
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly resolved, label-free nucleic acid sensing at solid–liquid interface using non-ionic DNA analogues †
Tanushree Manaa,
Jayanta Kundub,
Hiya Lahiri‡
a,
Sudipta Bera§
a,
Jayeeta Kolaya,
Surajit Sinhab and
Rupa Mukhopadhyay *a
*a
aSchool of Biological Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India. E-mail: bcrm@iacs.res.in; Fax: +91 33 2473 2805; Tel: +91 33 2473 4971 extn 1506
bSchool of Applied and Interdisciplinary Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700 032, India
First published on 24th March 2022
Abstract
Nucleic acid-based biosensors, where the capture probe is a nucleic acid, e.g., DNA or its synthetic analogue xeno nucleic acid (XNA), offer interesting ways of eliciting clinically relevant information from hybridization/dehybridization signals. In this respect, the application of XNA probes is attractive since the drawbacks of DNA probes might be overcome. Within the XNA probe repertoire, peptide nucleic acid (PNA) and morpholino (MO) are promising since their backbones are non-ionic. Therefore, in the absence of electrostatic charge repulsion between the capture probe and the target nucleic acid, a stable duplex can be formed. In addition, these are nuclease-resistant probes. Herein, we have tested the molecularly resolved nucleic acid sensing capacity of PNA and MO capture probes using a fluorescent label-free single molecule force spectroscopy approach. As far as single nucleobase mismatch discrimination is concerned, both PNA and MO performed better than DNA, while the performance of the MO probe was the best. We propose that the conformationally more rigid backbone of MO, compared to the conformationally flexible PNA, is an advantage for MO, since the probe orientation can be made more upright on the surface and therefore MO can be more effectively accessed by the target sequences. The performance of the XNA probes has been compared to that of the DNA probe, using fixed nucleobase sequences, so that the effect of backbone variation could be investigated. To our knowledge, this is the first report on molecularly resolved nucleic acid sensing by non-ionic capture probes, here, MO and PNA.
Introduction
The development of assays for target-specific detection of nucleic acid sequences, especially with the capacity of single nucleobase mismatch discrimination, is necessary for a number of applications, for example, in diagnosis of genetic diseases.1,2 The single nucleobase mismatches or the point mutations within the gene stretches can be the basis for a range of human diseases, for example, sickle-cell anemia. The point mutations are also known to be responsible for developing susceptibility to cancer, tuberculosis and Alzheimer's disease. Detection of the disease-relevant gene mutations is desirable for early disease diagnosis and several approaches have been proposed towards this direction.3 However, the classical detection methods, for example, microarray-based techniques, mostly consist of multiple steps, or require fluorescent labeling. These methods can also be time-consuming, and suffer from non-linear amplification problems. This has encouraged research activities on development of fluorescent label-free technologies so that sample preparation time is reduced, sample integrity is not interfered with, reliable data that is free from non-specific signals is acquired, and data analysis is made simpler. In this direction, some progress using a fluorescent label-free assay based on the single molecule force spectroscopy (SMFS) approach and synthetic xeno nucleic acid (XNA) capture probes has been made.4,5 Apart from being fluorescent label-independent, this assay allows molecularly resolved detection and is potentially a polymerase chain reaction (PCR)-amplification-free approach.The SMFS approach is a valuable methodology for estimating intra- and intermolecular forces,6–9 as operative in bio-molecular recognition processes, with high sensitivity and in near-physiological condition. In this approach, specific interactions are studied by functionalizing the surface and the SMFS tip with the desired molecules of interest.10–13 This approach allows sequence-specific recognition of nucleic acid sequences through measurement of the unbinding force values that are linked with dehybridization of the surface-anchored target-capture probe duplexes.4 SMFS requires low concentration of target nucleic acid (10–20 nM or 80–160 pg μL−1), and each recognition event takes place at the millisecond time scale.14 Here, SMFS is possible due to the tip geometry that can ensure that only a very few number of DNA molecules on the tip can interact with the XNA-modified surface. Importantly, it has been employed for quantitative estimation of the rupture force (at pN level) that is applied for unbinding DNA duplexes consisting of short oligonucleotides (10–30 bp),15,16 at single base pair resolution.17 This has been achieved by modifying the tip with ssDNA strands and functionalizing the surface with the complementary ssDNA strands. When the tip comes close to the surface during the tip approach step, hybridization occurs and a duplex is formed. The duplex is then dehybridized via force-induced rupture of the duplex by withdrawing the tip from the surface, where the tip withdrawal force needed to unbind the duplex is a measure of duplex dehybridization. In such an assay, detection of a specific target sequence requires oriented immobilization of the capture probe, where the capture probe density is optimal for achieving 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 target–capture interaction.18,19 Since the efficiency of hybridization depends on the level of complementarity between the nucleobases in the probe and the target nucleic acid sequences, the rupture force as measured by SMFS is directly influenced by the extent of complementarity in the target and the capture probe sequences.
:1 target–capture interaction.18,19 Since the efficiency of hybridization depends on the level of complementarity between the nucleobases in the probe and the target nucleic acid sequences, the rupture force as measured by SMFS is directly influenced by the extent of complementarity in the target and the capture probe sequences.
In this study, the amine-terminated ssDNA target sequences (Table 1) were immobilized onto the SMFS tip surface and the thiol-terminated capture probes were anchored onto the silicon surface. Both the capture probe and the target sequences were kept short in length (10–12 mer) as short oligonucleotide capture probes tend to arrange in end-anchored and extended configuration,20 and duplex stabilization is achieved best with the short sequences. While it has recently been shown that longer capture probes (15 mer) can be used to detect target sequences as long as 45 mer with overhang region,5 herein we used short 10–12 mer capture probes so that the best possible duplex stabilization could be achieved in this first SMFS-based nucleic acid detection study using PNA and MO capture probes. Since the duplex stability can be influenced by different factors such as the position of mismatch14 and mismatch type,21 we kept the mismatch type fixed and the mismatch site centrally positioned in both capture probe and target sequences as this site has been shown to be more effective in mismatch discrimination than the terminally positioned sites.14
| Target/capture probe | Nucleic acid sequences |
|---|---|
| DNA-1 | 5′-HS-C6-CTA-TGT-CAG-CAC-3′ |
| DNA-2 | 5′-HS-C6-CTA-TGT-AAG-CAC-3′ |
| DNA-3 | 5′-HS-C6-CGA-TCT-GCT-AAC-3′ |
| PNA-1 | N-ter-HS-C6-CTA-TGT-CAG-CAC-CONH2-C-ter |
| PNA-2 | N-ter-HS-C6-CTA-TGT-AAG-CAC-CONH2-C-ter |
| PNA-3 | N-ter-HS-C6-CGA-TCT-GCT-AAC-CONH2-C-ter |
| PNA-4 | N-ter-SH-C6-CT CT CT CT CT-CONH2-C-ter |
| MO-1 | 3′-HS-C6-CTA-TGT-CAG-CAC-5′ |
| MO-2 | 3′-HS-C6-CTA-TGT-AAG-CAC-5′ |
| T-DNA-1 | 5′-H2N-C6-GTG-CTG-ACA-TAG-3′ |
| T-DNAnc | 5′-H2N-C6-CGA-TCT-GCT-AAC-3′ |
| T-DNA-2 | 3′-H2N-C7-GTG-CTG-ACA-TAG-5′ |
| T-DNA-3 | 3′-H2N-C7-CGA-TCT-GAT-AGC-5′ |
| T-DNA-4 | 5′-NH2-C6-AG AG AG AG AG-3′ |
| T-DNA-5 | 3′-TC TC TC TC TC-5′ |
It has been observed before that when DNA is used as the capture probe, sensing efficiency may reduce due to non-specific interactions between DNA and substrate through the DNA nucleobases,22 and due to DNA degradation by the nuclease enzyme.23 Recently, the XNAs, which are the synthetic analogues of DNA differing in the sugar backbone compared to DNA/RNA, have been introduced as alternative capture probes to detect target DNA strands at the solid–liquid interface with greater sensitivity and target specificity compared to the DNA probes.4 Among these analogues, peptide nucleic acid or PNA is particularly an interesting DNA analogue since it is non-ionic in nature due to its polyamide backbone that consists of N-(2-aminoethyl)-glycine units that are linked by the peptide bonds24 (Fig. 1). Such structural feature provides PNA high chemical as well as thermal stability,24,25 and resistance to enzymatic degradation as the achiral backbone of PNA is not easily recognized by the nuclease and protease enzymes.26 An important property of PNA is sequence-specific binding with complementary DNA through Watson–Crick type base paring. The PNA has been proposed as a capture probe with clear advantages over the ssDNA capture probe in a number of studies.27–30 Another interesting non-ionic DNA analogue is morpholino (MO) having morpholine rings (Fig. 1), phosphorodiamidates and nucleobases.31 MOs are highly soluble in water,32 are resistant to the action of nuclease enzyme33 and can form thermally stable duplexes with DNA and RNA.32 In a recent work, the formation of morpholino monolayer on gold surface and morpholino's hybridization affinity towards target DNA sequences has been reported.34 The sequence-specific DNA detection using MO-functionalized silicon chip35 and nanowire36 has also been reported. These reports indicate that MO could be an interesting capture probe for nucleic acid sensing.
