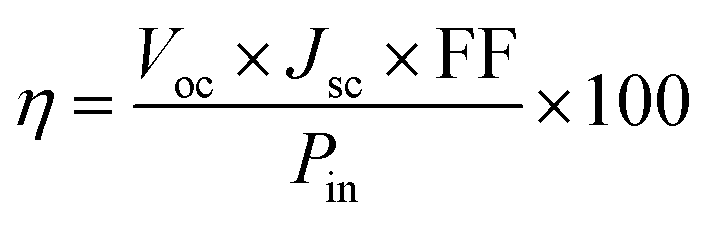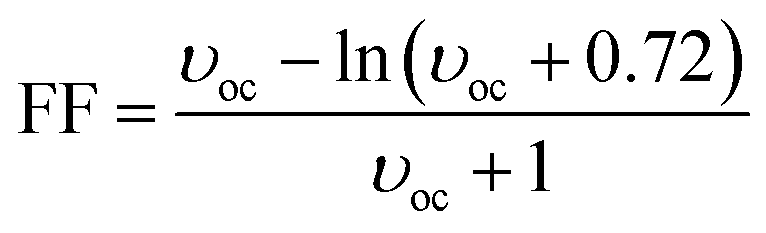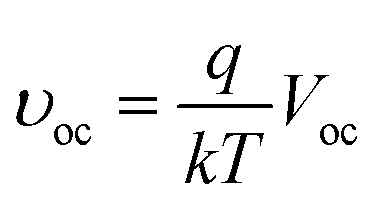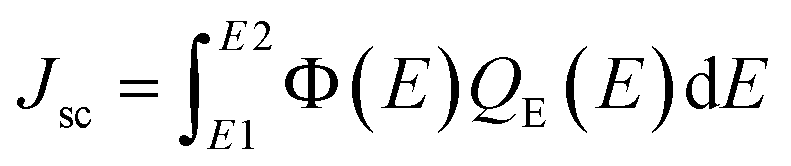DOI:
10.1039/D2RA00345G
(Paper)
RSC Adv., 2022,
12, 8945-8952
Theoretical investigation of FAPbSnGeX3 efficiency
Received
17th January 2022
, Accepted 5th March 2022
First published on 22nd March 2022
Abstract
The use of hybrid lead halide perovskites as light absorbers in photovoltaic cells have gained large interest due to their optoelectronic properties and high efficiency. However, most hybrid perovskites contain toxic lead which has a negative impact on the environment. In this work, we systematically study the structural, electronic, and optical properties of lower lead halide perovskites FAPb0.5Sn0.25Ge0.25X3 (X = I, Br, Cl), as well as discussing their photovoltaic performance (open circuit voltage (Voc), the short circuit current density (Jsc), and the power conversion efficiency (η)) using density functional theory (DFT), and we compare these with FAPbX3 (X = I, Br, Cl) frameworks. The compounds show a suitable band gap for photovoltaic applications, in which iodine has a lower gap value compared to chlorine. It is noteworthy that we found that lead doping by both germanium and tin in the FAPb0.5Sn0.25Ge0.25X3 (X = I, Br, Cl) materials significantly improves the adsorption coefficient and the stability of these systems compared to the FAPbX3 (X = I, Br, Cl) systems. The calculated Jsc shows a monotonical decrease from FAPb0.5Sn0.25Ge0.25I3 to FAPbCl3, which represents the lowest Jsc. Results reveal that FAPb0.5Sn0.25Ge0.25Cl3 demonstrates promising potential for photovoltaic application as it shows the highest efficiency. This study can help reduce the toxicity of hybrid lead halide perovskites and also raises their experimental power conversion efficiency.
1. Introduction
The energy requirement has grown during recent years with the increase of human population and elevated consumption.1–3 Solar energy represents an alternative to limit the use of fossil fuels and is a fast growing energy technology abundantly available.4,5 Recently, organic–inorganic hybrid lead halide perovskite materials with the general formula ABX3 (A = MA, FA, EA; B= Pb, Sn, Ge, Be; X= I, Br, Cl) have demonstrated great potential in different applications, especially photovoltaics.6–9 The high efficiency represented by these materials has reached a value of 25.6% in a few years.10 This rise has come from the unique properties of hybrid lead halide perovskites, such as a high absorption coefficient in the visible range, tunable bandgap, low effective mass, high carrier mobility.11–14 Currently, a lot of parameters reduce the hybrid perovskite efficiency performance in solar cells. The defects are in the hybrid perovskite structure, toxicity, and their degradation in the presence of ultraviolet light, humidity and oxygen.15–18
Iodine migration in hybrid perovskites creates structure defects which can be the cause of the current voltage hysteresis.19 Also, hybrid perovskite degradation under ultraviolet light has a direct relation to iodine oxidation; it has been demonstrated that iodine doping by bromine is a solution to limit this degradation.20,21 The choice of the organic cation in the hybrid perovskite also plays a major role in the material performance.22 Oranskaia et al. have demonstrated that ions have a lot more space to migrate in the MAPbBr3 structure than the FAPbBr3,23 as the H-bonding in MA and FA has an effect on the stabilization of the halogen vacancies. Other work has demonstrated that hydrogen vacancies can be present in MAPbI3 with high densities, in contrast with FAPbI3 where they are difficult to form and make the FAPbI3 more stable and higher performing in photovoltaic applications.24 On the other hand, the high efficiency achieved by hybrid perovskites shown by lead halide perovskites is counteracted by their toxicity,25,26 which blocks the commercialization of these compounds. The substitution of lead by tin in both FAPbI3 and MAPbI3 has demonstrated an efficiency of 5.51% and 3.13%, respectively,27 while the substitution of lead by Ge in MAPbI3 shows an efficiency of 0.2% and FAGeI3 does not show any photocurrent.28 A theoretical study has demonstrated that the partial substitution of lead by both tin and germanium shows a high performance in photovoltaic applications.29 So far no substitution of lead has demonstrated better performance than MAPbI3. In this work, we studied the lower levels of free lead halide perovskite FAPb0.5Sn0.25Ge0.25X3 (X= I, Cl and Br) using density functional theory, where the lead was doped with both Sn and Ge, and compared with the photovoltaic results of the FAPbX3 (X = I, Br, Cl) compounds. The structural, electronic and optical properties of these compounds were investigated, and we discuss their power conversion efficiency. For this purpose, the photovoltaic parameters, open circuit voltage and the short circuit current density were investigated.
