, 1H), 1.07 (s, 9H), 1.05–1.00 (m, 1H), 0.80–0.72 (m, 1H), 0.66 (d, J = 7.2 Hz, 3H), 0.54 (d, J = 6.6 Hz, 3H), 0.47–0.40 (m, 1H); 13C {1H}NMR (151 MHz, CDCl3): δ 178.2, 158.9, 158.9, 155.6, 148.4, 147.1, 147.0, 140.3, 136.6, 136.1, 135.8, 134.0, 133.0, 130.1, 130.0, 130.0, 129.9, 128.0, 127.9, 127.8, 127.7, 127.2, 123.2, 113.3, 86.8, 86.4, 80.4, 74.6, 71.6, 58.8, 55.4, 55.4, 39.6, 36.1, 27.1, 24.5, 19.5, 18.6, 18.0, 17.9; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C57H63F3N6NaO9Si 1083.4277, found 1083.4249.
(S)-5′-O-(4,4′-dimethoxytrityl)-2-N-isobutyryl-2′-O-methyl-5′-C-trifluoroacetylaminopropyl-guanosine (32). n-Tetrabuthylammonium fluoride (TBAF, 1.39 mL of a 1 M solution in THF) was added to a solution of compound 32 (0.98 g, 0.92 mmol) in THF (10 mL) at room temperature under argon atmosphere. After being stirred at room temperature for 19 hours, the mixture was concentrated. The residue was purified by column chromatography (100% ethyl acetate) to afford desired product 33 as a white solid (0.46 g, 0.56 mmol, 61%). 1H NMR (600 MHz, CDCl3): δ 12.11 (s, 1H), 8.43 (s, 1H), 7.72 (s, 1H), 7.54 (d, J = 6.6 Hz, 2H), 7.39 (d, J = 9.0 Hz, 4H), 7.25–7.18 (m, 3H), 6.79–6.76 (m, 4H), 6.37 (t, J = 5.4 Hz, 1H), 5.61 (d, J = 6.0 Hz, 1H), 4.90 (t, J = 6.0 Hz, 1H), 4.41–4.39 (m, 1H), 4.03 (t, J = 4.2 Hz, 1H), 3.77 (s, 3H), 3.77 (s, 3H), 3.44 (s, 3H), 3.36–3.33 (m, 1H), 3.12–3.06 (m, 1H), 2.98–2.93 (m, 1H), 2.70 (d, J = 3.6 Hz, 1H), 1.88–1.83 (m, 1H), 1.57–1.54 (m, 1H), 1.37–1.31 (m, 2H), 1.19–1.15 (m, 1H), 0.93 (d, J = 7.2 Hz, 3H), 0.84 (d, J = 7.2 Hz, 3H); 13C {1H}NMR (151 MHz, DMSO-d6): δ 180.7, 158.6, 158.6, 156.6, 156.4, 155.3, 149.5, 148.8, 146.8, 137.8, 137.0, 136.8, 130.8, 130.7, 128.5, 128.1, 127.1, 121.0, 113.4, 113.4, 86.5, 86.2, 84.2, 82.3, 79.7, 73.5, 68.6, 58.2, 55.5, 55.5, 35.3, 28.2, 24.6, 19.4, 19.3; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C41H45F3N6NaO9 845.3098, found 845.3108.
3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-(S)-5′-O-(4,4′-dimethoxytrityl)-2-N-isobutyryl-2′-O-methyl-5′-C-trifluoroacetylaminopropylguanosine (33). Under argon atmosphere, DIPEA (0.50 mL, 2.81 mmol), 2-cyanoethyl-N,N-diisopropylchlorophosphoroamidite (0.25 mL, 1.12 mmol) were added to a solution of 33 (0.46 g, 0.56 mmol) in THF (4.6 mL) at room temperature, and the mixture was stirred for 1.5 h. The mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (66% ethyl acetate in hexane) to afford desired product 34 as a white solid (0.38 g, 0.37 mmol, 65%). 31P NMR (162 MHz, CDCl3): δ 152.4, 150.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C50H63F3N8O10P 1023.4357, found 1023.4334.