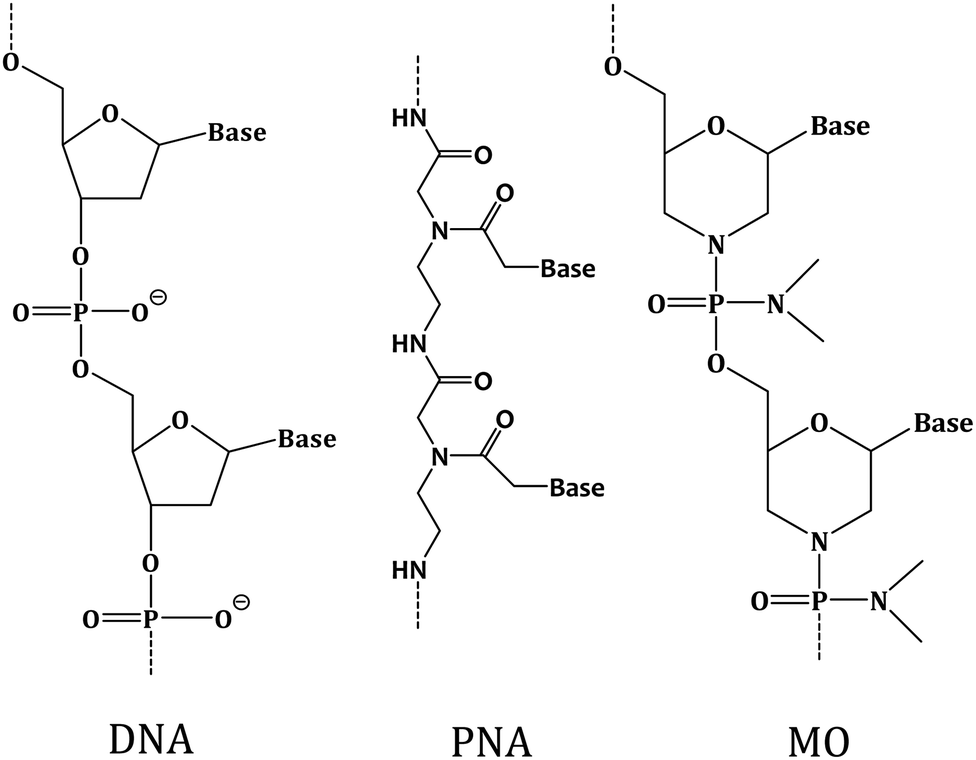 | ||
| Fig. 1 Chemical structures of deoxyribo nucleic acid (DNA), peptide nucleic acid (PNA) and morpholino (MO). | ||
Herein, we applied PNA and MO, the two different non-ionic XNAs with modified backbones, as the capture probes to develop an XNA-based nucleic acid detection method and exemplified how molecularly resolved, fluorescent label-free detection of ssDNA sequences can be performed by using the SMFS approach. The nucleobase sequences were kept fixed for these two XNA probes, and for the corresponding DNA capture probe, so that the effect of backbone variation could be investigated. The XNA-modified sensing layer was prepared by covalently immobilizing the probes onto silicon substrate, where the layer was found to be of monolayer thickness from an atomic force microscopy (AFM) investigation. The XNA attachment onto silicon was examined by X-ray photoelectron spectroscopy or XPS. Finally, the sequence-specific sensing ability of the XNA-modified surface was tested by using the SMFS approach. It has been observed that both PNA and MO are capable of sequence-specific detection including single nucleobase mismatch discrimination better than the DNA probe. MO appeared to be the most efficient capture probe as far as duplex stability and single nucleobase mismatch discrimination is concerned. We propose that the non-ionic character of PNA and MO is an advantage over the negatively charged DNA capture probe in detection of charged nucleic acids. We further propose that the conformationally rigid backbone of the MO probe is an advantage for MO over PNA and DNA since it allows more upright probe orientation which is conducive for sequence-specific nucleic acid detection.4
Materials and methods
Chemicals and materials
Filtered and autoclaved Milli-Q water (resistivity: 18.2 MΩ cm, Millipore) was used for preparing all the buffers and nucleic acid solutions. Sodium chloride (NaCl), disodium hydrogen phosphate (Na2HPO4) and sodium dihydrogen phosphate (NaH2PO4) were purchased from Merck (purity ≥ 99%). 1,4-Dithiothreitol (DTT), (3-mercaptopropyl) trimethoxysilane (MPTMS) and (3-aminopropyl) triethoxysilane (APTES) were procured from Sigma-Aldrich (purity ≥ 99%).Preparation of DNA solutions
The 12-mer ssDNA capture probe samples [DNA-1, DNA-2 and DNA-3 (Table 1), all having a hexyl thiol [–(CH2)6SH] group at the 5′ position] (Alpha DNA, Canada) were dissolved in sodium citrate buffer (0.3 M Na citrate·2H2O, 3 M NaCl, pH 4.5). DNA-1 sequence was used for full match and DNA-2 for single mismatch study, while DNA-3 was the fully mismatched sequence used for control experiments. The concentrations of the DNA solutions were determined by UV-visible spectrophotometry (Fig. S1A†) at room temperature, for the absorbance value at 260 nm, where the ε260 (L (mol cm)−1) values for DNA-1, DNA-2 and DNA-3 were 1.23 × 105, 1.31 × 105 and 1.23 × 105, respectively.Preparation of PNA solutions
The solutions of the ssPNA capture probes (10/12 mer) PNA-1, PNA-2, PNA-3 and PNA-4 (Table 1) (Panagene, Korea), all having a [–(CH2)6SH] group at N-ter position, were prepared in Milli-Q water of resistivity 18.2 MΩ cm. The concentrations of the PNA solutions were determined by UV-visible spectrophotometry (Fig. S1B†) for the absorbance value at 260 nm, where the ε260 (L (mol cm)−1) values for PNA-1, PNA-2, PNA-3 and PNA-4 are 1.17 × 105, 1.24 × 105, 1.17 × 105 and 0.77 × 105, respectively. The PNA-1 and PNA-2 were used for full match and for single nucleobase mismatch situations, respectively. The PNA-3 was the fully mismatched capture probe used in the control experiments. PNA-4 was used for the ssPNA:dsDNA triplex formation experiment.Preparation of MO solutions
The MO sequences were synthesized (the MO synthesis cycle and the MALDI-TOF mass spectrum for both MO-1 and MO-2 are provided in Fig. S2 and S3,† respectively) following the published protocols37–39 and HPLC purified (Fig. S4†). The solutions of the thiolated, fully modified MO sequences (12 mer) (Table 1) were made in Milli-Q water (resistivity: 18.2 MΩ cm, Millipore). The MO concentrations were verified by UV-visible spectrophotometry (Fig. S1C†) using ε260 (L (mol cm)−1) values for MO-1 and MO-2 as 1.22 × 105 and 1.41 × 105, respectively.Preparation of target DNA (NH2-terminated) solutions
The solutions of the NH2-terminated target DNA sequences (10/12-mer) (Sigma and GCC Biotech) (Table 1) were prepared in 20 mM Na-phosphate, 100 mM NaCl, pH 7.0 buffer. The concentrations of the DNA solutions were verified by using ε260 (L (mol cm)−1) values for T-DNA-1, T-DNAnc, T-DNA-2, T-DNA-3, T-DNA-4 and T-DNA-5 as 1.19 × 105, 1.11 × 105, 1.22 × 105, 1.18 × 105, 1.182 × 105 and 0.764 × 105, respectively.Modification of silicon substrate by the thiolated XNA capture probes
Application of the XNA-based SMFS assay has been tested earlier using silicon substrate – a substrate which is proven to be useful in a number of technological applications.40 The silicon wafers were cut into 10 × 10 mm2 pieces and cleaned by using ethyl acetate, acetone and last by ethanol by bath sonication for 2 min in each solvent. Then the wafer was further cleaned by treatment with piranha solution (7:3 v/v of H2SO4/H2O2) for 30 min at 80 °C and then washed in Milli-Q water and dried under soft nitrogen jet. The cleaned silicon substrate (Fig. S5A†) was then immersed into 10 mM 3-mercaptopropyl trimethoxysilane (3-MPTMS) solution, which was prepared in 2-propanol in presence of 5 mM DTT. After incubation for 12 h, the modified substrate was subjected to bath sonication in acetone for 2 min, and then rinsing in ethanol. Then to immobilize the PNA/MO probes, the 3-MPTMS-modified silicon substrate was kept immersed into the PNA/MO solution of desired concentration (Table 2), as the case may be, for 12 h. It is known that the concentration of the capture probe influences formation of self-assembled layer of the capture probes, and also the probe backbone orientation on surface, which affects target recognition ability.18,41 The probe concentration is one of the factors by controlling which the surface coverage can be optimized. The concentrations of the capture probes (here, DNA, PNA and MO) were optimized in such a manner that an ideal surface coverage of the capture probes could be generated and force curves with single unbinding events could be obtained by minimizing multiple binding events and/or non-specific interactions. The optimized concentrations as mentioned in Table 2 for the different capture probes are of different values because the structures of DNA, PNA and MO are different and therefore, their interactions (with substrate and with the adjacent probes) are also of different nature and degree. In general, we found that the non-ionic probes PNA and MO needed lower concentration than the negatively charged DNA probe, most likely because molecular crowding at the solid–liquid interface was necessary in case of DNA to force DNA layer formation in the presence of the inhibiting factor of electrostatic repulsion between the adjacent DNA probe sequences. The self-assembled XNA layer was prepared by the simple immersion approach41 so that the nucleic acid strands could be kept solvated during the preparative stage and an optimal surface coverage could be attained. The sample was finally washed with Milli-Q water. It was then dried under a gentle stream of nitrogen jet.
