
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAluminium alkyl complexes supported by imino-phosphanamide ligand as precursors for catalytic guanylation reactions of carbodiimides†
Himadri Karmakar a,
Srinivas Angab,
Tarun K. Panda*a and
Vadapalli Chandrasekhar*bc
a,
Srinivas Angab,
Tarun K. Panda*a and
Vadapalli Chandrasekhar*bc
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy 502285, Telangana, India. E-mail: tpanda@iith.ac.in; Fax: +91 40 2301 6032; Tel: +91 40 2301 6036
bTata Institute of Fundamental Research Hyderabad, Gopanpally, 500107, Hyderabad, India. E-mail: vc@tifrh.res.in
cDepartment of Chemistry, IIT Kanpur, Kanpur 208016, India. E-mail: vc@iitk.ac.in
First published on 3rd February 2022
Abstract
Herein, we report the synthesis, characterisation, and application of three aluminium alkyl complexes, [κ2-{NHIRP(Ph)NDipp}AlMe2] (R = Dipp (2a), Mes (2b); tBu (2c), Dipp = 2,6-diisopropylphenyl, Mes = mesityl, and tBu = tert-butyl), supported by unsymmetrical imino-phosphanamide [NHIRP(Ph)NDipp]− [R = Dipp (1a), Mes (1b), tBu (1c)] ligands as molecular precursors for the catalytic synthesis of guanidines using carbodiimides and primary amines. All the imino-phosphanamide ligands 1a, 1b and 1c were prepared in good yield from the corresponding N-heterocyclic imine (NHI) with phenylchloro-2,6-diisopropylphenylphosphanamine, PhP(Cl)NHDipp. The aluminium alkyl complexes 2a, 2b and 2c were obtained in good yield upon completion of the reaction between trimethyl aluminium and the protic ligands 1a, 1b and 1c in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in toluene via the elimination of methane, respectively. The molecular structures of the protic ligands 1b and 1c and the aluminium complexes 2a, 2b and 2c were established via single-crystal X-ray diffraction analysis. Complexes 2a, 2b and 2c were tested as pre-catalysts for the hydroamination/guanylation reaction of carbodiimides with aryl amines to afford guanidines at ambient temperature. All the aluminium complexes exhibited a high conversion with 1.5 mol% catalyst loading and broad substrate scope with a wide functional group tolerance during the guanylation reaction. We also proposed the most plausible mechanism, involving the formation of catalytically active three-coordinate Al species as the active pre-catalyst.
:1 molar ratio in toluene via the elimination of methane, respectively. The molecular structures of the protic ligands 1b and 1c and the aluminium complexes 2a, 2b and 2c were established via single-crystal X-ray diffraction analysis. Complexes 2a, 2b and 2c were tested as pre-catalysts for the hydroamination/guanylation reaction of carbodiimides with aryl amines to afford guanidines at ambient temperature. All the aluminium complexes exhibited a high conversion with 1.5 mol% catalyst loading and broad substrate scope with a wide functional group tolerance during the guanylation reaction. We also proposed the most plausible mechanism, involving the formation of catalytically active three-coordinate Al species as the active pre-catalyst.
Introduction
Metal-catalysed C–N bond formation is an intense area of research. One of the convenient strategies to achieve the C–N bond is the catalytic guanylation reaction.1 This reaction is a hydroamination reaction, where the N–H bond of an amine adds across carbodiimide, which produces guanidine derivatives.2 The metal-catalysed guanylation reaction is a 100% atom economy reaction and a straightforward method to prepare nitrogen-rich compounds.3 Guanidine derivatives are important due to their multipurpose usage in organic synthesis,4 coordination chemistry,5 pharmaceuticals,6 and the agrochemical industry. They are also important structural motifs of many biologically and pharmaceutically active molecules. They can serve as building blocks in various pharmaceutical and natural products.7 Due to their low nucleophilicity, it is difficult for arylamines to participate in guanylation reactions with unactivated carbodiimides in the absence of a catalyst.8 In 2003, Richeson et al. reported the catalytic guanylation reaction between primary arylamines and unactivated carbodiimides for the first time using a titaniumimido complex as a catalyst at elevated temperatures.9 Further, the guanylation reaction of primary arylamines and carbodiimides has been investigated using transition-,10 rare-earth-,11 and main group metal complexes.12 Recently, we reported various titanium(IV) metal complexes and zinc(II) metal complex as precursors for the catalytic guanylation reaction of carbodiimides.13Recent trends in synthetic chemistry demonstrate the increasing use of synthetic methods in multicomponent reactions and catalysis using earth-abundant metals. Multicomponent reactions emphasize the progress of reactions using fewer (compared to conventional reactions) steps, while the choice of earth-abundant metals is driven by their non-toxicity and economy. Therefore, aluminium, as the most abundant metal on earth and due to its non-toxic nature, has been used in complexes as a preeminent catalyst in several important multicomponent reactions. In 2009, the commercially available AlMe3 was utilised as a catalyst for the guanylation reaction by Zhang and co-workers.14 Bergman's group and Zhou's group also used aluminium alkyl complexes as catalysts for guanylation reactions.15 Recently, Mandal et al. reported the use of a cyclic (alkyl) amino carbene complex of AlMe3 as a catalyst for the guanylation reaction.16
There have been numerous studies on nitrogen-based electron rich ligands such as amidinate,17,18 guanidinate,18,19 iminophosphonamide,20 and boraamidinate8–21 ligands. Accordingly, some of these ligands have already found wide applications in the formation of a range of metal complexes including the stabilisation of unusual oxidation states of main-group elements.18 Among them, the amidinate and guanidinate ligands have received significant attention by many research groups.17–19
Monoanionic N-heterocyclic imines such as imidazolin-2-iminato ligands are often described by two limiting resonance structures, thus indicating that the ability of the imidazolium ring to stabilise a positive charge leads to highly basic ligands22 with strong electron-donating capacity toward early transition metals.23 Imidazolin-2-iminato ligands were successfully introduced in main group metals, transition metals, lanthanides, and more recently, actinide metals by Tamm and co-workers to achieve very short M–N bonds.24 Our group and others25 have already established that monoanionic imidazolin-2-iminato ligands are efficient systems for the preparation of catalytically active transition metal and rare earth metal complexes.26
Recently, the exceptional electron-donating ability and fine-tuning of the steric properties of imidazolin-2-iminato ligands were utilised to stabilise main group metal ions. The isolation of a carbene-stabilised phosphorus mononitride,27 monomeric phosphinyl radical,28 singlet phosphinonitrene,29 its protonated derivative30 and metal complexes was reported by Bertrand et al.31 The further utilisation of this ligand to stabilise several group 13- and group 16-metal ions was demonstrated by Inoue32–36 and Rivard.37,38 However, aluminium alkyl complexes with the imidiazoline-2-iminato ligand were only recently studied by the research groups of Inoue,35,36 Masuda39 and Eisen.40
Recently, we developed an N-heterocyclic imine-based imino-phosphanamide ligand-supported yttrium complex [{NHIRP(Ph)N-Dipp}Y{NSiMe3}2] (iPr, Me), where the ligand upon metalation is bonded with the yttrium ion in a monoanionic fashion similar to that of amidinate ligands.41 The two donor nitrogen atoms attached to the phosphorus atom are completely different and it is interesting to note that this system is amenable to stereoelectronic modulation through substitution at the phosphorus and nitrogen centers.42 Currently, we were interested in the interaction of the unsymmetrical imino-phosphanamide ligands with p-block elements such as aluminium and the exploration of the complexes formed in the catalytic guanylation reaction. Accordingly, herein, we report the synthesis and characterisation of imino-phosphanamine ligands [NHIRP(Ph)NHDipp] [R = Dipp (1a), Mes (1b), tBu (1c)] and the corresponding aluminium alkyl complexes [{NHIRP(Ph)NDipp}AlMe2] (R = Dipp (2a), Mes (2b); tBu (2c)), respectively, together with the catalytic applications of the latter towards the guanylation of primary arylamines with unactivated carbodiimides.
