
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(II)-thiophene-2,5-bis(amino-alcohol) mediated asymmetric Aldol reaction and Domino Knoevenagel Michael cyclization: a new highly efficient Lewis acid catalyst†
Abdullah Mohammed Al-Majid*a,
Abdullah Saleh Alammaria,
Saeed Alshahrani a,
Matti Haukkab,
Mohammad Shahidul Islam*a and
Assem Barakata
a,
Matti Haukkab,
Mohammad Shahidul Islam*a and
Assem Barakata
aDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: 436106737@student.ksu.edu.sa; alamary1401@yahoo.com; 436106738@student.ksu.edu.sa; chemistry99y@gmail.com; ambarakat@ksu.edu.sa; amajid@ksu.edu.sa; mislam@ksu.edu.sa; Fax: +966-61-1467-5992
bDepartment of Chemistry, University of Jyväskylä, P. O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: matti.o.haukka@jyu.fi
First published on 21st February 2022
Abstract
The highly efficient Lewis acid-catalytic system Cu(II)-thiophene-2,5-bis(amino-alcohol) has been developed for enantioselective Aldol reaction of isatin derivatives with ketones. The new catalytic system also proved to be highly enantioselective for the one pot three-component Domino Knoevenagel Michael cyclization reaction of substituted isatin with malononitrile and ethylacetoacetate. The chiral ligand (2S,2′S)-2,2′-((thiophene-2,5-diylbis(methylene))bis(azanediyl))bis(3-phenylpropan-1-ol) (L1) in combination with Cu(OAc)2·H2O employed as a new Lewis acid catalyst, furnished 3-substituted-3-hydroxyindolin-2-ones derivatives (3a–s) in good to excellent yields (81–99%) with high enantioselectivities (up to 96% ee) and spiro[4H-pyran-3,3-oxindole] derivatives (6a–l) in excellent yields (89–99%) with high ee (up to 95%). These aldol products and spiro-oxindoles constitute a core structural motif in a large number of pharmaceutically active molecules and natural products.
Introduction
The chiral ligand–Lewis acid–metal catalysed carbon–carbon bond construction process has become increasingly interesting to synthetic chemists as well as pharmacists, for carrying out various crucial organic transformations. In particular, chiral versions of these catalytic processes are one of the prominent strategies to access several enantiopure bioactive molecules.1–5 Among them, the asymmetric Aldol reaction and Domino Knoevenagel Michael cyclization reaction are the most powerful and efficient approaches for accessing various enantiomerically enriched biomolecules.6,7The core framework ‘3-alkyl-3-hydroxyindolin-2-ones’ is present in a large number of natural products8–14 and drug molecules15–19 such as maremycins,17 arundaphine,20 donaxaridine,21 paratunamide,22, (R)-convolutamydines A, B and E,23–27 flustraminol,28 diazonamides,29–33 leptosin D,34 3′-hydroxyglucoisatisin,35 CPC-1,36 3-hydroxy welwitindolinones C,37 TMC-95 (A–D),38–40 celogentin K,41 dioxibrassinin42 (Fig. 1).
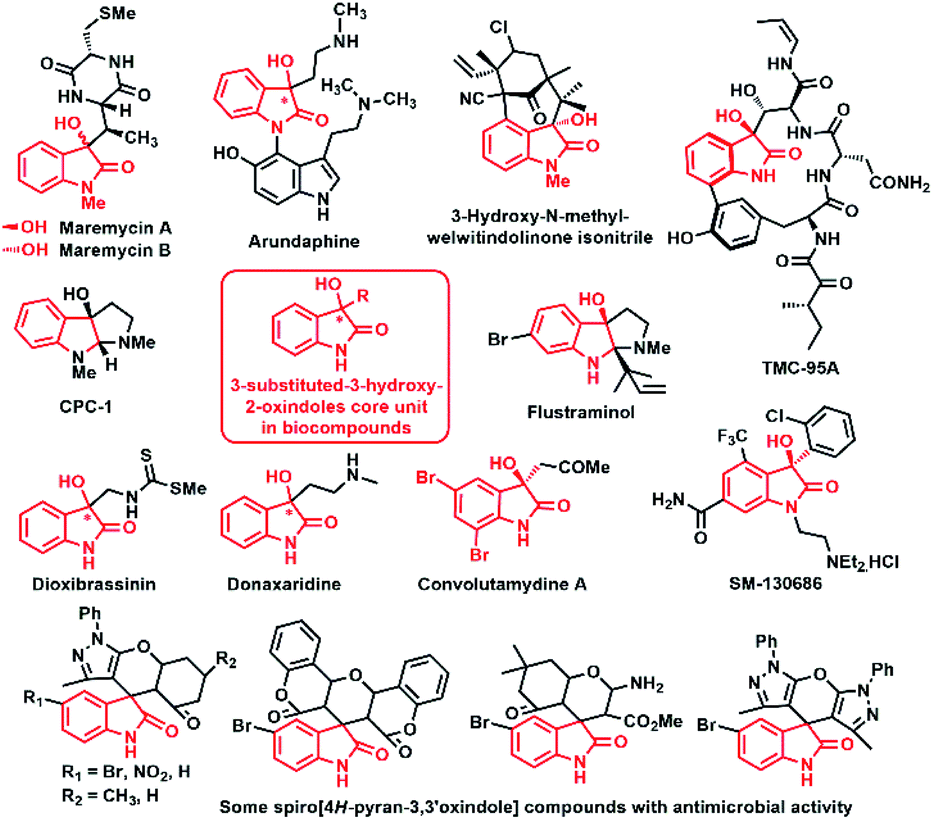 | ||
| Fig. 1 Prominent bioactive compounds based 3-substituted-3-hydroxy-2-oxindoles and spiro[4H-pyran-3,3′-oxindole] motifs. | ||
Therefore, the development of efficient and practical methods to synthesize this kind of building block is of great importance and is currently an open area of research in asymmetric catalysis.43–45 A representative example has been developed recently for asymmetric Aldol reaction of ketones with various isatins which is one of the most straightforward and powerful methods in order to achieve highly enantiopure biologically active core framework ‘3-alkyl-3-hydroxyindolin-2-ones’ with new chiral stereocenters.46–52 Ever since, proline catalysed cross-Aldol reaction of aldehydes and ketones first reported by List and Barbas in 2000,53 since then, lot of chiral amine ligands have been used as organo-catalyst for the cross-Aldol asymmetric reaction.47,54 In 2005, for the first time asymmetric Aldol reaction of isatin with acetone was first developed by Tomasini et al. employing dipeptide-based ligand as organocatalyst.55–59 However, significant efforts have been made for developing efficient asymmetric Aldol reactions of isatins with ketones mostly using organocatalysts such as proline,60–62 prolinamides,63–69 sulphonamides,70–72 amines,73,74 quinidine-thioureas,75–77 vicinal amino-alcohol,78 enzymes,79 4-hydroxydiarylprolinol80 and amino acid salts.81
The spiro-heterocyclic oxindoles are the eye-catching targets to the synthetic chemist and biologist because of their biological significance to a wide-range of synthetic utility such as alkaloids intermediates, drug candidates and clinical pharmaceuticals agents.82–89 In particular, ‘spiro[4H-pyran-3,3′-oxindole]’ moiety is the most important heterocyclic skeleton that could be found in spirooxindoles framework complex molecules of several alkaloids and drug agents.82,83 Over the couple years, sizable number of investigation have been carried out successfully for the construction of spiro[4H-pyran-3,3′-oxindole] framework as a core skeleton of spirooxindole structures.90–92 Domino Knoevenagel Michael cyclization is one of the highly successful procedures for the stereoselective synthesis of spirooxindole derivatives derived spiro[4H-pyran-3,3′-oxindole] moiety.93,94 To construct this interesting scaffold, Yuan and co-workers in 2010 for the first time, utilized cupreine-(60-hydroxycinchonidine) as organocatalyst for accessing chiral spiro[4H-pyran-3,3′-oxindole] derivatives from multi component reactions of N-alkylated isatins.95 Recently, Nakano et al. reported hybrid squaramide amino alcohol organo-catalyzed asymmetric Domino Michael cyclization reactions of oxoindolines with cyclic 1,3-diketones, affording chiral spiro-conjugated oxindoles tethering 2-aminopyrans heterocyclic ring systems with excellent yield and enantioselectivity.96 In 2019, Swapna Konda and co-workers synthesized chiral spiro[4H-pyran-3,3′-oxindole] derivatives with good chemical yield (up to 92%) and enantioselectivity (up to 87% ee) using cinchona alkaloid thioureas as an organocatalysts.97 However, till date very limited efforts have been made for the construction of stereoselective spiro-heterocyclic oxindole, engrafting 2-amino-4H-pyran-3-carbonitrile ring at the C3 position of oxindole via asymmetric Domino Michael cyclization reaction.
However, very limited findings have been reported for the application of Lewis acid catalyzed asymmetric Aldol reaction98 of ketones with isatin in order to excess derivative of ‘3-alkyl-3-hydroxyindolin-2-ones’ and asymmetric Domino Michael cyclization reaction99–101 of isatin with malononitrile and ethylacetoacetate to furnish enantiopure spiro[4H-pyran-3,3′-oxindole] derivatives. Therefore, further investigations are required to produce bioactive motif like ‘3-alkyl-3-hydroxyindolin-2-ones’ and spiro[4H-pyran-3,3′-oxindole] using Lewis acid catalyst, that remains a great challenge.
Recently, a series of new chiral bis(amino-alcohol)thiophene ligands-Cu(OAc)2·H2O as a Lewis acid catalytic system have been developed by our group and their catalytic application was successfully applied for enantioselective Henry reaction to afford the corresponding chiral Henry product with satisfactory yield and enantioselectivity.5 On observing their good catalytic enantioselective induction in the Henry reaction, we decided to explore the utility of those chiral ligands for different applications such as enantioselective Aldol reaction as well as Domino Michael cyclization reaction.
As per the best of our knowledge, we are reporting for the first time chiral bis(amino-alcohol)thiophene ligand–Lewis acid catalysed enantioselective Aldol reaction of isatins with substituted acetone to produce enantiopure 3-substituted-3-hydroxy-2-oxindoles derivatives as well as Domino Michael cyclization reaction of isatins with malononitrile and ethylacetoacetate to afford the chiral oxindoles spiro[4H-pyran-3,3′-oxindole] fused with 2-aminopyrans heterocyclic ring systems.
Results and discussion
Catalytic asymmetric studies of Aldol reaction
The following ligands as shown in Fig. 2 reported recently by our research group as an examples of C2-symmetric chiral thiophene-2,5-bis(amino-alcohol) ligands (L1–L5) and successfully applied them in catalytic asymmetric Henry reaction of nitromethane with substituted aldehydes in excellent yield and enantioselectivity (yield up to 99% and ee up to 95%).5 (Fig. 2). We further decided to examine their asymmetric catalytic efficiency and therefore enantioselective Aldol reaction of isatin with substituted acetone as well as three-component chiral Domino Michael cyclization reaction of isatin with malononitrile and ethylacetoacetate have been explored successfully.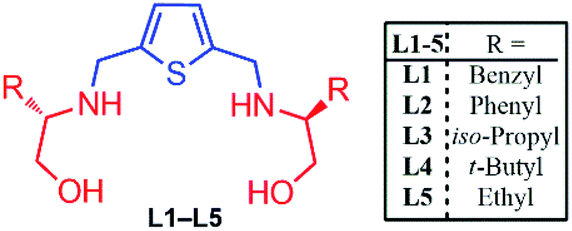 | ||
| Fig. 2 C2-symmetric chiral thiophene-2,5-bis(amino-alcohol) ligands (L1–L5) used for Aldol reaction and Domino Michael addition reaction. | ||
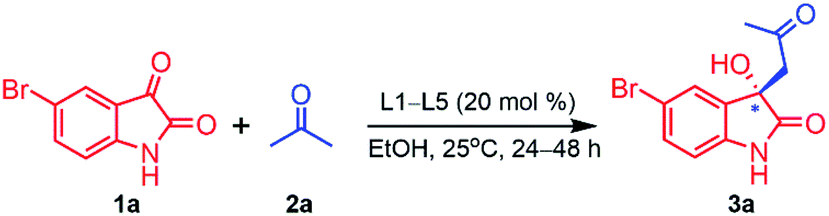
Initially, Aldol reaction of 5-bromoisatin (1a) with acetone (2a) as model substrate were carried out for accessing 3-substituted-3-hydroxy-2-oxindoles derivatives, using 20 mol% chiral thiophene-2,5-bis(amino-alcohol) ligands (L1–L5) as organocatalysts in ethanol (2 mL) at room temperature (rt) under inert atmosphere for 24–48 h and we observe that, all the ligands were found to be efficient to furnish aldol product 3a (89–99% yield) in excellent yield but unfortunately failed to induce enantioselectivity (16–21% ee) effectively (Table 1).
