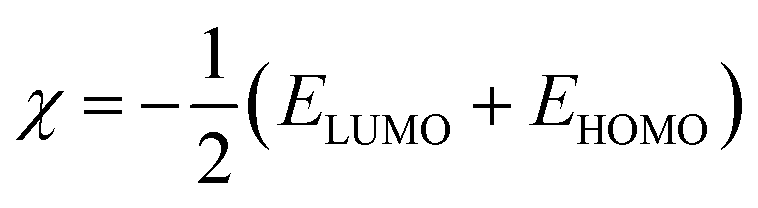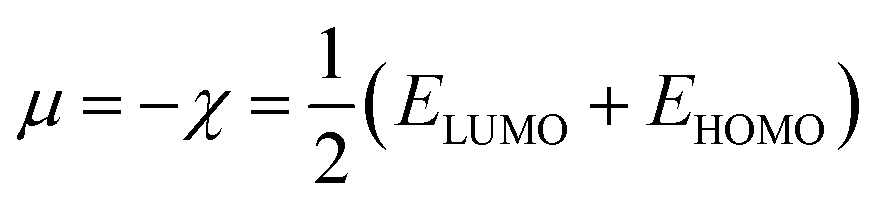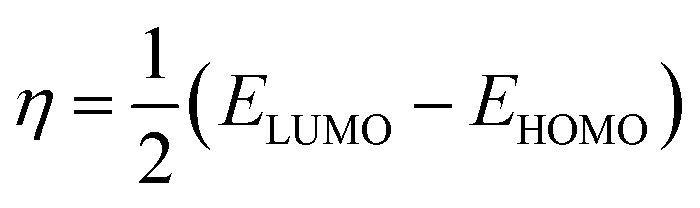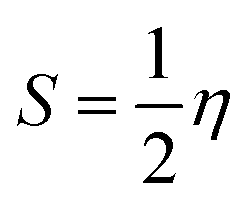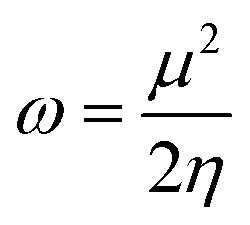DOI:
10.1039/D2RA00274D
(Paper)
RSC Adv., 2022,
12, 8520-8529
Selective separation of uranyl ions from some lanthanide elements using a promising β-enaminoester ligand by cloud point extraction†
Received
14th January 2022
, Accepted 14th March 2022
First published on 17th March 2022
Abstract
For uranyl extraction, a distinctive chelating ligand, namely ethyl 2-amino-6-hydroxy-5-(4-methoxyphenyldiazenyl)-4-phenyl-4H-benzo[f]chromene-3-carboxylate, has been synthesized and characterized using FT-IR, NMR, and ESI-MS. Subsequently, a cloud point extraction (CPE) protocol has been developed for the selective separation of the trace amounts of uranyl ions from some lanthanide ions after being captured by the ligand in the presence of non-ionic surfactant (Triton X-114). The extraction procedure has been optimized based on the concentration of the complexing agent and the non-ionic surfactant, phase separation temperatures, pH, and ionic strength. The developed CPE procedure exhibited a relatively low detection limit of 0.5 ng mL−1 in the linear range from 3 ng mL−1 to 250 ng mL−1. Furthermore, interference studies have been carried out to study the selectivity of our protocol. These studies revealed that the recoveries of uranyl ions were in the range from 96.1% to 99.9% in the presence of some lanthanide ions such as Th4+, Gd3+, and Sm3+. It is worth mentioning that the geometry optimization, reactivity, and molecular electrostatic potential maps of the ligand and the proposed UO22+ complex were acquired via DFT calculations to study their stabilities based on the geometry and binding affinity. The theoretical data confirmed the octahedral geometry of the UO22+ complex with the lowest energy and excellent stability. The robustness of the proposed methodology was evaluated by the detection of uranyl ions in different environmental samples and synthetic mixtures.
1. Introduction
Among the naturally occurring actinides, uranium is one of the most ubiquitous trace radioactive elements and has received much attention. Its commercial interest arises from its applications in nuclear power reactors, electrical power production, and the medical field.1–3 The main sources of uranium are seawater and oceans which are estimated to contain 4.5 billion tons.4,5 In addition, lanthanide and actinide ions are always coexisting with each other. Thus, the separation and preconcentration of uranium get great attention for nuclear energy development and the protection of the ecological environment. Up to now, several techniques such as solid-phase extraction, co-precipitation, and flotation,6–9 have been introduced globally for separation and pre-concentration of different metal ions. Compared with these techniques, cloud point extraction (CPE), and aqueous biphasic systems are commonly used for metal ion extraction owing to their eco-friendliness.10,11
However, the CPE has been reported as a more powerful protocol for the selective extraction and separation of trace organic and/or inorganic species owing to its simplicity, high concentration factor, good efficiency, low cost, and little organic solvent required.7–9,12–14 The CPE method is essentially based on the formation of hydrophobic complexes, for inorganic species, between target metal ions and chelating agents. Thanks to ionic and/or non-ionic surfactants, hydrophobic complexes were separated and preconcentrated above their critical micelle concentrations (CMC). On the other hand, below the CMC point, the surfactant molecules act as monomers in the solution. After heating the solution above the critical point, these monomers associate with each other to form micellar aggregates of colloidal dimensions. Different electrolytes have been introduced directly during the extraction process to reach cloud point at room temperature, depending on the salting-out effect.12 For the enhancement of the extraction efficiency of uranyl ions, different chelating agents, such as 4-(2-pyridylazo)resorcinol,15 dibenzoylmethane,16 alizarin red S,17 bis(2,4,4-trimethylpentyl)dithiophosphate18 and celestine blue19 have been used. However, the extraction processes showed low efficiencies, narrow linear ranges, high detection limits, and consumption of large organic solvents.
β-Enaminones are compounds containing N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CO sequence which have a propensity for chelation with various metal ions.20 The presence of oxygen and nitrogen electron-rich sites promotes the affinity toward diverse electrophiles.21–23 Cyclic primary β-enaminones can be represented in two geometric forms Z, s-Z and Z, s-E, based on the solvent nature and the steric properties, as depicted in Fig. S1 (ESI†).24 The spectroscopic investigations have shown that, in polar solvents, the Z, s-Z configuration is more predominated due to the system rigidity via the intramolecular hydrogen bond formation. On the other hand, using protic polar solvents increases the contribution of E form.25,26 Moreover, the diazo dyes are a very interesting compounds that are frequently used in the spectrophotometric determination of different metal ions.27–29 Due to the high potential of cyclic primary β-enaminones for the separation of metals, a distinctive diazo decorated β-enaminoester has been elaborated in this study for the extraction and preconcentration of uranyl ions.
C–CO sequence which have a propensity for chelation with various metal ions.20 The presence of oxygen and nitrogen electron-rich sites promotes the affinity toward diverse electrophiles.21–23 Cyclic primary β-enaminones can be represented in two geometric forms Z, s-Z and Z, s-E, based on the solvent nature and the steric properties, as depicted in Fig. S1 (ESI†).24 The spectroscopic investigations have shown that, in polar solvents, the Z, s-Z configuration is more predominated due to the system rigidity via the intramolecular hydrogen bond formation. On the other hand, using protic polar solvents increases the contribution of E form.25,26 Moreover, the diazo dyes are a very interesting compounds that are frequently used in the spectrophotometric determination of different metal ions.27–29 Due to the high potential of cyclic primary β-enaminones for the separation of metals, a distinctive diazo decorated β-enaminoester has been elaborated in this study for the extraction and preconcentration of uranyl ions.
In this work, we developed a Triton X-114 based CPE process with ethyl 2-amino-6-hydroxy-5-(4-methoxyphenyldiazenyl)-4-phenyl-4H-benzo[f]chromene-3-carboxylate as a chelating agent for the separation of uranyl ions from some lanthanide elements. The optimization of the experimental conditions (e.g.; pH, temperature, salting out effect, etc.) which affect the CPE efficiency were examined. Furthermore, the analytical figures of merit and interfering ions tolerance were assessed and listed. Finally, the optimized analytical procedure has been applied to determine uranyl ions in different real samples.
2. Experimental section
2.1. Chemicals and reagents
All chemicals and reagents were of analytical grade and used as received. Uranyl acetate, Triton X-114 (TX-114), potassium dihydrogen phosphate (KH2PO4), dipotassium hydrogen phosphate (K2HPO4), hydrochloric acid, 4-methoxyaniline, sodium nitrite, and potassium iodide were purchased from Sigma-Aldrich. In addition, ethanol (EtOH), methanol (MeOH), dimethylformamide (DMF), acetone, acetic acid (AcOH), pyridine, sodium acetate (AcONa), and sodium hydroxide (NaOH) were obtained from Merck. It is worth mentioning that all solutions were freshly prepared using ultrapure water (18.2 MΩ cm−1). Moreover, all the glasswares were stored in a bath of nitric acid (10%) for 48 h and then washed with ultrapure water before usage.
