
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2-Assisted asymmetric hydrogenation of prochiral allylamines†
Tamara M. de Winter ,
Jaddie Ho,
Christopher J. Alridge and
Philip G. Jessop*
,
Jaddie Ho,
Christopher J. Alridge and
Philip G. Jessop*
Department of Chemistry, Queen's University, Kingston, ON K7L 3N6, Canada. E-mail: jessop@queensu.ca
First published on 28th February 2022
Abstract
A new methodology for the asymmetric hydrogenation of allylamines takes advantage of a reversible reaction between amines and carbon dioxide (CO2) to suppress unwanted side reactions. The effects of various parameters (pressure, time, solvent, and base additives) on the enantioselectivity and conversion of the reaction were studied. The homogeneously-catalyzed asymmetric hydrogenation of 2-arylprop-2-en-1-amine resulted in complete conversion and up to 82% enantiomeric excess (ee). Added base, if chosen carefully, improves the enantioselectivity and chemoselectivity of the overall reaction.
Introduction
Optically active amines are used as pharmaceuticals, agrochemicals and resolving agents or chiral auxiliaries.1–6 Many efforts have been directed towards the enantioselective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN double bonds for the synthesis of chiral amines.1,3,4,7–13
C and CN double bonds for the synthesis of chiral amines.1,3,4,7–13
We sought an asymmetric hydrogenation of prochiral allylamines, in the expectation that it would represent a more direct, efficient and greener synthesis of chiral amines than current asymmetric hydrogenations of N-protected allylamines. Until today, the direct hydrogenation of unprotected allylamines has been largely ignored; the few examples are shown in Scheme 1. Botteghi et al.14 reported that the hydrogenation gave low yields due to the unwanted hydrogenolysis of the C–N bond. Fahrang et al.15 strategically hydrogenated the hydrogen chloride salt of their allylamine but did not comment on yield or purity of the product. However, both groups reported only moderate enantioselectivity. Yamashita and Yamano16 screened multiple Josiphos ligands to find one with good enantioselectivity for the hydrogenation of a precursor of Ramelteon, a melatonin receptor agonist.
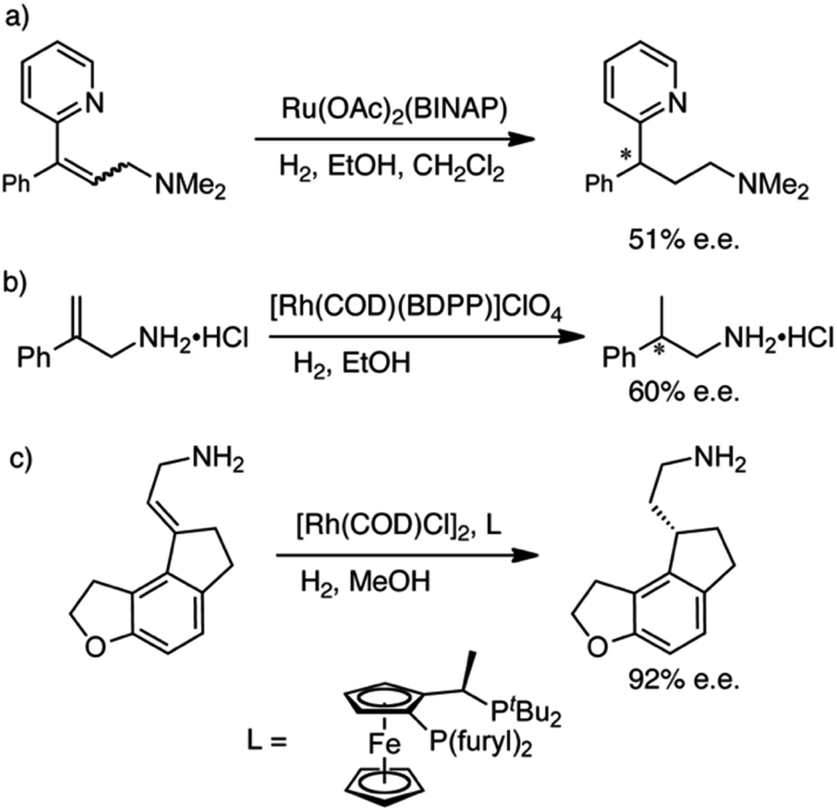 | ||
| Scheme 1 Literature examples of the hydrogenation of allyamines. (a) Botteghi et al.14 (b) Fahrang et al.15 (c) Yamashita and Yamano.16 | ||
We hypothesized that CO2 could act as an in situ protecting group in a way that protects the amine from undesired side reactions. This could potentially replace the N-acetyl protecting group that is currently used for asymmetric hydrogenation of protected allylamines. The CO2 would reversibly convert the allylamine substrate and/or the chiral amine product into a carbamate or carbamic acid (solid arrows in Scheme 2), which would circumvent additional steps of amine protection and deprotection, ultimately giving a more economical synthesis. In separate reports by Chatterjee