Results and discussion
Ligand synthesis
The protic ligands 1a and 1b were synthesised according to the protocol involving the reaction of imidazolin-2-imine [ImRNH] (R = Dipp and Mes) with PhP(Cl)NH(Dipp) (Dipp = 2,6-diisopropylphenyl) (1:1 equiv.) in the presence of triethylamine (1.5 equiv.) at room temperature, as previously reported by us.41 The ligand 1c was obtained in 80% yield by the reaction between 1,3-di-tert-butyl-2-(trimethylsilylimino)imidazoline and PhP(Cl)NH(Dipp) in a 1:1 molar ratio at 85 °C in toluene via the elimination of trimethylsilyl chloride (Scheme 1). All three ligands, 1a, 1b and 1c, were characterised using 1H, 13C{1H} and 31P{1H} NMR spectroscopy, and elemental analysis.
 | ||
| Scheme 1 Synthesis of the protic ligands 1a, 1b, and 1c. | ||
The multinuclear NMR spectra of ligand 1a and its molecular structure in the solid-state were previously reported by us.22 The solid-state structures of 1b and 1c were established by single-crystal X-ray diffraction analysis. In the 1H NMR spectrum, the signal for the amine NH hydrogen atom appears at δH = 3.98 ppm as a broad singlet for 1b, whereas the signal for a similar NH hydrogen atom of 1c appears at δH = 4.59 ppm as a doublet due to its coupling with the phosphorus atom [2JHP of 3 Hz (1c)]. The two olefinic protons of the imidazole ring display a singlet resonance at δH = 5.61 ppm (1b) and 6.07 ppm (1c). Three singlet peaks at δH = 2.26, 2.14 and 1.96 ppm appear in the 1H NMR spectrum of 1b due to the three methyl groups of the mesityl unit and a sharp singlet at δH = 1.46 ppm in the 1H NMR spectrum of 1c appears due to the 18 methyl protons of its two tert-butyl groups. Further, a septet signal centered at δH = 3.45 ppm (1b) and 3.52 ppm (1c) and two doublets at δH = 1.13 and 1.06 ppm (1b) and 1.27 and 1.18 ppm (1c) were observed due to the protons of the isopropyl groups attached to the Dipp moiety. In the 31P{1H} NMR spectra, compounds 1b and 1c exhibit a sharp singlet at δP = 73.13 ppm (1b) and 78.2 ppm (1c).
Compound 1b crystallises in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, whereas 1c crystallises in the monoclinic P21/n space group. The molecular structures of 1b and 1c in the solid-state are given in Fig. 1 and 2, respectively. The P–N1 and P–N2 bond distances of 1.7346(11) Å and 1.6844(12) Å (1b) and 1.729(2) Å and 1.652(2) Å (1c) are similar. Similar P–N bond distances were also observed in compound 1a (1.670(1) Å and 1.741(1) Å).22 The N1–P1–N2 bond angles in 1b (102.91(6)°) and 1c (102.63(12)°) are much wider than that in 1a (93.79°) due to the less steric crowding in compounds 1b and 1c.
space group, whereas 1c crystallises in the monoclinic P21/n space group. The molecular structures of 1b and 1c in the solid-state are given in Fig. 1 and 2, respectively. The P–N1 and P–N2 bond distances of 1.7346(11) Å and 1.6844(12) Å (1b) and 1.729(2) Å and 1.652(2) Å (1c) are similar. Similar P–N bond distances were also observed in compound 1a (1.670(1) Å and 1.741(1) Å).22 The N1–P1–N2 bond angles in 1b (102.91(6)°) and 1c (102.63(12)°) are much wider than that in 1a (93.79°) due to the less steric crowding in compounds 1b and 1c.
 | ||
| Fig. 1 ORTEP drawing of the molecular structure of 1b, where ellipsoids are drawn to encompass 30% probability, while hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and bond angles [°]: P1–N1 1.7346(11), P1–N2 1.6844(12), P1–C24 1.8390(13), N2–C13 1.2944(18), N3–C13 1.3853(18), N4–C13 1.3815(18), N1–P1–N2 102.91(6), N1–P1–C24 93.10(6), N2–P1–C24 104.11(6), N2–C13–N3 130.67(13), N2–C13–N4 125.01(13), N3–C13–N4 104.30(12). CCDC no. 2124041.† | ||
 | ||
| Fig. 2 ORTEP drawing of the molecular structure of 1c, where ellipsoids are drawn to encompass 30% probability, while hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and bond angles [°]: P1–N1 1.729(2), P1–N2 1.652(2), P1–C24 1.848(3), N2–C13 1.292(3), N3–C13 1.386(4), N4–C13 1.394(3), N1–P1–N2 102.63(12), N1–P1–C24 98.45(13), N2–P1–C24 99.86(13), N2–C13–N3 123.4(2), N2–C13–N4 131.6(3), N3–C13–N4 104.6(2). CCDC no. 2124043.† | ||
Aluminium bisalkyl complexes
Trimethyl aluminium (AlMe3) was reacted with one equivalent of protic ligands 1a, 1b, and 1c to afford, in good yield (Scheme 2), the corresponding aluminium-bisalkyl complexes [κ2-{NHIRP(Ph)-NDipp}AlMe2] (R = Dipp (2a), Mes (2b), tBu (2c)), respectively. These compounds are highly soluble in benzene, toluene, and THF, but partially soluble in n-hexane and diethyl ether. 2a, 2b, and 2c were characterised via 1H, 13C{1H} and 31P{1H} NMR spectral data and elemental analysis. Further, the solid-state structures of 2a, 2b, and 2c were established by single-crystal X-ray crystallography. | ||
| Scheme 2 Synthesis of Al-alkyl complexes 2a and 2b. | ||
Well-resolved NMR spectra measured in C6D6 were obtained for 2a, 2b, and 2c. In their 1H NMR spectra, the absence of resonance signals at δH = 3.73, 3.98, and 4.59 ppm confirmed the deprotonation of the NH proton of ligands 1a, 1b, and 1c, respectively. The six protons corresponding to the AlMe2 unit appeared as a sharp singlet at δH = −0.73 ppm for 2a, −0.57 and −0.60 ppm for 2b and −0.19 ppm for 2c in the 1H NMR spectra. Additionally, signals corresponding to the isopropyl groups of two Dipp moieties and methyl groups of two mesityl moieties were also observed for 2a and 2b, respectively. For 2c, a sharp singlet at δH = 0.99 ppm for the protons from the tert-butyl group was observed. In the 31P{1H} NMR spectra, 2a, 2b and 2c exhibit singlets at δP = 134.64 ppm, 130.60 ppm and 158.64 ppm, respectively, which are significantly shifted downfield compared to that of the corresponding ligands, which are δP = 71.79 ppm (1a), 73.13 ppm (1b) and 78.26 ppm (1c).