2.2. Apparatus
The structure elucidation of the enaminoester ligand was confirmed through the FT-IR spectrum (IR) (ν, cm−1) using the pressed KBr pellet method on a Mattson 5000 FT-IR spectrophotometer (Faculty of Pharmacy, Mansoura University). The 1H-NMR analysis (δ, ppm) was performed on Bruker Avance III 400 MHz (Faculty of Pharmacy, Mansoura University) using tetramethylsilane (TMS) as an internal reference and DMSO-d6 as a solvent. The exchangeable protons were detected via the D2O test. The multiplicities of the signals were reported as follows: s = singlet, t = triplet, and m = multiplet. The electron spray ionization-mass spectroscopy (ESI-MS) was determined on Advion compact mass spectrometer (CMS) (NAWAH Scientific Center). The elemental analyses (C, H, and N) were executed at the microanalytical lab at Cairo University.
The UV/VIS spectrophotometer, UV-7300 (Cole-Parmer Ltd., Staffordshire, UK) equipped with Xe lamp, was used for performing the spectra and measuring the absorbance. For uranyl determination, Agilent 5100 inductively coupled plasma-optical emission spectrometry (ICP-OES, Melbourne, Australia) was utilized and the instrumental parameters were summarized in Table S1 (ESI†). In contrast, the concentration of hydrogen ions was adjusted in the solutions using a digital pH/mV meter (Hanna model HI 8519). Moreover, the temperature of the reaction was controlled by using a thermostatic water bath.
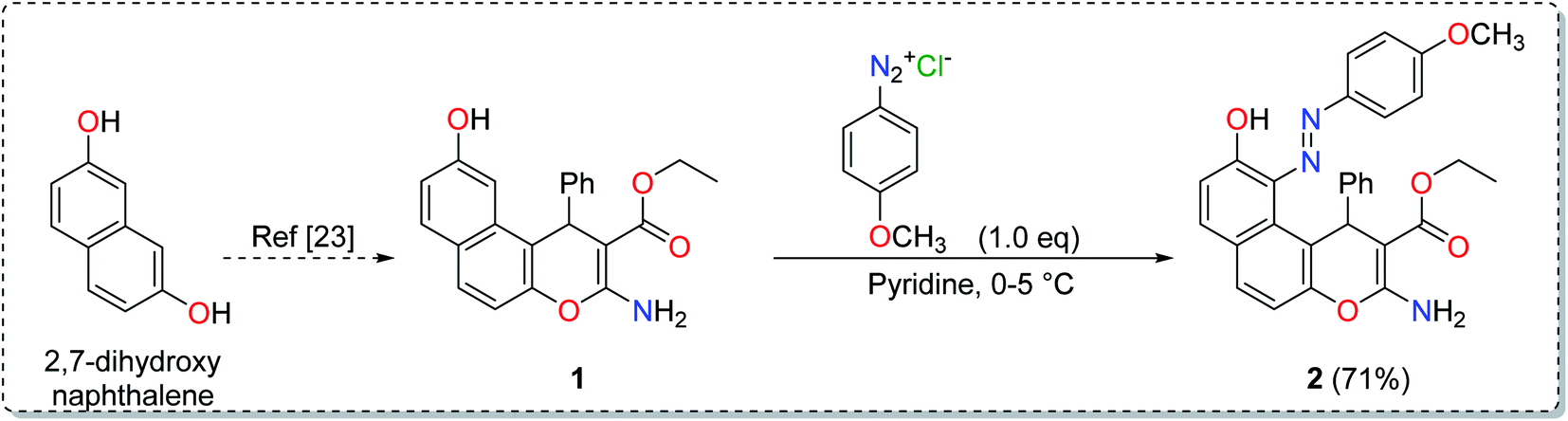
2.3. Synthesis of the chelating ligand
2.3.1. Ethyl 2-amino-6-hydroxy-5-(4-methoxyphenyldiazenyl)-4-phenyl-4H-benzo[f]chromene-3-carboxylate (2). A cold solution (temp. 0–5 °C) of freshly prepared 4-methoxybenzenediazonium chloride [prepared by mixing 4-methoxyaniline (123 mg, 1 mmol), sodium nitrite (138 mg, 2 mmol) and conc. HCl (0.4 mL, 4 mmol) in 4 mL water] was added dropwise to a cold solution of compound 1 (361 mg, 1 mmol) in 6 mL pyridine with stirring in an ice bath (40 g ice + 100 mL water) for 1 h. The formed precipitate was filtered off, washed well with water several times then recrystallized from EtOH (70%) to afford the desired diazo compound 2. Red powder; yield (355 mg, 71%); mp = 128–130 °C; IR (KBr, ν/cm−1): 3421 and 3313 (OH and NH2), 1674 (CO), 1633 (CC), 1217 (C–O); 1H-NMR (400.20 MHz, DMSO-d6): δ (ppm) = 1.25 (t, 3H, J = 7.2 Hz, 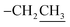 ), 2.51 (s, 3H, OCH3), 4.09 (m, 2H,
), 2.51 (s, 3H, OCH3), 4.09 (m, 2H, 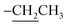 ), 5.29 (s, 1H, H-4), 6.94–7.79 (m, 13H, ArH), 8.33, 8.57, 9.89 (3H, 2NH, OH); ESI-MS (m/z) calcd for C29H25N3O5Na (M + Na)+ 518.50, found 518.20; anal. calcd for C29H25N3O5 (495.53): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.15; H, 5.22; N, 8.75%.
), 5.29 (s, 1H, H-4), 6.94–7.79 (m, 13H, ArH), 8.33, 8.57, 9.89 (3H, 2NH, OH); ESI-MS (m/z) calcd for C29H25N3O5Na (M + Na)+ 518.50, found 518.20; anal. calcd for C29H25N3O5 (495.53): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.15; H, 5.22; N, 8.75%.
2.4. Theoretical computation for geometry and binding affinity
The structures of the enaminoester ligand and its uranyl complex were optimized by employing DFT calculations using DMOL3 tool in the Material Studio (MS) package, with the generalized gradient approximation (GGA) using BLYP correlation functional method and double-numerical-basis set with polarization (DNP).30–33 Vibration frequency calculations were achieved to approve that each structure was the lowest on the potential energy surface. The HOMO/LUMO energies were evaluated at the most optimized geometries at the DFT/BLYP level. The energy gap (ΔE), electronegativity (χ), hardness (η), chemical potentials (μ), global softness (S), and electrophilicity index (ω) was calculated using subsequent equations.34–36
2.5. Cloud point extraction technique
An aliquot of the sample solution with a known concentration of uranyl ion was added into scaled-glass tubes with a conical bottom and a mixture of 350 μL β-enaminoester (5.0 × 10−3 mol L−1), 0.5 mL Triton X-114 (2% v/v), and 3 mL of acetate buffer (pH = 5) were pipetted to it. After that, it was diluted to the limited mark with ultrapure water. After gentle shaking, the centrifuge tube was transferred into a thermostatically controlled bath at 35 °C for 10 min. A cloudy solution was obtained, and the separation of the surfactant-rich phase and the aqueous phase was accomplished via centrifugation at 3500 rpm for 5 min. The viscosity of the surfactant-rich phase was also increased after cooling in an ice bath and the supernatant of the aqueous phase was decanted through the overturned glass tube. Subsequently, the concentrated uranyl complex in the rich phase layer was dissolved by using 0.5 mL of DMF for measuring the optical density at a specific wavelength.
2.6. Application
The performance of the developed CPE method was assessed for practical analytical applications in different environmental samples. Rock specimens were obtained from the mountains of Um Safi region at the central part of the Egyptian Eastern Desert and the Abu Tartur Phosphate project. The collected samples were incubated in pre-cleaned plastic bottles till transferred into the laboratory. After that, they were ground and homogenized by a friction machine, followed by digestion in Teflon tubes by using 6 mL of a mixture of HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3:DDW (1:1:1). Then, the containers were transferred into the microwave system with the subsequent parameters: two minutes at 8 atm and 600 W; three minutes at 12 atm and 800 W; eight minutes at 16 atm and 800 W. After cooling, 20 mL of 5% w/v boric acid was added to neutralize excess HF and then the volume was diluted to 50 mL using DDW.
:HNO3:DDW (1:1:1). Then, the containers were transferred into the microwave system with the subsequent parameters: two minutes at 8 atm and 600 W; three minutes at 12 atm and 800 W; eight minutes at 16 atm and 800 W. After cooling, 20 mL of 5% w/v boric acid was added to neutralize excess HF and then the volume was diluted to 50 mL using DDW.