et al.17 and Xie et al.18 CO2 was used in the hydrogenation of nitriles and imines to protect the desired amine products, by the formation of a carbamic acid, from undergoing undesired further reactions. Thus the carbamic acid acts as an in situ protecting group for the kinetic product during the hydrogenation.17,18 Fortunately, the reaction of allylamines with CO2 to form carbamic acids or carbamate anions is known, but in the context of synthesizing cyclic carbamate esters.19–25
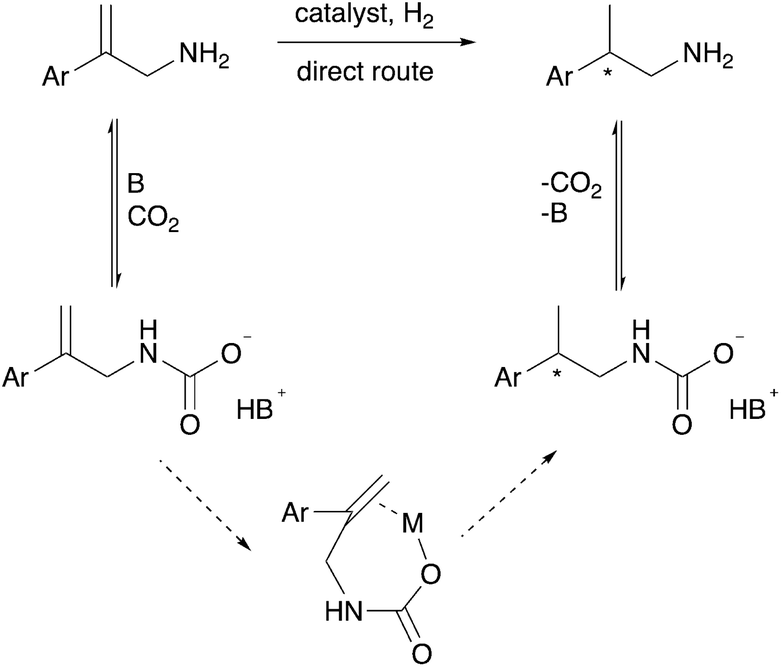 | ||
| Scheme 2 Upper route with solid arrows: the yield of the direct hydrogenation could be increased by the starting allylamine and/or the product being stabilized as the carbamate, even though the hydrogenation step itself involves the allylamine rather than the carbamate. Lower route with dotted arrows: alternatively, the yield and enantioselectivity could both be improved by the allylcarbamate binding to the metal centre, allowing chelation during the hydrogen transfer step. | ||
In addition to serving as a protecting group, the carbamic acid or carbamate anion produced by the reaction of CO2 with the amine might serve as a better metal-binding functional group (lower part of Scheme 2), allowing chelation in the hydrogenation transition state as occurs when unsaturated carboxylic acids are asymmetrically hydrogenated.26 The olefin binding step in the catalytic cycle, in which the CC double bond is bound to the metal centre prior to insertion into the M–H bond, would thereby become an intramolecular rather than intermolecular step, which would quite feasibly enhance enantioselectivity.
To explore these two intriguing hypotheses, we chose to study the asymmetric hydrogenation of a primary allylamine with and without CO2, and with and without added base. The option of adding a base was included in order to promote the formation of carbamate anions rather than carbamic acids.
Results and discussion
Five commercially available catalysts (Scheme 3) were chosen based upon their ability to asymmetrically hydrogenate prochiral unsaturated carboxylic acids. The catalysts were diacetato[(R)-(+)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl]ruthenium(II), 1,26 dichloro[(S)-(−)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl]ruthenium(II), 2, diacetato{(R)-(+)-2,2′-bis[di(3,5-xylyl)phosphino]-1,1′-binaphthyl}ruthenium(II), 3, diacetato{(R)-(+)-5,5′-bis[di(3,5-xylyl)phosphino]-4,4′-bi-1,3-benzodioxole} ruthenium(II), 4,27 and (−)-4,5-bis[(2R,5R)-2,5-dimethylphospholanyl](1,2-dimethyl-1,2′-dihydropyridazine-3,6-dione)(1,5-cyclooctadiene)rhodium(I) tetrafluoroborate, 5.10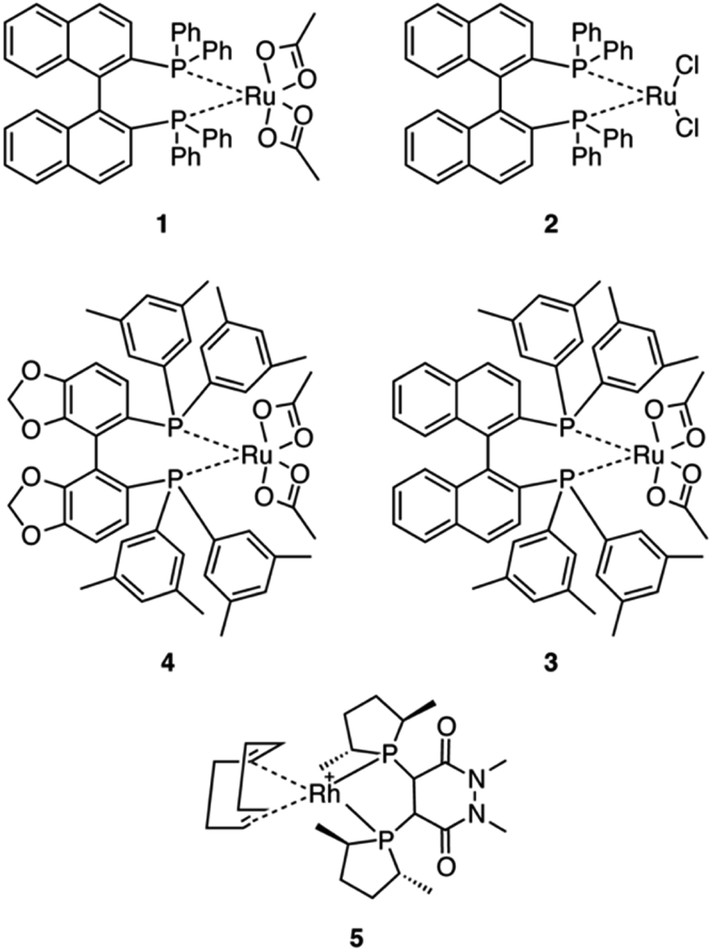 | ||
| Scheme 3 The catalysts initially tested for the asymmetric hydrogenation of prochiral allylamines. | ||
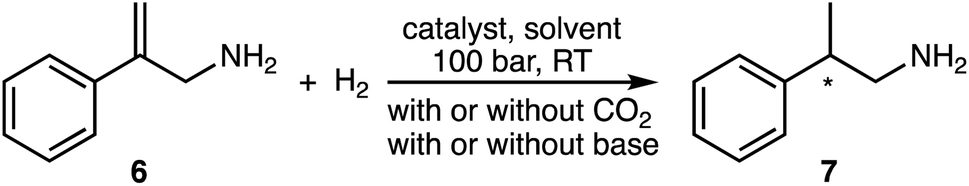