The solid-state structures of complexes 2a, 2b and 2c were determined by single-crystal X-ray diffraction analysis, which confirmed the attachment of the unsymmetrical imino-phosphanamide [NHIRP(Ph)NDipp] ligand to the aluminium ion. Complexes 2a and 2c crystallise in the monoclinic space group P21/n and P21/c, respectively, and complex 2b crystallises in the triclinic space group P. The crystallographic details and refinement parameters are given in Table TS1 in the ESI.† The molecular structures of 2a, 2b and 2c in their solid state are shown in Fig. 3, 4 and 5 respectively.
 | ||
| Fig. 3 ORTEP drawing of the molecular structure of 2a, where ellipsoids are drawn to encompass 30% probability, while hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Al1–N1 1.8642(19), Al1–N2 2.0022(18), Al1–C1 1.974(3), Al1–C2 1.952(3), N2–C15 1.316(2), N3–C15 1.380(3), N4–C15 1.372(2), N1–P1–N2 88.67(8), N1–Al1–N2 76.96(7), N1–Al1–C1 115.89(11), N1–Al1–C2 114.02(13), C1–Al1–C2 107.79(17), N2–Al1–C1 117.37(13), N2–Al1–C2 122.14(13), N2–C15–N3 130.04(18), N2–C15–N4 124.93(18), N3–C15–N4 105.02(16). CCDC no. 2124040.† | ||
 | ||
| Fig. 4 ORTEP drawing of the molecular structure of 2b, where ellipsoids are drawn to encompass 30% probability, while hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Al1–N1 1.8679(11), Al1–N2 1.9903(11), Al1–C1 1.9672(17), Al1–C2 1.9698(17), N2–C15 1.3177(17), N3–C15 1.3714(16), N4–C15 1.3688(17), N1–P1–N2 88.41(5), N1–Al1–N2 77.16(5), N1–Al1–C1 114.64(7), N1–Al1–C2 114.21(7), C1–Al1–C2 112.97(8), N2–Al1–C1 117.61(6), N2–Al1–C2 115.69(7), N2–C15–N3 130.10(12), N2–C15–N4 125.01(11), N3–C15–N4 104.89(11). CCDC no. 2124042.† | ||
 | ||
| Fig. 5 ORTEP drawing of the molecular structure of 2c, where ellipsoids are drawn to encompass 30% probability, while hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Al1–N1 1.8979(12), Al1–N2 1.9401(12), Al1–C1 1.987(2), Al1–C2 1.968(2), N2–C15 1.3479(18), N3–C15 1.3722(18), N4–C15 1.372(2), N1–P1–N2 89.26(6), N1–Al1–N2 78.36(5), N1–Al1–C1 120.36(8), N1–Al1–C2 115.83(8), C1–Al1–C2 108.41(10), N2–Al1–C1 115.77(7), N2–Al1–C2 115.77(8), N2–C15–N3 125.48(13), N2–C15–N4 128.37(13), N3–C15–N4 106.06(12). CCDC no. 2124039.† | ||
2a, 2b, and 2c are monomeric and their coordination polyhedrons are formed by the chelation of two nitrogen atoms from the monoanionic imino-phosphanamide [NHIRP(Ph)NDipp]− moiety. The aluminium ions are asymmetrically attached to the ligand backbone, as indicated by the Al1–N1 bond distances of 2a, 2b, and 2c [1.8642(19) Å, 1.8679(11) Å and 1.8979(12) Å, respectively] and the Al1–N2 bond distances of 2a, 2b and 2c [2.0022(18) Å, 1.9903(11) Å and 1.9401(12) Å, respectively]. The anionic amido nitrogen atoms are better electron donors towards the Al(III) cation than the imino-phosphanamide nitrogen atoms. In all the complexes, the central aluminium ion is four-fold coordinated due to the coordination of two nitrogens from the imino-phosphanamide ligand and two methyl groups are attached to the metal ion in each case. The geometry around the aluminium ion in each complex can best be described as distorted tetrahedral. In each complex, one four-membered metallacycle Al1–N1–P1–N2 is formed, suggesting molecular strain.
Catalytic study
At the outset of our study, the efficiency of a 2a, 2b, and 2c towards the catalytic hydroamination/guanylation of various aryl amines with carbodiimides was studied. The catalytic experiments were performed using equimolar amounts of carbodiimides and primary amines. The benchmark guanylation reaction of 2-bromoaniline with N,N′-diisopropylcarbodiimide (DIC) was chosen to check the efficiency of 2a and 2b as precatalysts. In the case of amine addition, due to the fact that –NH2 is a diprotic nucleophile, isomerisation of the initial product occurs, leading to guanidine derivatives (3a–3v) as the major products. In each case, the corresponding guanidine products were isolated and purified by washing with n-pentane and the results are given in Table 1. As a control experiment, a blank reaction was carried out at 60 °C for 20 h in the presence of only diisopropyl carbodiimide (DIC) and 2-bromoaniline, which showed that no guanidine product was formed (Table 1, entry 1). However, in the presence of 0.5 mol% 2a as a precatalyst, 71% of substituted guanidine 3a was achieved as the product at room temperature within a reaction time of 6 h (Table 1, entry 2). Under the same reaction conditions, 2b yielded 72% of product 3a (Table 1, entry 3). When the catalyst loading was increased to 1.0 mol%, and finally to 1.5 mol% for 2a, a substantial increase in the product yield up to 89% was observed within 2 h at room temperature (Table 1, entries 4 and 6). However, under the same conditions, complex 2b did not show much improvement in the product yield (Table 1, entries 5 and 7). The use of precatalyst 2c gave 83% of product 3a under similar conditions (Table 1, entry 8). To optimise the solvent effect, the catalytic reactions were carried out in different solvents such as n-hexane and THF as well as in neat conditions at room temperature. However, we did not observe any major effect of solvent on the catalytic reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Mol% | Solvent | Time (h) | Yield (%) |
| a Reaction conditions: Al complex 2a (1.5 mol%), toluene solvent, room temperature, 2 h (optimised reaction time).b Compounds were isolated and purified by washing with n-pentane.c Yields were calculated from isolated pure products. | |||||
| 1 | Toluene | 20 | 0 | ||
| 2 | 2a | 0.5 | Toluene | 6 | 71 |
| 3 | 2b | 0.5 | Toluene | 6 | 72 |
| 4 | 2a | 1 | Toluene | 2 | 88 |
| 5 | 2b | 1 | Toluene | 2 | 85 |
| 6 | 2a | 1.5 | Toluene | 2 | 89 |
| 7 | 2b | 1.5 | Toluene | 2 | 86 |
| 8 | 2c | 1.5 | Toluene | 2 | 83 |
| 9 | 2a | 1.5 | Hexane | 2 | 82 |
| 10 | 2b | 1.5 | Hexane | 2 | 81 |
| 11 | 2a | 1.5 | THF | 2 | 85 |
| 12 | 2b | 1.5 | THF | 2 | 86 |
| 13 | 2a | 1.5 | Neat | 2 | 88 |
| 14 | 2b | 1.5 | Neat | 2 | 87 |

After the preliminary evaluation of the catalysts for the hydroamination of DIC, we concluded that complex 2a is more efficient than 2b at room temperature and in toluene solvent. With room temperature and toluene as the solvent being the optimal reaction settings for the hydroamination of carbodiimides, we set out to examine the scope of various substituted aryl amine and carbodiimide substrates in the presence of 1.5 mol% of aluminium complex 2a as the precatalyst. The results of the catalytic hydroamination reactions are displayed in Table 2. All the products 3a–3v were fully characterised with the help of NMR spectroscopy and the yields were calculated from the isolated yields.