3. Results and discussion
3.1. Synthesis of the ligand
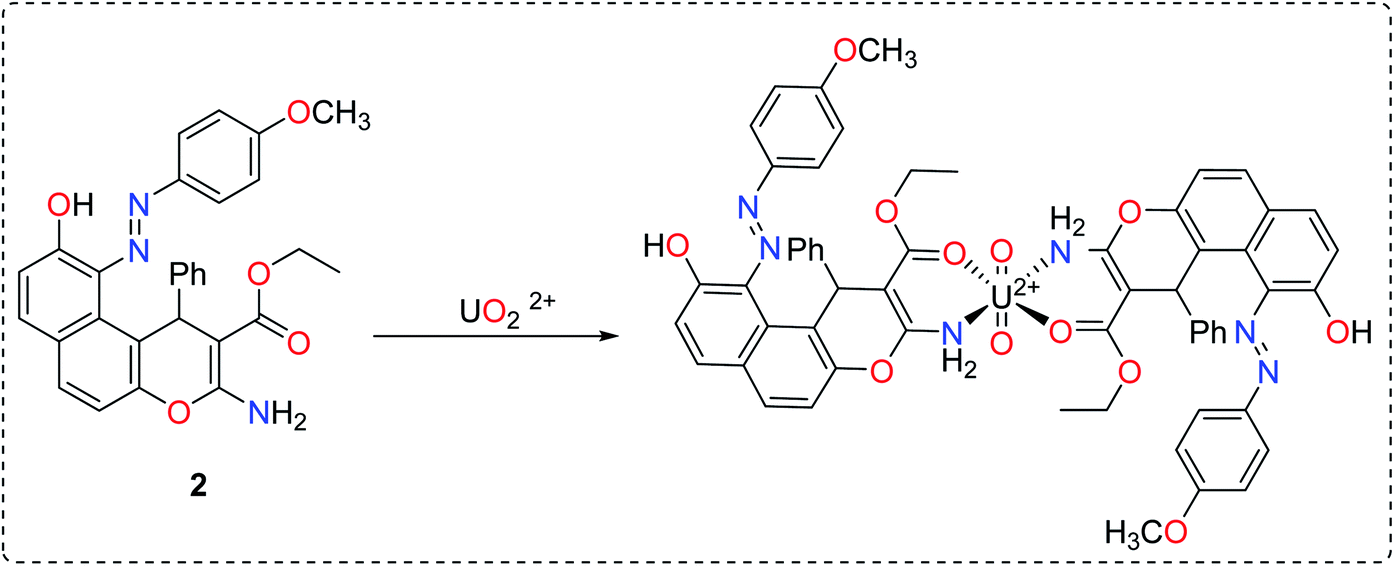
The synthesis of the enaminoester 1 was previously reported by our group starting with 2,7-dihydroxynaphthalene via a highly yielded protocol.23 Starting with enaminoester 1, the promising diazinylenaminoester ligand 2 has been synthesized through the insertion of the diazinyl chromophore at the C-5 position of the enaminoester 1 via its coupling with 4-methoxybenzenediazonium chloride in an equimolar ratio (Scheme 1).37,38 The coupling process acquired the enaminoester ligand a bright red color which could easily be detected with the spectrophotometer in the cloud point extraction method.
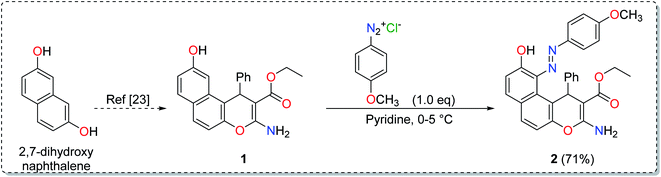 |
| | Scheme 1 Synthesis of the diazinylenaminoester ligand. | |
The IR spectrum of compound 2 showed the overlapped stretching bands of OH and NH2 groups at 3421 and 3313 cm−1, while the ester CO band appeared at 1674 cm−1. Moreover, the 1H-NMR spectrum showed two signals at δ 8.33 and 8.57 ppm which were attributed to the two magnetically non-equivalent protons of the NH2 group while the signals at δ 1.25 ppm and 4.09 ppm were assignable to the ester methyl and methylene groups, respectively. The elemental analysis (C, H, N) and the electron spray ionization-mass spectrum (ESI-MS) were in complete agreement with the desired structure.
3.2. Geometry optimization
The input files for DFT calculations were constructed and the optimized geometries for the ligand and its investigated UO22+ complex with numbering are shown in Fig. S4 (ESI†). While Table S2 (ESI†) shows the selected bond lengths and angles of enaminoester ligand that experience a modification upon complexation with the metal ion. For the UO22+ complex, metal ions are hexacoordinated in an octahedral geometry, in which O69 and O68 are in axial positions (angle = 179.78°–180°). Moreover, O32, N33, O65, and N66 atoms are located almost in one plane with angles (O32–U–N33 = 87.89°, O32–U–N66 = 91.69°, N33–U–O65 = 91.99°, and N66–U–O65 = 88.43°) ≈ 90°. In addition, angles of N66–U–N33 = 179.53°, and O65–U–O32 = 178.86°, were approximately at 180°.
3.3. Reactivity parameters
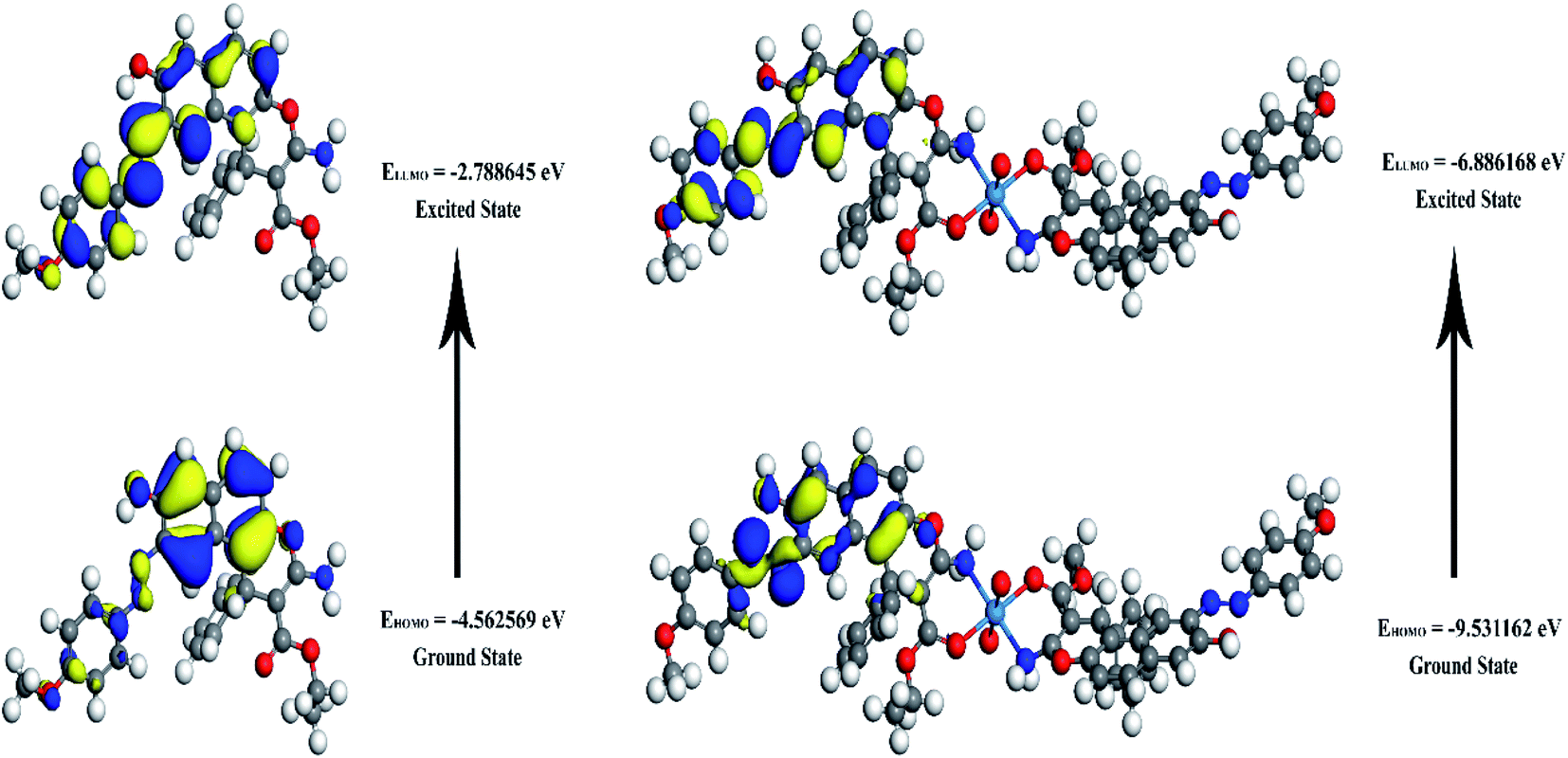
The energies of HOMO, LUMO, and the energy gap for the enaminoester ligand and its metal complex were utilized to inspect the stability, reactivity, chemical softness, and hardness of these compounds (Fig. 1). Furthermore, the energies values of FO's evaluate the capability of molecules to give electrons. In addition, the energy gap values designate the reactivity of the compound for the metal surface (as the energy required for the transition from HOMO to LUMO increase, the reactivity decrease). As shown in Table 1, the positive value of the global hardness indicates that the back donation is favored.