The prochiral allylamine substrate that was chosen for initial screening of catalysts and conditions was 2-phenylprop-2-en-1-amine, 6 (Scheme 4). Four hydrogenation conditions were investigated; each catalyst listed above was tested with (a) only H2, (b) H2 and base, (c) H2 and CO2, and lastly (d) H2, CO2(g) and base. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was the base chosen as it is more basic than the allylic amine. Initial experiments were conducted at 24 h reaction time in order to maximize the chances that at least one catalyst would be able to give a significant yield.
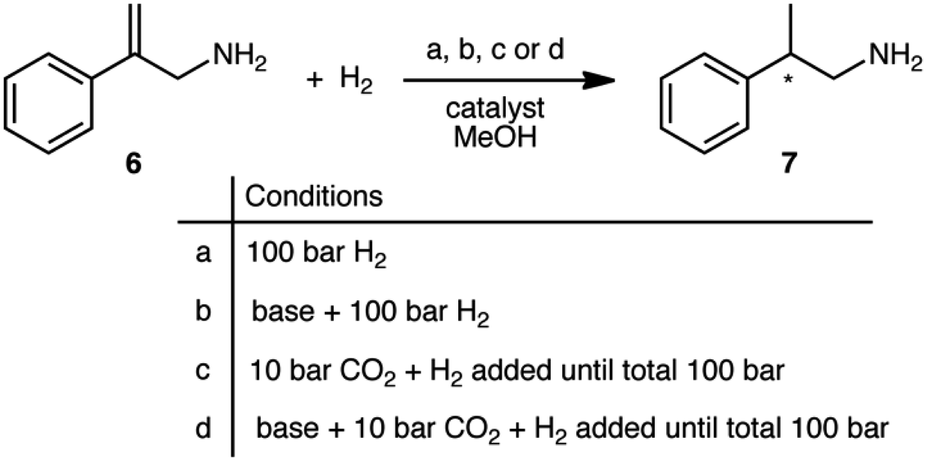 | ||
| Scheme 4 The four reaction conditions used for the asymmetric hydrogenation scheme of 2-phenylprop-2-en-1-amine, 6. | ||
The results show that the conditions greatly affect the asymmetric hydrogenation of 6 (Table 1, 24 h). With solely H2, all catalysts produced 2-phenylpropan-1-amine, 7, in low to moderate yields and enantioselectivity (ee), consistent with the findings of Botteghi et al.14 Catalyst 1 gave the highest yield, 72%, and catalyst 5 gave the best ee, 74%.
|
|||
|---|---|---|---|
| Additive | Cat. | % Yield (% ee) | |
| 24 h | 14–15 h | ||
| a Experiments were done in triplicate and at RT in a 160 mL stainless steel vessel containing 10 mg 6 and 2 mL methanol in a 1 dram vial under 100 bar total pressure. Conversions for all reactions above were >95% and the experimental error for % yield and % ee were ±10 and ±4, respectively. Catalysts 2 and 5 produced (S)-7, while catalysts 1, 3, and 4 produced (R)-7. Yields are 1H NMR values measured with an internal standard (1,3,5-trimethoxybenzene). Enantiomeric excess was determined by HPLC.b 100 bar H2.c 100 bar H2, 1 eq. DBU added (relative to 6).d 10 bar CO2(g) added, followed by enough H2(g) to bring the total pressure to 100 bar.e 10 bar CO2(g) added, followed by enough H2(g) to bring the total pressure to 100 bar, 1 eq. DBU added (relative to 6). | |||
| Noneb | 1 | 72 (33) | 79 (33) |
| 2 | 32 (30) | 72 (31) | |
| 3 | 35 (48) | 66 (39) | |
| 4 | 38 (60) | 67 (57) | |
| 5 | 29 (74) | 57 (68) | |
| DBUc | 1 | 21 (50) | 48 (45) |
| 2 | 38 (52) | 60 (46) | |
| 3 | 38 (49) | 52 (42) | |
| 4 | 48 (49) | 58 (48) | |
| 5 | 54 (31) | 50 (26) | |
| CO2d | 1 | 64 (26) | 70 (23) |
| 2 | 85 (26) | 72 (25) | |
| 3 | 45 (36) | 82 (36) | |
| 4 | 56 (41) | 84 (37) | |
| 5 | 64 (77) | 84 (75) | |
| CO2 + DBUe | 1 | 68 (25) | 73 (25) |
| 2 | 96 (31) | 92 (25) | |
| 3 | 64 (31) | 69 (36) | |
| 4 | 50 (40) | 62 (49) | |
| 5 | 96 (75) | 94 (73) | |

The addition of DBU made minor improvements to the enantioselectivity of catalysts 1 and 2, but was otherwise unhelpful. The addition of CO2 without base increased the yield (Table 1) and the purity of the product (by suppressing side products, Fig. S4†) but not the enantioselectivity for catalysts 2–5. The addition of CO2 with base dramatically increased the yield with all catalysts except 4. The enantioselectivity of the hydrogenation was adversely affected for all catalysts except 5. The best result, with high yield (96%) and reasonable ee (75%), was obtained with catalyst 5 in the presence of both CO2 and base.