(S)-5-C-Allyl-1,2-O-isopropylidene-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxandiyl)-α-D-ribose (34). To a solution of 6 in THF (2.0 mL) was added n-tetrabuthylammonium fluoride (TBAF, 0.52 mL of a 1 M solution in THF) under argon atmosphere. After being stirred at room temperature for 3.5 h, the mixture was concentrated. After concentration, the residue was purified by a silica gel column chromatography (CHCl3:CH3OH = 20:1). After the purified material was dissolved in pyridine (5.0 mL). 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2, 0.16 mL, 0.50 mmol) was added to the reaction mixture in an ice bath under argon atmosphere. After being stirred at room temperature for 21 h, the mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution; the organic layer was washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (2–5% ethyl acetate in hexane) to afford desired product 34 as a colorless oil (0.13 g, 0.28 mmol, 81% in 2 steps). 1H NMR (400 MHz, CDCl3): δ 5.87–5.77 (m, 1H), 5.72 (d, J = 3.6 Hz, 1H), 5.16 (dd, J = 17.0 Hz, 2.0 Hz, 1H), 5.07 (dd, J = 10.2 Hz, 2.0 Hz, 1H), 4.55 (t, J = 3.6 Hz, 1H), 4.06 (dd, J = 9.2 Hz, 3.6 Hz, 1H), 3.97–3.93 (m, 1H), 3.88 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 2.57–2.41 (m, 2H), 1.56 (s, 3H), 1.36 (s, 3H), 1.11–1.04 (m, 28H); 13C {1H}NMR (151 MHz, CDCl3): δ 134.4, 117.8, 113.1, 103.1, 79.4, 78.6, 71.5, 68.2, 38.4, 26.7, 26.6, 17.6, 17.5, 17.5, 17.4, 17.3, 17.2, 17.0, 13.7, 13.3, 13.3, 12.8, 12.7; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C23H44NaO6Si2 495.2574, found 495.2564.
(R)-5-C-Allyl-1,2-O-isopropylidene-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxandiyl)-α-D-ribose (35). To a solution of 7 in THF (2.5 mL) was added TBAF (0.77 mL of a 1 M solution in THF) under argon atmosphere. After being stirred at room temperature for 1 h, the mixture was concentrated. After concentration, the residue was purified by a silica gel column chromatography (CHCl3:CH3OH = 20:1). After the purified material was dissolved in pyridine (10 mL). 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl2, 0.32 mL, 1.02 mmol) was added to the reaction mixture in an ice bath under argon atmosphere. After being stirred at room temperature for 21 h, the mixture was extracted with ethyl acetate and saturated NaHCO3 aqueous solution; the organic layer was washed with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude material was purified by column chromatography (2% ethyl acetate in hexane) to afford desired product 35 as a colorless oil (0.19 mg, 0.40 mmol, 79% in 2 steps). 1H NMR (400 MHz, CDCl3): δ 5.97–5.87 (m, 1H), 5.68 (d, J = 4.0 Hz, 1H), 5.11 (dd, J = 17.2 Hz, 1.2 Hz, 1H), 5.06 (d, J = 10.0 Hz, 1H), 4.52 (t, J = 4.0 Hz, 1H), 4.02 (dd, J = 7.8 Hz, 4.8 Hz, 1H), 3.81–3.70 (m, 1H), 3.73 (t, J = 8.0 Hz, 1H), 2.54–2.50 (m, 1H), 2.33–2.26 (m, 1H), 1.55 (s, 3H), 1.36 (s, 3H), 1.13–1.04 (m, 28H); 13C {1H}NMR (101 MHz, CDCl3): δ 134.8, 117.5, 112.9, 103.2, 81.2, 81.0, 76.0, 75.6, 40.5, 27.4, 27.0, 18.1, 17.8, 17.8, 17.7, 17.5, 17.3, 17.2, 13.7, 13.3, 13.1, 13.0; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C23H44NaO6Si2 495.2574, found 495.2556.
Solid-phase oligonucleotide synthesis. The oligonucleotide synthesis was carried out with a DNA/RNA synthesizer by the phosphoramidite method. After the synthesis, the CPG beads were treated with 10% dimethylamine in acetonitrile (CH3CN) for 5 min followed by a rinse with CH3CN to selectively deprotect cyanoethyl groups. Then, the oligonucleotides were deprotected and cleaved from CPG beads by incubated in the mixture of concentrated NH3 aqueous solution/40% methylamine (1:1, v/v) for 10 min at 65 °C. Next, 2′-O-TBDMS groups were deprotected by treatment with Et3N·3HF (125 μL) in DMSO (100 μL) at 65 °C for 1.5 h. The reaction was quenched with 0.1 M TEAA buffer (pH 7.0), and the mixture was desalted by a Sep-Pak C18 cartridge. The impure oligomers were purified using denaturing 20% PAGE containing 7 M urea to give highly purified oligonucleotides.