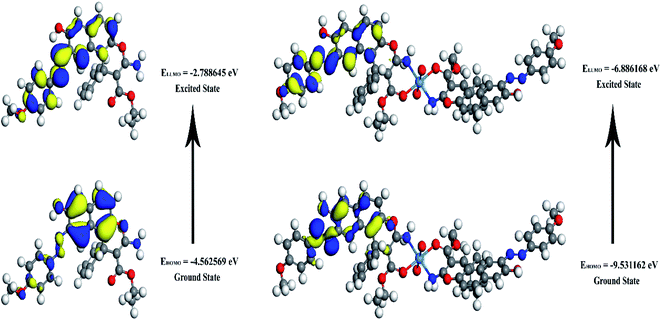 |
| | Fig. 1 3D plots frontier orbital energies using DFT method for the enaminoester ligand and UO22+ complex. | |
Table 1 Calculated EHOMO, ELUMO, energy band gap (EH–EL), chemical potential (μ), electronegativity (χ), global hardness (η), global softness (S) and global electrophilicity index (ω) for the ligand and its complex
| Compound |
EH (eV) |
EL (eV) |
(EH–EL) (eV) |
χ (eV) |
μ (eV) |
η (eV) |
S (eV−1) |
ω (eV) |
| Enaminoester ligand |
−4.562569 |
−2.788645 |
−1.77392 |
3.67561 |
−3.6756 |
0.88696 |
0.44348 |
7.615932 |
| UO22+ complex |
−9.531162 |
−6.886168 |
−2.64499 |
8.20867 |
−8.2087 |
1.32249 |
0.66125 |
25.47535 |
3.4. Dipole moment and other molecular properties
The energy components assessed by DFT were shown in Table 2. The increased value of the calculated binding energy of complex likened to that of the enaminoester signifies the higher stability of the designed complexes than the free ligand.
Table 2 Some energetic properties of the enaminoester and its uranyl complex calculated by DMOL3 using DFT-method
| The energetic properties |
Ligand |
UO22+ complex |
| Energy components (kcal mol−1) |
Sum of atomic energies |
−1035028.8 |
−2218810.5 |
| Kinetic energy |
−11341.5 |
−62858.9 |
| Electrostatic energy |
−1386.08 |
37668.05 |
| Exchange correlation energy |
2709.311 |
5914.362 |
| Spin polarization energy |
2394.886 |
3903.802 |
| Total energy |
−1042652 |
−2234183 |
| Binding energy (kcal mol−1) |
−7623.42 |
−15372.7 |
| Dipole moment (debye) |
μx |
0.999615 |
0.339863 |
| μy |
−0.01731 |
−2.03699 |
| μz |
−0.33377 |
−3.18124 |
| μTotal |
1.054009 |
3.792774 |
3.5. Molecular electrostatic potential (MEP) maps
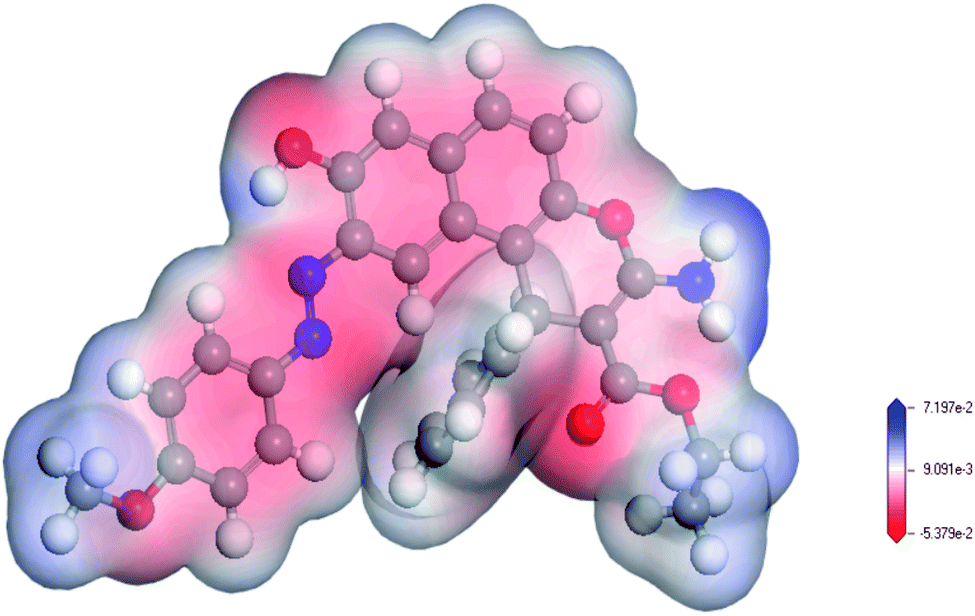
MEP maps are used to locate positive and negative regions and in the prediction of the reactive sites in molecule.39 As indicated in Fig. 2, the O32 and N33 in the ligand are electron-rich regions (donating sites) with electron densities of −0.409 e Å−3, and −0.648 e Å−3, respectively. These results confirmed that the involvement of sp2 ester's oxygen and sp3 nitrogen atoms in chelation with the target uranyl ion. Based on the geometry optimization and the DFT calculations, the chelation equation could be summarized in Scheme 2.
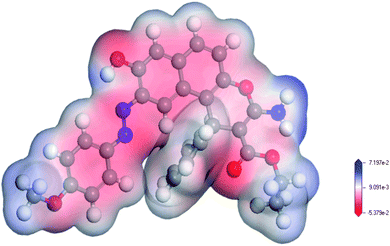 |
| | Fig. 2 MEP map of the enaminoester ligand. The red color indicates the negative zones (nucleophilic attack), and the blue color indicates the positive zones (electrophilic attack). | |
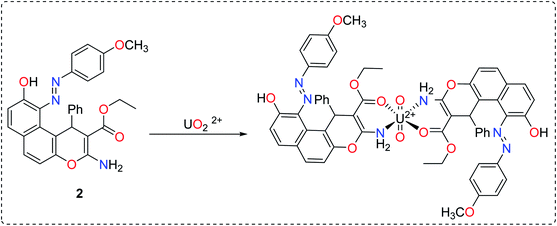 |
| | Scheme 2 The interaction between uranyl with enaminoester ligand based on the computational studies. | |
3.6. Preliminary investigation of the uranyl complex formation
A preliminary investigation of the uranyl–enaminoester complex showed the formation of a yellowish-colored complex in a wide pH range. Furthermore, the maximum absorbance of the proposed complex was recorded at pH 4.5 and wavelength 465 nm. In contrast, Gd3+, Sm3+, and Th4+ formed colored complexes with enaminoester ligand only in pH range from 6 to 7.3. It is worth mentioning that there is no overlap between the uranyl–enaminoester complex and other complexes. These results indicated that the uranyl complex could be separated easily at pH 4.5.