Following the positive results for the asymmetric hydrogenation at 24 h, the reaction time was investigated (Table 1, 14–15 h). With a decreased reaction time of 14–15, an increase in yield was observed for almost all catalysts and conditions, suggesting that extended reaction times allow the desired products to undergo further reactions giving unwanted products. However, enantioselectivities were not significantly changed by the decrease in reaction time. Even shorter reaction times give lower enantioselectivity (Table S2†).
The success of catalyst 5 suggests that Rh-based catalysts may be more suitable than classical Ru BINAP catalysts for the asymmetric hydrogenation of 2-phenylprop-2-en-1-amine, 6. This seems surprising if one considers allylamines to be close analogues of allylic alcohols, for which the classical Ru BINAP catalysts are known to be excellent hydrogenation catalysts.28 However, perhaps a better analogy would be to the β-ketoamines, for which cationic Rh complexes are better hydrogenation catalysts than the classical Ru BINAP complexes.29 In the proposed transition state for those hydrogenations, the amine group binds to the Rh centre and the CO double bond then undergoes Rh–H insertion leading to hydrogenation.29 A similar mechanism may operate for the asymmetric hydrogenation of allylamines, although it is worth noting that a DFT study of the mechanism for asymmetric isomerization of allylamines (for which Rh catalysts are again superior to Ru) shows that the nitrogen is not coordinated during the hydrogen transfer step.30
In light of the success of catalyst 5, three more Rh based catalysts were chosen from the catASium® family (Scheme 5) with the corresponding ligands (R,R)-Me-DUPHOS, 8, (R,R)-Me-BPE-Rh, 9,8 and 3,4-bis-[(R,R)-(2,5-dimethylphospholan-1-yl]maleic anhydride 10.8,31 Unfortunately, compared to catalyst 5, catalysts 8, 9 and 10 did not provide improved results, with catalyst 10 yielding similar results (Table S1†).
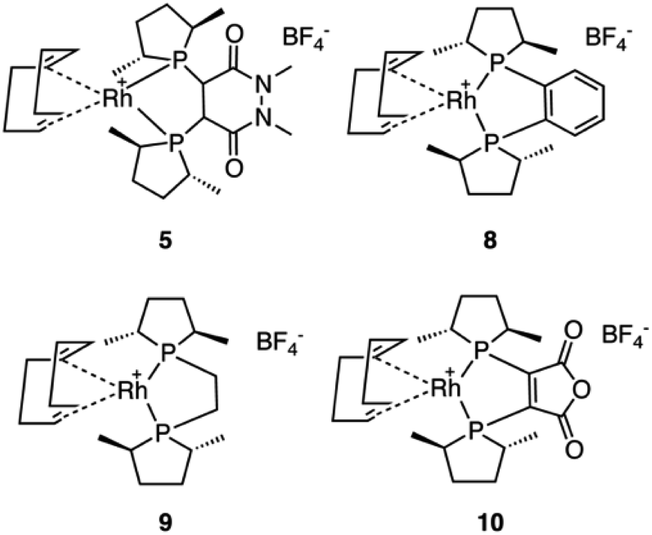 | ||
| Scheme 5 The four rhodium(I) catASium® catalysts applied to the asymmetric hydrogenation of 6. | ||
H2 pressure is known to affect hydrogenation enantioselectivity.26 To evaluate the effect of H2 pressure in the present system, a lower pressure was tested. The reaction time was increased to 24 h to compensate for the anticipated lower rate of reaction. Unfortunately, the lowered H2 pressure decreased the performance of catalyst 5 and caused no significant improvements with 8 and 9 (Table S1†).