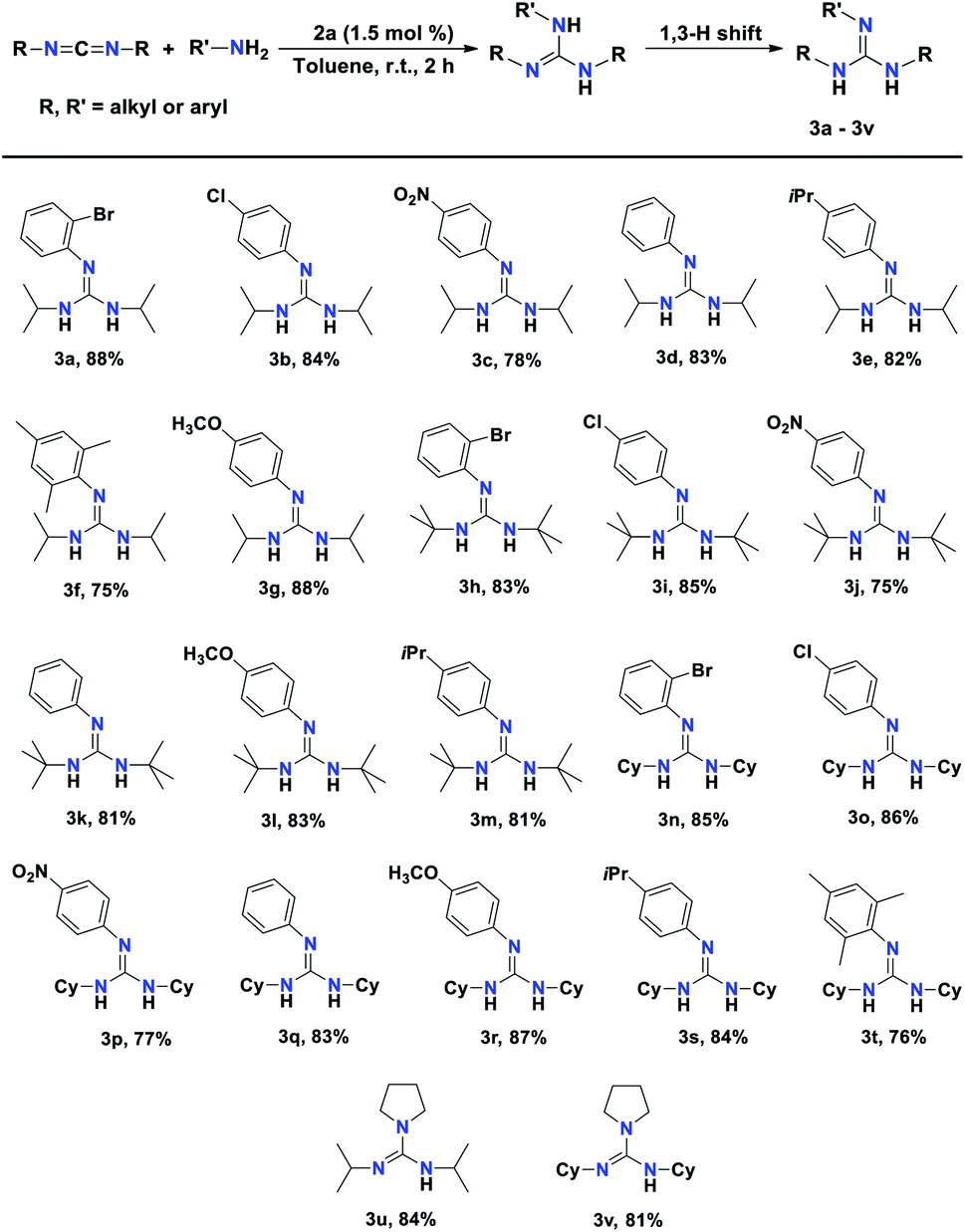
Substituted aryl amines containing electron-withdrawing groups, such as bromo, chloro, and nitro anilines reacted with DIC, affording very good yields (78–88%) of the desired guanidines within 2 h at room temperature (3a–3c), respectively. The unsubstituted aniline afforded 83% yield under the optimal conditions (Table 2, 3d). The aryl amines with electron-donating groups such as 4-isopropyl aniline, 2,4,6-trimethylaniline, and 4-methoxy aniline exhibited excellent conversion, affording the corresponding guanidines within 2 h at room temperature (Table 2, 3e–3g), respectively.
The substrate scope was further expanded by using N,N-di-tert-butylcarbodiimide and N,N-dicyclohexylcarbodiimide (DCC) with various arylamines in the presence of aluminium catalyst 2a. The reaction of aniline with N,N-di-tert-butylcarbodiimide afforded 81% yield of the corresponding guanidine product. Anilines with electron-releasing groups and deactivated anilines with electron-withdrawing groups reacted smoothly with two carbodiimides, N,N-di-tert-butylcarbodiimides and DCC, resulting in the corresponding guanidine products in excellent yields (Table 2, entries 3h–3t). This indicates the high efficiency of the aluminium complex 2a. Pyrrolidine, which is a secondary amine, also reacted smoothly with DIC under the optimised condition to afford N,N′-diisopropylpyrrolidine-1-carboximidamide (3u) and N,N′-dicyclohexylpyrrolidine-1-carboximidamide (3v) in 84% and 81% yield (Table 2, 3u and 3v), respectively.
To explore the conversion of amine to guanidine and the highest possible efficiency of catalyst 2a, the insertion of aniline into DIC was monitored using 1H NMR spectroscopy, showing the progress of the reaction as a function of DIC consumption followed by the formation of guanidine. The insertion of aniline into DIC was monitored at 10 minute intervals using 1H NMR spectroscopy, showing the formation of the guanidine product as a function of DIC consumption with time. The yield of the guanidine product was plotted against time, as shown in Fig. 6. It was observed that the formation of the guanidine product was very fast in the initial 20 min, and then it gradually slowed down after 2 h to attain saturation.
 | ||
| Fig. 6 Plot of the yield of the guanidine derivative (%) vs. time (min) as monitored by 1H-NMR spectroscopy for the reaction between 2-bromoaniline and DIC at room temperature. | ||
Most plausible mechanism
To explore the most plausible mechanism for the catalytic addition of N–H to carbodiimides, we performed some controlled reactions. Given that our ligand fragment contains a P-atom, it was easy for us to monitor the catalytic reaction through 31P{1H} NMR measurement. The stoichiometric reaction of catalyst 2a with DIC was carried out in benzene-d6 and the 1H and 31P{1H} NMR spectra of the reaction mixture suggested no reaction even at 70 °C. The Al-complex 2a was treated with primary arylamine p-anisidine in a 1:2 molar ratio at room temperature. In the product, we detected one sharp singlet at δP 71.79 ppm in the 31P{1H} NMR spectrum, and in the 1H NMR spectrum a singlet peak at δH = 3.73 ppm also appeared, which is ascribed to the NH proton of free ligand 1a (Fig. FS54 and FS55 in the ESI,† respectively). Analogously, the reaction of 2a with a secondary amine such as piperidine in a 1:2 molar ratio at 70 °C gave one sharp singlet at δP 71.79 ppm in the 31P{1H} NMR spectrum and δH = 3.73 ppm in the 1H NMR spectrum, confirming the formation of free ligand 1a (Fig. FS56 and FS57 in the ESI,† respectively). Thus, we easily observed the dissociation of the ligand moiety from the metal ion.