2. Computational methods
In this work, density functional theory implemented in the quantum espresso code was used to study the structural and electronic properties of FAPbX3 (X = I, Br, Cl), and FAPb0.5Sn0.25Ge0.25X3 (X= I, Cl and Br) compounds.30 The structure optimization of tetragonal FAPb0.5Sn0.25Ge0.25X3 was carried out using Normcons type pseudopotentials in which the Martins–Troullier method was employed, as well as a functional type Perdew–Burke–Ernzerhof (PBE) exchange correlation nonlinear non-relativistic core correction. A 65 Ry plane wave cut off with a 6 × 6 × 5 k-mesh was used to optimize the unit cell. The van der Waals-DFT (vdw-df-ob86) approximation was used for the exchange–correlation functional31 as it gives accurate results that match the experimental data.32 The optical properties of FAPbX3 and FAPb0.5Sn0.25Ge0.25X3 were calculated using the Yambo code.33
To estimate the photovoltaic performance of each compound, the conversion efficiency was calculated using the following equation:34
| |
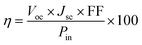 | (1) |
where the fill factor FF has been calculated using the following equation:
35| |
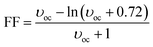 | (2) |
υoc is defined as a “normalized Voc”:
| |
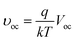 | (3) |
Pin represents the light power density of incident light on the solar cell in this work under the standard test conditions (T = 25°, AM1.5G) equal to 1000 W m−2, Voc represents the open circuit voltage that can be expressed for perovskites solar cells as follows:
| | |
eVoc = Eg − Eloss; Eloss = 0.7 or 0.5 eV
| (4) |
where
Eloss represents a variable parameter potential loss.
36
The short circuit current density Jsc was calculated using the following equation:37
| |
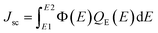 | (5) |
where Φ(
E) is the spectral irradiance (AM 1.5G) and
QE(
E) is the quantum efficiency calculated by the equation:
| | |
QE = (1 − R(E))(1 − e−α(E)t)
| (6) |
R is the reflectivity,
α(
E) is the absorption coefficient and
t is the thickness of the sample.
3. Results and discussion
3.1 Crystalline structure