|
||||
|---|---|---|---|---|
| Entriesa | Ligands | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed on 0.2 mmol of isatin and 10.0 mmol of acetone.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H (25 cm × 4.6 mm × 5 μm). | ||||
| 1 | L1 | 24 | 99 | 21 |
| 2 | L2 | 24 | 99 | 16 |
| 3 | L3 | 24 | 99 | 19 |
| 4 | L4 | 24 | 99 | 20 |
| 5 | L5 | 48 | 89 | 20 |
Then the catalytic efficacy of these ligands were examined in combination with metal salt like copper acetate monohydrate (Cu(OAc)2·H2O) generated a metal complex in situ as a Lewis acid catalytic system for asymmetric Aldol reaction of 5-bromoisatin (1a) with acetone (2a, 50 eq.). Subsequently, the reaction was performed in ethanol (2 mL) at 25 °C under inert atmosphere, using 10 mol% of ligands L1–L5 with 20 mol% of Cu(OAc)2·H2O for 24 h and the corresponding findings were documented in Table 2. Fortunately, all the ligands (L1–L5) were found to be capable of producing aldol product 3a with significantly enhanced enantioselectivity (44–58% ee) (Table 2, entries 1–5) but the chemical yields were significantly dropped (44–60%). Interestingly, ligand L1 and L4 bearing benzyl and t-butyl group respectively were found to be the most effective ligand in combination with Cu(OAc)2·H2O, furnishing highest yield (60% and 58%) accordingly. However, higher enantioselectivity (58% ee) was observed in presence of L1 as compared to L4 (48% ee) (Table 2, entries 1 and 4) and therefore L1 has been chosen for further optimization.
|
||||
|---|---|---|---|---|
| Entriesa | Ligands | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed on 0.2 mmol of isatin and 10.0 mmol of acetone.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H (25 cm × 4.6 mm × 5 μm). | ||||
| 1 | L1 | 24 | 60 | 58 |
| 2 | L2 | 24 | 44 | 47 |
| 3 | L3 | 24 | 48 | 45 |
| 4 | L4 | 24 | 58 | 48 |
| 5 | L5 | 24 | 45 | 44 |
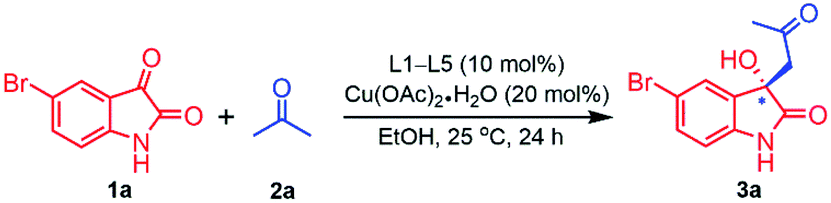
In order to improve the yield and enantioselectivity, asymmetric Aldol reaction of 5-bromoisatin (1a) with acetone (2a, 50 eq.) was further investigated. The effect of different metal salts such as Cu(OAc)2·H2O, Zn(OAc)2·2H2O, Zn(OTf)2, Mg(OTf)2, Er(OTf)3 and Yb(OTf)3 as a Lewis acid as well as various solvents system like EtOH, CH2Cl2, dioxane and THF were explored, using 10 mol% of ligand L1 with 20 mol% metal salts at rt over the period of 24–72 h and the summery of the results were shown in Table 3. It is evident from the results that, the catalytic system of L1-Cu(OAc)2·H2O in solvents like CH2Cl2 and dioxane, improved enantiomeric excess (ee) (74% & 76%) respectively as compared to ethanol (yield 60%, ee 58%) but the chemical yields were still low (58% and 45%) (Table 3, entries 1–3). However, in case of catalytic system L1-Cu(OAc)2·H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mol%) in THF as solvent was found to be the best choice as Lewis acid catalyst for the asymmetric Aldol reaction of 5-bromoisatin (1a) with acetone (2a, 50 eq.), furnishing the best yield and enantioselectivity (65% yield, 82% ee) (Table 3, entry 4). Nevertheless, L1-Zn(OAc)2·2H2O in THF produce 50% yield with very poor enantioselectivity (17% ee) in 72 h (Table 3, entry 4). Moreover, ligand L1 in combination with metal salts like Zn(OTf)2, Mg(OTf)2, Er(OTf)3 and Yb(OTf)3 in THF were found to be completely ineffective for the asymmetric Aldol reaction (Table 3, entries 6–9). The poor efficiency of Zn+2 complex as compared to Cu+2 complex could be attributed to the higher electronegativity [EN of (Cu2+) = 1.90 > EN of (Zn2+) = 1.65] and greater charge to radius ratio of Cu+2 than Zn+2 ion, those are two factors perhaps help Cu2+ to form covalent bond with O and N atoms of ligand stronger than Zn2+ during the complex formation.
:20 mol%) in THF as solvent was found to be the best choice as Lewis acid catalyst for the asymmetric Aldol reaction of 5-bromoisatin (1a) with acetone (2a, 50 eq.), furnishing the best yield and enantioselectivity (65% yield, 82% ee) (Table 3, entry 4). Nevertheless, L1-Zn(OAc)2·2H2O in THF produce 50% yield with very poor enantioselectivity (17% ee) in 72 h (Table 3, entry 4). Moreover, ligand L1 in combination with metal salts like Zn(OTf)2, Mg(OTf)2, Er(OTf)3 and Yb(OTf)3 in THF were found to be completely ineffective for the asymmetric Aldol reaction (Table 3, entries 6–9). The poor efficiency of Zn+2 complex as compared to Cu+2 complex could be attributed to the higher electronegativity [EN of (Cu2+) = 1.90 > EN of (Zn2+) = 1.65] and greater charge to radius ratio of Cu+2 than Zn+2 ion, those are two factors perhaps help Cu2+ to form covalent bond with O and N atoms of ligand stronger than Zn2+ during the complex formation.
|
|||||
|---|---|---|---|---|---|
| Entriesa | Solvents | Metal salts | Time (h) | Yieldb (%) | eec (%) |
| a Reactions were performed on 0.2 mmol of isatin and 10.0 mmol of acetone.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H (25 cm × 4.6 mm × 5 μm). | |||||
| 1 | EtOH | Cu(OAc)2·H2O | 24 | 60 | 58 |
| 2 | CH2Cl2 | Cu(OAc)2·H2O | 24 | 58 | 74 |
| 3 | Dioxane | Cu(OAc)2·H2O | 24 | 45 | 76 |
| 4 | THF | Cu(OAc)2·H2O | 24 | 65 | 82 |
| 5 | THF | Zn(OTf)2 | 72 | Trace | — |
| 6 | THF | Zn(OAc)2·2H2O | 72 | 50 | 17 |
| 7 | THF | Mg(OTf)2 | 72 | — | — |
| 8 | THF | Yb(OTf)3 | 72 | — | — |
| 9 | THF | Er(OTf)3 | 72 | — | — |
From these several attempts to achieve acceptable yield and high enantioselectivity, it can be concluded that, L1-Cu(OAc)2·H2O (10:20 mol%) as Lewis acid catalyst, in THF at rt for 24 h able to afford better yield and enantioselectivity for the asymmetric Aldol reaction of 5-bromoisatin (1a) with acetone (2a, 50 eq.). However, further optimizations are required as the results were not up to the marks as far as yields and enantioselectios are concerned. Therefore, other crucial parameters such as loading of catalyst, equivalent of acetone, time variation and temperature control were further screened to obtain the best optimized condition for the asymmetric Aldol reaction.
To find out the best optimized parameters, initially Aldol reaction were performed using 5 mol% and 15 mol% of ligand L1 with 20 mol% of Cu(OAc)2·H2O in THF at rt for 24 h and it can be seen from the results that neither the yields (52% & 68%) nor enantioselectivities (74% & 70% ee) were improved (Table 4, entries 1 & 2). Then the loading of Cu(OAc)2·H2O as metal salt were increased from 20 to 30 and 40 mol%, keeping the other parameters unchanged (10 mol% of ligand L1 in THF at rt for 24 h). We observed that, both the corresponding yields (88% and 88%) as well as enantiomeric excess (96% & 86% ee) were improved significantly (Table 4, entries 3 & 4), although 10:30 mol% ratio of L1-Cu(OAc)2·H2O was found to be the best choice for the asymmetric Aldol reaction as it has furnished the final product 3a in excellent yield and enantioselectivity (88% yield, 96% ee) (Table 4, entry 3). Next, may be the temperature has an impact on catalytic efficiency of this new catalytic system. Therefore, the reaction was further performed, using 10:30 mol% of L1-Cu(OAc)2·H2O in THF at rt for 24 h at lower temperature 10 °C as well as 0 °C, but unfortunately results indicated that, the yields (69% & 53%) and enantioselectivities (73%, 71% ee) both were significantly dropped (Table 4, entries 5 & 6) although prolong reaction time (48 h) was given. We further investigated the reaction with different molar ratio of acetone (10, 20, 40, 60 eq.) under the best optimum parameters and the findings are reported in Table 4 entries 7–10, still 50 eq. of acetone is the best choice to get better yield and enantioselectivity. From the overall investigation, we came to conclusion that, 10 mol% of ligand L1 with 30 mol% of Cu(OAc)2·H2O in THF at rt for 24 h produced best result as we obtained aldol product in high yield (88%) and enantioselectivity (96%).
|
||||||
|---|---|---|---|---|---|---|
| #a | L1/Cu(OAc)2·H2O (mol%) | Acetone (eq.) | Time (h) | Temp (oC) | Yieldb (%) | eec (%) |