| Capture probe | Concentration of capture probe (μM) | Concentration of target DNA (nM) | Cantilever retraction speed (μm s−1) |
|---|---|---|---|
| DNA | 0.5 | 20 | 0.5 |
| PNA | 0.001 | 10 | 0.5 |
| MO | 0.005 | 10 | 0.5 |
The steps for immobilization of the thiolated PNA and MO probes onto silicon substrate are shown in Fig. 2A. The anchoring is covalent in nature since a disulfide (–S–S–) bond is formed between 3-MPTMS and the thiolated XNA probe. Co-immobilization of a spacer was avoided because being a small molecule the spacer has a propensity to compete out some of the capture probes leading to lower capture probe density and therefore loss in signal strength. In all the capture probe sequences, a –(CH2)6– linkage was incorporated at the 5′/3′-end to keep the nucleic acid part away from the surface so that non-specific adsorption could be avoided and the target could access the capture probe. After cleaning silicon wafer with piranha solution (7:3 v/v of H2SO4/H2O2) at 80 °C for 30 min, the resulting surface has surface-exposed –OH groups.42 As this surface is treated with 3-MPTMS, nucleophilic attack occurs by the lone pair of electrons on oxygen atom of –OH group on silicon surface to the central Si atom of 3-MPTMS. Immobilization of 3-MPTMS on silicon occurs via –O–Si– bond formation by removal of the –OCH3 group of 3-MPTMS.43 After 3-MPTMS modification, the –SH groups of 3-MPTMS are available for interaction. When the 3-MPTMS modified surface is treated with the thiol-terminated capture probes (SH–XNA), disulfide (3-MPTMS–S–S–XNA) bond is formed as a result of oxidative coupling of the two thiol groups.44 Effective surface modification by the XNA molecules has been indicated in Fig. 2B–G.
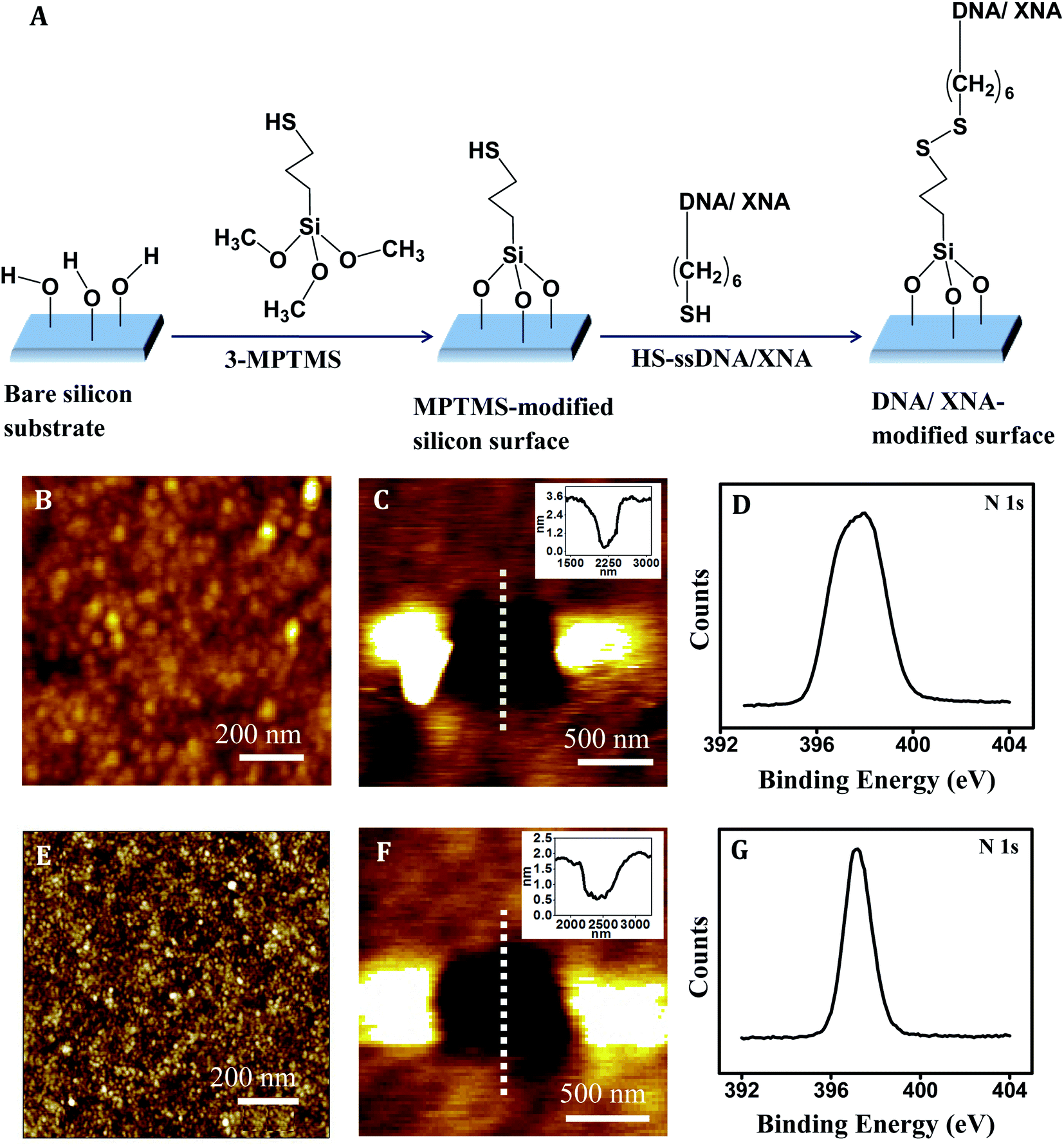 | ||
| Fig. 2 (A) Schematic representation of DNA and XNA (PNA/MO) immobilization steps on silicon surface. AFM topography image of (B) PNA film [Z-range 0–2.5 nm], (C) scratched region on PNA film, and inset figure corresponds to the cross-sectional line profile over the scratched region [Z-range: 0–3 nm]. (D) XPS of core-level peak of N(1s) for PNA on silicon substrate. AFM topography image of (E) MO film [Z-range: 0–4 nm], (F) scratched region on MO film, inset figure corresponds to the cross-sectional line profile over the scratched region [Z-range: 0–2 nm]. (G) XPS of core-level peak of N(1s) for MO on silicon substrate. | ||
AFM characterization by imaging
The AFM experiments were carried out with a picoLE AFM equipment (Agilent Corp., USA) using a 10 μm × 10 μm scanner and MFP3D AFM equipment (Asylum Research) using a 90 μm × 90 μm scanner. The intermittent contact mode (acoustic alternating current or AAC) was applied for topographic imaging. The cantilevers (μmasch, Estonia) that were used for all the imaging experiments had their backside coated with Al, were of the frequencies within 130–232 kHz, and had spring constant values within the range 3.5–12.5 N m−1. The AFM tips were cleaned immediately before imaging using a UV-ozone cleaner (Bioforce, Nanosciences). All the AFM images were acquired at the ambient condition, and at least from six different areas of each sample to confirm reproducibility of the observed surface characteristics.Nanoshaving using AFM contact mode scratching procedure
The nanoshaving experiment was performed in aqueous medium (Milli-Q water) using contact mode AFM. A 600 × 600 nm2 area of the DNA/XNA film was scanned under a large tip force of ∼150–200 nN using a triangular silicon nitride cantilever (Asylum Research) having tip radius < 10 nm and spring constant 2 N m−1. The applied force was sufficiently large to scratch away the DNA/XNA film (Fig. 2B, E and S5B in ESI†) only, leading to generation of scratched areas (Fig. 2C, F and S5C in ESI†) but not too large (i.e., of the μN order45) to scratch the underlying substrate surface (Fig. S5A†). The force applied in our present study is suitable for removal of DNA/XNA molecules from silicon substrate without any damage of the substrate. Furthermore, the spring constant of the tip used for scratching silicon surface in ref. 45 is 48 N m−1, indicating that this tip has a high wear resistance, which is much higher than that of a softer cantilever, for example, as in our case (spring constant 2 N m−1). Since we could repeatedly use the same probe for scratching on different places of a sample and on different samples prepared on different days, we do not think that the softer cantilever that we used scratched the underlying silicon substrate. Had it scratched the silicon substrate its sharpness would not remain unchanged during scratching and it could not be reused for scratching a number of times or be used for imaging after scratching. Following the scratching step, scanning was resumed over a large area under low force, centering on the scratched region, and the AFM topographic image of the scratched area under the same tip was obtained (Fig. 2C, F and S5C†).X-ray photoelectron spectroscopy (XPS) measurement