The FAPb0.5Sn0.25Ge0.25X3 structures were created by the substitution of 50% of the lead (Pb) in FAPbI3 with 25% of tin (Sn) and 25% of germanium (Ge). The halogens used were iodine, bromine, and chlorine. The optimized tetragonal structures and lattice parameters calculated for the MAPbI3, FAPbX3 and FAPb0.5Sn0.25Ge0.25X3 compounds are represented in Fig. 1 and Table 1, respectively. Table 1 shows clearly that the substitution of MA with FA changes the lattice from a = 8.75 Å to a = 8.99 Å due to the geometry of FA. The lattice parameters have been reduced with the substitution of half of the lead by tin and germanium, and further reduced with the use of bromine or chlorine. The calculated lattice parameters of MAPbI3, and FAPbX3 are in good agreement with previous experimental and theoretical reports.38–41
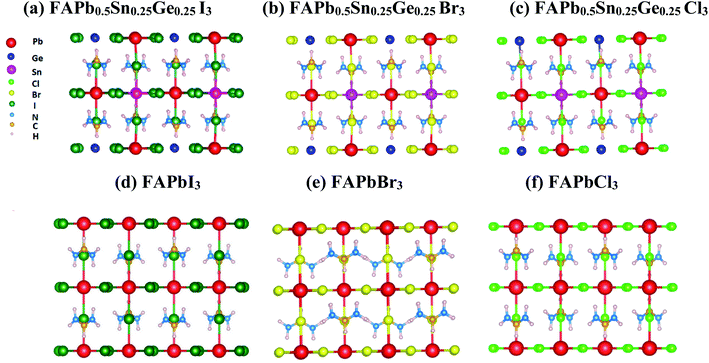 |
| | Fig. 1 Optimized tetragonal structures of FAPb0.5Sn0.25Ge0.25X3 and FAPbX3. | |
Table 1 Lattice parameters and formation energy of MAPbI3, FAPb0.5Sn0.25Ge0.25X3, and FAPbX3 structures
| Compounds |
a (Å) |
b (Å) |
c (Å) |
Eformation (eV Å−1) |
| MAPbI3 |
8.75 |
8.77 |
12.89 |
−4.26 |
| FAPb0.5Sn0.25Ge0.25I3 |
8.89 |
8.84 |
12.34 |
−4.66 |
| FAPb0.5Sn0.25Ge0.25Br3 |
8.42 |
8.36 |
11.58 |
−4.8 |
| FAPb0.5Sn0.25Ge0.25Cl3 |
8.10 |
8.04 |
11.14 |
−4.93 |
| FAPbI3 |
8.99 |
8.99 |
12.72 |
−4.66 |
| FAPbBr3 |
8.47 |
8.47 |
11.98 |
−4.76 |
| FAPbCl3 |
8.17 |
8.17 |
11.26 |
−4.93 |
3.2 Electronic structure
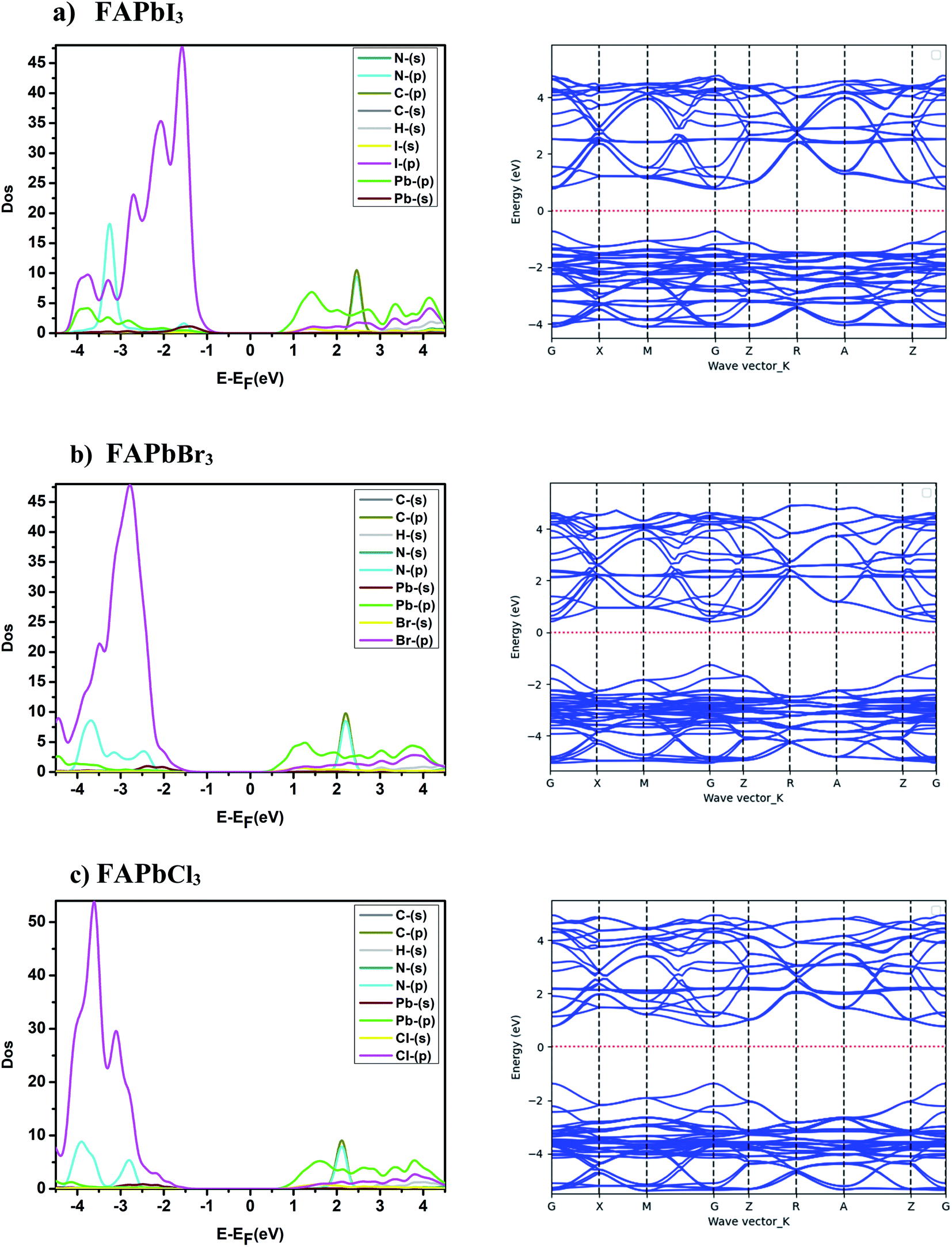
The electronic structure study of the compounds consists of analyzing the partial density of states, the band structure and the effective mass. The band structures and the partial state density of the FAPbX3, and FAPb0.5Sn0.25Ge0.25X3 compounds are presented in Fig. 2 and 3. The band structures show semiconductor character with a direct bandgap in the gamma points of the Brillouin zone. The difference between the band structures appears in the gap between the valence band maximum and the conduction band minimum. The calculated energy gap of the FAPbX3 frameworks are 1.5 eV, 1.67 eV, and 2.14 eV for FAPbI3, FAPbBr3, and FAPbCl3, respectively. The valence of FAPbX3 is formed by the p-orbital of the halogen (I, Br, Cl) while the p-orbital of Pb formed the conduction band. The calculated effective mass of each structure is listed in Table 2. FAPbCl3 presents a larger gap compared to the other compounds, a larger effective mass of electrons (Table 2) and therefore a low mobility. The latter improves its performance for photovoltaic applications through the doping of Pb by Sn and Ge, which modified the electronic structures and electronic properties, and slightly altered the energy gap. The above will be discussed in the following.