| a Reactions were performed on a 0.2 mmol of isatin and 10–60 eq. of acetone.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H (25 cm × 4.6 mm × 5 μm). | ||||||
| 1 | 05/20 | 50 | 24 | 25 | 52 | 74 |
| 2 | 15/20 | 50 | 24 | 25 | 68 | 70 |
| 3 | 10/30 | 50 | 24 | 25 | 88 | 96 |
| 4 | 10/40 | 50 | 24 | 25 | 88 | 86 |
| 5 | 10/30 | 50 | 48 | 10 | 69 | 73 |
| 6 | 10/30 | 50 | 48 | 0 | 53 | 71 |
| 7 | 10/30 | 10 | 24 | 25 | 65 | 82 |
| 8 | 10/30 | 20 | 24 | 25 | 72 | 85 |
| 9 | 10/30 | 40 | 24 | 25 | 79 | 86 |
| 10 | 10/30 | 60 | 24 | 25 | 74 | 85 |
After establishing standard reaction parameters, we extended the substrate scope for the asymmetric Aldol reaction under the optimized reaction condition, using Lewis acid catalyst L1-Cu(OAc)2·H2O (10:30 mol%) in THF at rt for 24 h. A variety of substituted isatin (1a–l) with several acetone derivatives (2a–h) were screened for the asymmetric Aldol reaction using optimized reaction parameter and the corresponding aldol chiral derivatives of 3-hydroxyindolin-2-one (3a–s) were obtained in good to excellent yields (81–99%) with moderate to high enantiomeric excess (35–96% ee) and the results are summarized in Table 5. Our optimized catalytic system worked efficiently in case of isatin derivatives bearing electron-donating groups (5-Me and N-Me) which afforded aldol products 3h (99% yield, 93% ee) and 3j (91% yield, 89% ee) respectively (Table 5, entries 8 & 10). Exceptionally, 5-bromoisatin afforded best enantioselectivity (96% ee) and good yield (88%) (Table 5, entry 1). While, in case of isatin derivatives containing electron-withdrawing groups such as 5-Cl, N-benzyl and N-ethylbromide, this catalytic system also worked remarkable well and afforded respective aldol products (3c, 3k and 3l) with excellent yields (99%, 91% and 98%) and very good enantiomeric excess (76%, 78% and 71% ee) (Table 5, entries 3, 11 & 12). However, this catalytic system furnished aldol products 3b, 3e, 3f and 3i with very good yields (99%, 99%, 88% and 97%) and moderate enantioselectivities (62%, 55%, 44% and 53% ee) when isatin, 5-methoxy, 5-nitro and 5-fluoro isatin were used as nucleophilic centre for Aldol reaction (Table 5, entries 2, 5, 6 & 9). Noteworthy to mention that, while using 6-chloro and 5,7-dibromo isatin for the Aldol reaction, our catalytic system performed under expectation in inducing chirality in the aldol products and afforded 3d and 3g with poor enantioselectivities (35% & 36% ee), however quantitative chemical yields were obtained (99% & 90%) (Table 5, entries 4 & 7).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entriesa | R1 | R2 | R3 (2a–g) | 3a–s | Time [h] | Yieldb [%] | eec [%] | Abs. conf.d |
| a Reactions were performed on a 0.2 mmol of isatin and 10.0 mmol of acetone or cyclohexanone in 2 mL of THF.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H/OD-H column (25 cm × 4.6 mm × 5 μm).d Absolute configurations were determined by crystal structure and retention time matched with the literature. | ||||||||
| 1 | 5-Br | H | CH3 | 3a | 24 | 88 | 96 | (S)d67 |
| 2 | H | H | CH3 | 3b | 30 | 99 | 62 | (S)d67 |
| 3 | 5-Cl | H | CH3 | 3c | 30 | 99 | 76 | (S)d67 |
| 4 | 6-Cl | H | CH3 | 3d | 30 | 99 | 35 | (S)d |
| 5 | 5-OMe | H | CH3 | 3e | 30 | 99 | 55 | (S)d67 |
| 6 | 5-NO2 | H | CH3 | 3f | 30 | 88 | 44 | (S)d67 |
| 7 | 5,7-Br | H | CH3 | 3g | 30 | 90 | 36 | (S)d67,78 |
| 8 | 5-CH3 | H | CH3 | 3h | 35 | 99 | 93 | (S)d67 |
| 9 | 5-F | H | CH3 | 3i | 35 | 97 | 53 | (S)d67 |
| 10 | H | CH3 | CH3 | 3j | 38 | 91 | 89 | (S)d67,78 |
| 11 | H | Bn | CH3 | 3k | 38 | 98 | 78 | (S)d67 |
| 12 | H | C2H4–Br | CH3 | 3l | 35 | 99 | 71 | (S)d |
| 13 | 5-Br | H | Ph | 3m | 15 | 93 | 77 | (S)d |
| 14 | 5-Br | H | 2-NO2Ph | 3n | 15 | 96 | 70 | (S)d |
| 15 | 5-Br | H | 4-FPh | 3o | 15 | 93 | 81 | (S)d |
| 16 | 5-Br | H | 4-BrPh | 3p | 15 | 92 | 81 | (S)d |
| 17 | 5-Br | H | 4-NO2Ph | 3q | 15 | 88 | 54 | (S)d |
| 18 | 5-Br | H | 4-OHPh | 3r | 15 | 81 | 50 | (S)d |
| 19 | 5-Br | H | Cyclohexanone | 3s | 35 | 99 | 85 | (S,R)d72 |
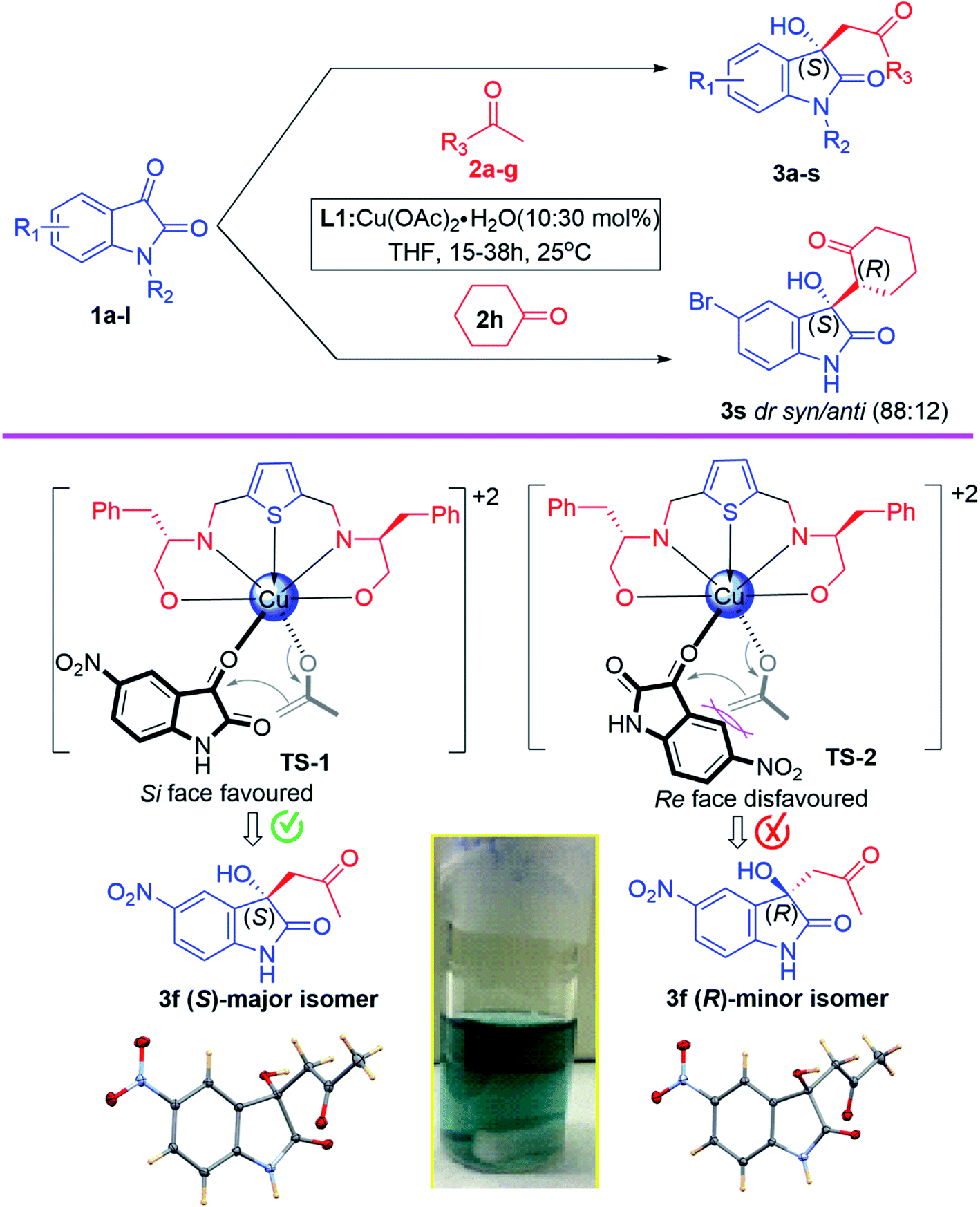
In view of the above findings, we further investigated the efficiency of our best catalytic system L1-Cu(II) as a Lewis acid catalyst under the optimized reaction parameters. Subsequently, the asymmetric Aldol reaction of 5-bromo isatin (1a) with substituted acetone (2b–e, 2h) were carried out and the corresponding aldol product 3m–p and 3s were obtained in excellent yields (93%, 96%, 93%, 92% and 99%) with very good enantiomeric excess (77%, 70%, 81%, 81% and 85% ee) (Table 5, entries 13–16, 19). However, aldol product 3q and 3r were furnished in good yields (88% and 81%) with moderate enantiomeric excess (54% and 50% ee) while using p-nitro and p-hydroxy acetophenone (2f, 2g) as substrate for the Aldol reaction under optimized reaction conditions for 15 h and the findings are reported in Table 5, entries 13–19.
Noteworthy to mention that, the (S)-enantiomer eluted faster than (R)-enantiomer in the HPLC chromatogram for aldol product 3a–c, 3e–l, while for the N-substituted aldol product 3j, 3k and 3l, reverse trends were followed, those are reported in the literature.67,78 For aldol product 3s, retention time of syn-major/minor and anti-major/minor were well matched with the data available in the literature.72 Therefore, all the aldol products are predominantly found to be enriched with (S)-enantiomer as their retention time and optical rotations were absolutely in agreement with the literature data.67,78 The absolute configuration of compound 3f was further unambiguously confirmed by single-crystal X-ray analysis (CCDC number-2143162, see ESI† S-64). In case of aldol products 3m–r the absolute configurations were also assigned as (S)-enantiomer, assuming that the reaction took place by following uniform mechanistic pathway (Fig. 3).
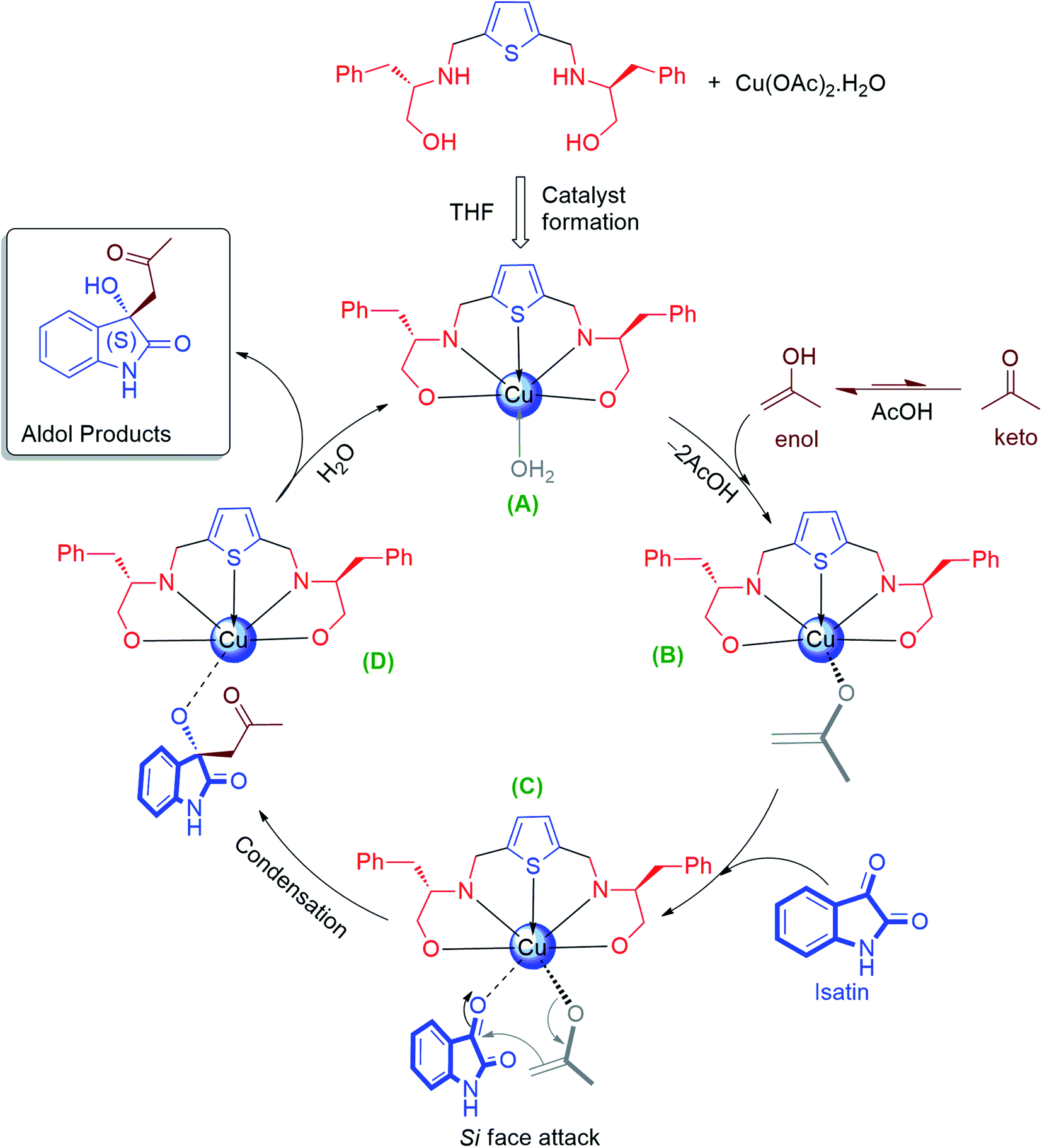 | ||
| Fig. 3 Proposed mechanism of Lewis acid catalysis for asymmetric Aldol condensation reaction. | ||
Catalytic asymmetric studies of Domino Knoevenagel Michael cyclization
To illustrate the generality for catalytic asymmetric application of our Lewis acid catalytic system (10:30 mol% of L1-Cu(II) in THF at room temperature for 5–15 h), we extended the reaction scope and therefore catalytic asymmetric Domino Knoevenagel Michael cyclization reactions were performed using the same optimized parameters and the summary of the findings are documented in Table 6. From the results of Domino Knoevenagel Michael reaction, it can be clearly observed that the substituted isatins (1a–l) reacted very well with malononitrile (4) and ethylacetoacetate (5) in order to furnished another set of biologically important oxindoles derivatives (6a–l) in excellent yields (89–99%) and moderate to high enantiomeric excess (24–95% ee), those constitutes of spiro[4H-pyran-3,3′-oxindole] motif fused with 2-aminopyrans heterocyclic ring. It can be noticed that this catalytic system produced best yields (98%, 93% & 97%) and enantiomeric excess (91%, 87% & 95% ee) when 5-bromo, 5-methoxy and 5-methyl isatins were subsequently used as substrate (Table 5, entries 1, 5 & 8); while in case of 5-nitro and N-benzyl isatins, our catalytic system L1-Cu(OAc)2·H2O are also found to be quite capable of inducing pretty good chirality (80% & 81% ee) with high yields (89% & 98%) accordingly (Table 6, entries 6 & 11). In case of isatin and its derivatives such as 5-Cl, 6-Cl, 5,7-dibromo, 5-F and N-Me isatins, the catalyst L1-Cu(OAc)2·H2O performed very well in terms of chemical yields (92–99%) to produce corresponding spiro[4H-pyran-3,3′-oxindole] derivatives 6b–d, 6g, 6i and 6j with moderate enantioselectivities (41.7%, 57.1%, 47%, 50%, 39%, 63% ee) (Table 6, entries 2–4, 7, 9 & 10). Nevertheless, this catalytic system performed poorly in prompting chiral induction when N-ethylbromide isatin was used as substrate perhaps due to the steric hindrance caused by the two long chain of N-ethylbromide of substrate (1l) and enol-ester (5) (Fig. 4) and the rate of reaction is comparatively high as it has taken 5 h only (89% yield, 24% ee). However chemical conversion is very good (Table 6, entry 12). All the Domino Knoevenagel Michael products are found to be enriched with (S)-enantiomer predominantly.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | R1 | R2 | 6a–l | Time [h] | Yieldb [%] | eec [%] | Abs. conf.d |