MALDI-TOF/MS analysis of ONs. The spectra were retrieved with a matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse). A mixture of 3-hydroxypicolinic acid (3-HPA) and diammonium hydrogen citrate in H2O was used as the matrix. The data of synthetic oligonucleotides: RNA 1 m/z = 6504.12 (calcd for C194H244N65O150P20 [M–H]−, 6506.43); RNA 2 m/z = 6545.59 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 3 m/z = 6718.21 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 4 m/z = 6546.83 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 5 m/z = 6717.47 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 6 m/z = 6546.03 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 7 m/z = 6718.32 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 8 m/z = 6814.75 (calcd for C203H247N86O144P20 [M–H]−, 6815.76); RNA 9 m/z = 6546.55 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 10 m/z = 6718.28 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 11 m/z = 6546.58 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 12 m/z = 6717.51 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 13 m/z = 6547.07 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 14 m/z = 6717.66 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 15 m/z = 6547.62 (calcd for C197H250N65O150P20 [M–H]−, 6548.52); RNA 16 m/z = 6720.78 (calcd for C206H271N68O150P20 [M–H]−, 6719.85); RNA 17 m/z = 6590.81 (calcd for C200H256N65O150P20 [M–H]−, 6590.61); RNA 18 m/z = 6932.71 (calcd for C218H298N71O150P20 [M–H]−, 6933.27); RNA 19 m/z = 6619.17 (calcd for C202H260N65O150P20 [M–H]−, 6618.67); RNA 20 m/z = 7076.39 (calcd for C226H316N73O150P20 [M–H]−, 7075.55); RNA 21 m/z = 6647.38 (calcd for C204H264N65O150P20 [M–H]−, 6646.73); RNA 22 m/z = 7218.06 (calcd for C234H334N75O150P20 [M–H]−, 7217.83); RNA 23 m/z = 6829.30 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 24 m/z = 6886.14 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 25 m/z = 6830.45 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 26 m/z = 6885.07 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 27 m/z = 6828.58 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 28 m/z = 6883.91 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 29 m/z = 6829.21 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 30 m/z = 6885.63 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 31 m/z = 6828.19 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 32 m/z = 6885.74 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 33 m/z = 6827.76 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 34 m/z = 6885.46 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 35 m/z = 6829.69 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 36 m/z = 6886.34 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 37 m/z = 6831.79 (calcd for C204H249N86O144P20 [M–H]−, 6829.79); RNA 38 m/z = 6886.96 (calcd for C207H256N87O144P20 [M–H]−, 6886.90); RNA 39 m/z = 7447.08 (calcd for C232H275N87O148P21FS4 [M–H]−, 7448.71).
Dual-luciferase assay. HeLa cells were transfected with the psiCHECK-2 vector (Promega) and the pcDNA3.1 containing a hygromycin resistance gene (Thermo Fisher Scientific). HeLa Cells were grown in the presence of 0.5 mg mL−1 hygromycin for 1 week. Stable HeLa-psiCHECK-2 cells expressing both firefly and Renilla luciferases were grown in Dulbecco's Modified Eagle Medium (D-MEM) supplemented with 0.25 mg mL−1 hygromycin and 10% bovine serum (BS) at 37 °C. 24 hours prior to transfection of siRNAs, HeLa-psiCHECK-2 cells (8.0 × 104 mL−1) were grown in a 96-well plate (100 μL per well). The cells were transfected with siRNAs targeting the Renilla luciferase gene using lipofectamine RNAiMAX in opti-MEM reduced serum medium. Transfection without siRNAs was used as a control. The cells were incubated for 1 hour, D-MEM (50 μL) containing 10% BS was added to each well and cells were further incubated for another 24 hours. The activities of firefly and Renilla luciferases in the cells were measured using the Dual-Luciferase Reporter Assay System (Promega) according to a manufacture's protocol. The activity of Renilla luciferase was normalized by the firefly luciferase activity. The results were confirmed by at least three independent transfection experiments with two cultures each and are expressed as the average from four experiments as mean ± SD.
Thermal stability of siRNA duplexes. The solution containing 3.0 μM passenger strand and guide strand of siRNA in a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl was heated at 100 °C and then cooled gradually to room temperature and used for the UV melting experiment. Thermally induced transitions were recorded at 260 nm with a UV/vis spectrometer fitted with temperature controller in quartz cuvettes with a path length of 1.0 cm. The sample temperature was increased by 0.5 °C min−1.