Based on these experiments, the catalytic cycle is proposed (Scheme 3) to proceed via amine exchange with the supporting ligand to form a catalytically active three-coordinate Al species, which can subsequently add to carbodiimides to form Al guanidinate species. The catalytically active species can then be regenerated by another amine exchange reaction with free aniline. A similar mechanism was also reported by other research groups.15 The first step in this mechanism involves the rapid protonolysis of the ligand fragment of the aluminium catalyst 2a by the N–H moiety of ArNH2, with the displacement of free ligand 1a, producing an active three-coordinate Al species (I). In the next step, the carbodiimide molecule is bonded to Al species I to form Al guanidinate species III. Further, species III undergoes an amine exchange reaction with the free aniline in the final step to regenerate active species I.
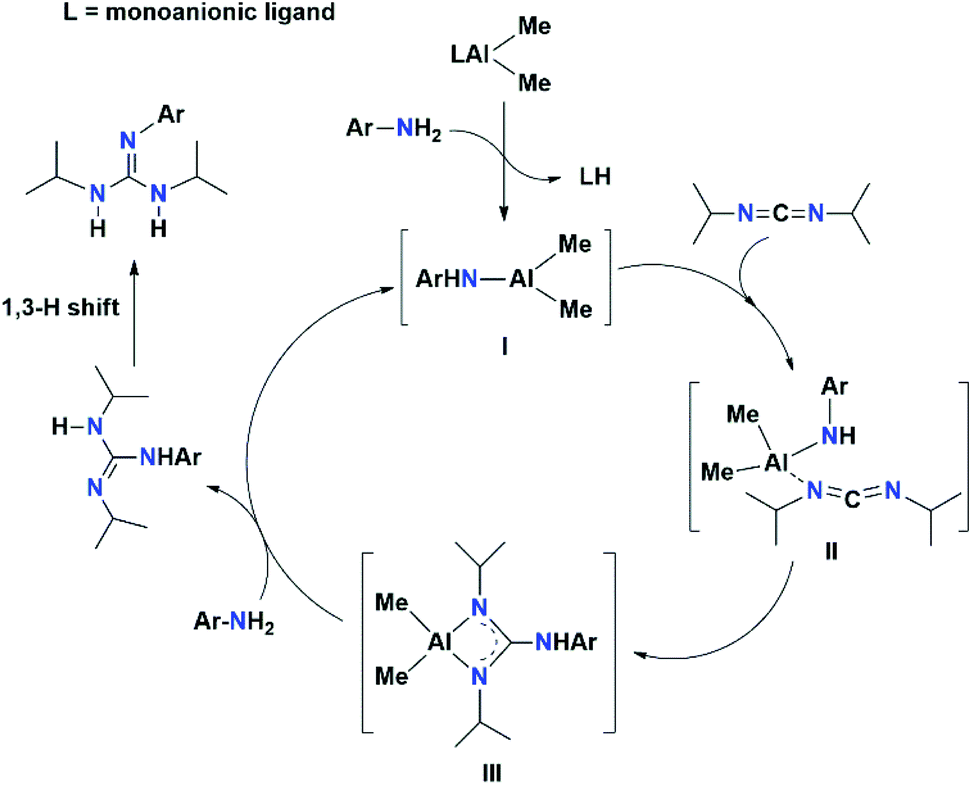 | ||
| Scheme 3 Most plausible mechanism for guanylation reaction between carbodiimide and aryl amines mediated by aluminium complex 2a as a precatalyst. | ||
Conclusions
In summary, we synthesised and characterised three unsymmetrical imino-phosphanamide-supported aluminium alkyl complexes and established their solid-state structures. The aluminium complex 2a acted as an efficient catalyst for the guanylation reaction of arylamines and carbodiimides at room temperature. The three different carbodiimides underwent guanylation reactions to provide near-quantitative yields of the corresponding guanidines at ambient temperature within short reaction times. The most plausible mechanism for the catalytic addition of the N–H bond of arylamine to carbodiimides was also described. The key features of this protocol include the high catalytic efficiency of the aluminium complex 2a with a wider substrate scope of carbodiimides and aryl amines at ambient temperature.General experimental procedures
All manipulations involving air- and moisture-sensitive compounds were carried out under argon, using the standard Schlenk technique or argon-filled M. Braun glove box. Hydrocarbon solvents (n-hexane, toluene, and benzene) were distilled under a nitrogen atmosphere from LiAlH4 and stored inside the glove box. THF was dried and deoxygenated by distillation over sodium benzophenone under argon and then distilled and dried over CaH2 before being stored in the glove box. 1H NMR (300 MHz), 31P{1H} (121.5 MHz) and 13C{1H} (75 MHz) spectra were recorded on a Nano Bay 300 MHz spectrometer. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad. Imidazolin-2-imines [ImRNH] (R = Dipp, Mes) or their silylated derivative [ImRNSiMe3] (R = tBu) prepared according to the published procedure.17 The protic ligand 1a was synthesised according to our previously reported method.22 Trimethylaluminium, all amines, diisopropyl-carbodiimide, N,N-di-tert-butylcarbodiimide, and N,N-dicyclohexylcarbodiimide were purchased from Sigma Aldrich. Amines were distilled over CaH2 before use. NMR spectroscopy solvent C6D6 was purchased from Sigma Aldrich, India and dried over Na/K, distilled, and stored in a glove box.Preparation of [ImMesNP(Ph)NH(Dipp)] (1b)
In a flame-dried 50 ml Schlenk flask, 500 mg (1.56 mmol) 1,3-bis(mesityl)imidazolin-2-imine and triethylamine (0.22 ml, 1.56 mmol) were dissolved in 20 ml toluene. Then PhP(Cl)NH(Dipp) (Dipp = 2,6-diisopropylphenyl) (500 mg, 1.56 mmol) in 20 ml toluene was added dropwise to the mixture. The reaction mixture was allowed to stir for 3 h at room temperature. Subsequently, the reaction mixture was filtered to obtain a clear solution and the solvent was evaporated from the filtrate. A white solid powder was obtained, which was further washed with hexane. The compound was recrystallised from a toluene/hexane mixture at room temperature. White crystals were obtained after 2 days.1b: yield: 858 mg, 91%. 1H NMR (300 MHz, C6D6, 298 K): δH 7.53 (t, J = 6 Hz, 2H, Ar–H), 7.13–6.98 (m, 6H, Ar–H), 6.77 (s, 2H, Ar–H), 6.64 (s, 2H, Ar–H), 5.61 (s, 2H, NCH), 3.98 (bs, 1H, NH), 3.49–3.40 (m, 2H, CH(CH3)2), 2.26 (s, 6H, CH3), 2.14 (s, 6H, CH3), 1.96 (s, 6H, CH3), 1.13 (d, J = 9 Hz, 6H, CH3), 1.06 (d, J = 6 Hz, 6H, CH3) ppm. 13C{1H} NMR (75 MHz, C6D6, 298 K): δC 150.38, 150.13, 145.70, 145.37, 140.97, 140.85, 138.33, 137.80, 137.10, 134.15, 129.93, 129.62, 129.22, 129.05, 123.31, 122.71, 113.79, 28.43, 28.35, 24.12, 24.09, 23.98, 21.12, 18.55, 18.49, 18.00 ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δP 73.13 ppm. Elemental analysis for C39H47N4P (602.7). Calcd for C 77.71, H 7.86, N 9.29; found C 77.63, H 7.58, N 9.03.