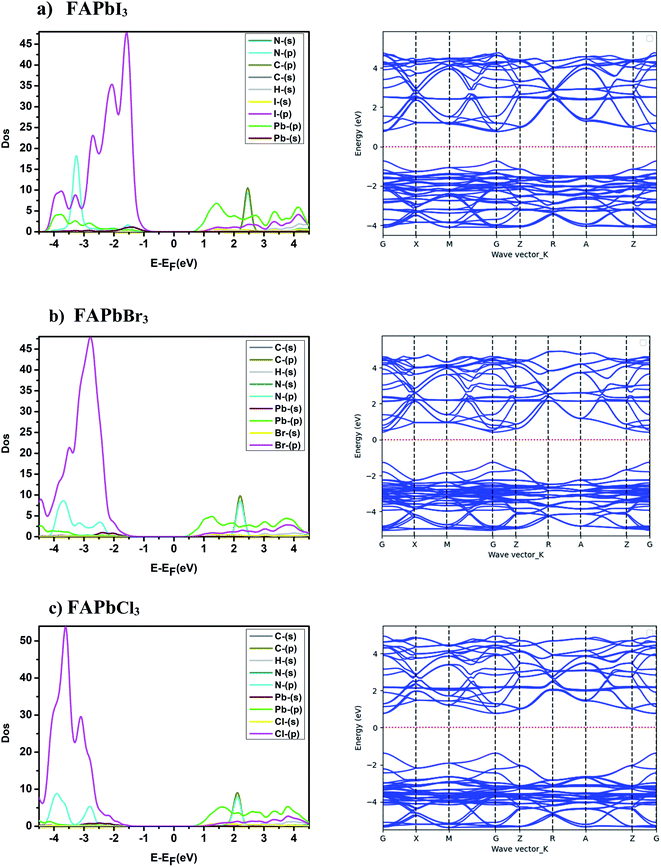 |
| | Fig. 2 Band structure and partial state density of FAPbX3. | |
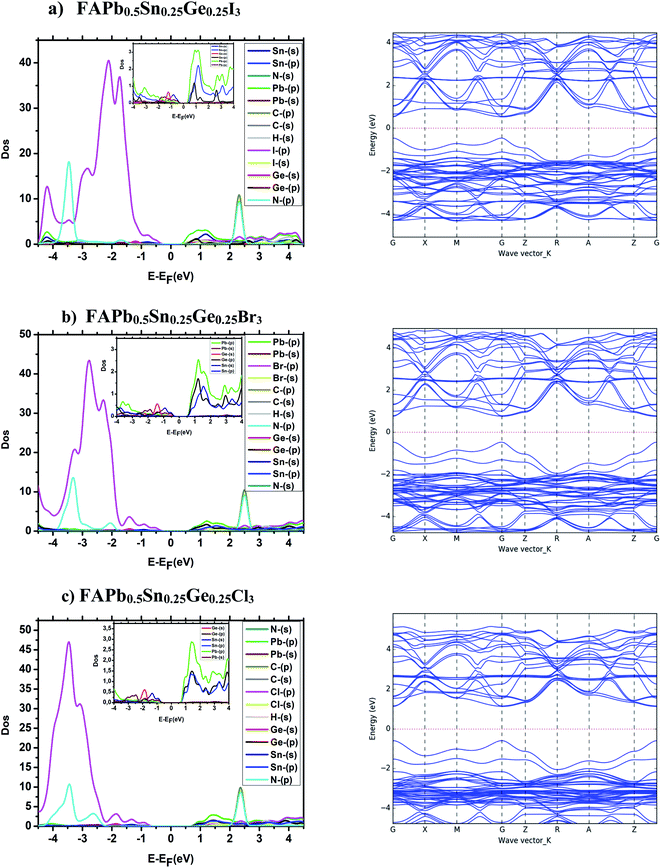 |
| | Fig. 3 Band structure and partial state density of FAPb0.5Sn0.25Ge0.25X3. | |
Table 2 Effective mass of MAPbI3, FAPb0.5Sn0.25Ge0.25X3, and FAPbX3 structures
| Compounds |
Eg (eV) |
me |
mh |
| MAPbI3 |
1.6 |
0.19 |
0.34 |
| FAPb0.5Sn0.25Ge0.25I3 |
0.98 |
0.29 |
0.07 |
| FAPb0.5Sn0.25Ge0.25Br3 |
1.22 |
0.24 |
0.08 |
| FAPb0.5Sn0.25Ge0.25Cl3 |
1.7 |
1.13 |
0.36 |
| FAPbI3 |
1.5 |
0.44 |
0.19 |
| FAPbBr3 |
1.67 |
0.35 |
0.11 |
| FAPbCl3 |
2.14 |
0.72 |
0.19 |
The electronic properties of the lower free lead halide perovskite FAPb0.5Sn0.25Ge0.25X3 where the lead was doped with both Sn and Ge, are depicted in Fig. 3. The band structures represent a band gap of 0.98 eV, 1.22 eV and 1.7 eV for FAPb0.5Sn0.25Ge0.25I3, FAPb0.5Sn0.25Ge0.25Br3 and FAPb0.5Sn0.25Ge0.25Cl3, respectively. The valence band maximum (VBM) and the conduction band minimum (CBM) for both compounds FAPb0.5Sn0.25Ge0.25I3 and FAPb0.5Sn0.25Ge0.25Br3 shows a curvature indicating the low effective mass of carriers (electron–hole). In contrast the bands of the FAPb0.5Sn0.25Ge0.25Cl3 structure are almost flattened, thus the effective mass is greater when compared to the two other compounds as represented in Table 2. The FAPb0.5Sn0.25Ge0.25I3, FAPb0.5Sn0.25Ge0.25Br3 structures have an effective hole mass, lower than MAPbI3. The effective mass is inversely proportional to the mobility of the carriers, therefore the mobility of the carriers of FAPb0.5Sn0.25Ge0.25I3 and FAPb0.5Sn0.25Ge0.25Br3 are large which will allow the electrons to easily cross the band gap. The CBM is mainly contributed by the p orbital of Pb in the FAPb0.5Sn0.25Ge0.25I3 structure, while it is dominated by the p orbital of the three elements Pb, Ge and Sn for the FAPb0.5Sn0.25Ge0.25Br3 and FAPb0.5Sn0.25Ge0.25Cl3. The VBM of the FAPb0.5Sn0.25Ge0.25X3 structures do not show any obvious difference, it is mainly derived from the p orbital of I, whereas the deep valence band is mainly contributed by the p orbital of N and the s orbital of Pb, Ge and Sn. The organic cation is positioned far from the VBM and the CBM. We notice that the co-doping of both Sn, and Ge elements shift the p states of halogen (I, Br, Cl) towards the lowest levels, which has reduced the gap energies, this doping also adds additional energy levels which will allow more optical transitions.
3.3 Optical properties
The absorption coefficient determines how far into a material light of a particular wavelength can penetrate before it is absorbed. The absorption coefficient and the reflectivity of the three compounds were calculated from the dielectric constant ε = ε1+ iε2, where ε1 and ε2 represent the real and imaginary part of the dielectric function, respectively.