Nuclease resistance of siRNA. Fluorescein labeled siRNAs (600 pmol) were dissolved in 20 μL of a buffer of 10 mM sodium phosphate (pH 7.0) containing 100 mM NaCl. The samples were hybridized by heating 100 °C and then cooling gradually to room temperature to use for the serum stability test. 45 μL of opti-MEM and 60 μL of bovine serum were added, and the solution was incubated at 37 °C for the required time. Aliquots of 6.7 μL were diluted with a stop solution (65 mM EDTA, 15% glycerol, 6.0 μL). Samples were subjected to electrophoresis in nondenaturing 15% polyacrylamide-TBE and analyzed by a Luminescent Image analyzer LAS-4000 (Fujifilm).
Quantitative reverse-transcriptional PCR (RT-qPCR) analysis. Human colon cancer HCT116 cells (2.0 × 105 mL−1) were plated in a 12-well plate (1 mL per well) before transfection. The cells were transfected with siRNAs using Lipofectamine RNAiMAX. After a 24 h incubation, cells were replated in fresh cell culture media and were further incubated for another 24 hours. Then, total RNA of the cells was extracted using an NucleoSpin RNA Plus Kit (Takara Bio, Shiga, Japan). cDNA was synthesized using a PrimeScript RT Master Mix (Takara Bio) and qRT-PCR was performed using a TB Green Premix Ex Taq II (Takara Bio) with a Thermal Cycler Dice Real Time System (Takara Bio). Primer sequences were for human ACTB: forward 5′-GGAGCAATGATCTTGATCTT-3′, reverse 5′-CCTTCCTGGGCATGGAGTCCT-3′ and for human KNTC2: 5′-CCTCTCCATGCAGGAGTTAAGA-3′, reverse 5′-GGTCTCGGGTCCTTGATTTTCT-3′. All reactions were run in duplicate, and the relative expression levels were determined by the ΔΔCT method.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Japan Agency for Medical Research and Development (AMED) through its Funding Program for the research project number: 21ae0121029h0001. This work was also supported by JST SPRING, grant number JPMJSP2125. The authors thank Professor Yoko Hirata (Gifu University) for supplying the cells and providing technical advice. The authors would like to thank Ms Miwa Kawade (GF Mille Co., Ltd) for donating the modified KNTC2-siRNA (siRNA 49) containing (S)-5′-C-aminopropyl-2′-O-methyluridine. The authors also would like to thank Dr Yasuko Kohda (BIKAKEN) for her technical assistance.
References
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Potent and specific genetic interference by double stranded RNA in Caenorhabditis elegans, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Duplexes of 21 ± nucleotide RNAs mediate RNA interference in cultured mammalian cells, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- S. M. Elbashir, W. Lendeckel and T. Tuschl, RNA interference is mediated by 21-and 22-nucleotide RNAs, Genes Dev., 2001, 15, 188–200 CrossRef CAS PubMed.
- S. F. Dowdy, Overcoming cellular barriers for RNA therapeutics, Nat. Biotechnol., 2017, 35, 222–229 CrossRef CAS PubMed.
- K. Garber, Worth the RISC?, Nat. Biotechnol., 2017, 35, 198–202 CrossRef CAS PubMed.
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Advances in oligonucleotide drug delivery, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- S. Ly, D. M. Navaroli, M. C. Didiot, J. Cardia, L. Pandarinathan, J. F. Alterman, K. Fogarty, C. Standley, L. M. Lifshitz, K. D. Bellve, M. Prot, D. Echeverria, S. Corvera and A. Khvorova, Visualization of self-delivering hydrophobically modified siRNA cellular internalization, Nucleic Acids Res., 2017, 45, 15–25 CrossRef CAS PubMed.
- C. R. Allerson, N. Sioufi, R. Jarres, T. P. Prakash, N. Naik, A. Berdeja, L. Wanders, R. H. Griffey, E. E. Swayze and B. Bhat, Fully 2'-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA, J. Med. Chem., 2005, 48, 901–904 CrossRef CAS PubMed.
- X. Shen and D. R. Corey, Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs, Nucleic Acids Res., 2018, 46, 1584–1600 CrossRef CAS PubMed.
- S. Ly, D. Echeverria, J. Sousa and A. Khvorova, Single-stranded phosphorothioated regions enhance cellular uptake of cholesterol-conjugated siRNA but not silencing efficacy, Mol. Ther., 2020, 21, 991–1005 CAS.