Preparation of [ImtBuNP(Ph)NH(Dipp)] (1c)
In a flame-dried 50 ml Schlenk flask, 332 mg (1.24 mmol) 1,3-di-tert-butyl-2-(trimethylsilylimino)imidazoline was added and dissolved in 25 ml toluene. Then PhP(Cl)NH(Dipp) (Dipp = 2,6-diisopropylphenyl) (396.16 mg, 1.24 mmol) in 20 ml toluene was added dropwise to the mixture. The reaction mixture was allowed to stir for 12 h at 85 °C. Subsequently, the reaction mixture was filtered to obtain a clear solution and the solvent was evaporated from the filtrate. A white solid powder was obtained, which was further washed with hexane. The compound was recrystallised from a toluene/hexane mixture at room temperature. White crystals were obtained after 2 days.1c: yield: 475 mg, 80%. 1H NMR (300 MHz, C6D6, 298 K): δH 7.90 (t, J = 6 Hz, 2H, Ar–H), 7.26 (t, J = 6 Hz, 2H, Ar–H), 7.18–7.07 (m, 4H, Ar–H), 6.07 (s, 2H, NCH), 4.59 (d, J = 3 Hz, 1H, NH), 3.57–3.48 (m, 2H, CH(CH3)2), 1.46 (s, 18H, CH3), 1.27 (s, 6H, CH3), 1.18 (s, 6H, CH3) ppm. 13C{1H} NMR (75 MHz, C6D6, 298 K): δC 149.35, 149.13, 147.11, 146.67, 141.21, 140.47, 130.09, 129.80, 123.81, 122.50, 108.78, 55.75, 29.61, 28.69, 24.45, 24.24 ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δP 78.26 ppm. Elemental analysis for C31H47N4P (506.7). Calcd for C 73.48, H 9.35, N 11.06; found C 73.31, H 9.21, N 10.93.
Preparation of [κ2-{ImRNP(Ph)NDipp}AlMe2] (R = Dipp (2a), Mes (2b), tBu (2c))
In a flame-dried 25 ml Schlenk flask, ligand 1a (200 mg, 0.29 mmol) or 1b (179 mg, 29 mmol) or 1c (134 mg, 0.29 mmol) was added and dissolved in 15 ml toluene. Then trimethylaluminium solution (2 M) in toluene (0.15 ml, 0.29 mmol) was added to it under ice-cold conditions and stirred at room temperature for 18 h. Next, the solvent was evaporated, and the residue was washed with hexane. The crude product was recrystallised from a toluene/hexane mixture at room temperature.2a: yield: 177.3 mg, 82%. 1H NMR (300 MHz, C6D6, 298 K): δH 7.34 (t, J = 6 Hz, 3H, Ar–H), 7.21–7.13 (m, 3H, Ar–H), 7.07–7.02 (m, 5H, Ar–H), 6.93 (d, J = 9 Hz, 3H, Ar–H), 5.89 (s, 2H, NCH), 3.28–3.19 (m, 4H, CH(CH3)2), 2.81–2.72 (m, 2H, CH(CH3)2), 1.64 (d, J = 6 Hz, 6H, CH3), 1.04 (d, J = 6 Hz, 18H, CH3), 0.94 (d, J = 9 Hz, 12H, CH3), −0.73 (s, 6H, Al–CH3) ppm. 13C{1H} NMR (75 MHz, C6D6, 298 K): δC 150.57, 149.76, 149.16, 149.00, 147.92, 146.79, 141.32, 141.11, 133.00, 131.40, 129.88, 129.54, 124.93, 124.80, 122.84, 118.39, 29.61, 29.43, 26.71, 26.35, 25.93, 22.47, 22.13, −3.50, −5.01 ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δP 134.64 ppm. Elemental analysis for C47H64AlN4P (743.0) (2a). Calcd for C 75.98, H 8.68, N 7.54; found C 75.63, H 8.39, N 7.32.
2b: yield: 162 mg, 85%. 1H NMR (300 MHz, C6D6, 298 K): δH 7.45 (s, 2H, Ar–H), 7.07 (s, 6H, Ar–H), 6.83 (s, 2H, Ar–H), 6.61 (s, 2H, Ar–H), 5.33 (s, 2H, NCH), 4.07 (bs, 1H, CH(CH3)2), 3.65 (bs, 1H, CH(CH3)2), 2.32 (s, 6H, CH3), 2.12 (s, 6H, CH3), 1.66 (s, 6H, CH3), 1.54 (bs, 6H, CH3), 1.17 (bs, 3H, CH3), 0.40 (bs, 3H, CH3), −0.57 (s, 3H, Al–CH3), −0.60 (s, 3H, Al–CH3) ppm. 13C{1H} NMR (75 MHz, C6D6, 298 K): δC 151.06, 150.27, 145.73, 145.58, 142.15, 141.96, 140.37, 138.46, 138.39, 137.08, 131.62, 131.60, 131.40, 131.04, 129.85, 129.56, 129.34, 123.08, 123.04, 115.82, 21.11, 18.47, 18.41, 17.87, −4.36, −4.67 ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δP 130.60 ppm. Elemental analysis for C41H52AlN4P (658.8) (2b). Calcd for C 74.74, H 7.96, N 8.50; found C 74.58, H 7.83, N 8.42.
2c: yield: 112 mg, 75%. 1H NMR (300 MHz, C6D6, 298 K): δH 8.11 (q, J = 7 Hz, 2H, Ar–H), 7.16–7.10 (m, 6H, Ar–H), 5.82 (s, 2H, NCH), 4.20–4.06 (m, 1H, CH(CH3)2), 3.98–3.84 (m, 1H, CH(CH3)2), 1.58 (d, J = 6 Hz, 3H, CH3), 1.47 (d, J = 6 Hz, 3H, CH3), 1.19 (d, J = 6 Hz, 3H, CH3), 0.99 (s, 18H, CH3), 0.83 (d, J = 6 Hz, 3H, CH3), −0.19 (s, 6H, Al–CH3) ppm. 13C{1H} NMR (75 MHz, C6D6, 298 K): δC 150.80, 150.64, 139.38, 139.24, 137.80, 134.53, 134.32, 131.16, 124.71, 123.87, 115.21, 61.83, 31.97, 31.04, 29.06, 25.30, 25.02, 23.06, 14.37, −4.13 ppm. 31P{1H} NMR (121.5 MHz, C6D6, 298 K): δP 158.46 ppm. Elemental analysis for C31H48AlN4P (534.6) (2c). Calcd for C 69.63, H 9.05, N 10.48; found C 69.54, H 8.87, N 10.29.