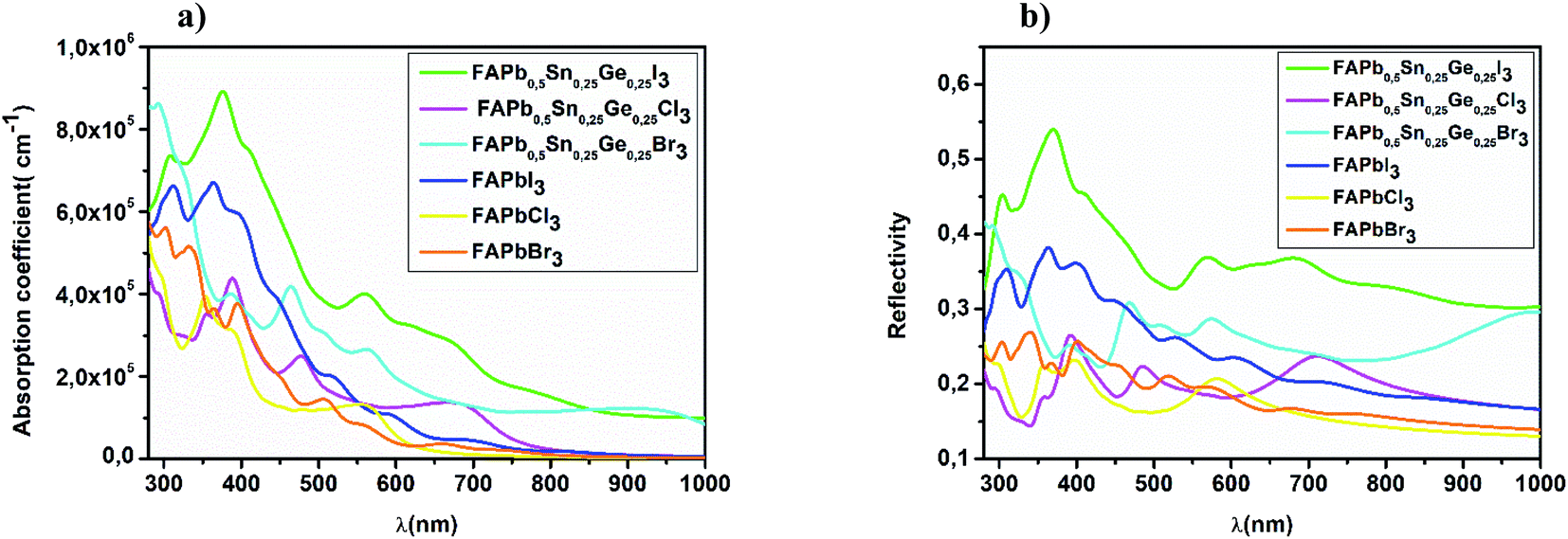
As Shown in Fig. 4, FAPbI3 has a larger absorption coefficient in the visible spectra than FAPbCl3 and FAPbBr3, the co-doping with Sn and Ge improves the optical properties. As can be seen from the same figure, the three FAPb0.5Sn0.25Ge0.25X3 compounds have a high absorption coefficient in the visible range where the FAPb0.5Sn0.25Ge0.25I3 structure shows the highest absorption; while the FAPb0.5Sn0.25Ge0.25Br3 and FAPb0.5Sn0.25Ge0.25Cl3 structures revealed the best absorption coefficient in the ultraviolet region. In physics and electrical engineering, the reflection coefficient describes how much of a wave is reflected by an impedance discontinuity in the medium transmission. According to Fig. 4b, all the investigated perovskite materials show a medium reflectivity in the IR-visible-UV region of the spectra. This reflectivity suggests that the materials have high absorptivity and/or transmissivity. Absorption coefficient and the reflectivity are the main parameters for the calculation of the short circuit current density.
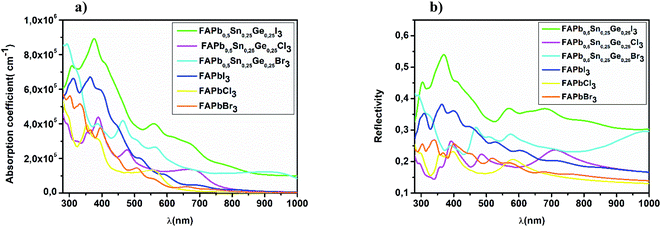 |
| | Fig. 4 Spectral (a) absorption coefficient and (b) reflectivity of FAPb0.5Sn0.25Ge0.25X3. | |
3.4 Power conversion efficiency
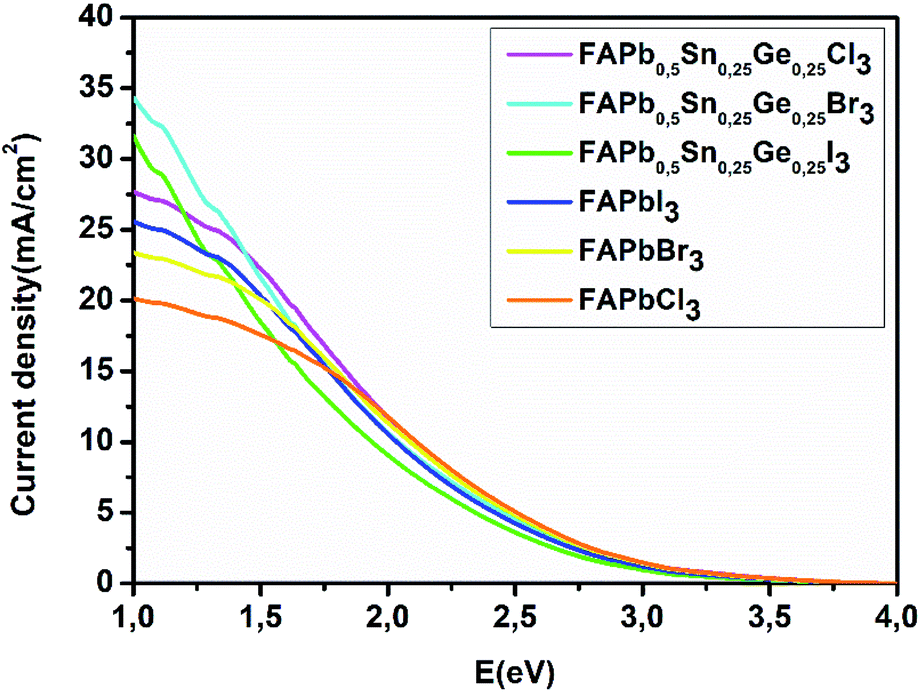
The power conversion efficiency of solar cells is related to the material’s electronic and optical properties. In order to calculate the efficiency of these compounds we first start by determining the photovoltaic parameters: short-circuit current density and open circuit voltage. The calculated power conversion efficiency (η) of MAPbI3 with an open circuit voltage (Voc) of 1.1 V, a Jsc of 17.14 mA cm−2, and a fill factor (FF) of 60% gives a value of η = 11%, this value matches the experimental results.42,43 The current density characteristics of the FAPbX3, and the FAPb0.5Sn0.25Ge0.25X3 compounds under AM1.5G solar irradiation are displayed in Fig. 5. FAPb0.5Sn0.25Ge0.25I3 has the highest current density with a value of 31.5 mA cm−2. The short-circuit current density shows a monotonical decrease from FAPb0.5Sn0.25Ge0.25I3 to FAPbCl3 that represents the lowest Jsc. In particular, the calculated short-circuit current density for the FAPbX3 shows lower values regarding those of FAPb0.5Sn0.25Ge0.25X3 compounds, revealing that the doping of lead has largely improved the Jsc. On the other hand, the halogen (I, Br, Cl) variation increases the band gap enhancing the rise in open circuit voltage. The calculated open circuit voltage and the efficiency Eloss of 0.7 and 0.5 are listed in Table 3. Despite the high current density of FAPb0.5Sn0.25Ge0.25I3, this compound demonstrates the lowest efficiency among the three compounds due to its lower Voc that can be improved by reducing the Eloss. Thus, as we can conclude from Fig. 5 and Table 3, a high short circuit current density is not the only parameter that determines the power of producing high efficiency. To summarize, Fig. 6 shows the variation of the efficiency as a function of the FAPbX3, and FAPb0.5Sn0.25Ge0.25X3 bandgaps. FAPb0.5Sn0.25Ge0.25Cl3 reaches a high efficiency of 19.3% proving that this compound is a promising material for photovoltaic applications with low toxicity.