- D. J. Foster, C. R. Brown, S. Shaikh, C. Trapp, M. K. Schlegel, K. Qian, A. Sehgal, K. G. Rajeev, V. Jadhav, M. Manoharan, S. Kuchimanchi, M. A. Maier and S. Milstein, Advanced siRNA designs further improve in vivo performance of GalNAc-siRNA conjugates, Mol. Ther., 2018, 26, 708–717 CrossRef CAS PubMed.
- C. R. Brown, S. Gupta, J. Qin, T. Racie, G. He, S. Lentini, R. Malone, M. Yu, S. Matsuda, S. S. Morskaya, A. V. Nair, C. S. Theile, K. Schmidt, A. Shahraz, V. Goel, R. G. Parmar, I. Zlatev, M. K. Schlegel, J. K. Nair, M. Jayaraman, M. Manoharan, D. Brown, M. A. Maier and V. Jadhav, Investigating the pharmacodynamic durability of GalNAc–siRNA conjugates, Nucleic Acids Res., 2020, 48, 11827–11844 CrossRef CAS PubMed.
- E. Malek-Adamian, D. C. Guenther, S. Matsuda, S. Martínez-Montero, I. Zlatev, J. Harp, M. B. Patrascu, D. J. Foster, J. Fakhoury, L. Perkins, N. Moitessier, R. M. Manoharan, N. Taneja, A. Bisbe, K. Charisse, M. Maier, K. G. Rajeev, M. Egli, M. Manoharan and M. J. Damha, 4'-C-Methoxy-2'-deoxy-2'-fluoro modified ribonucleotides improve metabolic stability and elicit efficient RNAi-mediated gene silencing, J. Am. Chem. Soc., 2017, 139, 14542–14555 CrossRef CAS PubMed.
- E. Malek-Adamian, M. B. Patrascu, S. K. Jana, S. Martínez-Montero, N. Moitessier and M. J. Damha, Adjusting the structure of 2'-modified nucleosides and oligonucleotides via C4'-α-F or C4'-α-OMe substitution: synthesis and conformational analysis, J. Org. Chem., 2018, 83, 9839–9849 CrossRef CAS PubMed.
- J. M. Harp, D. C. Guenther, A. Bisbe, L. Perkins, S. Matsuda, G. R. Bommineni, I. Zlatev, D. J. Foster, N. Taneja, K. Charisse, M. A. Maier, K. G. Rajeev, M. Manoharan and M. Egli, Structural basis for the synergy of 4'- and 2'-modifications on siRNA nuclease resistance, thermal stability and RNAi activity, Nucleic Acids Res., 2018, 46, 8090–8104 CrossRef CAS PubMed.
- E. Malek-Adamian, J. Fakhoury, A. E. Arnold, S. Martínez-Montero, M. S. Shoichet and M. J. Damha, Effect of sugar 2', 4'-modifications on gene silencing activity of siRNA duplexes, Nucleic Acid Ther., 2019, 29, 187–194 CrossRef CAS PubMed.
- A. V. Kel'in, I. Zlatev, J. Harp, M. Jayaraman, A. Bisbe, J. O'Shea, N. Taneja, R. M. Manoharan, S. Khan, K. Charisse, M. A. Maier, M. Egli, K. G. Rajeev and M. Manoharan, Structural basis of duplex thermodynamic stability and enhanced nuclease resistance of '-C-methyl pyrimidine-modified oligonucleotides, J. Org. Chem., 2016, 81, 2261–2279 CrossRef PubMed.
- A. Mikami, N. Erande, S. Matsuda, A. Kel’in, L. B. Woods, T. Chickering, P. S. Pallan, M. K. Schlegel, I. Zlatev, M. Egli and M. Manoharan, Synthesis, chirality-dependent conformational and biological properties of siRNAs containing 5'-(R)- and 5'-(S)-C-methyl-guanosine, Nucleic Acids Res., 2020, 48, 10101–10124 CrossRef CAS PubMed.
- R. Kajino, Y. Maeda, H. Yoshida, K. Yamagishi and Y. Ueno, Synthesis and biophysical characterization of RNAs containing (R)- and (S)-5'-C-aminopropyl-2'-O-methyluridines, J. Org. Chem., 2019, 84, 3388–3404 CrossRef CAS PubMed.