General procedure for catalytic guanylation reaction
In a 25 ml Schlenk flask, aromatic primary amine (1 mmol) and carbodiimide (1 mmol) were added and 2 ml toluene was added to form a homogeneous mixture. Then complex 2a (1.5 mol%) was added to the reaction mixture as a precatalyst. The mixture was allowed to stir at room temperature for 2 h. Then the solvent was evaporated, and the residue was extracted by ether. After evaporation of the ether, the final product was obtained by washing with n-pentane. The products were characterised by 1H and 13C{1H} NMR spectroscopy and details are given in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. K. thanks CSIR India for his Ph.D. fellowships (09/1001(0038)/2018-EMR-I). Instrumental support was provided by the Indian Institute of Technology Hyderabad and TIFR Hyderabad.Notes and references
- (a) C. Alonso-Moreno, A. Antiñolo, F. Carrillo-Hermosilla and A. Otero, Chem. Soc. Rev., 2014, 43, 3406 RSC; (b) S. Raoufmoghaddam, Org. Biomol. Chem., 2014, 12, 7179 RSC; (c) W.-X. Zhang and Z. Hou, Org. Biomol. Chem., 2008, 6, 1720 RSC; (d) W.-X. Zhang, L. Xu and Z. Xi, Chem. Commun., 2015, 51, 254 RSC.
- F. Montilla, D. del Río, A. Pastor and A. Galindo, Organometallics, 2006, 25, 4996 CrossRef CAS.
- (a) S. Zhou, S. Wang, G. Yang, Q. Li, L. Zhang, Z. Yao, Z. Zhou and H.-B. Song, Organometallics, 2007, 26, 3755 CrossRef CAS; (b) H. Shen, H.-S. Chan and Z. Xie, Organometallics, 2006, 25, 5515 CrossRef CAS; (c) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497 CrossRef CAS.
- (a) X. Fu and C.-H. Tan, Chem. Commun., 2011, 47, 8210 RSC; (b) J. C. McManus, T. Genski, J. S. Carey and R. J. K. Taylor, Synlett, 2003, 3, 369 Search PubMed; (c) T. Ishikawa and T. Isobe, Chem.–Eur. J., 2002, 8, 553 CrossRef; (d) B. Kovačević and Z. B. Maksić, Org. Lett., 2001, 3, 1523 CrossRef PubMed.
- (a) P. M. Coles, Dalton Trans., 2006, 8, 985 RSC; (b) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC.
- (a) D. Castagnolo, S. Schenone and M. Botta, Chem. Rev., 2011, 111, 5247 CrossRef CAS PubMed; (b) R. G. S. Berlinck, A. E. Trindade-Silva and M. F. C. Santos, Nat. Prod. Rep., 2012, 29, 1382 RSC; (c) R. G. S. Berlinck, A. C. B. Burtoloso, A. E. Trindade-Silva, S. Romminger, R. P. Morais, K. Bandeira and C. M. Mizuno, Nat. Prod. Rep., 2010, 27, 1871 RSC.
- (a) E. J. Corey, B. Czakó and L. Kürti, Molecules and medicine, Wiley, Hoboken, NJ, 1st edn, 2007 Search PubMed; (b) C. Anselmi, A. Ettorre, M. Andreassi, M. Centini, P. Neri and A. Di Stefano, Biochem. Pharmacol., 2002, 63, 437 CrossRef CAS PubMed; (c) G. Byk, J. Soto, C. Mattler, M. Frederic and D. Scherman, Biotechnol. Bioeng., 1998, 61, 81 CrossRef CAS.
- T. P. Hopkins, J. M. Dener and A. M. Boldi, J. Comb. Chem., 2002, 4, 167 CrossRef CAS.
- T.-G. Ong, G. P. A. Yap and D. S. Richeson, J. Am. Chem. Soc., 2003, 125, 8100 CrossRef CAS PubMed.
- (a) F. Montilla, A. Pastor and A. Galindo, J. Organomet. Chem., 2004, 689, 993 CrossRef CAS; (b) D. Li, J. Guang, W.-X. Zhang, Y. Wanga and Z. Xi, Org. Biomol. Chem., 2010, 8, 1816 RSC; (c) H. Shen and Z. Xie, J. Organomet. Chem., 2009, 694, 1652 CrossRef CAS.
- (a) X. Zhu, Z. Du, F. Xu and Q. Shen, J. Org. Chem., 2009, 74, 6347 CrossRef CAS PubMed; (b) Q. Li, S. Wang, S. Zhou, G. Yang, X. Zhu and Y. Liu, J. Org. Chem., 2007, 72, 6763 CrossRef CAS; (c) Z. Du, W. Li, X. Zhu, F. Xu and Q. Shen, J. Org. Chem., 2008, 73, 8966 CrossRef CAS; (d) W.-X. Zhang, M. Nishiura and Z. Hou, Chem.–Eur. J., 2007, 13, 4037 CrossRef CAS; (e) Y. Wu, S. Wang, L. Zhang, G. Yang, X. Zhu, C. Liu, C. Yin and J. Rong, J. Organomet. Chem., 2009, 362, 2814 CAS.
- (a) C. Alonso-Moreno, F. Carrillo-Hermosilla, A. Garcés, A. Otero, I. López-Solera, A. M. Rodríguez and A. Antiñolo, Organometallics, 2010, 29, 2789 CrossRef CAS; (b) J. R. Lachs, A. G. M. Barrett, M. R. Crimmin, G. Kociok-Köhn, M. S. Hill, M. F. Mahon and P. A. Procopiou, Eur. J. Inorg. Chem., 2008, 26, 4173 CrossRef; (c) T.-G. Ong, J. S. O'Brien, I. Korobkov and D. S. Richeson, Organometallics, 2006, 25, 4728 CrossRef CAS; (d) L. Xu, Y.-C. Wang, W. Ma, W.-X. Zhang and Z. Xi, J. Org. Chem., 2014, 79, 12004 CrossRef CAS PubMed.
- (a) S. Das, J. Bhattacharjee and T. K. Panda, Dalton Trans., 2019, 48, 7227 RSC; (b) J. Bhattacharjee, S. Das, R. K. Kottalankaa and T. K. Panda, Dalton Trans., 2016, 45, 17824 RSC; (c) J. Bhattacharjee, M. Sachdeva, I. Banerjee and T. K. Panda, J. Chem. Sci., 2016, 128, 875 CrossRef CAS.
- W.-X. Zhang, D. Li, Z. Wang and Z. Xi, Organometallics, 2009, 28, 882 CrossRef CAS.
- (a) J. Koller and R. G. Bergman, Organometallics, 2010, 29, 5946 CrossRef CAS; (b) Y. Wei, S. Wang, S. Zhou, Z. Feng, L. Guo, X. Zhu, X. Mu and F. Yao, Organometallics, 2015, 34, 1882 CrossRef CAS.
- P. K. Vardhanapu, V. Bheemireddy, M. Bhunia, G. Vijaykumar and S. K. Mandal, Organometallics, 2018, 37, 2602 CrossRef CAS.
- (a) P. C. Junk and M. L. Cole, Chem. Commun., 2007, 1579–1590 RSC; (b) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC; (c) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC; (d) J. A. R. Schmidt and J. J. Arnold, J. Chem. Soc., Dalton Trans., 2002, 3454–3461 RSC; (e) C. A. Nijhuis, E. Jellema, T. J. J. Sciarone, A. Meetsma, P. H. M. Budzelaar and B. Hessen, Eur. J. Inorg. Chem., 2005, 2089–2099 CrossRef CAS; (f) D. A. Kissounko, M. V. Zabalov, G. P. Brusova and D. A. Lemenovskii, Russ. Chem. Rev., 2006, 75, 351–374 CrossRef CAS.