3.7. Stoichiometry between ligand and metal
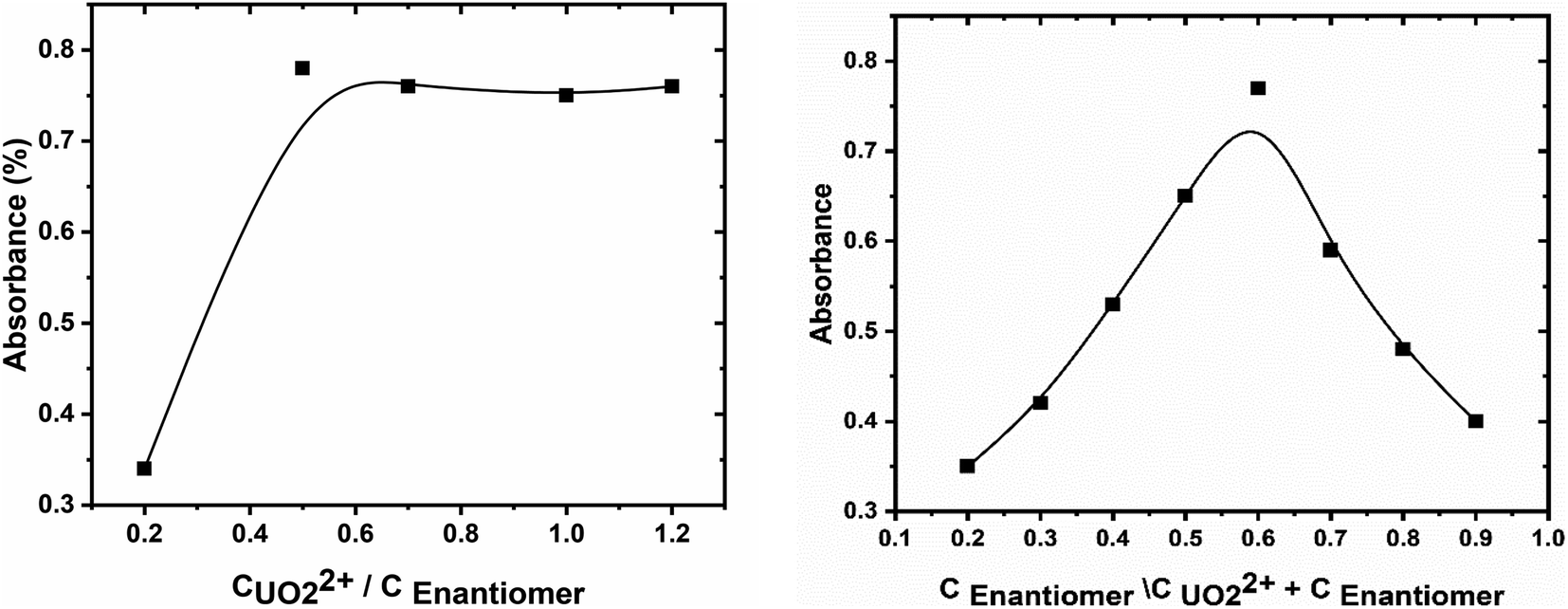
The stoichiometry of the formed uranyl–enaminoester complex, at the optimized conditions, was investigated by using continuous variation (Job method) and molar ratio methods.40 The plot of absorbance against the molar ratio of enaminoester to uranyl ion, obtained by varying the concentration of the chelating compound, showed inflection at molar ratio 2.0, indicating 1:2 metal to ligand complex formed (Fig. 3A). This result was emphasized by the continuous variation method (Fig. 3B). It is worth mentioning that the stability of the proposed complex has been studied and the stability constant was calculated spectrophotometry depending on the Harvey and Manning method and was found to be 8.26.41
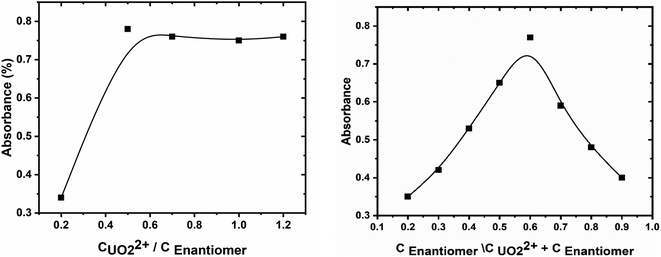 |
| | Fig. 3 (A) The mole-ratio plot and (B) continuous variation plot for enaminoester and uranyl interaction. | |
3.8. Optimization of the cloud point extraction
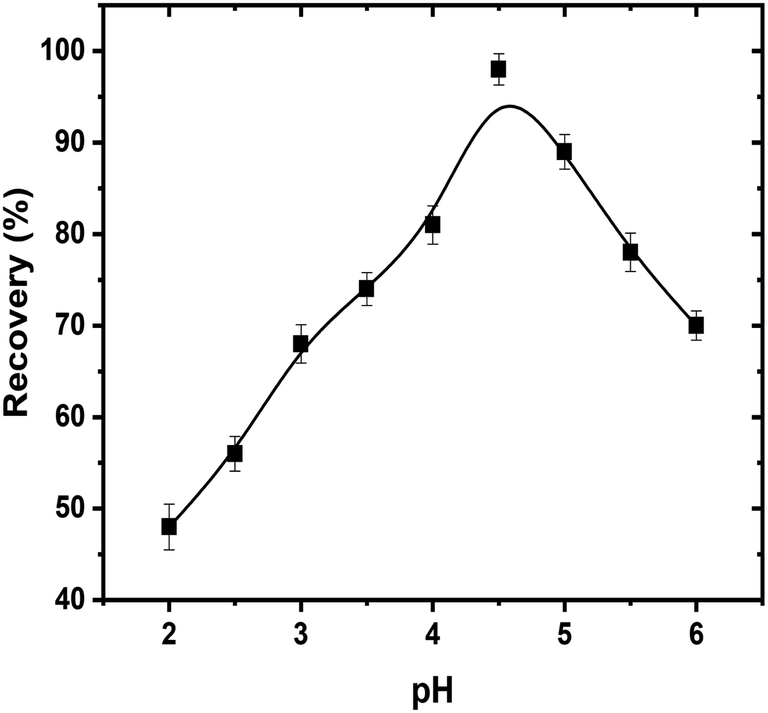
3.8.1. pH. The separation and extraction of uranyl ions by using surfactant micelles are usually executed after the complex formation with sufficient hydrophobicity. The pH is a critical parameter that plays a vital role not only in uranyl–enaminoester complex formation but also in the coacervation of the micelles. So, it was the first parameter, which optimized in the CPE process. The effect of pH upon the recovery of uranyl ions was investigated in the pH range from 2 to 8. As exhibited in Fig. 4, the high extraction efficiency was obtained at pH 4.5. At lower pH, the protonation of the sp3 amino group in the enaminoester ligand hindered the complex formation, while above pH 4.5 the complex hydrolyzed resulting in the low efficiency in the extraction of uranyl ions. Owing to this behavior, 2.0 mL of acetate buffer solution pH 4.5 was selected as the optimum dosage for subsequent experiments.
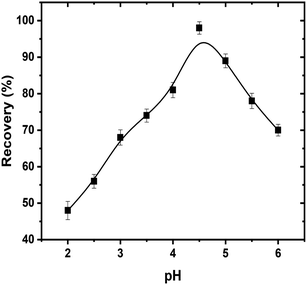 |
| | Fig. 4 Effect of hydrogen ion concentration on CPE extraction efficiencies of uranyl ions by using enaminoester ligand. Optimized conditions: volume of sample, 50 mL; pH, 4.5; UO22+, 100 ng mL−1; Triton X-114, 5.0 × 10−4 mol L−1; enaminoester, 5.0 × 10−3 mol L−1; KI, 0.1 mol L−1. | |
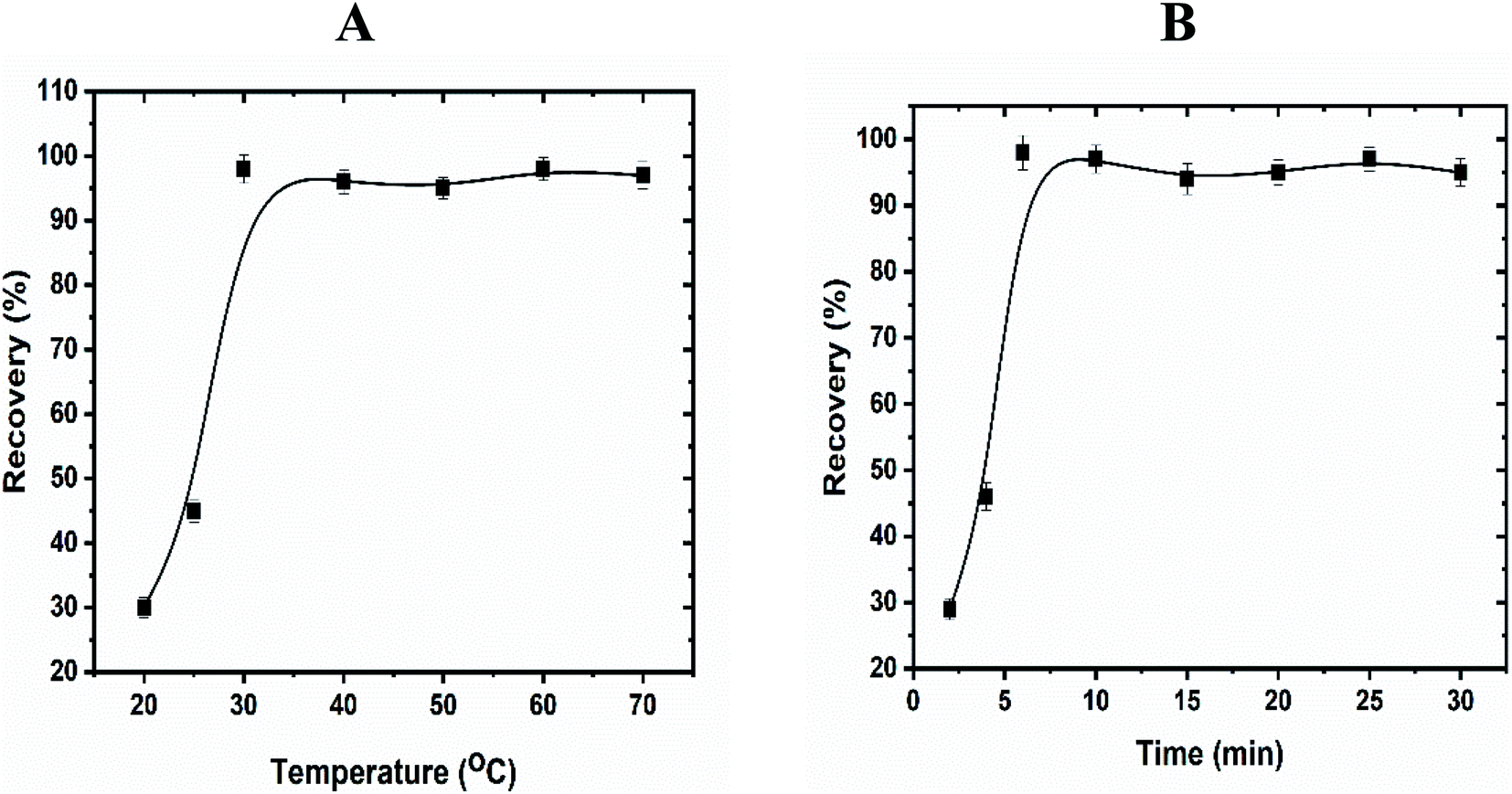
3.8.2. Effect of time, temperature, and salt. To achieve high separation and efficient pre-concentration of uranyl ions, the equilibrium temperature and incubation time are essential to be investigated. Thus, these parameters were tested carefully in the range 25–70 °C and 4–30 min and the results were presented in Fig. 5. Depending on the obtained absorbance values, the highest recoveries were achieved with increasing the temperature and incubation time till reached to 30 °C and 6 min. Furthermore, the extraction efficiency decreased dramatically above 30 °C, while the incubation time kept constant after 6 min. The most likely explanation of these results is that the rate of the interaction between enaminoester and uranyl ions is controlled by the extraction temperature. The decreased recoveries at low temperature may be attributed to the hydrogen bond formation between the carbonyl and amine groups of the functionalized ligand which inhibited the uranyl–enaminoester complex formation. On the other hand, at high temperatures, the micelles tend to coacervate, consequently, the viscosity of the surfactant-rich phase is sharply elevated and decreases the interaction between the metal ion and the chelating ligand. It was noted that the segregation of the micelles after the complex formation is the key point to achieve the CPE process. As reported in the literature, the addition of salt to the sample solution not only enhancement the micelles' segregation but also decreases the equilibrium temperature. Therefore, different inorganic salts (e.g.: KI, NaCl, Na2SO4, KCl, K2SO4.) were tested and potassium iodide (0.1 mol L−1) was found as the best electrolyte, which improves the recovery of the uranyl ions and the equilibrium temperature becomes to be 25 °C. So, the optimum temperature of 25 °C and 6 min time were chosen for further study after the addition of the KI.
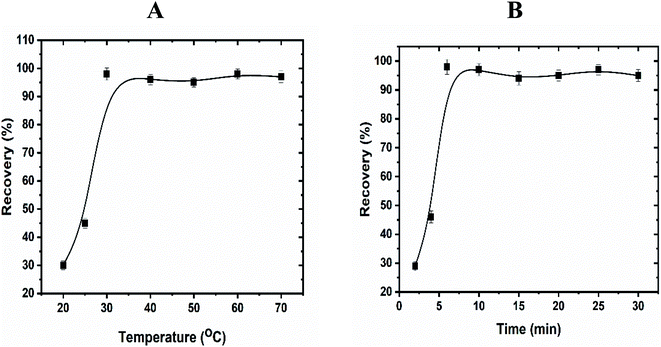 |
| | Fig. 5 Effect of equilibration temperature (A) and time (B) on the extraction of uranyl ions by using CPE. Optimized conditions: volume of sample, 50 mL; pH, 4.5; UO22+, 100 ng mL−1; Triton X-114, 5.0 × 10−4 mol L−1; enaminoester, 5.0 × 10−3 mol L−1. | |