- R. Kajino and Y. Ueno, (S)-5'-C-Aminopropyl-2'-O-methyl nucleosides enhance antisense activity in cultured cells and binding affinity to complementary single-stranded RNA, Bioorg. Med. Chem., 2021, 30, 115925 CrossRef CAS PubMed.
- S. Knapp, M. R. Madduru, Z. Lu, G. J. Morriello, T. J. Emge and G. A. Doss, Synthesis of griseolic acid B by π-face-dependent radical cyclization, Org. Lett., 2001, 3, 3583–3585 CrossRef CAS PubMed.
- R. D. Giacometti, J. C. Salinas, M. E. Østergaard, E. E. Swayze, P. P. Seth and S. Hanessian, Design, synthesis, and duplex-stabilizing properties of conformationally constrained tricyclic analogues of LNA, Org. Biomol. Chem., 2016, 14, 2034–2040 RSC.
- A. M. Sørensen, K. E. Nielsen, B. Vogg, J. P. Jacobsen and P. Nielsen, Synthesis and NMR-studies of dinucleotides with conformationally restricted cyclic phosphotriester linkages, Tetrahedron, 2001, 57, 10191–10201 CrossRef.
- Y. Zhou, R. Kajino, S. Ishii, K. Yamagishi and Y. Ueno, Synthesis and evaluation of (S)-5'-C-aminopropyl and (S)-5'-C-aminopropyl-2'-arabinofluoro modified DNA oligomers for novel RNase H-dependent antisense oligonucleotides, RSC Adv., 2020, 10, 41901–41914 RSC.
- S. Furegati, A. J. P. White and A. D. Miller, Observation of a 1,5-silyl-migraftion on fructose, Synlett, 2005, 15, 2385–2387 Search PubMed.
- M. Govindarajan, Protecting group migrations in carbohydrate chemistry, Carbohydr. Res., 2020, 497, 108151 CrossRef CAS PubMed.
- S. De, S. D. Jonghe and P. Herdewijn, Synthesis of a 3'-fluoro-3'-deoxytetrose adenine phosphonate, J. Org. Chem., 2017, 82, 9464–9478 CrossRef CAS PubMed.
- V. Korotkovs, L. F. Reichenbach, C. Pescheteau, G. A. Burley and R. M. J. Liskamp, Molecular construction of sulfonamide antisense oligonucleotides, J. Org. Chem., 2019, 84, 10635–10648 CrossRef CAS PubMed.
- H. Blade, D. Bradley, L. Diorazio, T. Evans, B. R. Hayter and G. P. Howell, Modular Synthesis of constrained ethyl (cEt) purine and pyrimidine nucleosides, J. Org. Chem., 2015, 80, 5337–5343 CrossRef CAS PubMed.
- T. A. Rand, S. Petersen, F. Du and X. Wang, Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation, Cell, 2005, 123, 621–629 CrossRef CAS PubMed.
- J. J. Song, S. K. Smith, G. J. Hannon and L. Joshua-Tor, Crystal structure of argonaute and its implications for RISC slicer activity, Science, 2004, 305, 1434–1437 CrossRef CAS PubMed.
- S. Harikrishna and P. I. Pradeepkumar, Probing the binding interactions between chemically modified siRNAs and human Argonaute 2 using microsecond molecular dynamics simulations, J. Chem. Inf. Model., 2017, 57, 883–896 CrossRef CAS PubMed.
- J. J. Turner, S. W. Jones, S. A. Moschos, M. A. Lindsay and M. J. Gait, MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity, Mol. BioSyst., 2007, 3, 43–50 RSC.
- N. Kaneko, K. Miura, Z. Gu, H. Karasawa, S. Ohnuma, H. Sasaki, N. Tsukamoto, S. Yokoyama, A. Yamamura, H. Nagase, C. Shibata, I. Sasaki and A. Horii, siRNA-mediated knockdown against CDCA1 and KNTC2, both frequently overexpressed in colorectal and gastric cancers, suppresses cell proliferation and induces apoptosis, Biochem. Biophys. Res. Commun., 2009, 390, 1235–1240 CrossRef CAS PubMed.
- Y. Makita, S. Murata, Y. Katou, K. Kikuchi, H. Uejima, M. Teratani, Y. Hoashi, E. Kenjo, S. Matsumoto, M. Nogami, K. Otake and Y. Kawamata, Anti-tumor activity of KNTC2 siRNA in orthotopic tumor model mice of hepatocellular carcinoma, Biochem. Biophys. Res. Commun., 2017, 493, 800–806 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abcd
*abcd