The XPS spectra were acquired using an Omicron system (model: 1712-62-11) with an anode source providing Al-Kα radiation (1486.6 eV). The radiation was focused on the sample at an electron take-off angle (TOA) of 45 ± 3° relative to the substrate and the N(1s), O(1s) and C(1s) spectra were acquired. The slit width and the TOA were kept fixed for each sample in order to probe each of the samples at the same depth.AFS probe modification for SMFS experiments
The Si3N4 AFM tip (Bruker) was cleaned in a UV-ozone cleaner (Bioforce, Nanosciences) for 15 min immediately followed by silanization. Silanization was carried out with 5% solution of 3-aminopropyltriethoxysilane (3-APTES) in 5% ethanol/95% water mixture. Next, the tip was rinsed with 5% ethanol/95% water solution, and air dried for 15 min. The tip was then immersed in a 2.5% glutaraldehyde solution in 100 mM Na-phosphate buffer (pH 7.0) for 45 min and then thoroughly rinsed with Milli-Q water. In the next step, 10–20 nM of the target DNA solution was deposited onto the AFM tip and incubated for 30 min at room temperature. The probe was finally rinsed with buffer (same as used for preparing target DNA solution), followed by washing with Milli-Q water.Single molecule force spectroscopy (SMFS) measurement
When the target DNA-modified tip comes close to the XNA-modified surface, XNA–DNA hybridization occurs and the XNA–DNA duplex is formed. During retraction of the tip, unbinding or dehybridization of the XNA–DNA duplex takes place and the unbinding peak in the retraction force curve is generated due to application of a pulling force (also called unbinding force) as required to dissociate the stable hybridization interaction between the XNA capture sequence (attached onto substrate surface) and the DNA target sequence (attached onto AFS probe). The magnitude of the unbinding peak is characteristic of the XNA–DNA sequence combination since a fully matched combination would require the greatest amount of force for duplex dissociation, while the fully mismatched sequence should not require application of any force in absence of any duplex formation. The duplex dissociation force as required for unbinding the singly or other partially mismatched XNA–DNA duplexes would need application of intermediate force values. The unbinding force values were elicited only from those force–distance curves that showed single unbinding peak with a slope change in the retrace curve, and a sharp unbinding event that ended at the zero deflection line (Fig. S6A†). Greater than 70% of the force curves displayed such features and the rest of the curves were discarded as they did not fulfill the above-mentioned criteria. Experimental conditions such as the contact force as well as the cantilever retraction speed were kept constant (Table 2) throughout the study.The force measurement experiments were carried out using PicoLE AFM (Agilent Corp., USA) and a 10 μm × 10 μm scanner and Picoview 1.12.2 software at room temperature (24 ± 1 °C). The force curves were acquired with freshly functionalized AFS tip and substrate in 20 mM sodium phosphate buffer (pH 7.0), and having NH2-terminated target ssDNA sequences on the AFS tip and capture probes immobilized onto the silicon substrate. For triplex formation experiment, 10 mM Na-phosphate, 150 mM NaCl buffer (pH 5.0) was used, having the ssPNA capture probe anchored onto silicon substrate and –NH2 terminated dsDNA onto the AFS tip. In all cases, only the retrace curves were considered to measure the unbinding force values, as the unbinding event occurred during the cantilever retraction step. Few hundred (300–500) force curves were recorded for each type of experiment. Each set of experiment was repeated at least thrice with different batches of substrates at a constant contact force value of 30 pN in order to avoid sample damage and constant cantilever retraction speed 0.5 μm s−1. The cantilever spring constant was calibrated by thermal fluctuation method46,47 using the Thermal K software (in-built in Picoview 1.12.2 software). The spring constants of the modified cantilevers were found to be within the range 0.02–0.06 N m−1. The unbinding force that is needed to overcome tip–sample interaction was obtained from the product of cantilever deflection (nm) at the rupture point or jump-off point and the calibrated cantilever spring constant (N m−1). For each type of measurement, the unbinding force was calculated from each individual force curve of the data set. The most probable unbinding force value was then found by Gaussian fitting to the distribution of the unbinding force values. The statistical error was estimated by 2σ/√N, where σ is the distribution width of the N rupturing events in the histogram.