Next, the effect of solvent on the reaction was examined (Table 2). For catalyst 8 it was reported that the best solvents for the asymmetric hydrogenations of α-aminomethylacrylates,26 ene-carbonates,3 β-acylamido acrylates,3 and enamides4 were isopropanol (IPA), methanol (MeOH), and tetrahydrofuran (THF). For this reason, the asymmetric hydrogenation of 6 in the presence of CO2 was tested in these solvents with catalysts 5 and 8 but no significant improvement was obtained relative to the results with catalyst 5 in MeOH.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
||||
|---|---|---|---|---|
| Additive | Cat. | % Yield (% ee) | ||
| MeOH | IPA | THF | ||
| a Experiments were done in triplicate and at RT in a 160 mL stainless steel vessel containing 10 mg 6 and 2 mL of the indicated solvent in a 1 dram vial under 100 bar total pressure. Reaction time was 14–15 h. Conversions for all reactions above were ≥93% and the experimental error for % yield and % ee were ±10 and ±4, respectively. Catalysts 5 and 8 produced (S)-7. Catalyst 10 produced (R)-7. Yields are 1H NMR values measured with an internal standard (1,3,5-trimethoxybenzene). Enantiomeric excess determined by HPLC.b 100 bar H2.c 100 bar H2, 1 eq. of base added (relative to 6).d 10 bar CO2(g) added, followed by enough H2(g) to bring the total pressure to 100 bar.e 10 bar CO2(g) added, followed by enough H2(g) to bring the total pressure to 100 bar, 1 eq. of base added (relative to 6).f Conversion% 70–76%.g Conversion% 83%. | ||||
| Noneb | 5 | 57 (68) | 65 (62) | 52 (3) |
| 8 | 66 (61) | 62 (64) | 52 (7) | |
| DBUc | 5 | 50 (26) | 73 (17) | 69 (4) |
| 8 | 71 (69) | 71 (25) | 58 (1) | |
| CyNMe2c | 5 | 58 (64) | 62 (44) | — |
| 8 | — | 60 (63) | — | |
| iPr2NEtc | 5 | 66 (59) | 56 (64) | — |
| 8 | — | 48 (65) | — | |
| CO2d | 5 | 84 (75) | 79 (60) | 40f (14) |
| 8 | 54 (65) | 83 (69) | 46 (43) | |
| CO2 + DBUe | 5 | 94 (73) | 96 (50) | 36f (22) |
| 8 | 72 (69) | 80 (70) | 31f (6) | |
| 10 | 69 (76) | — | — | |
| CO2 + CyNMe2e | 5 | 77 (71) | 80 (55) | — |
| 8 | — | 78 (72) | — | |
| 10 | 95 (71) | — | — | |
| CO2 + iPr2NEte | 5 | >99 (69) | 75 (72) | — |
| 8 | — | 83g (72) | — | |
| 10 | 85 (73) | — | — | |
Despite the complete conversion of 6 in all runs, yields were low in many instances and unidentified peaks were observed in the 1H NMR spectra. Even though the overall yields were found to be higher with the addition of DBU, we suspected that the use of DBU as the base may be leading to or assisting the decomposition of either the starting material or product. Therefore, we investigated the use of weaker bases (Table 2). N,N-Dimethylcyclohexylamine (CyNMe2), and N,N-diisopropylethylamine (iPr2NEt) were tested with catalysts 5 and 8 in the solvent that provided the best results for each (catalyst 5 with MeOH and IPA, and catalyst 8 with IPA). The best condition for catalyst 5 were found in the presence of H2, CO2, MeOH and DBU. However, CyNMe2 produced the cleanest reaction by 1H NMR spectroscopy with comparable enantioselectivity. For catalyst 8, the addition of CO2, IPA and iPr2NEt resulted in the highest yield (83%) and ee (72%). Catalyst 10 gave decent results in methanol with CO2 and the weaker bases, but the best overall result is still with catalyst 5 in the presence of CO2 and DBU. Using chiral bases (Scheme 6) caused modest improvements in the enantioselectivity with catalyst 8 but not catalyst 5 (Table S3†). The enantioselectivity was not affected by the chirality of the base, presumably because the chiral bases, in their cationic form, were not close enough to the catalytic centre to induce a chiral environment. Therefore, the success of these chiral bases at mildly improving the enantioselectivity is due to their weaker basicity rather than their chirality.
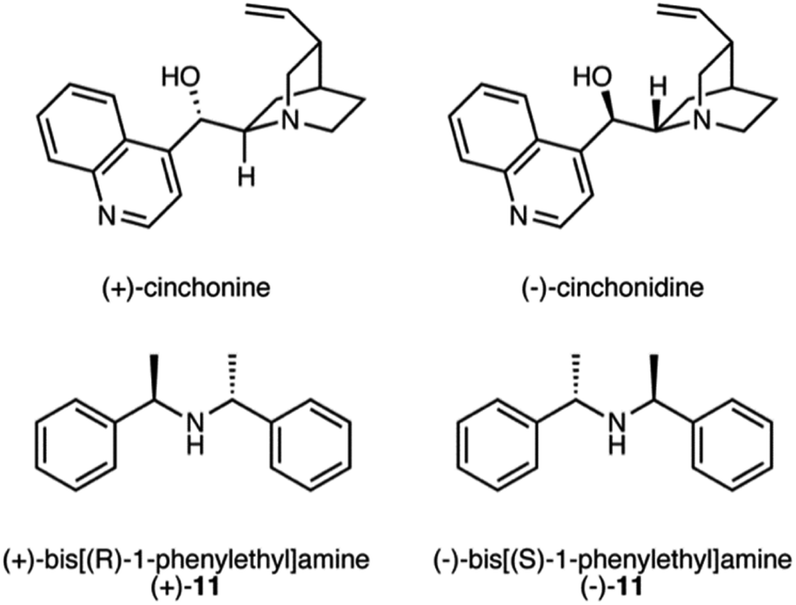 | ||
| Scheme 6 Chiral bases used in the asymmetric hydrogenation of 6. | ||
Using the best hydrogenation conditions (CO2 + DBU in MeOH for 24 h) with catalyst 5, the reaction was scaled to 650 mg to obtain an isolated yield of 416 mg (64%, Fig. S5†).