3.8.3. Effect of surfactant. The selection of a proper surfactant (e.g.; cetyltrimethylammonium bromide, sodium lauryl sulfate, Triton X-100, Triton X-114) is pivotal for the extraction analysis which directly influences on the accuracy and precision of the final analytical results. The non-ionic surfactant (Triton X-114) was selected for CPE due to its high density that facilitates phase separation and its low cloud point temperature (25 °C). Furthermore, the effect of the Triton X-114 concentration on the separation of uranyl ions was investigated in the range from 1 × 10−5 to 1 × 10−2 mol L−1. As shown in Fig. S5 (ESI†), the absorbance of the samples elevated progressively with increasing Triton X-114 concentration up to 5 × 10−4 mol L−1 and remained almost constant beyond this concentration. A possible explanation, at low concentration, might be due to the insufficient non-ionic surfactant to entrap the hydrophobic complex quantitatively. Consequently, with increasing the amount of the Triton X-114, the number of formed micelles increased rapidly. Therefore, a concentration of 5 × 10−4 mol L−1 was chosen as an optimum concentration of Triton X-114 to determine uranyl ions in the samples.
3.8.4. Effect of enaminoester concentration. To achieve the effective complexation between the uranyl ions with the enaminoester ligand and consequently high extraction efficiency; a proper amount of complexing agent should be prudently utilized. Therefore, the effect of various quantities of enaminoester on the sensitivity of the extraction was examined in a range of 0.6–7.0 mmol L−1. As can be seen in Fig. S6 (ESI†), the recovery percentage of uranyl ions increased gradually with the chelating agent concentration increment until 5.0 × 10−3 mol L−1 and then slightly decreased. The lowering in extraction efficiency, at higher concentrations, may be attributed to the self-aggregation of ion-pair reagent.15 As a result, a concentration of 5.0 × 10−3 mol L−1 enaminoester was applied for the subsequent experiments.
3.8.5. Back extraction. To restore the uranyl ions from the complex, nitric acid, hydrochloric acid, and sodium hydroxide were tested for extracting these ions only from the surfactant-rich phase that contains uranyl–β-enaminones complex. It is worth mentioning that the complete elution of the proposed ions was accomplished via using hydrochloric acid (0.1 mol L−1).
3.8.6. Effect of concomitants. A serious problem that hindered the determination of uranyl ions by different techniques is the matrix effect. Generally, the influence of the matrix may be introduced from ions of the sample matrix or by the sample preparation process. Therefore, the relative extraction and separation of the target ion are also required for direct detection. For this reason, the effect of different cations and anions on the determination of the uranyl ion was studied by the proposed method. Briefly, under experimental condition, 50 mL of an aliquot aqueous solution (50 ng mL−1) and other interfering ions (e.g.; Na+, K+, Ca2+, Al3+, Fe3+, Cu2+, Zn2+, Cd2+, Pb2+, Gd3+, Sm3+, Th4+, Cl−, SO42−, PO43−) with known concentrations were taken into a centrifuge tube and followed by CPE procedure. As shown in Table 3, there is no significant effect from the diverse metal ions on the quantitative determination of uranyl ions. Furthermore, the tolerance limit, the ratio of the maximum concentration of the diverse metal ions to the uranyl ions, was tested at a 5.0% error in the analytical signals. It is worth mentioning that the tolerance limits were established at, diverse metal ion/uranyl ion ratios, 1000 for Gd3+, Sm3+, Th4+, Na+, K+ and 250 for Cu2+, Zn2+, Cd2+, Ca2+, Al3+, Fe3+, Pb2+, Cl−, SO42− and PO43−. Therefore, the CPE method can easily be applied for the determination of uranyl ions in different real samples.
Table 3 Influence of interfering ions on the determination of the uranyl ions
| Interfering ion |
Tolerance limit |
Recovery (%) |
| Gd3+ |
1000 |
99.2 |
| Sm3+ |
1000 |
98.8 |
| Th4+ |
1000 |
97.9 |
| Na+ |
1000 |
98.9 |
| K+ |
1000 |
99.1 |
| Cu2+ |
250 |
98.6 |
| Zn2+ |
250 |
98.9 |
| Cd2+ |
250 |
99.3 |
| Ca2+ |
250 |
98.4 |
| Al3+ |
250 |
98.6 |
| Fe3+ |
250 |
99.4 |
| Pb2+ |
250 |
98.7 |
| Cl− |
250 |
97.9 |
| SO42− |
250 |
98.1 |
| PO43− |
250 |
98.4 |
3.8.7. Analytical figure of merit. Under optimized conditions, the analytical performances (e.g.; calibration linear range, reproducibility, repeatability, the limit of detection,… etc.) of the proposed CPE for the detection of uranyl ions were performed and summarized in Table S3 (ESI†). Good linearity (R2 = 0.996, N = 9) was noticed over the concentration range from 3 ng mL−1 to 250 ng mL−1, and the linear regression equation was expressed as (Y = 0.0024X + 0.0064), where X is the uranyl concentration and Y is the analytical signal. Furthermore, the limit of detection (LOD) and limit of quantification (LOQ), calculated as three and ten times the standard deviation of the blank signals divided by the calibration slope, were 0.5 ng mL−1 and 1.6 ng mL−1, respectively. In addition, the precision of the proposed method in terms of reproducibility and repeatability, expressed as relative standard deviation (RSD), were found to be 2.4% for reproducibility and 1.9% for repeatability. Depending on the initial volume and final surfactant-rich phase volume, the pre-concentration factor was observed at 63. Additionally, the proposed method exhibited satisfactory sensitivity and selectivity superior to previous studies concerning the determination and quantification of uranyl ions (Table 4).