| a Reactions wer performed on a 0.2 mmol of isatin in 2 mL of THF.b Isolated yields after column purification.c Enantiomeric excess (ee) was determined by chiral HPLC using a Daicel Chiralpak AD-H column (25 cm × 4.6 mm × 5 μm).d Absolute configurations were determined by crystal structure and retention time matched with the literature. | |||||||
| 1 | 5-Br | H | 6a | 15 | 98 | 91 | (S)97 |
| 2 | H | H | 6b | 13 | 99 | 42 | (S)97 |
| 3 | 5-Cl | H | 6c | 8 | 96 | 57 | (S)97 |
| 4 | 6-Cl | H | 6d | 15 | 92 | 47 | (S) |
| 5 | 5-OCH3 | H | 6e | 15 | 93 | 87 | (S)97 |
| 6 | 5-NO2 | H | 6f | 15 | 89 | 80 | (S) |
| 7 | 5,7-Br | H | 6g | 15 | 96 | 50 | (S) |
| 8 | 5-CH3 | H | 6h | 15 | 97 | 95 | (S)97 |
| 9 | 5-F | H | 6i | 15 | 98 | 39 | (S)97 |
| 10 | H | CH3 | 6j | 15 | 99 | 63 | (S)97 |
| 11 | H | Bn | 6k | 15 | 98 | 81 | (S)95,97 |
| 12 | H | C2H4–Br | 6l | 5 | 89 | 24 | (S) |
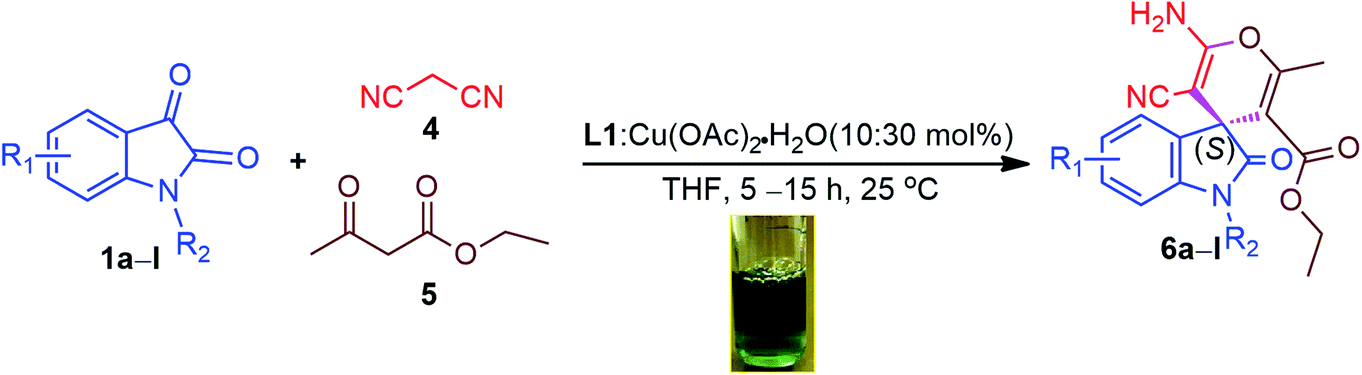
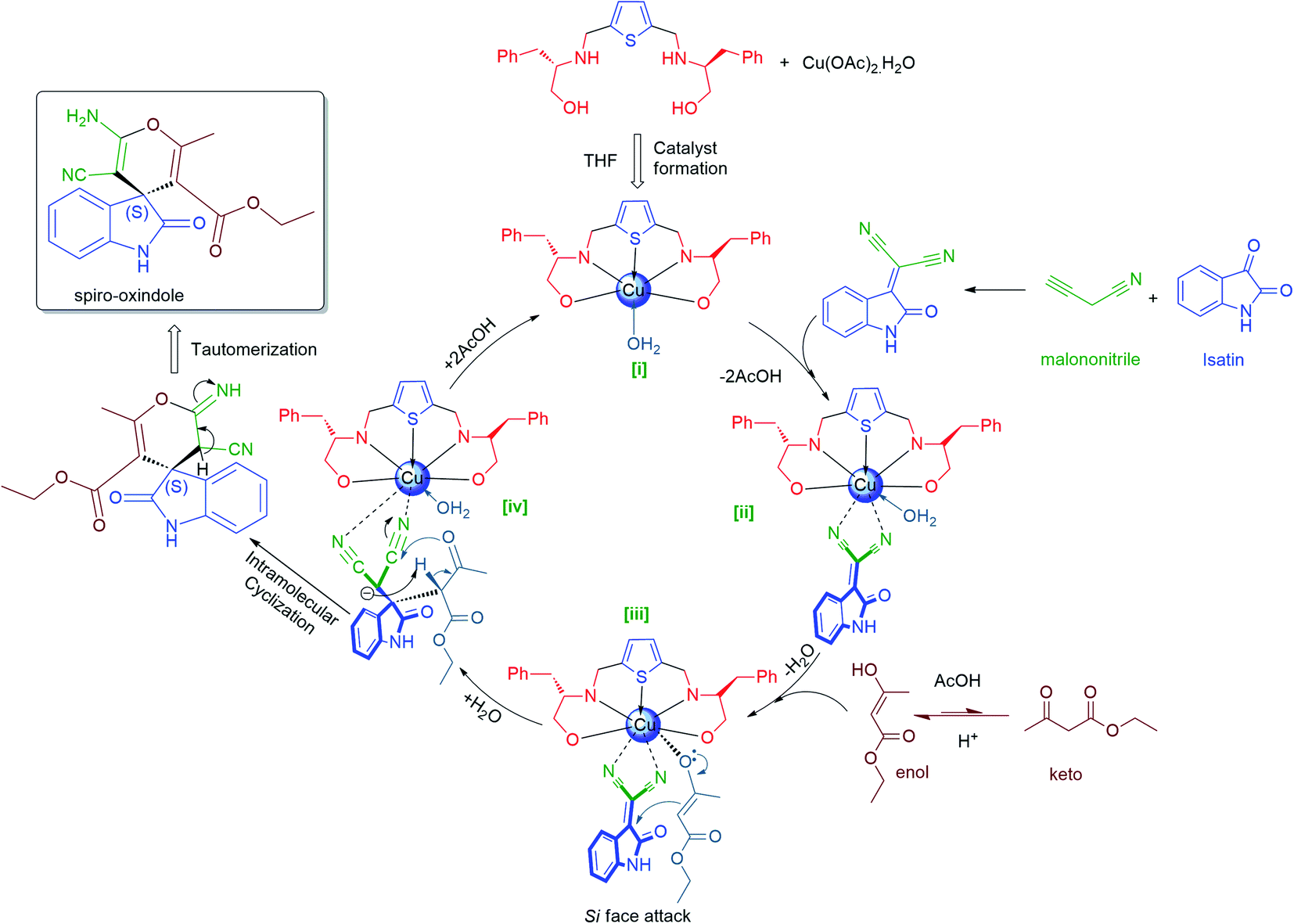 | ||
| Fig. 4 Proposed mechanism of Lewis acid catalysis for asymmetric Domino Michael cyclization reaction. | ||
(R)-enantiomer eluted faster than (S)-enantiomer in the HPLC chromatogram for Domino Knoevenagel Michael cyclized spiro-oxindole products 6a–c, 6e, 6h and 6i, while for the N-substituted spiro-oxindole 6j, 6k and 6l, reverse trends were followed, those are well reported in the literature.97 All the spiro-oxindole products are predominantly found to be enriched with (S)-enantiomer as their retention time and optical rotation were well in agreement with the literature data.97 The absolute configuration for spiro-oxindole products 6d, 6f and 6g were also assigned as (S)-enantiomer as their retention time followed the similar trends as of compound 6a–c, assuming that the reaction took place by following uniform mechanistic pathway (Fig. 4).
Experimental
General
All the chemicals (isatin, derivatives of isatin, acetone, acetone derivatives, metal salts) and solvents were purchased from Sigma-Aldrich and used as received. Thiophene-2,5-bis(β-amino-alcohol) ligands (L1–L5) were freshly synthesized from commercially available thiophene-2,5-dicarbaldehyde and chiral β-amino alcohols in a well dried flask under a static pressure of nitrogen. Standard procedures were followed for solvents drying before usage. Reactions were monitored by thin layer chromatography using Merck silica gel 60 Kieselgel F254 TLC (Merck, Kenilworth, NJ, USA) and column chromatography was performed on silica gel 100–200 (40–63 μm, ASTM) from Merck using the proper solvents. 1H and 13C-NMR spectra were recorded in CDCl3 and DMSO-d6 on a Jeol Spectrometer (Jeol, Tokyo, Japan) (500 MHz). The chemical shifts are reported in ppm. All the racemic products were freshly prepared as per the method reported in the literature.83 Thermo Scientific Nicolet iS10 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) has been used for Infrared spectra recording. Enantiomeric ratios were determined by analytical chiral HPLC analysis on a Shimadzu LC-20A (Shimadzu, Kyoto, Japan) Prominence instrument with a chiral stationary phase using Daicel OD-H columns (Chiral Technologies Europe, Illkirch-Graffenstaden, France) and 70–75% n-hexane/isopropanol as eluents (ESI†). PerkinElmer-343 Polarimeter (PerkinElmer, Waltham, MA, USA) has been used for optical rotations measurement. Melting points (m.p.) were taken on a Thomas-Hoover capillary melting point apparatus (Thomas-Hoover, Texas City, USA) and were not corrected. Agilent Technologies 6410-triple quad LC/MS instrument (Agilent, Santa Clara, CA, USA) has been used for mass spectrometric analysis. PerkinElmer PE-2400 CHN Elemental Analyzer with autosampler has been used for elemental analysis only in CHN mode. X-ray diffraction data were collected on a Rigaku Oxford Diffraction Supernova diffractometer and processed with CrysAlisPro software v. 1.171.41.93a (Rigaku Oxford Diffraction, Yarnton, UK, 2020) using Cu K_ radiation”.General procedure (GP1) for the catalytic asymmetric aldol condensation reaction (3a–s)
(S)-5-Bromo-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3a). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to afford product 3a as pale yellow solid; m.p.: 180–182 °C; isolated yield (47.2 mg, 83%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1.; tmajor = 9.64 min; tminor = 13.91 min; λ = 254 nm; 96% ee; [α]20D = −9.57° (c 0.05, CH3OH); [ref. 67 [α]25D = −7.5° (c 0.48, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.35 (s, 1H, NH), 7.43 (s, 1H, Ar–H), 7.35 (dd, J = 8.3, 2.1 Hz, 1H, Ar–H), 6.74 (d, J = 8.2 Hz, 1H, Ar–H), 6.09 (s, 1H, OH), 3.39 (d, J = 17.3 Hz, 1H, CH2(a)), 3.06 (d, J = 17.3 Hz, 1H, CH2(b)), 2.01 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.33, 177.76, 142.02, 134.17, 131.53, 126.67, 112.92, 111.38, 72.58, 49.96, 30.31. All the analytical data are well in agreement with the reported literature.67
(S)-3-Hydroxy-3-(2-oxopropyl)indolin-2-one (3b). Isatin 1b (29.4 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3b as pale yellow solid; m.p.: 169–171 °C; isolated yield (40.6 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 9.88 min; tminor = 13.29 min; λ = 254 nm; 62% ee; [α]20D = −12.65° (c 0.04, CH3OH); [ref. 67 [α]25D = −31.6° (c 1.37, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.21 (s, 1H, NH), 7.24 (d, J = 7.4 Hz, 1H, Ar–H), 7.17 (td, J = 7.6, 1.3 Hz, 1H, Ar–H), 6.90 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 6.78 (d, J = 7.6 Hz, 1H, Ar–H), 5.97 (s, 1H, OH), 3.27 (d, J = 16.5 Hz, 1H, CH2(a)), 3.00 (d, J = 16.5 Hz, 1H, CH2(b)), 2.00 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.18, 178.17, 142.55, 131.53, 128.99, 123.70, 121.24, 109.43, 72.66, 50.28, 30.58. All the analytical data are well in agreement with the reported literature.67
(S)-5-Chloro-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3c). 5-Chloroisatin 1c (47.9 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3c as pale yellow solid; m.p.: 155–157 °C; isolated yield (47.5 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 8.98 min; tminor = 12.94 min; λ = 254 nm; 76% ee; [α]20D = −5.57° (c 0.04, CH3OH); [ref. 67 [α]25D = −9.3° (c 0.9, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.34 (s, 1H, NH), 7.31 (d, J = 2.2 Hz, 1H, Ar–H), 7.23–7.20 (m, 1H, Ar–H), 6.87 (d, J = 8.2 Hz, 1H, Ar–H), 6.09 (s, 1H, OH), 3.38 (d, J = 17.3 Hz, 1H, CH2(a)), 3.06 (d, J = 17.3 Hz, 1H, CH2(b)), 2.01 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.31, 177.90, 141.60, 133.76, 128.68, 125.21, 123.98, 110.81, 72.62, 49.96, 30.32. All the analytical data are well in agreement with the reported literature67
(S)-6-Chloro-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3d). 6-Chloroisatin 1d (47.9 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3d as pale yellow solid; m.p.: 170–172 °C; isolated yield (47.5 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 9.92 min; tminor = 13.48 min; λ = 254 nm; 35% ee; [α]20D = −9.99° (c 0.05, CH3OH); IR (KBr): 3364, 3242, 2924, 2853, 1712, 1620, 1484, 1448, 1362, 1339, 1227, 1185, 1072, 825, 780, 732, 654, 587, 565, 551 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.38 (s, 1H, NH), 7.25 (d, J = 7.9 Hz, 1H, Ar–H), 6.95 (dd, J = 7.9, 2.0 Hz, 1H, Ar–H), 6.79 (d, J = 1.9 Hz, 1H, Ar–H), 6.06 (s, 1H, OH), 3.34 (d, J = 16.8 Hz, 1H, CH2(a)), 3.06 (d, J = 17.1 Hz, 1H, CH2(b)), 2.00 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.30, 178.15, 144.26, 133.18, 130.57, 125.16, 120.89, 109.48, 72.17, 50.06, 30.39; LC/MS (ESI): found 240.1 [M(35Cl) + H]+; 242.0 [M(37Cl) + H]+, C11H10ClNO3 requires 239.03; anal. calcd for C11H10ClNO3: C, 55.13; H, 4.21; N, 5.84; found: C, 54.96; H, 4.15; N, 6.06.