The asymmetric hydrogenation was also tested on three other allylamine substrates using catalysts 5, 8 and 10 (Scheme 7). For catalyst 5 the asymmetric hydrogenation was performed with CO2 and CyNMe2, whereas catalyst 8 was utilized with CO2 and (−)-11. For 10, both sets of conditions used for 5 and 8 were applied and found to be equally successful (Table 3).
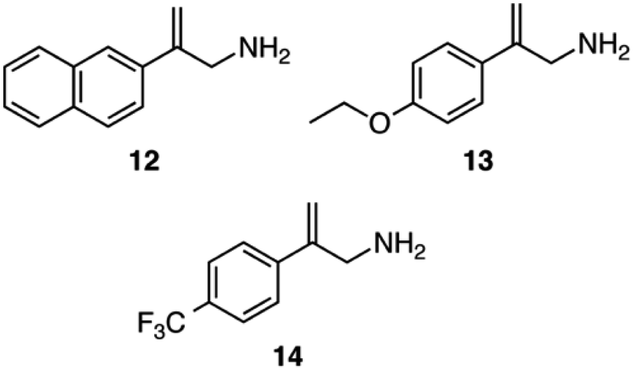 | ||
| Scheme 7 Three additional allylamines employed as substrates for asymmetric hydrogenation: 2-(naphthalene-2-yl)prop-2-en-1-amine, 12, 2-(4-ethoxyphenyl)prop-2-en-1-amine, 13, and 2-[4-(trifluoromethyl)phenyl]prop-2-en-1-amine, 14. | ||
|
||
|---|---|---|
| Cat. (base + solvent) | % Yield (% ee) | |
| 6 | 12 | |
| a Experiments were done in triplicate and at RT. Conversions for all reactions above was >95%, except as noted. The experimental error for yield% and ee% were ±10 and ±4, respectively. Reaction conditions: 160 mL stainless steel pressure vessel, 10 mg of allylamine, 10 bar CO2 followed by enough H2 pressure to bring the total pressure to 100 bar H2(g), 1 eq. base, 2 mL solvent in a 1 dram vial. The reaction was stopped after 6 h. Yields are 1H NMR values, internal standard was 1,3,5-trimethoxybenzene. Enantiomeric excess was determined by HPLC. Catalysts 5 and 8 produced (S)-7, while catalyst 10 produced (R)-7.b Conversion only 90–93%. | ||
| 5 (CyNMe2 + MeOH) | 94 (73) | 84 (51) |
| 8 ((−)-11 + IPA) | 88 (76) | 80 (67) |
| 10 (CyNMe2 + MeOH) | 95 (71) | 77 (62) |
| 10 ((−)-11 + MeOH) | 96 (71) | 85 (66) |
Changing the phenyl ring of substrate 6 to a larger naphthyl ring in substrate 12 lowered the enantioselectivity by about 10% (Table 3). Adding electron-donating and electron-withdrawing groups to the para position of substrate 6 affected both the yield and the enantioselectivity of the allylamine. Adding an electron-donating ethoxy group (substrate 13) increased the ee to 81–82%. However, adding an electron-withdrawing trifluoromethyl group on the para position (substrate 14) had the opposite effect where yields and enantioselectivity both decreased; the 1H NMR spectrum of the reaction mixture after the reaction appeared clean and showed that the reaction was incomplete after 6 h. While the amount of product from substrate 14 might improve if the reaction time were longer, the enantioselectivity is clearly poor for this substrate.
Conclusions
A new methodology has been developed for the asymmetric hydrogenation of allylamines. It was found that the Rh-based catASium® catalysts resulted in higher conversion and enantiomeric excess values than the Ru-binap based catalysts. Furthermore, by employing CO2 and an added base in the asymmetric hydrogenation of 2-phenylprop-2-en-1-amine, 6, a clean reaction was obtained, probably due to CO2 acting as a temporary protecting group for the amine functionality and ultimately increasing the yield of the reaction up to 94–96%. Nonetheless, the enantioselectivity of the reaction was not affected by the addition of CO2 and DBU. This demonstrates that the CO2 is not affecting the enantioselectivity-determining step and therefore the allylamine is not bound to the catalyst in the form of a carbamate ligand. The CO2 helps by acting as a protecting group and not by causing the allylamines to bind as carbamates to the metal centre.Four 2-arylprop-2-en-1-amines were asymmetrically hydrogenated with the best catalysts from the above study at their optimal reaction conditions. Allylamine 2-(4-ethoxyphenyl)prop-2-en-1-amine, with an electron donating group, was hydrogenated with the greatest enantioselectivity (82% ee) and good yield (93%).