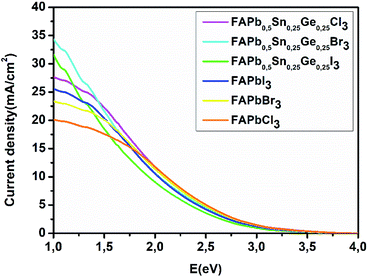 |
| | Fig. 5 Short-circuit current density of FAPbX3, and FAPb0.5Sn0.25Ge0.25X3. | |
Table 3 The calculated bandgap Eg, short-circuit current density Jsc, open circuit voltage Voc and power conversion efficiency η of FAPb0.5Sn0.25Ge0.25X3 structures
| Compounds |
Eg |
Voc |
Jsc |
η% |
| Eloss (0.7 eV) |
Eloss (0.5 eV) |
Eloss (0.7 eV) |
Eloss (0.5 eV) |
| FAPb0.5Sn0.25Ge0.25I3 |
0.98 |
0.28 |
0.48 |
31.5 |
6.17 |
12 |
| FAPb0.5Sn0.25Ge0.25Br3 |
1.22 |
0.52 |
0.72 |
29 |
12 |
17.5 |
| FAPb0.5Sn0.25Ge0.25Cl3 |
1.7 |
1 |
1.2 |
18 |
15.8 |
19.3 |
| FAPbI3 |
1.5 |
0.8 |
1 |
20.3 |
14 |
17.8 |
| FAPbBr3 |
1.67 |
0.97 |
1.17 |
17.3 |
14.6 |
18 |
| FAPbCl3 |
2.14 |
1.44 |
1.64 |
9.6 |
12.5 |
14.3 |
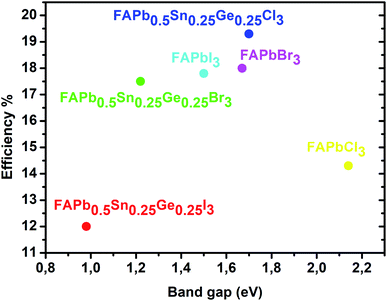 |
| | Fig. 6 Efficiency as a function of the FAPbX3, and FAPb0.5Sn0.25Ge0.25X3 bandgaps. | |
4. Conclusion
In order to reduce the toxicity of the hybrid lead halide perovskite, the structural, electronic and optical properties of hybrids with less lead, perovskite FAPb0.5Sn0.25Ge0.25X3 (X = I, Br, Cl), are investigated using density functional theory and compared with those of FAPbX3 (X = I, Br, Cl) compounds. The results show that the compounds have a suitable bandgap for photovoltaic cells. The evaluation of the photovoltaic parameters (Jsc, Voc, FF, Pin) was carried out for the power conversion efficiency calculation. Results show that half doping of lead by both Ge and Sn, improves the stability of the system and enhances its performance for photovoltaic application. The adsorption coefficient and short-circuit current density have also been improved significantly with the doping effect, where the Jsc shows a monotonical increase from the FAPbCl3 to FAPb0.5Sn0.25Ge0.25I3. We notice that, although the FAPb0.5Sn0.25Ge0.25I3 represents a high absorption coefficient in the visible range and the highest short-circuit current density, it represents the lowest power conversion efficiency due to its low open circuit voltage. FAPb0.5Sn0.25Ge0.25Cl3 has the highest power conversion efficiency demonstrating that this compound is promising for photovoltaic applications with low toxicity. This study can be a good reference for the development of new absorber materials in photovoltaic cells with lower toxicity and more efficiency.
Conflicts of interest
There are no conflicts to declare.
References
- R. Ayres and V. Voudouris, The Economic Growth Enigma: Capital, Labour and Useful Energy?, Energy Policy, 2014, 64(C), 16–28 CrossRef.
- J. Eggoh, C. Bangaké and C. Rault, Energy Consumption and Economic Growth Revisited in African Countries, Energy Policy, 2011, 39(11), 7408–7421 CrossRef.