Results and discussion
Characterization of XNA-modified surface
The XNA film as developed by immobilization of the ssXNA molecules onto MPTMS-modified silicon substrate (Fig. 2A) was characterized by AFM imaging. A homogeneous coverage was generated in case of PNA (Fig. 2B). Monolayer formation was reflected in the depth value of ∼2.0 nm as evident in the cross-sectional profile, which was derived from the AFM nanoshaving experiment (Fig. 2C). As PNA is made of polyamide backbone linked to nucleobases, the presence of a sharp primary peak of N(1s) (at 398.2 eV) in the XPS spectrum (Fig. 2D) confirms attachment of PNA on MPTMS-modified silicon substrate. In case of MO, a homogeneous coverage (Fig. 2E) with monolayer thickness (as evident from the cross-sectional depth value of ∼1.4 nm) (Fig. 2F) could be observed. The presence of a sharp primary peak of N(1s) (at 397.3 eV) in the XPS spectrum (Fig. 2G) confirms attachment of MO on MPTMS-modified silicon substrate. Since the maximum depth of the MPTMS layer can be 0.8 ± 0.1 nm,48,49 the observed depth values of 2.0 nm and 1.4 nm for the PNA and the MO layer, respectively, must be primarily for the XNA layer that corresponds well to the formation of self-assembled XNA monolayer on silicon. Once the capture probe concentrations were optimized (Table 2) to generate XNA SAMs as described above, the force measurement experiments were performed (Fig. 3–5).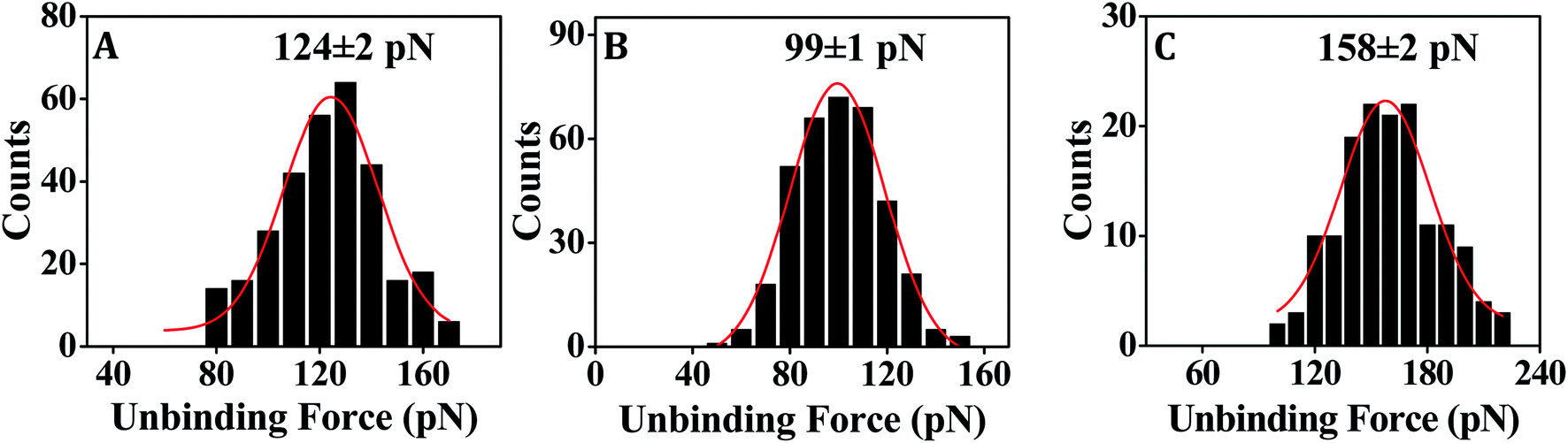 | ||
| Fig. 3 Representative force distribution histograms showing the most probable unbinding force values for dissociation of the fully matched duplexes (A) DNA–DNA (using DNA-1 and T-DNA-1), (B) PNA–DNA (using PNA-1 and T-DNA-1) and (C) MO–DNA (using MO-1 and T-DNA-2). | ||
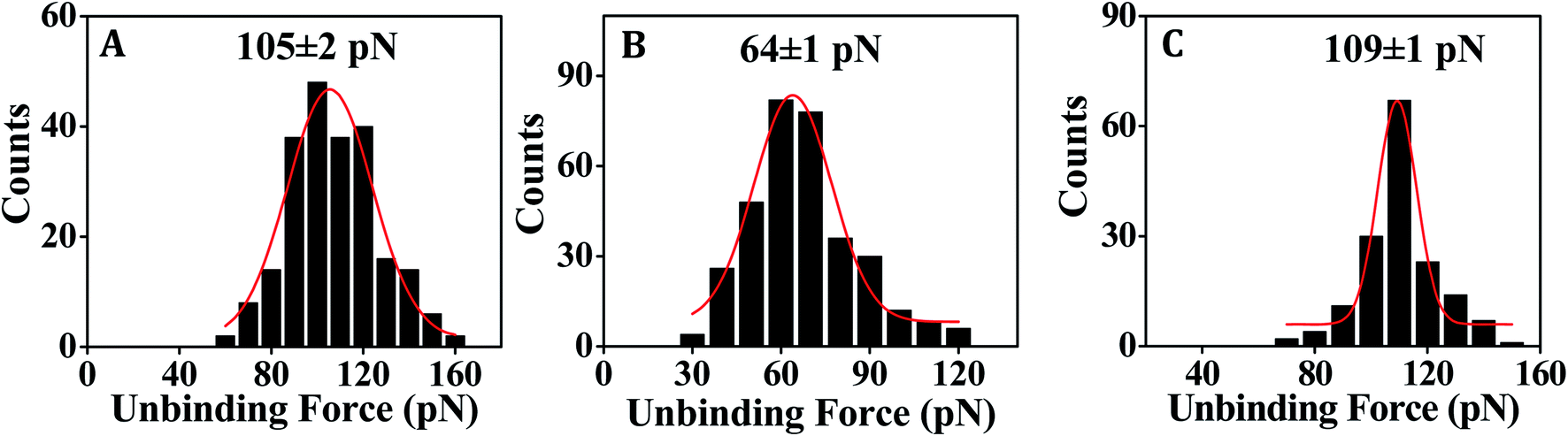 | ||
| Fig. 4 Representative force distribution histograms showing the most probable unbinding force values for dissociation of the singly mismatched duplexes (A) DNA–DNA (using DNA-2 and T-DNA-1), (B) PNA–DNA (using PNA-2 and T-DNA-1) and (C) MO–DNA (using MO-2 and T-DNA-2). | ||
 | ||
| Fig. 5 Representative force distribution histogram for PNA–DNA fully matched (A) duplex (ssPNA–ssDNA) using PNA-4 with T-DNA-4 and (B) triplex (ssPNA–dsDNA) using PNA-4, T-DNA-4 and T-DNA-5. Triplex experiment was done in 10 mM sodium phosphate, 150 mM NaCl buffer at pH = 5.0. | ||
Nucleic acid detection by PNA capture probe
In order to assess the applicability of PNA capture probe in molecularly resolved detection of nucleic acid target sequences, the unbinding force was measured for each case of fully matched, fully mismatched and singly mismatched PNA–DNA duplexes. Where DNA-1 and PNA-1 capture probe sequences were fully complementary with T-DNA-1 target sequence (Table 1) and therefore used to study the fully matched duplexes, the DNA-3 and PNA-3 were fully mismatched with T-DNAnc (Table 1), and were therefore used for the control experiments. In order to test the capability of single nucleobase mismatch detection by the surface-anchored PNA/DNA capture probe, the PNA-2 and DNA-2 capture probe sequences that form singly mismatched duplex with T-DNA-1 were used. The DNA–DNA duplex hybridization event was studied using 20 mM sodium phosphate, 150 mM NaCl (pH 7.0) buffer. As DNA contains negatively charged phosphate backbone, salt was added to stabilize duplex formation via minimizing the electronic charge repulsion between the two DNA strands. The PNA–DNA hybridization events were studied using 20 mM sodium phosphate buffer (pH 7.0) without salt addition since PNA is non-ionic in nature.In case of both the DNA–DNA and the PNA–DNA fully matched duplexes, the force distribution histograms exhibit single peak (Fig. 3A and B), meaning single unbinding event, which is reflective of 1:1 complex dissociation.50–54 The XNA probe density that was pre-optimized for 1:1 capture–target duplex formation, is in the order of 1014 probes per cm2.28 The observed variation in the unbinding force values could be due to heterogeneity in formation of the duplexes, small differences in the relative arrangements of the participating strands, orientation of the immobilized molecules, and thermal fluctuations etc.9,55,56 In some cases, the duplex might not have formed completely (if the target did not enter completely inside the film and access the total stretch of the capture probe) resulting in lower unbinding force value. Also, the duplex during its formation stage might have been compressed inside the film requiring an extra amount of force to stretch the compressed form to the nearly straightened form by the tip during ‘pulling’ or the duplex unbinding step. Since, we compare between the force distribution peak values, for comparing performance between the different capture probes or different experimental conditions, the effect of the variations during the unbinding events as mentioned above should be minimal. The peak force value is found to be clearly greater for DNA–DNA duplex (Fig. 3A) in comparison to that for the PNA–DNA duplex (Fig. 3B). When the singly mismatched duplexes were studied, a similar trend, i.e., greater unbinding force value for the DNA–DNA duplex compared to that for the PNA–DNA duplex was observed (Fig. 4A and B). In order to check whether the unbinding peak, as observed in the force–distance curve, was due to dehybridization of the complementary sequences, a control experiment was performed by monitoring association between the target DNA-modified tip and the substrate modified with the fully non-complementary capture probe sequences. Few hundred curves were recorded and among them 93% of the force curves did not display any unbinding peak (Fig. S6B†). So, we conclude that sequence-specific detection of target DNA by the PNA capture probe has been performed using the SMFS approach where the PNA probe is able to discriminate the fully matched from the fully mismatched and the singly mismatched target sequences.