(S)-3-Hydroxy-5-methoxy-3-(2-oxopropyl)indolin-2-one (3e). 5-Methoxyisatin 1e (35.4 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3e as pale yellow solid; m.p.: 165–167 °C; isolated yield (46.6 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 13.04 min; tminor = 18.74 min; λ = 254 nm; 55% ee; [α]20D = −3.45° (c 0.06, CH3OH); [ref. 67 [α]25D = −3.9° (c 1.17, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.04 (s, 1H, NH), 6.91 (d, J = 2.6 Hz, 1H, Ar–H), 6.74 (dd, J = 8.4, 2.6 Hz, 1H, Ar–H), 6.69 (d, J = 8.4 Hz, 1H, Ar–H), 5.97 (s, 1H, OH), 3.69 (s, 3H, OCH3), 3.27 (d, J = 16.6 Hz, 1H, CH2(a)), 3.00 (d, J = 16.6 Hz, 1H, CH2(b)), 2.01 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.17, 178.11, 154.68, 135.74, 132.81, 113.36, 111.01, 109.72, 73.09, 55.44, 50.26, 30.59. All the analytical data are well in agreement with the reported literature.67
(S)-3-Hydroxy-5-nitro-3-(2-oxopropyl)indolin-2-one (3f). 5-Nitroisatin 1f (38.4 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3f as pale yellow solid; m.p.: 178–180 °C; isolated yield (44.1 mg, 88%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 12.11 min; tminor = 15.78 min; λ = 254 nm; 44% ee; [α]20D = −2.32° (c 0.04, CH3OH); [ref. 67 [α]25D = −21.7° (c 1.10, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.96 (s, 1H, NH), 8.18–8.16 (m, 2H, Ar–H), 6.99 (d, J = 9.2 Hz, 1H, Ar–H), 6.28 (s, 1H, OH), 3.61 (d, J = 17.8 Hz, 1H, CH2(a)), 3.18 (d, J = 17.9 Hz, 1H, CH2(b)), 2.02 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.62, 178.59, 149.49, 141.93, 132.79, 126.42, 119.50, 109.54, 72.09, 49.88, 30.04. All the analytical data are well in agreement with the reported literature.67
(S)-5,7-Dibromo-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3g). 5,7-Dibromoisatin 1g (61.0 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3g as pale yellow solid; m.p.: 182–184 °C; isolated yield (65.3 mg, 90%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 8.14 min; tminor = 10.02 min; λ = 254 nm; 36% ee; [α]20D = −4.41° (c 0.05, CH3OH); [ref. 67 [α]25D = −9.4° (c 2.14, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.70 (s, 1H, NH), 7.61 (d, J = 1.9 Hz, 1H, Ar–H), 7.47 (d, J = 1.9 Hz, 1H, Ar–H), 6.24 (s, 1H, OH), 3.46 (d, J = 17.7 Hz, 1H, CH2(a)), 3.14 (d, J = 17.7 Hz, 1H, CH2(b)), 2.02 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.48, 177.65, 141.79, 135.48, 133.40, 125.81, 113.41, 102.53, 73.34, 49.99, 30.10. All the analytical data are well in agreement with the reported literature.67
(S)-3-Hydroxy-5-methyl-3-(2-oxopropyl)indolin-2-one (3h). 5-Methylisatin 1h (32.2 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3h as white solid; m.p.: 165–167 °C; isolated yield (43.9 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 24.88 min; tminor = 31.93 min; λ = 254 nm; 93% ee; [α]20D = −5.10° (c 0.05, CH3OH); [ref. 67 [α]25D = −2.8° (c 0.81, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.12 (s, 1H, NH), 7.07 (s, 1H, Ar–H), 6.99–6.95 (m, 1H, Ar–H), 6.67 (d, J = 7.8 Hz, 1H, Ar–H), 5.94 (s, 1H, OH), 3.24 (d, J = 16.6 Hz, 1H, CH2(a)), 3.00 (d, J = 16.6 Hz, 1H, CH2(b)), 2.23 (s, 1H,CH3), 2.01 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.21, 178.23, 140.10, 131.65, 130.03, 129.17, 124.41, 109.21, 72.80, 50.36, 30.60, 20.73. All the analytical data are well in agreement with the reported literature.67
(S)-5-Fluoro-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3i). 5-Fluoroisatin 1i (33.0 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3i as pale yellow solid; 184–186 °C; isolated yield (43.3 mg, 97%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 9.21 min; tminor = 12.36 min; λ = 254 nm; 53% ee; [α]20D = −11.49° (c 0.06, CH3OH); [α]25D = −19.0° (c 0.97, MeOH);67 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.25 (s, 1H, NH), 7.15 (dd, J = 8.2, 2.7 Hz, 1H, Ar–H), 7.02–7.98 (m, 1H, Ar–H), 6.76 (dd, J = 8.5, 4.3 Hz, 1H, Ar–H), 6.10 (s, 1H, OH), 3.34 (d, J = 17.0 Hz, 1H, CH2(a)), 3.06 (d, J = 17.0 Hz, 1H, CH2(b)), 2.01 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.28, 178.23, 158.83 & 156.95 (C1–F, JC–F = 236.75 Hz), 138.82, 133.45 & 133.39 (C2–F, JC–F = 7.56 Hz), 115.14 & 114.96 (C3–F, JC–F = 23.44 Hz), 111.78 & 111.58 (C5–F, JC–F = 24.44 Hz), 110.15 & 110.09 (C6–F, JC–F = 7.81 Hz), 72.92, 50.06, 30.44. All the analytical data are well in agreement with the reported literature.67
(S)-3-Hydroxy-1-methyl-3-(2-oxopropyl)indolin-2-one (3j). 1-Methylisatin 1j (32.2 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3j as pale yellow solid; m.p.: 154–156 °C; isolated yield (43.0 mg, 98%). Enantiomeric excess (ee) was determined by chiral HPLC [Chiralcel OD-H column], 90.0% n-hexane/i-PrOH, 1.0 mL min−1; tminor = 19.93 min; tmajor = 21.09 min; λ = 254 nm; 87% ee; [α]20D = −21.94° (c 0.05, CH3OH); [ref. 67 [α]25D = −22.3° (c 1.59, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.31–7.25 (m, 2H, Ar–H), 6.99 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 6.96 (d, J = 7.7 Hz, 1H, Ar–H), 6.06 (s, 1H, OH), 3.34 (d, J = 16.8 Hz, 1H, CH2(a)), 3.09 (s, 3H, NCH3), 3.05 (d, J = 16.8 Hz, 1H, CH2(b)), 1.98 (s, 1H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.13, 176.50, 144.05, 130.90, 129.14, 123.25, 121.94, 108.26, 72.33, 50.42, 30.47, 25.89. All the analytical data are well in agreement with the reported literature.67
(S)-1-Benzyl-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3k). 1-Benzylisatin 1k (47.5 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3k as pale yellow solid; m.p.: 170–173 °C; isolated yield (57.9 mg, 98%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 85.0% n-hexane/i-PrOH, 1.0 mL min−1; tminor = 16.93 min; tmajor = 17.82 min; λ = 254 nm; 78% ee; [α]20D = −16.23° (c 0.06, CH3OH); [ref. 67 [α]25D = −17.8° (c 1.53, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.48–7.43 (m, 2H, Ar–H(Bn)), 7.38–7.32 (m, 3H, Ar–H(Bn)), 7.29–7.24 (m, 1H, Ar–H), 7.17 (td, J = 7.7, 1.3 Hz, 1H, Ar–H), 6.98 (td, J = 7.4, 1.0 Hz, 1H, Ar–H), 6.77–6.73 (m, 1H, Ar–H), 6.26 (s, 1H, OH), 4.93 (d, J = 16.1 Hz, 1H, Ph-CH2(a)), 4.85 (d, J = 16.0 Hz, 1H, Ph-CH2(b)), 3.47 (d, J = 16.9 Hz, 1H, CH2(a)), 3.21 (d, J = 16.9 Hz, 1H, CH2(b)), 2.04 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.32, 176.75, 143.18, 136.40, 130.99, 129.03, 128.49, 127.26, 127.23, 123.45, 122.07, 108.99, 72.41, 50.36, 42.73, 30.44. All the analytical data are well in agreement with the reported literature.67
(S)-1-(2-Bromoethyl)-3-hydroxy-3-(2-oxopropyl)indolin-2-one (3l). 1-(2-Bromoethyl)isatin 1l (50.8 mg, 0.2 mmol) and acetone 2a (581.0 mg, 10.0 mmol) were reacted according to the GP1 to yield product 3l as pale yellow solid; m.p.:161–162 °C; isolated yield (61.8 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 1.0 mL min−1; tminor = 16.07 min; tmajor = 17.39 min; λ = 254 nm; 71% ee; [α]20D = −19.86° (c 0.04, CH3OH); IR (KBr): 3350, 2925, 2853, 1709, 1610, 1488, 1469, 1454, 1432, 1416, 1375, 1359, 1336, 1183, 1179, 1163, 1132, 1006, 795, 751, 736, 708, 697, 563, 496, 476, 455 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.32 (dt, J = 7.3, 0.9 Hz, 1H, Ar–H), 7.29–7.26 (m, 1H, Ar–H), 7.11 (dt, J = 7.8, 0.8 Hz, 1H, Ar–H), 7.02–6.99 (m, 1H, Ar–H), 6.14 (s, 1H, OH), 4.08–4.05 (m, 2H, NCH2), 3.63 (td, J = 7.1, 3.5 Hz, 2H, CH2–Br), 3.36 (d, J = 16.9 Hz, 1H, CH2(a)), 3.08 (d, J = 16.9 Hz, 1H, CH2(b)), 2.00 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 205.24, 176.69, 142.68, 130.70, 129.17, 123.63, 122.16, 108.70, 72.25, 50.66, 40.97, 30.45, 28.66; LC/MS (ESI): found 390.01 [M(79Br) + H]+; 391.90 [M(81Br) + H]+, C13H13Br2NO3 requires 388.93; anal. calcd for C13H13Br2NO3: C, 39.93; H, 3.35; N, 3.58; found: C, 40.14; H, 3.27; N, 3.49.
(S)-5-Bromo-3-hydroxy-3-(2-oxo-2-phenylethyl)indolin-2-one (3m). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and acetophenone 2b (240.0 mg, 2.0 mmol) were reacted according to the GP1 to yield product 3m as white solid; m.p.:179–180 °C; isolated yield (64.4 mg, 93%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 0.8 mL min−1; tmajor = 14.98 min; tminor = 32.84 min; λ = 254 nm; 77% ee; [α]20D = −55.21° (c 0.05, CH3OH); IR (KBr): 3350, 3218, 1731, 1698, 1672, 1617, 1595, 1472, 1448, 1402, 1352, 1278, 1176, 1094, 1065, 991, 880, 824, 764, 686, 637, 591, 534, 478 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.44 (s, 1H, NH), 7.89 (d, J = 7.6 Hz, 2H, Ar–H), 7.63 (t, J = 7.5 Hz, 1H, Ar–H), 7.51 (d, J = 7.8 Hz, 3H, Ar–H), 7.35 (d, J = 8.3 Hz, 1H, Ar–H), 6.80 (d, J = 8.3 Hz, 1H, Ar–H), 6.23 (s, 1H, OH), 4.17 (d, J = 17.9 Hz, 1H, CH2(a)), 3.65 (d, J = 18.1 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 196.64, 178.00, 142.38, 135.95, 134.42, 133.59, 131.55, 128.78, 127.98, 126.71, 112.94, 111.43, 73.05, 45.77; LC/MS (ESI): found 346.1 [M(79Br) + H]+; 348.1 [M(81Br) + H]+, C16H12BrNO3 requires 345.0; anal. calcd for C16H12BrNO3: C, 55.51; H, 3.49; N, 4.05; found: C, 55.43; H, 3.44; N, 4.13.
(S)-5-Bromo-3-hydroxy-3-(2-(2-nitrophenyl)-2-oxoethyl)indolin-2-one (3n). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and 1-(2-nitrophenyl)ethan-1-one 2c (330.3 mg, 2.0 mmol) were reacted according to the GP1 to yield product 3n as white solid; m.p.:188–190 °C; isolated yield (75.1 mg, 96%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 70.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 11.98 min; tminor = 31.04 min; λ = 254 nm; 70% ee; [α]20D = −75.51° (c 0.08, CH3OH); IR (KBr): 3337, 3390, 1738, 1702, 1615, 1573, 1530, 1471, 1441, 1388, 1314, 1216, 1180, 1130, 1059, 998, 895, 838, 754, 744, 731, 699, 672, 542 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.46 (s, 1H, NH), 8.04 (d, J = 8.0 Hz, 1H, Ar–H), 7.78 (dt, J = 32.4, 7.8 Hz, 2H, Ar–H), 7.60 (d, J = 7.6 Hz, 1H, Ar–H), 7.54 (s, 1H, Ar–H), 7.38 (d, J = 8.3 Hz, 1H, Ar–H), 6.78 (d, J = 8.4 Hz, 1H, Ar–H), 6.30 (s, 1H, OH), 3.91 (d, J = 17.5 Hz, 1H, CH2(a)), 3.53 (d, J = 17.6 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 198.01, 177.39, 146.19, 142.04, 134.51, 133.97, 133.37, 132.14, 131.82, 128.53, 127.29, 124.27, 113.11, 111.50, 72.87, 48.42; LC/MS (ESI): found 391.1 [M(79Br) + H]+; 393.1 [M(81Br) + H]+, C16H11BrN2O5 requires 389.99; anal. calcd for C16H11BrN2O5: C, 49.13; H, 2.83; N, 7.16; found: C, 49.27; H, 2.89; N, 7.09.