- J. Asafu-Adjaye, The Relationship between Energy Consumption, Energy Prices and Economic Growth: Time Series Evidence from Asian Developing Countries, Energy Econ., 2000, 22(6), 615–625 CrossRef.
- R. York, Do Alternative Energy Sources Displace Fossil Fuels?, Nat. Clim. Change, 2012, 2(6), 441–443, DOI:10.1038/nclimate1451.
- N. Kannan and D. Vakeesan, Solar Energy for Future World: – A Review, Renewable Sustainable Energy Rev., 2016, 62, 1092–1105, DOI:10.1016/j.rser.2016.05.022.
- K. Frohna and S. D. Stranks, 7 – Hybrid Perovskites for Device Applications, in Handbook of Organic Materials for Electronic and Photonic Devices, ed. Ostroverkhova, O., Woodhead Publishing Series in Electronic and Optical Materials, Woodhead Publishing, 2nd edn, 2019, pp. 211–256, DOI:10.1016/B978-0-08-102284-9.00007-3.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Halide Perovskite Photovoltaics: Background, Status, and Future Prospects, Chem. Rev., 2019, 119(5), 3036–3103, DOI:10.1021/acs.chemrev.8b00539.
- H. B. Lee, N. Kumar, B. Tyagi, S. He, R. Sahani and J.-W. Kang, Bulky Organic Cations Engineered Lead-Halide Perovskites: A Review on Dimensionality and Optoelectronic Applications, Mater. Today Energy, 2021, 21, 100759, DOI:10.1016/j.mtener.2021.100759.
- A. Boubekraoui, H. Moatassim, A. Al-Shami and H. Ez-Zahraouy, DFT Study of Structural, Electronic, and Thermoelectric Properties of Cs2PdX(X=Br2Be2Te2) Compound, Comput. Condens. Matter, 2021, 29, e00600, DOI:10.1016/j.cocom.2021.e00600.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Pseudo-Halide Anion Engineering for α-FAPbI 3 Perovskite Solar Cells, Nature, 2021, 592(7854), 381–385, DOI:10.1038/s41586-021-03406-5.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber, Science, 2013, 342(6156), 341–344, DOI:10.1126/science.1243982.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, Organometallic Halide Perovskites: Sharp Optical Absorption Edge and Its Relation to Photovoltaic Performance, J. Phys. Chem. Lett., 2014, 5(6), 1035–1039, DOI:10.1021/jz500279b.
- G. E. Eperon, S. D. Stranks, C. Menelaou, M. B. Johnston, L. M. Herz and H. J. Snaith, Formamidinium Lead Trihalide: A Broadly Tunable Perovskite for Efficient Planar Heterojunction Solar Cells, Energy Environ. Sci., 2014, 7(3), 982–988, 10.1039/C3EE43822H.
- S. R. Kumavat, Y. Sonvane, D. Singh and S. K. Gupta, Two-Dimensional CH3NH3PbI3 with High Efficiency and Superior Carrier Mobility: A Theoretical Study, J. Phys. Chem. C, 2019, 123(9), 5231–5239, DOI:10.1021/acs.jpcc.8b11427.
- T.-Y. Zhu and D.-J. Shu, Polarization-Controlled Surface Defect Formation in a Hybrid Perovskite, J. Phys. Chem. Lett., 2021, 12(16), 3898–3906, DOI:10.1021/acs.jpclett.1c00702.
- A. Babayigit, A. Ethirajan, M. Muller and B. Conings, Toxicity of Organometal Halide Perovskite Solar Cells, Nat. Mater., 2016, 15(3), 247–251, DOI:10.1038/nmat4572.
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Impact of H2O on Organic–Inorganic Hybrid Perovskite Solar Cells, Energy Environ. Sci., 2017, 10(11), 2284–2311, 10.1039/C7EE01674C.
- A. Fakharuddin, F. De Rossi, T. M. Watson, L. Schmidt-Mende and R. Jose, Research Update: Behind the High Efficiency of Hybrid Perovskite Solar Cells, APL Mater., 2016, 4(9), 091505, DOI:10.1063/1.4962143.
- C. Li, S. Tscheuschner, F. Paulus, P. E. Hopkinson, J. Kießling, A. Köhler, Y. Vaynzof and S. Huettner, Iodine Migration and Its Effect on Hysteresis in Perovskite Solar Cells, Adv. Mater., 2016, 28(12), 2446–2454, DOI:10.1002/adma.201503832.
- M. Ouafi, B. Jaber, L. Atourki, R. Bekkari and L. Laânab, Improving UV Stability of MAPbI3 Perovskite Thin Films by Bromide Incorporation, J. Alloys Compd., 2018, 746, 391–398, DOI:10.1016/j.jallcom.2018.02.240.
- H. Moatassim, A. El Kenz, A. Benyoussef, M. Loulidi and O. Mounkachi, Degradation Mechanism of CH3NH3PbI3 and Enhancing Its Optical Absorption through Variety of Doping Sites, Comput. Condens. Matter, 2021, 29, e00611, DOI:10.1016/j.cocom.2021.e00611.
- C. Wu, D. Guo, P. Li, S. Wang, A. Liu and F. Wu, A Study on the Effects of Mixed Organic Cations on the Structure and Properties in Lead Halide Perovskites, Phys. Chem. Chem. Phys., 2020, 22(5), 3105–3111, 10.1039/C9CP06182G.
- A. Oranskaia, J. Yin, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, Halogen Migration in Hybrid Perovskites: The Organic Cation Matters, J. Phys. Chem. Lett., 2018, 9(18), 5474–5480, DOI:10.1021/acs.jpclett.8b02522.
- X. Zhang, J.-X. Shen, M. E. Turiansky and C. G. Van de Walle, Minimizing Hydrogen Vacancies to Enable Highly Efficient Hybrid Perovskites, Nat. Mater., 2021, 20(7), 971–976, DOI:10.1038/s41563-021-00986-5.