Table 4 Comparative data from some recent studies on CPE and our proposed technique for uranyl separation
| Target ion |
Chelating agent |
Surfactant |
Linear range |
LOD |
Repeatability (RSD%) |
Enrichment factor |
Application |
Ref. |
| Uranium(IV) |
4-(2-Pyridylazo)resorcinol |
Triton X-114 |
100–750 μg L−1 (U(IV)) |
17.03 μg L−1 (U(IV)) |
6.84 (U(IV)) |
11.81 (U(IV)) |
Tap water and standard mix |
15 |
| Vanadium(V) |
50–600 μg L−1 (V(V)) |
5.51 μg L−1 (V(V)) |
2.43 (V(V)) |
15.0 (V(V)) |
| Uranium(VI) |
Dibenzoylmethane |
Triton X-114 |
15–300 ng mL−1 |
11 ng mL−1 |
3.7 |
62 |
Water sample and reference material |
16 |
| Uranium(VI) |
Cyanex-301 |
Triton X-100 |
5–200 ng mL−1 |
0.57 ng mL−1 |
2.6 |
10 |
Different water sample |
4 |
| Uranium(VI) |
Trioctylphosphine oxide and N,N,N,N-tetraoctyldiglycolamide |
Triton X-114 |
15–1000 ng mL−1 (U(VI)) |
— |
1–3 (U(VI)) |
37.0 (U(VI)) |
Different water sample |
5 |
| Thorium(IV) |
10–1000 ng mL−1 (Th (IV)) |
2–5 (Th (IV)) |
43.6 (Th (IV)) |
| Uranium(VI) |
2-(5-Bromo-2-pyridylazo)-5-(diethylamino)-phenol |
Triton X-114 |
1.1–18.1 mg L−1 |
0.15 mg L−1 |
1.8–2.3 |
50.4 |
Natural water |
42 |
| Uranium(VI) |
Alizarin red S |
Triton X-114 |
0.01–3 mg L−1 |
2.0 μg L−1 (U(VI)) |
— |
20 |
Water samples |
17 |
| Zirconium(II) |
0.80 μg L−1 (Zr(II)) |
| Uranium(VI) |
Diglycolamide-capped quantum dot |
Triton X-114 |
1.0–100 ng mL−1 |
0.03 ng mL−1 |
≤5 |
— |
Natural water samples |
43 |
| Uranium(VI) |
Pyrocatechol violet |
Triton X-114 and cetyl trimethyl ammonium bromide |
0.20–10.00 ng mL−1 |
0.06 ng mL−1 |
3.0 |
14.3 |
Water samples |
44 |
| Uranium(VI) |
P,P-Di(2-ethylhexyl)methanediphosphonic acid |
Cetyl trimethyl ammonium bromide |
45–1000000 ng L−1 |
3 ng L−1 |
— |
62 |
Drinking water and wastewaters |
1 |
| Uranium(VI) |
4-(2-Pyridylazo)resorcinol |
Triton X-114 and cetyl trimethyl ammonium bromide |
2–30 μg |
2 μg |
3.9 |
— |
Plant samples |
45 |
| Uranium(VI) |
3-Mercaptopropionylamidoxime functionalized gold nanoparticles |
Triton X-114 and cetyl trimethyl ammonium bromide |
2–100 ng mL−1 |
0.3 ng mL−1 |
≤4 |
99 |
Water samples |
46 |
| Uranium(VI) |
Celestine blue |
Triton X-114 |
50–1500 ng mL−1 |
14.20 ng mL−1 |
3.25–4.16 |
42 |
Water samples and rock including radioactive elements |
19 |
| Uranyl ion(VI) |
Enaminoester |
Triton X-114 |
3–250 ng mL−1 |
0.5 ng mL−1 |
1.9 |
63 |
Different rock samples |
Present work |
3.9. Application
To assess the practical applicability and accuracy of the proposed method, different rock specimens were analyzed for uranyl determination under the optimized conditions. Moreover, each sample was spiked with different concentrations of uranyl standard solution directly to perform the recovery test. As seen in Table 5, the obtained extraction recoveries were in the range of 96.8% to 99.9%, which proves the capability of the proposed method for environmental samples with high precision and accuracy. Furthermore, the F-test and t-Student test at 95% confidence limits emphasized that there is no significant difference between the analysis results of the proposed method and the ICP-AES method.
Table 5 Detection of UO22+ in different real samples by the proposed CPE technique (n = 6)
| Samples |
Added (μg L−1) |
Found (μg L−1) |
Recovery of the proposed method (%) |
F-Test |
t-Test |
| Proposed |
ICP-AES |
| Sample 1 |
0 |
0.2 ± 0.12 |
0.18 ± 0.1 |
— |
1.44 |
1.3 |
| 5 |
4.98 ± 0.19 |
4.99 ± 0.15 |
96.1 |
1.6 |
0.9 |
| 10 |
9.97 ± 0.21 |
9.89 ± 0.16 |
97.7 |
1.7 |
1.4 |
| Sample 2 |
0 |
— |
— |
— |
— |
— |
| 5 |
4.96 ± 0.18 |
4.84 ± 0.2 |
99.2 |
1.2 |
1.2 |
| 10 |
9.89 ± 0.2 |
9.96 ± 0.15 |
98.9 |
1.7 |
1.4 |
| Sample 3 |
0 |
0.18 ± 0.12 |
0.21 ± 0.1 |
— |
1.4 |
0.9 |
| 5 |
5.12 ± 0.1 |
5.2 ± 0.13 |
98.8 |
1.69 |
1.3 |
| 10 |
10.1 ± 0.15 |
10.16 ± 0.14 |
99.2 |
1.14 |
1.6 |
| Sample 4 |
0 |
— |
— |
— |
— |
— |
| 5 |
4.99 ± 0.13 |
4.96 ± 0.1 |
99.8 |
1.69 |
1.4 |
| 10 |
10.1 ± 0.15 |
9.99 ± 0.12 |
101 |
1.56 |
1.2 |
| Sample 5 |
0 |
0.23 ± 0.14 |
0.2 ± 0.1 |
— |
1.96 |
1.4 |
| 5 |
5.1 ± 0.22 |
5.1 ± 0.2 |
97.5 |
1.2 |
0.9 |
| 10 |
10.2 ± 0.15 |
10.1 ± 0.1 |
99.7 |
2.25 |
1.3 |
In addition, the developed method was also tested for selective separation and detection of UO22+, Th4+, Gd3+, and Sm3+ in various synthetic mixtures (Table 6). High quantitative extraction of uranyl ions was obtained from other metal ions, which confirmed the reliability of the proposed method for the specific extraction of UO22+ from other metal ions.
Table 6 Separation of UO22+ (10 μg) from different synthetic mixtures using CPE (n = 6)
| Synthetic mixture |
Concentration of uranyl found (μg) |
Extraction (%) |
RSD (%) |
| UO22+ (10 μg), Gd3+ (100 μg), Sm3+ (100 μg), and Th4+ (100 μg) |
9.9 ± 0.05 |
99 |
2.1 |
| UO22+ (10 μg), Cd2+ (100 μg), Ni2+ (100 μg), Pb2+ (100 μg), Al3+ (100 μg), Zn2+ (100 μg), Cu2+ (100 μg), Fe3+ (100 μg), Mg2+ (100 μg), Na+ (1000 μg), and K+ (1000 μg) |
9.84 ± 0.1 |
98.4 |
1.9 |
| UO22+ (100 μg), Cl− (1000 μg), NO2− (1000 μg), SO42− (100 μg), CH3COO− (1000 μg), PO43− (100 μg), NO3− (1000 μg), and HCO3− (1000 μg) |
9.9 ± 0.1 |
99 |
2.8 |
4. Conclusion
In summary, a developed CPE using β-enaminoester chelating agent and Triton X-114 has been shown to be efficient for the selective separation and preconcentration of uranyl ions in presence of some lanthanide metal. Under the optimum conditions, the complexation between the chromogenic agent and uranyl ions takes place in an acidic medium at pH 4.5, which is a prerequisite for achieving a good selectivity and complete recovery of uranyl ions. Moreover, the mechanism of this reaction has been confirmed via the DFT calculations (e.g.; geometry optimization, reactivity, and MEP). Furthermore, from the experimental results, the molar ratio of enaminoester to uranyl ion was deduced by using molar ratio and Job methods. In addition, the experimental results demonstrated that the CPE not only has good sensitivity in the range of 3–250 ng mL−1 with the low detection limit (0.5 ng mL−1) but also increases the enrichment factor (63) of the target analyte. For these reasons, the utility of the proposed technique was successfully tested for detection and monitoring of uranyl ions in different real samples, satisfactory results in terms of the accuracy (96.8% to 99.9%) and the precision (RSD ≤ 4%) were noted.