(S)-5-Bromo-3-(2-(4-fluorophenyl)-2-oxoethyl)-3-hydroxyindolin-2-one (3o). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and 1-(4-fluorophenyl)ethan-1-one 2d (276.3 mg, 2.0 mmol) were reacted according to the GP1 to yield product 3o as white solid; m.p.:186–187 °C; isolated yield (67.7 mg, 93%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 70.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 8.96 min; tminor = 17.02 min; λ = 254 nm; 81% ee; [α]20D = −5.90° (c 0.04, CH3OH); IR (KBr): 3235, 1698, 1686, 1619, 1600, 1507, 1473, 1449, 1409, 1348, 1234, 1181, 1166, 1072, 991, 827, 732, 591, 559 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.44 (s, 1H, NH), 7.98 (s, 2H, Ar–H), 7.51 (s, 1H, Ar–H), 7.38–7.25 (m, 3H, Ar–H), 6.86–6.72 (m, 1H, Ar–H), 6.24 (s, 1H, OH), 4.16 (d, J = 17.9 Hz, 1H, CH2(a)), 3.63 (d, J = 18.2 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 195.28, 177.98, 166.25, 164.24, 142.35, 134.36, 132.76, 131.58, 131.26, 131.13, 131.06, 126.75, 115.88, 115.71, 112.96, 111.45, 73.06, 45.72; LC/MS (ESI): found 364.1 [M(79Br) + H]+; 366.1 [M(81Br) + H]+, C16H11BrFNO3 requires 362.99; anal. calcd for C16H11BrFNO3: C, 52.77; H, 3.04; 3.85; found: C, 52.61; H, 3.18; N, 3.95.
(S)-5-Bromo-3-(2-(4-bromophenyl)-2-oxoethyl)-3-hydroxyindolin-2-one (3p). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and 1-(4-bromophenyl)ethan-1-one 2e (398.1, 2.0 mmol) were reacted according to the GP1 to yield product 3p as white solid; m.p.:184–185 °C; isolated yield (78.2 mg, 92%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 70.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 12.55 min; tminor = 32.98 min; λ = 254 nm; 81% ee; [α]20D = −21.79° (c 0.03, CH3OH); IR (KBr): 3188, 1713, 1699, 1618, 1587, 1472, 1448, 1397, 1344, 1215, 1183, 1069, 1008, 989, 827, 810, 733, 647, 556 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.45 (s, 1H, NH), 7.76 (d, J = 54.0 Hz, 4H, Ar–H), 7.50 (s, 1H, Ar–H), 7.34 (s, 1H, Ar–H), 6.89–6.70 (m, 1H, Ar–H), 6.24 (s, 1H, OH), 4.15 (d, J = 18.1 Hz, 1H, CH2(a)), 3.61 (d, J = 18.2 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 195.91, 177.90, 142.30, 134.94, 134.25, 131.84, 131.60, 130.05, 127.77, 126.79, 112.97, 111.45, 73.02, 45.72; LC/MS (ESI): found 423.9 [M(79Br) + H]+; 425.9 [M(81Br) + H]+, C16H11Br2NO3 requires 422.91; anal. calcd for C16H11Br2NO3: C, 45.21; H, 2.61; N, 3.30; found: C, 42.16; H, 2.53; N, 3.27.
(S)-5-Bromo-3-hydroxy-3-(2-(4-nitrophenyl)-2-oxoethyl)indolin-2-one (3q). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and 1-(4-nitrophenyl)ethan-1-one 2f (330.3, 2.0 mmol) were reacted according to the GP1 to yield product 3q as white solid; m.p.:187–188 °C; isolated yield (68.8 mg, 88%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 70.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 22.05 min; tminor = 30.35 min; λ = 254 nm; 54% ee; [α]20D = −11.43° (c 0.04, CH3OH); IR (KBr): 3196, 1654, 1618, 1528, 1478, 1447, 1343, 1249, 1209, 1183, 1123, 1060, 1000, 903, 895, 855, 824, 742, 644, 595, 535 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.48 (s, 1H, NH), 8.31 (d, J = 8.8 Hz, 2H, Ar–H), 8.12 (d, J = 8.8 Hz, 2H, Ar–H), 7.52 (d, J = 2.1 Hz, 1H, Ar–H), 7.36 (dd, J = 8.2, 2.1 Hz, 1H, Ar–H), 6.79 (d, J = 8.2 Hz, 1H, Ar–H), 6.30 (s, 1H, OH), 4.23 (d, J = 18.0 Hz, 1H, CH2(a)), 3.70 (d, J = 17.9 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 196.01, 177.77, 150.10, 142.24, 140.46, 134.02, 131.71, 129.53, 126.93, 123.88, 113.03, 111.51, 73.04, 46.23; LC/MS (ESI): found 391.1 [M(79Br) + H]+; 393.1 [M(81Br) + H]+, C16H11BrN2O5 requires 389.99; anal. calcd for C16H11BrN2O5: C, 49.13; H, 2.83; N, 7.16; found: C, 49.23; H, 2.69; N, 7.24.
(S)-5-Bromo-3-hydroxy-3-(2-(4-hydroxyphenyl)-2-oxoethyl)indolin-2-one (3r). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and 1-(4-hydroxyphenyl)ethan-1-one 2g (272.3 mg, 2.0 mmol) were reacted according to the GP1 to yield product 3r as white solid; M.P.:191–192 °C; isolated yield (58.7 mg, 81%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 9.31 min; tminor = 23.06 min; λ = 254 nm; 50% ee; [α]20D = −78.91° (c 0.04, CH3OH); IR (KBr): 3230, 1714, 1710, 1698, 1651, 1619, 1605, 1582, 1479, 1446, 1356, 1290, 1225, 1182, 1171, 1063, 997, 825, 734, 612, 592, 558, cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.42 (s, 1H, NH), 10.37 (s, 1H, OH), 7.76 (d, J = 8.8 Hz, 2H, Ar–H), 7.47 (d, J = 2.1 Hz, 1H, Ar–H), 7.33 (dd, J = 8.2, 2.1 Hz, 1H, Ar–H), 6.82 (d, J = 8.8 Hz, 2H, Ar–H), 6.77 (d, J = 8.3 Hz, 1H, Ar–H), 6.15 (s, 1H, OH), 4.04 (d, J = 17.6 Hz, 1H, CH2(a)), 3.52 (d, J = 17.7 Hz, 1H, CH2(b)); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 194.61, 178.13, 162.35, 142.42, 134.66, 131.45, 130.59, 127.71, 126.59, 115.27, 112.86, 111.37, 73.12, 45.31; LC/MS (ESI): found 362.1 [M(79Br) + H]+; 364.1 [M(81Br) + H]+, C16H12BrNO4 requires 360.99; anal. calcd for C16H12BrNO4: C, 53.06; H, 3.34; N, 3.87; found: C, 52.92; H, 3.46; N, 3.97.
(S)-5-Bromo-3-hydroxy-3-((R)-2-oxocyclohexyl)indolin-2-one (3s). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol) and cyclohexanone 2h (196.3 mg, 0.21 mL, 2.0 mmol) were reacted according to the GP1 to yield product 3s as white solid; m.p.: 224–225 °C; isolated yield (64.2 mg, 99%), syn/anti = (88
:12). Enantiomeric excess (ee) of (syn, major) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 92.0% n-hexane/i-PrOH, 0.8 mL min−1; tsyn-major = 53.32 min; tsyn-minor = 74.56 min; tanti-major = 63.64 min; λ = 254 nm; syn/anti = 88:12; Enantiomeric excess (ee) of syn diastereomers = 85%; [α]20D = −46.97° (c 0.02, CH3OH); [ref. 72. [α]25D = −16.3° (c 0.1, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.37 (s, 1H, NH), 7.45–7.25 (m, 2H, Ar–H), 6.77 (s, 1H, Ar–H), 6.01 (s, 1H, OH), 3.10 (dt, J = 8.0, 6.1 Hz, 1H, COCH), 2.59–2.50 (m, 1H, COCH2), 2.33–2.30 (m, 1H, COCH2), 2.06–1.51 (m, 6H, (CH2)3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 209.34, 178.25, 142.79, 133.34, 131.30, 127.45, 112.52, 111.40, 73.93, 57.43, 41.45, 26.72, 26.66, 24.44. All the analytical data are well in agreement with the reported literature.72
General procedure (GP2) for synthesis of asymmetric spiro[4H-pyran-3,3-oxindole] derivatives (6a–l) of three components with one pot
Ethyl-(S)-2′-amino-5-bromo-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6a). 5-Bromoisatin 1a (45.2 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6a as white solid; m.p.: 259–260 °C; isolated yield (80.8 mg, 98%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 38.79 min; tmajor = 41.37 min; λ = 254 nm; 91% ee; [α]20D = +8.62° (c 0.07, CH3OH); [ref. 97 [α]24D = +34.3° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.55 (s, 1H, NH), 7.40–7.32 (m, 1H, Ar–H), 7.29 (s, 1H, Ar–H), 7.23 (s, 2H, NH2), 6.77 (d, J = 8.3 Hz, 1H, Ar-H), 3.82 (q, J = 8.0 Hz, 2H, CH2CH3), 2.33 (s, 3H, CH3), 0.84 (t, J = 7.4 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.27, 164.37, 159.56, 158.91, 141.44, 137.23, 131.29, 126.27, 117.38, 113.45, 111.31, 103.72, 60.42, 55.94, 49.24, 18.80, 13.06. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6b). Isatin 1b (29.4 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6b as white solid; m.p.: 253–254 °C; isolated yield (64.4 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 85.0% n-hexane/i-PrOH, 1.0 mL min−1; tminor = 19.12 min; tmajor = 19.89 min; λ = 254 nm; 42% ee; [α]20D = +21.83° (c 0.05, CH3OH); [ref. 97 [α]24D = +20.4° (c 1.4, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.39 (s, 1H, NH), 7.18 (td, J = 7.6, 1.3 Hz, 1H, Ar–H), 7.14 (s, 2H, NH2), 7.05 (dd, J = 7.4, 1.2 Hz, 1H, Ar–H), 6.93 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 6.79 (dt, J = 7.7, 0.8 Hz, 1H, Ar–H), 3.81–3.72 (m, 2H, CH2CH3), 2.31 (s, 3H, CH3), 0.78 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.60, 164.52, 158.95, 158.52, 142.17, 134.56, 128.56, 123.41, 121.86, 117.51, 109.33, 104.67, 60.26, 56.52, 48.96, 18.57, 12.99. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-5-chloro-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6c). 5-Chloroisatin 1c (36.3 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6c as white solid; m.p.: 247–248 °C; isolated yield (69.1 mg, 96%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 36.52 min; tmajor = 40.92 min; λ = 254 nm; 57% ee; [α]20D = −31.08° (c 0.05, CH3OH); [ref. 97 [α]24D = −21.7° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.56 (s, 1H, NH), 7.25 (s, 2H, NH2), 7.23 (dd, J = 8.2, 2.2 Hz, Ar–H), 7.19 (d, J = 2.2 Hz, 1H, Ar–H), 6.81 (d, J = 8.2 Hz, 1H, Ar–H), 3.81 (q, J = 7.1 Hz, 2H, CH2CH3), 2.33 (s, 3H, CH3), 0.83 (t, J = 7.1 Hz, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.45, 164.40, 159.57, 158.95, 141.06, 136.87, 128.47, 125.83, 123.65, 117.44, 110.80, 103.75, 60.46, 55.90, 49.31, 18.82, 13.08. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-6-chloro-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6d). 6-Chloroisatin 1d (36.3 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6d as white solid; m.p.: 258–259 °C; isolated yield (66.2 mg, 92%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 54.81 min; tmajor = 62.03 min; λ = 254 nm; 47% ee; [α]20D = +41.62° (c 0.04, CH3OH); IR (KBr): 3382, 3317, 3201, 2988, 2205, 1720, 1706, 1655, 1616, 1595, 1487, 1455, 1418, 1379, 1364, 1322, 1284, 1250, 1223, 1182, 1132, 854, 808, 776, 745, 719, 687, 633, 531 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.59 (s, 1H, NH), 7.24 (s, 2H, NH2), 7.18 (td, J = 7.6, 1.3 Hz, 1H, Ar–H), 7.14 (s, 2H, NH2), 7.10 (d, J = 7.9 Hz, 1H, Ar–H), 6.98 (dd, J = 7.9, 1.9 Hz, 1H, Ar–H), 6.81 (d, J = 1.9 Hz, 1H, Ar–H), 3.88–3.75 (m, 2H, CH2CH3), 2.32 (s, 3H, CH3), 0.84 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.62, 164.40, 159.20, 158.96, 143.69, 133.59, 132.74, 124.96, 121.63, 117.41, 109.40, 104.04, 60.48, 56.08, 48.75, 18.74, 13.11. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-3′-cyano-5-methoxy-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6e). 5-Methoxyisatin 1e (35.4 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6e as white solid; m.p.: 249–250 °C; isolated yield (66.1 mg, 93%). Enantiomeric excess (ee) was determined by chiral HPLC [Chiralpak IB column], 70.0% n-hexane/i-PrOH, 1.0 mL min−1; tminor = 15.36 min; tmajor = 33.13 min; λ = 254 nm; 87% ee; [α]20D = +11.08° (c 0.05, CH3OH); [ref. 97 [α]24D = +32.2° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.23 (s, 1H, NH), 7.15 (s, 2H, NH2), 6.75 (dd, J = 8.4, 2.6 Hz, 1H, Ar–H), 6.70 (d, J = 8.4 Hz, 1H, Ar–H), 6.68 (d, J = 2.6 Hz, 1H, Ar–H), 3.80–3.76 (m, 2H, CH2CH3), 3.67 (s, 3H, OCH3), 2.32 (s, 3H, CH3), 0.80 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.56, 164.55, 158.93, 158.67, 155.11, 135.90, 135.47, 117.60, 113.17, 110.28, 109.75, 104.56, 60.30, 56.60, 55.48, 49.46, 18.65, 13.05. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-3′-cyano-6′-methyl-5-nitro-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6f). 5-Nitroisatin 1f (38.4 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6f as white solid; m.p.: 253–254 °C; isolated yield (65.9 mg, 89%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 61.83 min; tmajor = 64.37 min; λ = 254 nm; 80% ee; [α]20D = +16.51° (c 0.05, CH3OH); IR (KBr): 3391, 3314, 3204, 2207, 1724, 1653, 1626, 1594, 1558, 1522, 1471, 1456, 1404, 1378, 1340, 1283, 1253, 1224, 1177, 1129, 1078, 844, 802, 756, 739, 690, 634, 557 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 11.18 (s, 1H, NH), 8.19 (dd, J = 8.6, 2.4 Hz, 1H, Ar–H), 8.05 (d, J = 2.4 Hz, 1H, Ar–H), 7.37 (s, 2H, NH2), 7.03 (d, J = 8.6 Hz, 1H, Ar–H), 3.86–3.78 (m, 2H, CH2CH3), 2.38 (s, 3H, CH3), 0.84 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 179.22, 164.26, 160.59, 159.04, 148.62, 142.50, 136.05, 126.09, 119.20, 117.26, 109.63, 103.00, 60.66, 55.18, 49.21, 19.02, 13.12.