These findings demonstrate that a direct asymmetric hydrogenation of prochiral allylamines, without prior derivatization or protection, is a viable strategy for preparing chiral amines. Further optimization of the catalyst and the conditions, including the beneficial effect of CO2, should be able to bring the enantioselectivity to industrially useable levels.
Experimental methods
Solvents were dried by standard distillation procedures before use. All reagents were purchased from chemical suppliers, Alfa Aesar, Sigma Aldrich, and Strem, and used as received unless otherwise specified. The four allylamines were synthesized as described in the ESI.† Glassware was dried in an oven at 110 °C before use. 1H NMR and 13C NMR spectra were recorded at 300 K on a Bruker AV-400 or AV-500 NMR spectrometer. Chemical shifts (δ) are expressed in ppm. Conversion and yield values were obtained through quantitative NMR spectroscopy, which was carried out using 1,3,5-trimethoxybenzene as the internal standard. Enantiomeric excess values were obtained by HPLC using Agilent Technologies 1260 Infinity with Chiralpak IA chiral column (25 cm × 0.46 cm i.d.) from Daicel. High resolution mass spectra (HRMS) ESI and EI were obtained on a Qstar XL QqTOF from Applied Biosystems/MDS Sciex.General racemic hydrogenation for allylamines
The allylamines were first hydrogenated with achiral catalysts in order to generate samples of the racemates for the development of instrumental methods capable of analyzing the enantiomeric mixture. The non-enantioselective hydrogenations were developed from a procedure by Hattori et al.32 The procedure below is the same for all substrates: the hydrogenation of the allylamines, Pd(5%)/CaCO3 was used. The hydrogenation of 2-phenylprop-2-en-1-amine, 6, is presented below and can be regarded as a general protocol for the procedure regardless of minor changes in the substrates.To a test tube, equipped with a magnetic stir bar, 2-phenylprop-2-en-1-amine (10 mg, 75.1 mmol) and catalyst (0.5 wt% of the weight of the substrate, 2.1 mg) was added. The test tube was sealed with a rubber septum and then evacuated. THF (1 mL) was added and then hydrogen was added via a syringe needle from a hydrogen-filled rubber balloon (1 atm). The reaction mixture was stirred at room temperature for 5–6 h. Upon completion, the catalyst was removed by filtration through a diatomaceous earth plug, after which the solvent was removed from the product by rotatory evaporation.
Asymmetric hydrogenation for allylamines
The following procedure was used to asymmetrically hydrogenate the allylamines. The study was completed with a variety of solvents (MeOH, IPA, and THF) with or without a non-chiral or chiral base (DBU, CyNMe2, iPr2NEt, (+)-cinchonine, (−)-cinchonidine, (+)-11, and (−)-11) with and without the presence of CO2, and with multiple catalysts. The hydrogenation presented below of 2-phenylprop-2-en-1-amine, 6, with (−)-4,5-bis[(2R,5R)-2,5-dimethylphospholanyl](1,2-dimethyl-1,2-dihydropyridazine-3,6-dione)(1,5-cyclooctadiene) rhodium(I) tetrafluoroborate can be regarded as a general procedure for the asymmetric hydrogenation of prochiral allylamines.Stock solutions of the allylamine, catalyst, and the optional base were prepared in dry methanol the same day as the planned hydrogenation to ensure no decomposition of the chemicals occurred. In a 160 mL stainless steel high pressure vessel, containing up to a dozen 1 dram glass vials, each containing a magnetic stir bar, 2-phenylprop-2-en-1-amine, 6, (10 mg, 0.075 mmol), catalyst, (−)-4,5-bis[(2R,5R)-2,5-dimethylphospholanyl](1,2-dimethyl-1,2-dihydropyridazine-3,6-dione)(1,5-cyclooctadiene) rhodium(I) tetrafluoroborate, (1 mg, 0.0015 mmol) and, if desired, the optional base (ca. 0.075 mmol) was added under a nitrogen atmosphere. Additional dry methanol was added to each vial to obtain a total volume of 2 mL and then the vessel was sealed. The vessel was flushed 3 times with H2 gas or (if CO2 use in the experiment was planned) CO2 gas, and pressurized at room temperature (22 °C) to either 100 bar H2 gas, or if the presence of CO2 is desired, the vessel was pressurized to 10 bar with CO2 gas and then H2 gas was added until the total pressure was 100 bar. It is not correct to assume that the partial pressure of the H2 gas was equal to the difference between the total pressure and the pressure of the CO2 gas, because of significant CO2–H2 interactions. Once the vessel was pressurized, the reaction mixture was stirred for 6–12 h at room temperature. Once the reaction time was complete, the vessel was slowly depressurized, the solutions were filtered through diatomaceous earth, and concentrated by rotatory evaporation. Enantiomeric excess was determined by HPLC and yield was determined by 1H-NMR spectroscopy using an internal standard, 1,3,5-trimethoxybenzene.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PGJ acknowledges the support of the Canada Research Chairs program and the NSERC Discovery grant program. TMD acknowledges the Ontario Graduate Scholarship program and the Walter C. Sumner Foundation.References
- G. Shang, W. Li and X. Zhang, in Catalytic Asymmetric Synthesis, ed. I. Ojima, John Wiley & Sons, Inc., New York, 2010, p. 343 Search PubMed.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- P. Dupau, A.-E. Hay, C. Bruneau and P. H. Dixneuf, Tetrahedron: Asymmetry, 2001, 12, 863–867 CrossRef CAS.