A reason behind the observation of lower unbinding force value in case of both the fully matched and the singly mismatched PNA–DNA duplexes than the corresponding DNA–DNA duplexes could be due to formation of generally weaker duplexes in case of the PNA capture probe. It is possible that since the molecular backbone of PNA is conformationally more flexible than the DNA backbone,57,58 PNA is less likely to adopt an upright orientation when anchored onto a solid substrate, especially at low concentrations.41 Thereby, the PNA probe becomes less accessible to the target sequences, leading to incomplete duplex formation. It is also possible that in a surface-assembled configuration, the PNA strand engages itself in intermolecular hydrogen bonding interaction with the adjacent PNA strands via the backbone peptide regions.29 In consequence, the PNA strand may face hindrance to establishing full interaction with the target DNA sequence. Also, the relatively flexible backbone of PNA could increase its propensity towards self-aggregation. However, in spite of these concerns, we observed that the solution phase capacity of mismatch discrimination of the PNA capture probe was retained on surface, and the single nucleobase mismatch discrimination by the PNA capture probe was found to be superior compared to the DNA capture probe (Table 3).
| Nucleic acid duplexes | Mean unbinding force (pN) | Mismatch discrimination (pN) | |
|---|---|---|---|
| Full match | Single base mismatch | ||
| DNA–DNA | 124 ± 3 | 105 ± 2 | ∼19 |
| PNA–DNA | 95 ± 2 | 64 ± 1 | ∼31 |
| MO–DNA | 157 ± 2 | 108 ± 1 | ∼49 |
In order to obtain a supportive evidence for the conformational flexibility of the surface-confined PNA, we performed an experiment on ssPNA:dsDNA triplex dissociation using target dsDNA [T-DNA-4 and T-DNA-5, see Table 1] that was anchored onto the SMFS tip. Molecular modeling studies suggested that triplex formation involving PNA (e.g., PNA/DNA/DNA) is favoured due to the conformational flexibility of PNA,59,60 where the relatively free rotation of the backbone bonds in PNA32 leads to its conformational flexibility. The propensity of triplex formation of PNA with target dsDNA due to the conformational flexibility of PNA32 has been demonstrated in solution phase61 as well. Given the present immobilization scheme (Fig. 2A), the PNA part should be sufficiently away from the surface due to presence of the intermediate 3-MPTMS layer, and the hexyl spacer that is integrated in the PNA capture probe. Therefore, the conformational flexibility of PNA is expected to remain unaltered on surface. Especially, the presence of the hexyl spacer should ensure that the behavior of the surface-confined PNA capture probe is similar to that of the PNA probe in solution phase. We showed earlier that the association constant obtained from an on-surface study using the PNA probe having hexyl spacer is comparable to that obtained from the solution phase study29 in support of this. The presence of an extra intermediate layer, i.e., 3-MPTMS, would further act in favour of an unrestricted (surface-independent) behavior of the PNA probe. The surface-anchored PNA capture probe's [PNA-4, see Table 1] capacity of stable triplex formation has been indicated in the SMFS-derived unbinding force value, which is higher than that for the ssPNA:ssDNA duplex (Fig. 5), reflecting the fact that the PNA backbone retains its conformational flexibility in surface-confined state.
Nucleic acid detection by MO capture probe
For fully matched MO–DNA duplex formation, we employed MO-1 and T-DNA-2 sequences, and for fully mismatched duplex formation, MO-1 and T-DNA-3 were used (Table 1). For formation of the singly mismatched duplex, MO-2 and T-DNA-2 were applied (Table 1). All the MO–DNA hybridization events were studied using 20 mM phosphate buffer (pH 7.0) without salt addition since MO is non-ionic in nature.From the SMFS experiments, it is revealed that the MO capture probe can clearly detect the fully matched target DNA (Fig. 3C) as well as the singly mismatched target DNA (Fig. 4C). The unbinding force value is found to be greater in case of the MO–DNA duplex for both the fully and the singly mismatched sequence combinations (Fig. 3C and 4C) in comparison to the analogous DNA–DNA duplexes (Fig. 3A and 4A) that indicates that the MO probe binds with target DNA sequence with greater affinity and forms stronger duplexes in comparison to the DNA capture probe. However, the dissimilarity in the unbinding force values for the singly mismatched MO–DNA and DNA–DNA duplexes is small (∼3 pN) (Table 3) compared to the case of the fully matched MO–DNA and DNA–DNA duplexes (∼33 pN) (Table 3). Consequently, the MO capture probe discriminates the singly mismatched sequence from the fully matched sequence better in comparison to the DNA capture probe (Table 3).
The orientational advantage that the MO capture probe may enjoy due to the presence of a conformationally more rigid backbone32 is not present in case of the DNA capture probe since ssDNA has a low persistence length62 and is therefore conformationally more flexible. Complete access of the capture probe by the target sequence is therefore more feasible in case of the MO capture probe, since MO backbone being more rigid should stand more upright on the substrate surface, compared to the DNA capture probe. Consequently, more complete formation of the duplexes is possible in case of the MO–DNA duplexes than the DNA–DNA duplexes. This results in stronger duplex formation and greater stability of the MO–DNA duplexes than the DNA–DNA duplexes, leading to greater unbinding force values for the MO–DNA duplexes (Table 3).
We considered the unbinding force values as an estimate for stability since it has been shown that the order of the unbinding force values for different cases is maintained in the order of the free energy values that are extracted using the Jarzinsky's equality treatment.5 Hummer and Szabo presented a theoretical proof in support of the validity of Jarzinsky's equality in case of the non-equilibrium single molecule pulling experiments.63 The improved capacity for single nucleobase mismatch discrimination by the MO capture probe, in comparison to the DNA capture probe, as demonstrated by the greater unbinding force values for mismatch discrimination in case of MO (Table 3), can be explained from high sequence selectivity of the morpholino.31,35,36
Performance of the PNA capture probe in comparison to the MO capture probe
It is clear from the unbinding force values that both the PNA and MO capture probes are capable of detection of target DNA in a sequence-specific manner including the singly mismatched sequences, and are therefore capable of single nucleobase mismatch discrimination (Table 3). However, a clear decrease in the unbinding force values for both the fully and singly mismatched duplexes has been observed in case of the PNA capture probe compared to the MO capture probe (Table 3). The surface-confined PNA–DNA duplexes are therefore formed as generally weaker duplexes than the MO–DNA duplexes. Another important observation is that the difference in the unbinding force value between the fully matched duplex and the singly mismatched duplex is greater in case of the MO capture probe than the PNA probe (Table 3). The MO capture probe is therefore capable of better single nucleobase mismatch discrimination than the PNA capture probe.As discussed in the earlier sections, the conformational flexibility of the PNA backbone57,58 can result in a molecular orientation that is not upright, thereby making full access of the capture probe sequence by the target nucleic acid difficult. On the contrary, MO backbone is relatively rigid32 and therefore its propensity for adopting an upright orientation in the surface-confined state is greater than PNA. The chances of formation of completely hybridized duplexes are therefore higher in case of MO, leading to more stable duplexes.
It has been reported that the structural change of oligonucleotides correlates with hybridization affinity.64 In this study, the only difference between the PNA and MO probes arises due to the backbone structure since the PNA and the MO probes were having the same nucleobase sequences. It has been demonstrated that the conformational rigidity has an impact in molecular recognition ability.65–67 The thermal stability of the duplexes could be increased65 and sequence selectivity could be enhanced66,67 by incorporating diethylene glycol (miniPEG) at the γ position of the peptide backbone. It is evident from the structure that the MO backbone is conformationally more rigid32 in comparison to PNA. In a study on PNA, it has been suggested that the backbone of PNA is more flexible than that of the native nucleic acid.68 Since the backbone conformation influences the base stacking interaction, thereby affecting duplex stabilization,69 the higher unbinding force value for MO–DNA duplex than that of PNA–DNA duplex could be related to the enhancement of base stacking interaction in case of MO.