Ethyl-(S)-2′-amino-5,7-dibromo-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6g). 5,7-Dibromoisatin 1g (61.0 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6g as white solid; m.p.: 250–251 °C; isolated yield (92.8 mg, 96%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 20.53 min; tmajor = 31.65 min; λ = 254 nm; 50% ee; [α]20D = +14.56° (c 0.04, CH3OH); IR (KBr): 3479, 3296, 3160, 3066, 2980, 2197, 1723, 1710, 1677, 1610, 1596, 1584, 1459, 1409, 1302, 1288, 1253, 1213, 1161, 1129, 1080, 779, 762, 744, 664, 867, 464 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.94 (s, 1H, NH), 7.65 (s, 1H, Ar–H), 7.39 (s, 1H, Ar–H), 7.35 (s, 2H, NH2), 3.83 (q, J = 9.3 Hz, 2H, CH2CH3), 2.35 (s, 3H, CH3), 0.86 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.34, 164.23, 160.20, 158.90, 141.15, 138.39, 133.18, 125.69, 117.32, 114.05, 103.26, 102.68, 60.62, 55.46, 50.40, 18.92, 13.01.
Ethyl-(S)-2′-amino-3′-cyano-5,6′-dimethyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6h). 5-Methylisatin 1h (32.2 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6h as white solid; m.p.: 255–256 °C; isolated yield (65.8 mg, 97%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tminor = 41.56 min; tmajor = 42.89 min; λ = 220 nm; 95% ee; [α]20D = −26.47° (c 0.06, CH3OH); [ref. 97 [α]24D = −24.0° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.28 (s, 1H, NH), 7.11 (s, 2H, NH2), 6.97 (d, J = 8.6 Hz, 1H, Ar–H), 6.87 (s, 1H, Ar–H), 6.68 (d, J = 7.9 Hz, 1H, Ar–H), 3.78 (q, J = 7.1 Hz, 2H, CH2CH3), 2.31 (s, 3H, CH3), 0.80 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.58, 164.55, 158.88, 158.51, 139.66, 134.73, 130.67, 128.78, 123.94, 117.57, 109.07, 104.71, 60.27, 56.75, 49.02, 20.63, 18.61, 13.00. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-3′-cyano-5-fluoro-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6i). 5-Fluoroisatin 1i (33.0 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6i as white solid; m.p.: 253–254 °C; isolated yield (65.9 mg, 98%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 90.0% n-hexane/i-PrOH, 0.8 mL min−1; tmajor = 39.61 min; tminor = 43.67 min; λ = 220 nm; 39% ee; [α]20D = −23.38° (c 0.08, CH3OH); [ref. 97 [α]24D = −18.9° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 10.44 (s, 1H, NH), 7.23 (s, 2H, NH2), 7.07–6.96 (m, 2H, Ar–H), 6.78 (dd, J = 8.5, 4.3 Hz, 1H, Ar–H), 3.87–3.73 (m, 2H, CH2CH3), 2.32 (s, 3H, CH3), 0.82 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 178.72, 164.42, 159.29, 159.20, 158.97, 157.31, 138.36, 136.48, 136.43, 117.44, 114.90, 114.71, 111.43, 111.23, 110.12, 110.06, 103.97, 60.40, 56.03, 49.53, 18.76, 13.06. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-3′-cyano-1,6′-dimethyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6j). 1-Methylisatin 1j (32.2 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6j as white solid; m.p.: = 257–258 °C, isolated yield (67.2 mg, 99%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 9.67 min; tminor = 11.24 min; λ = 254 nm; 63% ee; [α]20D = +26.29° (c 0.07, CH3OH); [ref. 97 [α]24D = +14.7° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.29 (td, J = 7.7, 1.4 Hz, 1H, Ar–H), 7.20 (s, 2H, NH2), 7.12 (dd, J = 7.4, 1.3 Hz, 1H, Ar–H), 7.04–6.99 (m, 2H, Ar–H), 3.72 (q, J = 3.0 Hz, 2H, CH2–CH3), 3.14 (s, 3H, NCH3), 2.33 (s, 3H, CH3), 0.72 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 176.96, 164.34, 158.98, 158.95, 143.50, 133.74, 128.76, 123.13, 122.62, 117.33, 108.25, 104.41, 60.19, 56.15, 48.51, 26.30, 18.62, 13.19. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-1-benzyl-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6k). 1-Benzylisatin 1k (47.5 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6k as white solid; m.p.: 251–252 °C; isolated yield (81.4 mg, 98%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 80.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 12.33 min; tminor = 20.59 min; λ = 254 nm; 81% ee; [α]20D = +19.55° (c 0.06, CH3OH); [ref. 97 [α]24D = +20.4° (c 1.0, MeOH)]; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.48–7.45 (m, 2H, Ar–H), 7.33–7.30 (m, 2H, Ar–H), 7.28 (s, 1H, Ar–H), 7.25 (s, 2H, NH2), 7.20 (td, J = 7.7, 1.3 Hz, 1H, Ar–H), 7.15 (dd, J = 7.4, 1.3 Hz, 1H, Ar–H), 7.00 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 6.86 (d, J = 7.7 Hz, 1H, Ar–H), 5.00 (d, J = 15.8 Hz, 1H, CH2(a)Bn), 4.78 (d, J = 15.8 Hz, 1H, CH2(b)Bn), 3.78 (q, J = 7.1 Hz, 1H, CH2CH3), 3.52 (dd, J = 10.8, 7.1 Hz, 1H, CH2CH3), 2.34 (s, 3H, CH3), 0.58 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 177.20, 164.38, 159.19, 158.86, 142.77, 136.19, 133.65, 128.40, 127.55, 123.34, 122.73, 117.57, 108.91, 104.55, 60.11, 56.12, 48.52, 43.39, 18.65, 13.08. All the analytical data are well in agreement with the reported literature.97
Ethyl-(S)-2′-amino-1-(2-bromoethyl)-3′-cyano-6′-methyl-2-oxospiro[indoline-3,4′-pyran]-5′-carboxylate (6l). 1-(2-Bromoethyl)isatin 1l (50.8 mg, 0.2 mmol), malononitrile 4 (13.2 mg, 0.2 mmol) and ethylacetoacetate 5 (26.0 mg, 0.2 mmol) were reacted according to the GP2 to yield product 6l as white solid; m.p.: 148–150 °C; isolated yield (76.9 mg, 89%). Enantiomeric excess (ee) was determined by chiral HPLC [Daicel Chiralpak AD-H column], 85.0% n-hexane/i-PrOH, 1.0 mL min−1; tmajor = 15.03 min; tminor = 24.53 min; λ = 254 nm; 23% ee; [α]20D = +5.34° (c 0.10, CH3OH); IR (KBr): 3382, 2924, 2854, 2191, 1709, 1682, 1613, 1599, 1486, 1466, 1419, 1380, 1348, 1282, 1253, 1211, 1149, 1077, 756, 682 cm−1; 1H-NMR (500 MHz, DMSO-d6): δ (ppm) = 7.29 (td, J = 7.6, 1.3 Hz, 1H, Ar–H), 7.23 (s, 2H, NH2), 7.18–7.13 (m, 2H, Ar–H), 7.03 (td, J = 7.5, 1.0 Hz, 1H, Ar–H), 4.16 (ddd, J = 14.2, 7.7, 6.5 Hz, 1H, CH3CH2(a)), 4.03 (ddd, J = 14.2, 7.7, 6.1 Hz, 1H, CH3CH2(b)), 3.82 (dq, J = 11.0, 7.1 Hz, 1H, NCH2(a)), 3.72 (qd, J = 7.1, 4.6 Hz, 1H, NCH2(b)), 3.63–3.57 (m, 2H, CH2Br), 2.33 (s, 3H, CH3), 0.71 (t, J = 7.1 Hz, 3H, CH2CH3); 13C-NMR (126 MHz, DMSO-d6): δ (ppm) = 177.03, 164.32, 158.99, 142.18, 133.54, 128.76, 123.41, 122.84, 117.14, 108.70, 104.36, 60.25, 56.08, 48.41, 41.57, 28.36, 18.67, 13.22.
Conclusions
In summary, we have developed very efficient enantioselective process for the asymmetric Aldol reaction of substituted isatin with several acetone derivatives using L1-Cu(II) as a new Lewis acid catalyst based on thiophene-2,5-bis(amino-alcohol) framework. Our Lewis acid catalytic system L1-Cu(II) acts brilliantly and furnished aldol products 3a–s in high yields and selectivity (up to 99% yield; up to 96% ee). This catalytic system L1-Cu(OAc)2·H2O was further utilized for asymmetric Domino Knoevenagel Michael cyclization reaction of substituted isatin with malononitrile and ethylacetoacetate and afforded chiral spiro[4H-pyran-3,3-oxindole] derivatives 6a–l with high yields and moderate to high enantioselectivities (up to 99% yield; up to 95% ee). Both the reactions were performed under mild reaction condition at room temperature.Author contributions
Conceptualization, A. M. A.-M. and A. B.; supervision, A. M. A.-M., A. B. and M. S. I; methodology, A. S. A., M. S. I. and A. M. A.-M.; validation, M. S. I., A. S. A., S. A. and A. M. A.-M.; formal analysis, A. S. A.; M. S. I. and S. A.; investigation, A. S. A.; M. S. I. and S. A.; resources, A. M. A.-M. and A. B.; data curation, A. S. A., M. S. I., and A. B.; writing—original draft preparation, M. S. I., A. B. and A. S. A.; writing—review and editing, M. S. I., A. B. and A. M. A.-M.; visualization, A. B., M. S. I and S. A.; project administration, A. M. A.-M. and A. B.; funding acquisition, A. M. A.-M. and A. B.; software A. S. A., M. S. I., M. A. and A. B.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to extend their sincere appreciation to the Researchers Supporting Project (RSP2022R427), King Saud University, Riyadh, Saudi Arabia.Notes and references
- P. W. V. Leeuwen, P. C. Kamer, C. Claver, O. Pamies and M. Dieguez, Chem. Rev., 2011, 111, 2077–2118 CrossRef PubMed.
- A. Barakat, M. S. Islam, A. M. Al Majid and Z. A. Al-Othman, Tetrahedron, 2013, 69, 5185–5192 CrossRef CAS.
- M. S. Islam, A. M. Al Majid, Z. A. Al-Othman and A. Barakat, Tetrahedron: Asymmetry, 2014, 25, 245–251 CrossRef CAS.
- M. S. Islam, A. S. Alammari, A. Barakat, S. Alshahrani, M. Haukka and A. M. Al-Majid, Molecules, 2021, 26, 7408 CrossRef CAS PubMed.
- A. S. Alammari, A. M. Al-Majid, A. Barakat, S. Alshahrani, M. Ali and M. S. Islam, Catalysts, 2021, 11, 1208 CrossRef CAS.
- T. Mukaiyama, Org. React., 1982 Search PubMed.
- Q. Guo, M. Bhanushali and C. G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 9460–9464 CrossRef CAS PubMed.
- T. Yan, X. Wang, H. Sun, J. Liu and Y. Xie, Molecules, 2013, 18, 14505–14518 CrossRef PubMed.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed.
- V. Raj, Int. J. Curr. Pharm. Res., 2012, 4, 1–9 CAS.
- C. Marti and E. M. Carreira, Eur. J. Org. Chem., 2003, 2003, 2209–2219 CrossRef.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20–38 CrossRef CAS.
- S. Mohammadi, R. Heiran, R. P. Herrera and E. Marques-Lopez, ChemCatChem, 2013, 5, 2131–2148 CrossRef CAS.
- R. B. Labroo and L. A. Cohen, J. Org. Chem., 1990, 55, 4901–4904 CrossRef CAS.
- P. Hewawasam, N. A. Meanwell, V. K. Gribkoff, S. I. Dworetzky and C. G. Boissard, Bioorg. Med. Chem. Lett., 1997, 7, 1255–1260 CrossRef CAS.
- Y. Q. Tang, I. Sattler, R. Thiericke, S. Grabley and X. Z. Feng, Eur. J. Org. Chem., 2001, 2001, 261–267 CrossRef.
- T. Tokunaga, W. E. Hume, T. Umezome, K. Okazaki, Y. Ueki, K. Kumagai, S. Hourai, J. Nagamine, H. Seki and M. Taiji, J. Med. Chem., 2001, 44, 4641–4649 CrossRef CAS PubMed.