- M. Lyu, J.-H. Yun, P. Chen, M. Hao and L. Wang, Addressing Toxicity of Lead: Progress and Applications of Low-Toxic Metal Halide Perovskites and Their Derivatives, Adv. Energy Mater., 2017, 7(15), 1602512, DOI:10.1002/aenm.201602512.
- J. Prakash, A. Singh, G. Sathiyan, R. Ranjan, A. Singh, A. Garg and R. K. Gupta, Progress in Tailoring Perovskite Based Solar Cells through Compositional Engineering: Materials Properties, Photovoltaic Performance and Critical Issues, Mater. Today Energy, 2018, 9, 440–486, DOI:10.1016/j.mtener.2018.07.003.
- L. Peng and W. Xie, Theoretical and Experimental Investigations on the Bulk Photovoltaic Effect in Lead-Free Perovskites MASnI3 and FASnI3, RSC Adv., 2020, 10(25), 14679–14688, 10.1039/D0RA02584D.
- T. Krishnamoorthy, H. Ding, C. Yan, W. L. Leong, T. Baikie, Z. Zhang, M. Sherburne, S. Li, M. Asta, N. Mathews and S. G. Mhaisalkar, Lead-Free Germanium Iodide Perovskite Materials for Photovoltaic Applications, J. Mater. Chem. A, 2015, 3(47), 23829–23832, 10.1039/C5TA05741H.
- D. Liu, Q. Li, J. Hu, R. Sa and K. Wu, Photovoltaic Performance of Lead-Less Hybrid Perovskites from Theoretical Study, J. Phys. Chem. C, 2019, 123(20), 12638–12646, DOI:10.1021/acs.jpcc.9b02705.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21(39), 395502, DOI:10.1088/0953-8984/21/39/395502.
- J. Klimeš, D. R. Bowler and A. Michaelides, Van Der Waals Density Functionals Applied to Solids, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(19), 195131, DOI:10.1103/PhysRevB.83.195131.
- Y. Wang, T. Gould, J. F. Dobson, H. Zhang, H. Yang, X. Yao and H. Zhao, Density Functional Theory Analysis of Structural and Electronic Properties of Orthorhombic Perovskite CH3NH3PbI3, Phys. Chem. Chem. Phys., 2013, 16(4), 1424–1429, 10.1039/C3CP54479F.
- D. Sangalli, A. Ferretti, H. Miranda, C. Attaccalite, I. Marri, E. Cannuccia, P. Melo, M. Marsili, F. Paleari, A. Marrazzo, G. Prandini, P. Bonfà, M. O. Atambo, F. Affinito, M. Palummo, A. Molina-Sánchez, C. Hogan, M. Grüning, D. Varsano and A. Marini, Many-Body Perturbation Theory Calculations Using the Yambo Code, J. Phys.: Condens. Matter, 2019, 31(32), 325902, DOI:10.1088/1361-648X/ab15d0.
- J. Wu, G. Yue, Y. Xiao, J. Lin, M. Huang, Z. Lan, Q. Tang, Y. Huang, L. Fan, S. Yin and T. Sato, An Ultraviolet Responsive Hybrid Solar Cell Based on Titania/Poly(3-Hexylthiophene), Sci. Rep., 2013, 3(1), 1283, DOI:10.1038/srep01283.
- M. A. Green, Solar Cell Fill Factors: General Graph and Empirical Expressions, Solid-State Electron., 1981, 24(8), 788–789, DOI:10.1016/0038-1101(81)90062-9.
- M. R. Filip, C. Verdi and F. Giustino, GW Band Structures and Carrier Effective Masses of CH3NH3PbI3 and Hypothetical Perovskites of the Type APbI3: A = NH4, PH4, AsH4, and SbH4, J. Phys. Chem. C, 2015, 119(45), 25209–25219, DOI:10.1021/acs.jpcc.5b07891.
- N. Baaalla, Y. Ammari, E. K. Hlil, R. Masrour, A. E. Kenz and A. Benyoussef, Study of Optical, Electrical and Photovoltaic Properties of CH3NH3PbI3 Perovskite: Ab Initio Calculations, Phys. Scr., 2020, 95(9), 095104, DOI:10.1088/1402-4896/abae1e.
- X.-F. Diao, Y. Tang, T. Tang, Q. Xie, K. Xiang and G. Liu, Study on the Stability of Organic–Inorganic Perovskite Solar Cell Materials Based on First Principle, Mol. Phys., 2020, 118(8), e1665200, DOI:10.1080/00268976.2019.1665200.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties, Inorg. Chem., 2013, 52(15), 9019–9038, DOI:10.1021/ic401215x.
- X. Qian, X. Gu and R. Yang, Lattice Thermal Conductivity of Organic–Inorganic Hybrid Perovskite CH3NH3PbI3, Appl. Phys. Lett., 2016, 108(6), 063902, DOI:10.1063/1.4941921.
- L. Leppert, S. E. Reyes-Lillo and J. B. Neaton, Electric Field- and Strain-Induced Rashba Effect in Hybrid Halide Perovskites, J. Phys. Chem. Lett., 2016, 7(18), 3683–3689, DOI:10.1021/acs.jpclett.6b01794.
- P. Fan, D. Gu, G.-X. Liang, J.-T. Luo, J.-L. Chen, Z.-H. Zheng and D.-P. Zhang, High-Performance Perovskite CH3NH3PbI3 Thin Films for Solar Cells Prepared by Single-Source Physical Vapour Deposition, Sci. Rep., 2016, 6(1), 29910, DOI:10.1038/srep29910.
- D. Liu, M. K. Gangishetty and T. L. Kelly, Effect of CH3NH3PbI3 Thickness on Device Efficiency in Planar Heterojunction Perovskite Solar Cells, J. Mater. Chem. A, 2014, 2(46), 19873–19881, 10.1039/C4TA02637C.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
H. Zaaria,
A. El Kenza,
A. Benyoussefb,
M. Loulidia and
O. Mounkachi*ac
*a,
H. Zaaria,
A. El Kenza,
A. Benyoussefb,
M. Loulidia and
O. Mounkachi*ac