Performance of the XNA capture probes compared to the DNA capture probe
In the present SMFS-based approach, the ability of the synthetic XNA probes for mismatch discrimination is found to be greater than that of DNA, where the order of the mismatch discrimination ability of these capture probes is found to be MO > PNA > DNA (Table 3). The ability for mismatch discrimination should be linked to duplex stabilization since the unbinding force value directly depends on duplex stability.5 For greater duplex stability, higher amount of force is needed to dehybridize the duplex, meaning higher will be the unbinding force. Since, two factors are mainly responsible for formation of the duplex, i.e., base pairing through hydrogen bonding between the complementary strands and base stacking interactions between the adjacent nucleobases, these unbinding force values are indicative of the presence and extent of inter-strand hydrogen bonding involving nucleobases and the base stacking interactions. An additional force contributing to holding the nucleic acid strands together can arise from the van der Waals attractive interactions involving the backbone conformation.70 In our case, since a clear variation in the unbinding force value, and therefore in duplex stability, was observed with a change in the nature of the capture probe, where all the capture probes DNA, PNA and MO had identical nucleobase sequences (Table 1), it is reasonable to propose that the different structural properties of the DNA, PNA and MO backbones must have led to their different binding affinities towards target nucleic acid, and therefore, to the differences in duplex stabilities and the variations in the unbinding force values.Although the basic structural features of DNA, i.e., phosphate group, sugar moiety and nucleobases have all been subjected to modifications in recent years,71 the most relevant modifications are probably those in the backbone.71 DNA contains deoxyribose sugar with phosphate group in its backbone whereas PNA has non-ionic, achiral, flexible, polyamide backbone composing of N-(2-aminoethyl)-glycine units connected by peptide bonds. In case of MO, the backbone is made of non-ionic phosphoroamidate with 6-membered morpholino ring that imparts conformational rigidity to the backbone. The role of the relatively rigid backbone of XNA probe in orienting the capture probe sufficiently upright at the solid–liquid interface and therefore enhancing the prospect of access of the capture probe by the target strands has been exemplified in SMFS-based nucleic acid sensing studies.4 We reported higher unbinding force value for the case of locked nucleic acid (LNA) capture probe, when compared between the LNA–DNA duplex and DNA–DNA duplex unbinding events,14 where the greater unbinding force value in case of LNA–DNA duplex could be due to enhancement of base stacking72 and a favourable orientation of the LNA probe due to its relatively rigid backbone.18 In the present study, we have observed that MO, having a conformationally more rigid backbone than PNA and DNA, offers the best duplex stability, followed by the case of DNA, whereas PNA having the conformationally most flexible backbone leads to the least duplex stability (Table 3, see Fig. S7 in ESI†). As the majority of the base-stacking interactions in nucleic acids are between nucleobases within a strand,73 the strengthening of stacking interactions leads to the tendency of pre-organization of the single-stranded oligonucleotides into a more regular helical conformation that favors duplex formation by lowering the entropic cost.73 Though increased base stacking results in backbone rigidity,72 which has an important role in increasing duplex stabilization, a certain degree of backbone flexibility is also necessary to conformationally adjust the two strands to each other. If there is no space for conformational adjustment, duplex formation becomes difficult. Since, too flexible or too rigid structures don't show affinity to target nucleic acid due to the unfavorable conformation, a constrained flexibility is necessary to attain effective DNA binding.74,75 Therefore, the observed order of unbinding force value for the DNA–DNA, PNA–DNA and MO–DNA duplexes could be due to a fine balance and combination of duplex formation and stabilization factors such as backbone rigidity, base stacking and pre-organization. In other words, a balance between the ability for adopting a conformation which is the most suitable for DNA binding, thereby minimizing the loss of entropy of binding process, and the backbone charge, since non-ionic backbone decreases the potential energy barrier for hybridization by eliminating the inter-strand electrostatic repulsion, is crucial.
The best capture probe for sensing nucleic acids
Among the various alternative probes that have been designed and synthesized for improved sensing of nucleic acids, both PNA and MO are potentially useful.4,76 Increased thermal stability of the MO-inclusive duplexes, solubility of MO in aqueous medium, its capacity of adopting upright orientation, and ability to resist nuclease- and protease-induced degradation, all point to potential diagnostic applications of the MO probe. Although PNA is nuclease-/protease-resistant too, and it offers nucleic acid detection capacity with high binding affinity and sequence specificity, it's lower water solubility than MO makes PNA a less attractive candidate than MO. When compared to DNA, both PNA and MO probes are more robust, and offer improved single nucleobase mismatch discrimination capacity, although DNA is freely soluble in water. Since in the SMFS approach, only 1–5 nM PNA/MO concentration is needed for preparing the sensing surface, the cost involved should be within a reasonable limit. For all these reasons, we propose MO to be the most attractive candidate, followed by PNA, and then DNA, for molecularly resolved SMFS-based nucleic acid detection.Conclusions
In conclusion, single nucleobase mismatch discrimination could be performed better by the non-ionic XNA probes MO and PNA compared to the DNA probe as revealed from the SMFS data. When compared amongst the three probes, the performance of the MO probe suggests this probe to be the most suitable for SMFS-based molecularly resolved nucleic acid sensing. This is primarily because the MO probe not only enjoys the advantage of being non-ionic, but its backbone being the most rigid, it can also be oriented the most upright, which is conducive for maximizing target access and therefore the capture probe's interaction with the target sequence. Since the nucleobase sequences were kept fixed for all the three probes, the effect of backbone variation could be elicited from the SMFS analyses in terms of an interplay of the backbone charge and rigidity. Apart from presenting a significant use of the non-ionic XNA probes, like MO and PNA, in nucleic acid sensing, the present work also adds to the repertoire of fluorescent label-free approaches for nucleic acid sensing. Since the present assay allows molecularly resolved sensing where low target concentration (nM level) is needed, the assay is potentially PCR-free, which needs to be explored and developed in future.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Rupa Mukhopadhyay acknowledges financial and infrastructural support from Indian Association for the Cultivation of Science (IACS), Kolkata; the fellowship of Tanushree Mana from IACS, Kolkata; and the fellowships of Hiya Lahiri and Sudipta Bera from DST-Inspire scheme of Department of Science and Technology (DST), Govt. of India. Surajit Sinha acknowledges research grant from Science and Engineering Research Board (grant No. EMR/2016/004563), New Delhi, Government of India; and the fellowship of Jayanta Kundu from IACS, Kolkata.References
- K. M. Abu-Salah, M. M. Zourob, F. Mouffouk, S. A. Alrokayan, M. A. Alaamery and A. A. Ansari, Sensors, 2015, 15, 14539–14568 Search PubMed.
- A. Sassolas, B. D. Leca-Bouvier and L. J. Blum, Chem. Rev., 2008, 108, 109–139 Search PubMed.
- P. Nollau and C. Wagener, Clin. Chem., 1997, 43, 1114–1128 Search PubMed.
- H. Lahiri, S. Mishra and R. Mukhopadhyay, Langmuir, 2019, 35, 8875–8888 Search PubMed.
- H. Lahiri, S. Banerjee and R. Mukhopadhyay, ACS Sens., 2019, 4, 2688–2696 Search PubMed.
- D. J. Müller and Y. F. Dufrêne, Nat. Nanotechnol., 2008, 3, 261–269 Search PubMed.
- G. Žoldák and M. Rief, Curr. Opin. Struct. Biol., 2013, 23, 48–57 Search PubMed.
- J. Zlatanova, S. M. Lindsay and S. H. Leuba, Prog. Biophys. Mol. Biol., 2000, 74, 37–61 Search PubMed.
- F. Kienberger, A. Ebner, H. J. Gruber and P. Hinterdorfer, Acc. Chem. Res., 2005, 39, 29–36 Search PubMed.
- C. D. Frisbie, L. F. Rozsnyai, A. Noy, M. S. Wrighton and C. M. Lieber, Science, 1994, 265, 2071–2074 Search PubMed.
- V. T. Moy, E. L. Florin and H. E. Gaub, Science, 1994, 266, 257–259 Search PubMed.
- H. Skulason and C. D. Frisbie, J. Am. Chem. Soc., 2002, 124, 15125–15133 Search PubMed.
- Y. Jiang, C. Zhu, L. Ling, L. Wan, X. Fang and C. Bai, Anal. Chem., 2003, 75, 2112–2116 Search PubMed.
- S. Mishra, H. Lahiri, S. Banerjee and R. Mukhopadhyay, Nucleic Acids Res., 2016, 44, 3739–3749 Search PubMed.
- T. Strunz, K. Oroszlan, R. Schäfer and H. J. Güntherodt, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11277–11282 Search PubMed.
- M. V. Wal, S. Kamper, J. Headley and K. Sinniah, Langmuir, 2006, 22, 882–886 Search PubMed.
- B. D. Sattin, A. E. Pelling and M. C. Goh, Nucleic Acids Res., 2004, 32, 4876–4883 Search PubMed.
- S. Mishra, S. Ghosh and R. Mukhopadhyay, Langmuir, 2012, 28, 4325–4333 Search PubMed.
- S. Mishra, S. Ghosh and R. Mukhopadhyay, Langmuir, 2014, 30, 10389–10397 Search PubMed.