Conflicts of interest
There are no conflicts to declare.
References
- C. Labrecque, S. Potvin, L. Whitty-Léveillé and D. Larivière, Talanta, 2013, 107, 284–291 CrossRef CAS PubMed.
- S. Gao, T. Sun, Q. Chen and X. Shen, J. Hazard. Mater., 2013, 263, 562–568 CrossRef CAS PubMed.
- J. Li, G. Li and Q. Han, Spectrochim. Acta, Part A, 2016, 169, 208–215 CrossRef CAS PubMed.
- Z. F. Akl, J. Radioanal. Nucl. Chem., 2018, 315, 21–28 CrossRef CAS.
- A. Saha, S. B. Deb, A. Sarkar, M. K. Saxena and B. Tomar, RSC Adv., 2016, 6, 20109–20119 RSC.
- S. L. Ferreira, J. B. de Andrade, A. K. Maria das Graças, M. d. G. Pereira, V. A. Lemos, W. N. dos Santos, F. de Medeiros Rodrigues, A. S. Souza, H. S. Ferreira and E. G. da Silva, J. Hazard. Mater., 2007, 145, 358–367 CrossRef CAS PubMed.
- G. Blanchet-Chouinard and D. Larivière, Appl. Radiat. Isot., 2021, 168, 109549 CrossRef CAS PubMed.
- M. A. Bezerra, U. M. Ferreira da Mata Cerqueira, S. L. C. Ferreira, C. G. Novaes, F. C. Novais, G. S. Valasques and B. Novaes da Silva, Appl. Spectrosc. Rev., 2021, 1–15 CrossRef.
- F. Genç, N. P. Milcheva, D. G. Hristov and K. B. Gavazov, Chem. Pap., 2020, 74, 1891–1901 CrossRef.
- P. Samaddar and K. Sen, J. Ind. Eng. Chem., 2014, 20, 1209–1219 CrossRef CAS.
- R. Karmakar and K. Sen, J. Radioanal. Nucl. Chem., 2019, 322, 57–66 CrossRef CAS.
- R. Kachangoon, J. Vichapong, Y. Santaladchaiyakit and S. Srijaranai, Microchem. J., 2020, 152, 104377 CrossRef CAS.
- Z. F. Akl and M. Hegazy, J. Environ. Chem. Eng., 2020, 8, 104185 CrossRef CAS.
- J. L. M. Viana, A. A. Menegário and A. H. Fostier, Talanta, 2021, 122119 CrossRef CAS PubMed.
- P. Pathak, S. Ansari, S. Kumar, B. Tomar and V. Manchanda, J. Colloid Interface Sci., 2010, 342, 114–118 CrossRef CAS PubMed.
- F. Shemirani, R. Rahnama Kozani, M. Reza Jamali, Y. Assadi and S. Mohammad Reza Milani, Sep. Sci. Technol., 2005, 40, 2527–2537 CrossRef CAS.
- J. B. Ghasemi, B. Hashemi and M. Shamsipur, J. Iran. Chem. Soc., 2012, 9, 257–262 CrossRef CAS.
- H. Liang, Q. Chen, C. Xu and X. Shen, Sep. Purif. Technol., 2019, 210, 835–842 CrossRef CAS.
- H. İ. Ulusoy, J. Radioanal. Nucl. Chem., 2014, 302, 497–504 CrossRef CAS.
- J. Greenhill, Chem. Soc. Rev., 1977, 6, 277–294 RSC.
- E. K. Varbanova, P. A. Angelov and V. M. Stefanova, Talanta, 2016, 160, 389–399 CrossRef CAS PubMed.
- E. Varbanova, P. Angelov, K. Simitchiev, L. Kaynarova and V. Stefanova, Bulg. Chem. Commun., 2017, 49, 49–54 Search PubMed.
- A. Abdallah, M. R. El-kholany, A. Molouk, T. A. Ali, A. El-Shafei and M. E. Khalifa, RSC Adv., 2021, 11, 30771–30780 RSC.
- C. M. Kascheres, J. Braz. Chem. Soc., 2003, 14, 945–969 CrossRef CAS.
- J. Dabrowski and K. Kamienska-Trela, J. Am. Chem. Soc., 1976, 98, 2826–2834 CrossRef CAS.
- L. Kozerski, K. Kamienska-Trela, L. Kania and W. Von Philipsborn, Helv. Chim. Acta, 1983, 66, 2113–2128 CrossRef CAS.
- A. S. Amin, Arabian J. Chem., 2016, 9, S326–S333 CrossRef CAS.
- A. S. Amin, Microchem. J., 2000, 65, 261–267 CrossRef CAS.
- M. Rana, H.-J. Cho, T. K. Roy, L. M. Mirica and A. K. Sharma, Inorg. Chim. Acta, 2018, 471, 419–429 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- X. Wu and A. K. Ray, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 085403 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413 CrossRef.
- J. Janak, Phys. Rev. B: Condens. Matter Mater. Phys., 1978, 18, 7165 CrossRef CAS.
- R. G. Parr, in Horizons of quantum chemistry, Springer, 1980, pp. 5–15 Search PubMed.
- A. M. Younis, M. M. El-Gamil, T. H. Rakha and G. M. Abu El-Reash, Appl. Organomet. Chem., 2021, 35, e6250 CAS.
- M. A. Abozeid, M. R. El-Kholany, L. A. Abouzeid, A. R. H. Abdel-Rahman and E. S. I. El-Desoky, J. Heterocycl. Chem., 2019, 56, 2922–2933 CrossRef CAS.
- M. A. Abozeid, M. R. El-Kholany, A.-R. H. Abdel-Rahman and E.-S. I. El-Desoky, Int. J. Mod. Org. Chem., 2018, 5, 1–11 CAS.
- A. M. Younis, T. H. Rakha, M. M. El-Gamil and G. M. A. El-Reash, J. Mol. Struct., 2021, 1245, 131110 CrossRef CAS.
- A. A. Gouda and A. S. Amin, Spectrochim. Acta, Part A, 2014, 120, 88–96 CrossRef CAS PubMed.
- A. E. Harvey and D. L. Manning, J. Am. Chem. Soc., 1950, 72, 4488–4493 CrossRef CAS.
- H. S. Ferreira, M. de Almeida Bezerra and S. L. C. Ferreira, Microchim. Acta, 2006, 154, 163–167 CrossRef CAS.
- A. Saha, T. Debnath, S. Neogy, H. N. Ghosh, M. K. Saxena and B. S. Tomar, Sens. Actuators, B, 2017, 253, 592–602 CrossRef CAS.
- T. Madrakian, A. Afkhami and A. Mousavi, Talanta, 2007, 71, 610–614 CrossRef CAS PubMed.
- C. Mukhopadhyay, M. Suba, D. Sivakumar, K. Dhamodharan and R. S. Rao, J. Radioanal. Nucl. Chem., 2019, 322, 743–750 CrossRef CAS.
- A. Saha, S. Neogy, D. R. M. Rao, S. B. Deb, M. K. Saxena and B. S. Tomar, Microchim. Acta, 2019, 186, 1–10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00274d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Adel M. Youins
*,
Adel M. Youins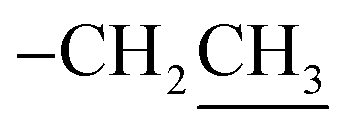 ), 2.51 (s, 3H, OCH3), 4.09 (m, 2H,
), 2.51 (s, 3H, OCH3), 4.09 (m, 2H, 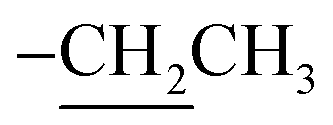 ), 5.29 (s, 1H, H-4), 6.94–7.79 (m, 13H, ArH), 8.33, 8.57, 9.89 (3H, 2NH, OH); ESI-MS (m/z) calcd for C29H25N3O5Na (M + Na)+ 518.50, found 518.20; anal. calcd for C29H25N3O5 (495.53): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.15; H, 5.22; N, 8.75%.
), 5.29 (s, 1H, H-4), 6.94–7.79 (m, 13H, ArH), 8.33, 8.57, 9.89 (3H, 2NH, OH); ESI-MS (m/z) calcd for C29H25N3O5Na (M + Na)+ 518.50, found 518.20; anal. calcd for C29H25N3O5 (495.53): C, 70.29; H, 5.09; N, 8.48%. Found: C, 70.15; H, 5.22; N, 8.75%.