- J. Holz, A. Monsees, H. Jiao, J. You, I. V. Komarov, C. Fischer, K. Drauz and A. Börner, J. Org. Chem., 2003, 68, 1701–1707 CrossRef CAS PubMed.
- X. Dai and D. Cahard, Adv. Synth. Catal., 2014, 356, 1317–1328 CrossRef CAS.
- A. Baeza and A. Pfaltz, Chem.–Eur. J., 2009, 15, 2266–2269 CrossRef CAS PubMed.
- D. Evans, J. A. Osborn, F. H. Jardine and G. Wilkinson, Nature, 1965, 208, 1203–1204 CrossRef CAS.
- M. J. Burk, Y. M. Wang and J. R. Lee, J. Am. Chem. Soc., 1996, 118, 5142–5143 CrossRef CAS.
- L. Qiu, M. Prashad, B. Hu, K. Prasad, O. Repic, T. J. Blacklock, F. Y. Kwong, S. H. L. Kok, H. W. Lee and A. S. C. Chan, Proc. Natl. Acad. Sci., 2007, 104, 16787–16792 CrossRef CAS PubMed.
- J. Almena, A. Monsees, R. Kadyrov, T. H. Riermeier, B. Gotov, J. Holz and A. Börner, Adv. Synth. Catal., 2004, 346, 1263–1266 CrossRef CAS.
- K. Junge, G. Oehme, A. Monsees, T. Riermeier, U. Dingerdissen and M. Beller, J. Organomet. Chem., 2003, 675, 91–96 CrossRef CAS.
- G. P. Aguado, A. G. Moglioni, E. García-Expósito, V. Branchadell and R. M. Ortuño, J. Org. Chem., 2004, 69, 7971–7978 CrossRef CAS PubMed.
- S. Enthaler, G. Erre, K. Junge, D. Michalik, A. Spannenberg, F. Marras, S. Gladiali and M. Beller, Tetrahedron: Asymmetry, 2007, 18, 1288–1298 CrossRef CAS.
- C. Botteghi, G. D. Ponte and C. Marchetti, J. Mol. Catal., 1993, 83, L1–L4 CrossRef CAS.
- R. Fahrang and D. Sinou, Bull. Soc. Chim. Belg., 1989, 98, 387–394 CrossRef CAS.
- M. Yamashita and T. Yamano, Chem. Lett., 2009, 38, 100–101 CrossRef CAS.
- M. Chatterjee, H. Kawanami, M. Sato, T. Ishizaka, T. Yokoyama and T. Suzuki, Green Chem., 2010, 12, 87–93 RSC.
- X. Xie, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2004, 43, 7907–7911 CrossRef CAS.
- T. Toda and Y. Kitagawa, Angew. Chem., Int. Ed. Engl., 1987, 26, 334–335 CrossRef.
- J.-H. Ye, L. Song, W.-J. Zhou, T. Ju, Z.-B. Yin, S.-S. Yan, Z. Zhang, J. Li and D.-G. Yu, Angew. Chem., Int. Ed. Engl., 2016, 55, 10022–10026 CrossRef CAS PubMed.
- M.-Y. Wang, Y. Cao, X. Liu, N. Wang, L.-N. He and S.-H. Li, Green Chem., 2017, 19, 1240–1244 RSC.
- L. Zhu, J.-H. Ye, M. Duan, X. Qi, D.-G. Yu, R. Bai and Y. Lan, Org. Chem. Front., 2018, 5, 633–639 RSC.
- Z.-B. Yin, J.-H. Ye, W.-J. Zhou, Y.-H. Zhang, L. Ding, Y.-Y. Gui, S.-S. Yan, J. Li and D.-G. Yu, Org. Lett., 2018, 20, 190–193 CrossRef CAS PubMed.
- L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018, 20, 3049–3052 CrossRef CAS PubMed.
- Z. Zhang, J.-H. Ye, D.-S. Wu, Y.-Q. Zhou and D.-G. Yu, Chem.–Asian J., 2018, 13, 2292–2306 CrossRef CAS PubMed.
- R. Noyori, Asymmetric Catalysis in Organic Synthesis, John Wiley and Sons, New York, 1994 Search PubMed.
- T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Miura and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264–267 CrossRef CAS.
- H. Takaya, T. Ohta, N. Sayo, H. Kumobayashi, S. Akutagawa, S.-i. Inoue, I. Kasahara and R. Noyori, J. Am. Chem. Soc., 1987, 109, 1597–1600 CrossRef.
- F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed.
- A. Nova, G. Ujaque, A. C. Albéniz and P. Espinet, Chem.–Eur. J., 2008, 14, 3323–3329 CrossRef CAS PubMed.
- J. Holz, O. Zayas, H. Jiao, W. Baumann, A. Spannenberg, A. Monsees, T. H. Riermeier, J. Almena, R. Kadyrov and A. Börner, Chem.–Eur. J., 2006, 12, 5001–5013 CrossRef CAS.
- K. Hattori, H. Sajiki and K. Hirota, Tetrahedron, 2000, 56, 8433–8441 CrossRef CAS.
- M. Szostak, B. Sautier, M. Spain and D. J. Procter, Org. Lett., 2014, 16, 1092–1095 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00263a |
| This journal is © The Royal Society of Chemistry 2022 |