- A. B. Steel, R. L. Levicky, T. M. Herne and M. J. Tarlov, Biophys. J., 2000, 79, 975–981 Search PubMed.
- H. Lahiri, S. Mishra, T. Mana and R. Mukhopadhyay, Analyst, 2016, 141, 4035–4043 Search PubMed.
- E. Casero, M. Darder, D. J. Díaz, F. Pariente, J. A. Martín-Gago, H. Abruña and E. Lorenzo, Langmuir, 2003, 19, 6230–6235 Search PubMed.
- M. P. Garilhe and M. Laskowski, J. Biol. Chem., 1955, 215, 269–276 Search PubMed.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 Search PubMed.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566–568 Search PubMed.
- V. V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskil, M. Egholm, O. Buchard, S. H. Sönnichsen and P. E. Nielsen, Biochem. Pharmacol., 1994, 48, 1310–1313 Search PubMed.
- S. Ghosh, S. Mishra, T. Banerjee and R. Mukhopadhyay, Langmuir, 2013, 29, 3370–3379 Search PubMed.
- S. Ghosh, S. Mishra and R. Mukhopadhyay, Langmuir, 2013, 29, 11982–11990 Search PubMed.
- S. Ghosh, S. Mishra and R. Mukhopadhyay, J. Mater. Chem. B, 2014, 2, 960–970 Search PubMed.
- L. Simon, G. Lautner and R. E. Gyurcsanyi, Anal. Methods, 2015, 7, 6077–6082 Search PubMed.
- J. Summerton and D. Weller, Antisense Nucleic Acid Drug Dev., 1997, 7, 187–195 Search PubMed.
- J. E. Summerton, Lett. Pept. Sci., 2003, 10, 215–236 Search PubMed.
- R. M. Hudziak, E. Barofsky, D. F. Barofsky, D. L. Weller, S. B. Huang and D. D. Weller, Antisense Nucleic Acid Drug Dev., 1996, 6, 267–272 Search PubMed.
- N. Tercero, K. Wang, P. Gong and R. Levicky, J. Am. Chem. Soc., 2009, 131, 4953–4961 Search PubMed.
- W. Hu, Q. Hu, L. Li, J. Kong and X. Zhang, Anal. Methods, 2015, 7, 2406–2412 Search PubMed.
- G. J. Zhang, Z. H. H. Luo, M. J. Huang, G. K. I. Tay and E. J. A. Lim, Biosens. Bioelectron., 2010, 25, 2447–2453 Search PubMed.
- J. Bhadra, S. Pattanayak and S. Sinha, Curr. Protoc. Nucleic Acid Chem., 2015, 62, 4.65.1–4.65.26 Search PubMed.
- S. Pattanayak and S. Sinha, Tetrahedron Lett., 2012, 53, 6714–6717 Search PubMed.
- J. Bhadra, J. Kundu, K. C. Ghosh and S. Sinha, Tetrahedron Lett., 2015, 56, 4565–4568 Search PubMed.
- K. I. Chen, B. R. Li and Y. T. Chen, Nano Today, 2011, 6, 131–154 Search PubMed.
- S. Ghosh and R. Mukhopadhyay, J. Colloid Interface Sci., 2011, 360, 52–60 Search PubMed.
- L. T. Zhuravlev, Langmuir, 1987, 3, 316–318 Search PubMed.
- S. Bera, J. Kolay, S. Banerjee and R. Mukhopadhyay, Langmuir, 2017, 33, 1951–1958 Search PubMed.
- D. Witt, Synthesis, 2008, 16, 2491–2509 Search PubMed.
- X. Jiang, G. Wu, J. Zhou, S. Wang, A. A. Tseng and Z. Du, Nanoscale Res. Lett., 2011, 6, 518 Search PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 Search PubMed.
- J. P. Cleveland, S. Manne, D. Bocek and P. K. Hansma, Rev. Sci. Instrum., 1993, 64, 403–405 Search PubMed.
- P. K. Gothe, D. Gaur and V. G. Achanta, J. Phys. Commun., 2018, 2, 035005 Search PubMed.
- D. K. Aswal, S. Lenfant, D. Guerin, J. V. Yakhmi and D. A. Vuillaume, Small, 2005, 1, 725–729 Search PubMed.
- A. R. Bizzarri and S. Cannistraro, Chem. Soc. Rev., 2010, 39, 734–749 Search PubMed.
- J. Fritz, A. G. Katopodis, F. Kolbinger and D. Anselmetti, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12283–12288 Search PubMed.
- P. Hinterdorfer, W. Baumgartner, H. J. Gruber, K. Schilcher and H. Schindler, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3477–3481 Search PubMed.
- Y. Jiang, F. Qin, Y. Li, X. Fang and C. Bai, Nucleic Acids Res., 2004, 32, e101 Search PubMed.
- J. Yu, Q. Wang, X. Shi, X. Ma, H. Yang, Y.-G. Chen and X. Fang, J. Phys. Chem. B, 2007, 111, 13619–13625 Search PubMed.
- I. Schumakovitch, W. Grange, T. Strunz, P. Bertoncini, H.-J. Güntherodt and M. Hegner, Biophys. J., 2002, 82, 517–521 Search PubMed.
- T. A. Sulchek, R. W. Friddle, K. Langry, E. Y. Lau, H. Albrecht, T. V. Ratto, S. J. DeNardo, M. E. Colvin and A. Noy, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16638–16643 Search PubMed.
- E. Hatcher, A. Balaeff, S. Keinan, R. Venkatramani and D. N. Beratan, J. Am. Chem. Soc., 2008, 130, 11752–11761 Search PubMed.
- R. Soliva, E. Sherer, F. J. Luque, C. A. Laughton and M. Orozco, J. Am. Chem. Soc., 2000, 122, 5997–6008 Search PubMed.
- G. C. Shields, C. A. Laughton and M. Orozco, J. Am. Chem. Soc., 1998, 120, 5895–5904 Search PubMed.
- H. J. Larsen and P. E. Nielsen, Norfolk, Horizon Scientific Press, 1999, pp. 221–240 Search PubMed.
- P. E. Nielsen, Curr. Med. Chem., 2001, 8, 545–550 Search PubMed.
- Q. Chi, G. Wang and J. Jiang, Phys. A, 2013, 392, 1072–1079 Search PubMed.
- G. Hummer and A. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3658–3661 Search PubMed.
- S. M. Freier and K.-H. Altmann, Nucleic Acids Res., 1997, 25, 4429–4443 Search PubMed.
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, J. Org. Chem., 2011, 76, 5614–5627 Search PubMed.
- V. Menchise, G. D. Simone, T. Tedeschi, R. Corradini, S. Sforza, R. Marchelli, D. Capasso, M. Saviano and C. Pedone, Proc. Natl. Acad. Sci. U. S. A., 2013, 100, 12021–12026 Search PubMed.
- S. Dutta, B. A. Armitage and Y. L. Lyubchenko, Biochemistry, 2016, 55, 1523–1528 Search PubMed.
- H. Rasmussen, J. S. Kastrup, J. N. Nielsen, J. M. Nielsen and P. E. Nielsen, Nat. Struct. Biol., 1997, 4, 98–101 Search PubMed.
- A. F. Ghobadia and A. Jayaraman, Soft Matter, 2016, 12, 2276–2287 Search PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, 4th edition, Section 4.1, Structure of Nucleic Acids, W. H. Freeman, New York, 2000 Search PubMed.
- K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18, 112.S Search PubMed.
- E. Kierzek, A. Pasternak, K. Pasternak, Z. Gdaniec, I. Yildirim, D. H. Turner and R. Kierzek, Biochemistry, 2009, 48, 4377–4387 Search PubMed.
- E. T. Kool, Chem. Rev., 1997, 97, 1473–1488 Search PubMed.
- K. C. Schneider and S. A. Benner, J. Am. Chem. Soc., 1990, 112, 453–455 Search PubMed.
- P. Nielsen, F. Kirpekar and J. Wengel, Nucleic Acids Res., 1994, 22, 703–710 Search PubMed.
- A. Bagi, S. D. Soelberg, C. E. Furlong and T. Baussant, Sensors, 2018, 18, 3259 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00386d |
| ‡ Present address: Hebrew University of Jerusalem, Israel. |
| § Present address: Weizmann Institute of Science, Israel. |
| This journal is © The Royal Society of Chemistry 2022 |