- P. Hewawasam, M. Erway, S. L. Moon, J. Knipe, H. Weiner, C. G. Boissard, D. J. Post-Munson, Q. Gao, S. Huang and V. K. Gribkoff, J. Med. Chem., 2002, 45, 1487–1499 CrossRef CAS PubMed.
- V. Khuzhaev, I. Zhalolov, K. Turgunov, B. Tashkhodzhaev, M. Levkovich, S. Aripova and A. Shashkov, Chem. Nat. Compd., 2004, 40, 269–272 CrossRef CAS.
- H. B. Rasmussen and J. K. MacLeod, J. Nat. Prod., 1997, 60, 1152–1154 CrossRef CAS.
- T. Kagata, S. Saito, H. Shigemori, A. Ohsaki, H. Ishiyama, T. Kubota and J. i. Kobayashi, J. Nat. Prod., 2006, 69, 1517–1521 CrossRef CAS PubMed.
- Y. Kamano, H.-p. Zhang, Y. Ichihara, H. Kizu, K. Komiyama and G. R. Pettit, Tetrahedron Lett., 1995, 36, 2783–2784 CrossRef CAS.
- C. Moody, I. Richards and A. Z. Slawin, J. Chem. Soc., Perkin Trans. 1, 1997, 2405–2412 Search PubMed.
- T. Nakamura, S.-i. Shirokawa, S. Hosokawa, A. Nakazaki and S. Kobayashi, Org. Lett., 2006, 8, 677–679 CrossRef CAS PubMed.
- S. Nakamura, N. Hara, H. Nakashima, K. Kubo, N. Shibata and T. Toru, Chem.–Eur. J., 2008, 14, 8079–8081 CrossRef CAS PubMed.
- G. Luppi, M. Monari, R. J. Corrêa, F. d. A. Violante, A. C. Pinto, B. Kaptein, Q. B. Broxterman, S. J. Garden and C. Tomasini, Tetrahedron, 2006, 62, 12017–12024 CrossRef CAS.
- J. S. Carle and C. Christophersen, J. Org. Chem., 1981, 46, 3440–3443 CrossRef CAS.
- K. Nicolaou, X. Huang, N. Giuseppone, P. Bheema Rao, M. Bella, M. V. Reddy and S. A. Snyder, Angew. Chem., Int. Ed., 2001, 40, 4705–4709 CrossRef CAS.
- K. Nicolaou, M. Bella, D. Y. K. Chen, X. Huang, T. Ling and S. A. Snyder, Angew. Chem., Int. Ed., 2002, 114, 3645–3649 CrossRef.
- K. Nicolaou, P. Bheema Rao, J. Hao, M. V. Reddy, G. Rassias, X. Huang, D. Y. K. Chen and S. A. Snyder, Angew. Chem., Int. Ed., 2003, 42, 1753–1758 CrossRef CAS PubMed.
- K. Nicolaou, S. A. Snyder, N. Giuseppone, X. Huang, M. Bella, M. V. Reddy, P. B. Rao, A. E. Koumbis, P. Giannakakou and A. O'Brate, J. Am. Chem. Soc., 2004, 126, 10174–10182 CrossRef CAS PubMed.
- K. Nicolaou, D. Y.-K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, J. Am. Chem. Soc., 2004, 126, 12888–12896 CrossRef CAS PubMed.
- C. Takahashi, A. Numata, Y. Ito, E. Matsumura, H. Araki, H. Iwaki and K. Kushida, J. Chem. Soc., Perkin Trans. 1, 1994, 1859–1864 RSC.
- A. Fréchard, N. Fabre, C. Péan, S. Montaut, M.-T. Fauvel, P. Rollin and I. Fourasté, Tetrahedron Lett., 2001, 42, 9015–9017 CrossRef.
- M. Kitajima, I. Mori, K. Arai, N. Kogure and H. Takayama, Tetrahedron Lett., 2006, 47, 3199–3202 CrossRef CAS.
- J. I. Jimenez, U. Huber, R. E. Moore and G. M. Patterson, J. Nat. Prod., 1999, 62, 569–572 CrossRef CAS PubMed.
- B. K. Albrecht and R. M. Williams, Org. Lett., 2003, 5, 197–200 CrossRef CAS PubMed.
- S. Lin, Z.-Q. Yang, B. H. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347–6355 CrossRef CAS PubMed.
- K. S. Feldman and A. G. Karatjas, Org. Lett., 2004, 6, 2849–2852 CrossRef CAS PubMed.
- H. Suzuki, H. Morita, M. Shiro and J. i. Kobayashi, Tetrahedron, 2004, 60, 2489–2495 CrossRef CAS.
- M. Suchý, P. Kutschy, K. Monde, H. Goto, N. Harada, M. Takasugi, M. Dzurilla and E. Balentova, J. Org. Chem., 2001, 66, 3940–3947 CrossRef PubMed.
- F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS.
- J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758–776 CAS.
- A. Kumar and S. S. Chimni, RSC Adv., 2012, 2, 9748–9762 RSC.
- B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2003, 42, 858–860 CrossRef CAS PubMed.
- B. List, L. Hoang and H. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5839–5842 CrossRef CAS.
- C. Allemann, R. Gordillo, F. R. Clemente, P. H.-Y. Cheong and K. Houk, Acc. Chem. Res., 2004, 37, 558–569 CrossRef CAS PubMed.
- S. Saito and H. Yamamoto, Acc. Chem. Res., 2004, 37, 570–579 CrossRef CAS PubMed.
- S. M. Dean, W. A. Greenberg and C. H. Wong, Adv. Synth. Catal., 2007, 349, 1308–1320 CrossRef CAS.
- M. Raj and V. K. Singh, Chem. Commun., 2009, 6687–6703 RSC.
- V. Bisai, A. Bisai and V. K. Singh, Tetrahedron, 2012, 68, 4541–4580 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- G. Luppi, P. G. Cozzi, M. Monari, B. Kaptein, Q. B. Broxterman and C. Tomasini, J. Org. Chem., 2005, 70, 7418–7421 CrossRef CAS PubMed.
- E. M. Beccalli, A. Marchesini and T. Pilati, J. Chem. Soc., Perkin Trans. 1, 1994, 579–587 RSC.
- S. J. Garden, R. B. da Silva and A. C. Pinto, Tetrahedron, 2002, 58, 8399–8412 CrossRef CAS.
- T. Kawasaki, M. Nagaoka, T. Satoh, A. Okamoto, R. Ukon and A. Ogawa, Tetrahedron, 2004, 60, 3493–3503 CrossRef CAS.
- W.-B. Chen, Y.-H. Liao, X.-L. Du, X.-M. Zhang and W.-C. Yuan, Green Chem., 2009, 11, 1465–1476 RSC.
- R. J. Corrêa, S. J. Garden, G. Angelici and C. Tomasini, Eur. J. Org. Chem., 2008, 2008, 736–744 CrossRef.
- G. Angelici, R. J. Corrêa, S. J. Garden and C. Tomasini, Tetrahedron Lett., 2009, 50, 814–817 CrossRef CAS.
- J. G. Hernández, V. García-López and E. Juaristi, Tetrahedron, 2012, 68, 92–97 CrossRef.
- M. Kinsella, P. G. Duggan and C. M. Lennon, Tetrahedron: Asymmetry, 2011, 22, 1423–1433 CrossRef CAS.
- J.-R. Chen, X.-P. Liu, X.-Y. Zhu, L. Li, Y.-F. Qiao, J.-M. Zhang and W.-J. Xiao, Tetrahedron, 2007, 63, 10437–10444 CrossRef CAS.
- C. Shen, F. Shen, H. Xia, P. Zhang and X. Chen, Tetrahedron: Asymmetry, 2011, 22, 708–712 CrossRef CAS.
- T. P. Kumar, N. Manjula and K. Katragunta, Tetrahedron: Asymmetry, 2015, 26, 1281–1284 CrossRef CAS.
- G. D. Yadav and S. Singh, Tetrahedron: Asymmetry, 2016, 27, 123–129 CrossRef CAS.
- A. J. Pearson and S. Panda, Org. Lett., 2011, 13, 5548–5551 CrossRef CAS PubMed.
- G. D. Yadav and S. Singh, Tetrahedron: Asymmetry, 2015, 26, 1156–1166 CrossRef CAS.
- N. Hara, S. Nakamura, N. Shibata and T. Toru, Adv. Synth. Catal., 2010, 352, 1621–1624 CrossRef CAS.
- N. Hara, S. Nakamura, N. Shibata and T. Toru, Chem.–Eur. J., 2009, 15, 6790–6793 CrossRef CAS PubMed.
- J. Wang, Q. Liu, Q. Hao, Y. Sun, Y. Luo and H. Yang, Chirality, 2015, 27, 314–319 CrossRef CAS PubMed.
- M. Raj, N. Veerasamy and V. K. Singh, Tetrahedron Lett., 2010, 51, 2157–2159 CrossRef CAS.
- Q. Guo and J. C.-G. Zhao, Tetrahedron Lett., 2012, 53, 1768–1771 CrossRef CAS.
- D. Ma, L. Ren, H. Yao, W. Tian, F. Chen, J. Zhang, Y. Liu and T. Mao, J. Orthop. Res., 2013, 31, 1082–1088 CrossRef CAS PubMed.
- H. Lu, J. Bai, J. Xu, T. Yang, X. Lin, J. Li and F. Ren, Tetrahedron, 2015, 71, 2610–2615 CrossRef CAS.
- S. Abbaraju and J. C. G. Zhao, Adv. Synth. Catal., 2014, 356, 237–241 CrossRef CAS PubMed.
- A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Org. Lett., 2007, 9, 5473–5476 CrossRef CAS PubMed.
- Z.-Q. Liu, Z.-W. Xiang, Z. Shen, Q. Wu and X.-F. Lin, Biochimie, 2014, 101, 156–160 CrossRef CAS PubMed.
- T. Itoh, H. Ishikawa and Y. Hayashi, Org. Lett., 2009, 11, 3854–3857 CrossRef CAS PubMed.
- G. Chen, Y. Ju, T. Yang, Z. Li, W. Ang, Z. Sang, J. Liu and Y. Luo, Tetrahedron: Asymmetry, 2015, 26, 943–947 CrossRef CAS.
- R. Singh and R. Vince, Chem. Rev., 2012, 112, 4642–4686 CrossRef CAS PubMed.
- Y.-T. Yang, J.-F. Zhu, G. Liao, H.-J. Xu and B. Yu, Curr. Med. Chem., 2018, 25, 2233–2244 CrossRef CAS PubMed.
- M. S. Islam, M. Ali, A. M. Al-Majid, A. S. Alamary, S. Alshahrani, S. Yousuf, M. I. Choudhary and A. Barakat, Molecules, 2021, 26, 2212 CrossRef CAS PubMed.
- M. S. Islam, A. M. Al-Majid, M. Azam, V. P. Verma, A. Barakat, M. Haukka, A. A. Elgazar, A. Mira and F. A. Badria, ACS Omega, 2021, 31539–31556 CrossRef CAS PubMed.
- M. S. Islam, A. M. Al-Majid, F. F. El-Senduny, F. A. Badria, A. Rahman, A. Barakat and Y. A. Elshaier, Appl. Sci., 2020, 10, 2170 CrossRef CAS.
- M. S. Islam, A. M. Al-Majid, M. Azam, V. Prakash Verma, A. Barakat, M. Haukka, L. R. Domingo, A. A. Elgazar, A. Mira and F. A. Badria, ChemistrySelect, 2021, 6, 14039–14053 CrossRef.
- A. Barakat, M. S. Islam, M. Ali, A. M. Al-Majid, S. Alshahrani, A. S. Alamary, S. Yousuf and M. I. Choudhary, Symmetry, 2021, 13, 1426 CrossRef CAS.
- A. M. Al-Majid, M. Ali, M. S. Islam, S. Alshahrani, A. S. Alamary, S. Yousuf, M. I. Choudhary and A. Barakat, Molecules, 2021, 26, 6305 CrossRef CAS PubMed.
- R. Dworczak, H. Sterk, C. Kratky and H. Junek, Chem. Ber., 1989, 122, 1323–1328 CrossRef CAS.
- S. J. Chai, Y. F. Lai, J. C. Xu, H. Zheng, Q. Zhu and P. F. Zhang, Adv. Synth. Catal., 2011, 353, 371–375 CrossRef CAS.
- G. Brahmachari and B. Banerjee, Asian J. Org. Chem., 2016, 5, 271–286 CrossRef CAS.
- D. Cheng, Y. Ishihara, B. Tan and C. F. Barbas III, ACS Catal., 2014, 4, 743–762 CrossRef CAS.
- G. M. Ziarani, R. Moradi and N. Lashgari, Tetrahedron, 2018, 74, 1323–1353 CrossRef.
- W.-B. Chen, Z.-J. Wu, Q.-L. Pei, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 3132–3135 CrossRef CAS PubMed.
- M. Chennapuram, I. A. Owolabi, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, ACS Omega, 2018, 3, 11718–11726 CrossRef CAS PubMed.
- S. Konda, S. Jakkampudi, H. D. Arman and J. C.-G. Zhao, Synth. Commun., 2019, 49, 2971–2982 CAS.
- S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402–3415 CrossRef CAS PubMed.
- W.-J. Hao, S.-Y. Wang and S.-J. Ji, ACS Catal., 2013, 3, 2501–2504 CrossRef CAS.
- A. Khalafi-Nezhad and S. Mohammadi, ACS Comb. Sci., 2013, 15, 512–518 CrossRef CAS PubMed.
- A. Mondal and C. Mukhopadhyay, ACS Comb. Sci., 2015, 17, 404–408 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2143162. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00674j |
| This journal is © The Royal Society of Chemistry 2022 |