- (a) P. J. Bailey and S. Pace, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS; (b) A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS; (c) S. T. Barry, Coord. Chem. Rev., 2013, 257, 3192–3201 CrossRef CAS.
- (a) R. Vollmerhaus, R. Tomaszewski, P. Shao, N. J. Taylor, K. J. Wiacek, S. P. Lewis, A. Al-Humydi and S. Collins, Organometallics, 2005, 24, 494–507 CrossRef CAS; (b) B. Nekoueishahraki, H. W. Roesky, G. Schwab, D. Stern and D. Stalke, Inorg. Chem., 2009, 48, 9174–9179 CrossRef CAS PubMed; (c) N. Nebra, C. Lescot, P. Dauban, S. Mallet-Ladeira, B. Martin-Vaca and D. Bourissou, Eur. J. Org. Chem., 2013, 2013, 984–990 CrossRef CAS; (d) A. Stasch, Angew. Chem., Int. Ed., 2014, 53, 10200–10203 CrossRef CAS PubMed; (e) B. Prashanth and S. Singh, Dalton Trans., 2014, 43, 16880–16888 RSC; (f) A. L. Hawley and A. Stasch, Eur. J. Inorg. Chem., 2015, 2015, 258–270 CrossRef CAS; (g) K. A. Rufanov, N. K. Pruss and J. Sundermeyer, Dalton Trans., 2016, 45, 1525–1538 RSC; (h) S. Wingerter, M. Pfeiffer, A. Murso, C. Lustig, T. Stey, V. Chandrasekhar and D. Stalke, J. Am. Chem. Soc., 2001, 123, 1381–1388 CrossRef CAS; (i) A. Stasch, Chem.–Eur. J., 2012, 12, 15105–15112 CrossRef PubMed; (j) T. J. Feuerstein, B. Goswami, P. Rauthe, R. Koppe, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. Sci., 2019, 10, 4742–4749 RSC.
- (a) C. Fedorchuk, M. Copsey and T. Chivers, Coord. Chem. Rev., 2007, 251, 897–924 CrossRef CAS; (b) A. M. Corrente, M. Andrea and T. Chivers, Inorg. Chem., 2010, 49, 2457–2463 CrossRef CAS PubMed; (c) S. Harder and D. Naglav, Eur. J. Inorg. Chem., 2010, 18, 2836–2840 CrossRef; (d) S. Harder, Dalton Trans., 2010, 39, 6677–6681 RSC; (e) S. Harder, B. Freitag, P. Stegner, J. Pahl and D. Naglav, Z. Anorg. Allg. Chem., 2015, 641, 2129–2134 CrossRef CAS; (f) T. Chivers, D. J. Eisler, C. Fedorchuk, G. Schatte, H. M. Tuononen and R. T. Boere, Inorg. Chem., 2006, 45, 2119–2131 CrossRef CAS PubMed; (g) T. Chivers, D. J. Eisler, C. Fedorchuk, G. Schatte, H. M. Tuononen and R. T. Boere, Chem. Commun., 2005, 3930–3932 RSC.
- (a) S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed; (b) S. J. Bonyhady, D. Collis, G. Frenking, N. Holzmann, C. Jones and A. Stasch, Nat. Chem., 2010, 2, 865–869 CrossRef CAS PubMed; (c) S. S. Sen, A. Jana, H. W. Roesky and C. Schulzke, Angew. Chem., Int. Ed., 2009, 48, 8536–8538 CrossRef CAS PubMed; (d) S. Nagendran, S. S. Sen, H. W. Roesky, D. Koley, H. Grubmüller, A. Pal and R. Herbst-Irmer, Organometallics, 2008, 27, 5459–5463 CrossRef CAS; (e) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS; (f) S. P. Green, C. Jones, P. C. Junk, K.-A. Lippert and A. Stasch, Chem. Commun., 2006, 3978–3980 RSC; (g) M. K. Bisai, V. S. V. S. N. Swamy, T. Das, K. Vanka, R. G. Gonnade and S. S. Sen, Inorg. Chem., 2019, 58, 10536–10542 CrossRef CAS PubMed; (h) V. S. V. S. N. Swamy, K. V. Raj, K. Vanka, S. S. Sen and H. W. Roesky, Chem. Commun., 2019, 55, 3536–3539 RSC; (i) S. Pahar, S. Karak, M. Pait, K. V. Raj, K. Vanka and S. S. Sen, Organometallics, 2018, 37, 1206–1213 CrossRef CAS.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523–530 RSC.
- (a) M. Tamm, S. Randoll, T. Bannenberg and E. Herdtweck, Chem. Commun., 2004, 876–877 RSC; (b) M. Tamm, S. Beer and E. Herdtweck, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1497–1504 CrossRef CAS; (c) M. Tamm, S. Randoll, E. Herdtweck, N. Kleigrewe, G. Kehr, G. Erker and B. Rieger, Dalton Trans., 2006, 459–467 RSC; (d) S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg and M. Tamm, Angew. Chem., Int. Ed., 2007, 46, 8890–8894 CrossRef CAS PubMed; (e) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones and M. Tamm, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed; (f) S. H. Stelzig, M. Tamm and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6064–6070 CrossRef CAS; (g) S. Beer, K. Brandhorst, C. G. Hrib, X. Wu, B. Haberlag, J. Grunenberg, P. G. Jones and M. Tamm, Organometallics, 2009, 28, 1534–1545 CrossRef CAS.
- (a) A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS; (b) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS; (c) T. K. Panda, S. Randoll, C. G. Hrib, P. G. Jones, T. Bannenberg and M. Tamm, Chem. Commun., 2007, 5007–5009 RSC; (d) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones and M. Tamm, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed; (e) D. Petrovic, L. M. R. Hill, P. G. Jones, W. B. Tolman and M. Tamm, Dalton Trans., 2008, 887–894 RSC; (f) D. Petrovic, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Organometallics, 2008, 27, 778–783 CrossRef CAS; (g) S. Beer, K. Brandhorst, C. G. Hrib, X. Wu, B. Haberlag, J. Grunenberg, P. G. Jones and M. Tamm, Organometallics, 2009, 28, 1534–1545 CrossRef CAS; (h) T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462–5472 CrossRef CAS PubMed; (i) A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed; (j) T. K. Panda, C. G. Hrib, P. G. Jones and M. Tamm, J. Organomet. Chem., 2010, 695, 2768–2773 CrossRef CAS; (k) M. Tamm, A. G. Trambitas, C. G. Hrib and P. G. Jones, Terrae Rarae, 2010, 7, 1–4 Search PubMed; (l) A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753–6761 CrossRef CAS PubMed; (m) I. Karmel, M. Botoshansky, M. Tamm and M. S. Eisen, Inorg. Chem., 2014, 53, 694–696 CrossRef CAS PubMed; (n) S. Das, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 16812 RSC.
- N. Kuhn, M. Göhner, M. Grathwohl, J. Wiethoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS.
- T. K. Panda, C. G. Hrib, P. G. Jones, J. Jenter, P. W. Roesky and M. Tamm, Eur. J. Inorg. Chem., 2008, 2008, 4270 CrossRef.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858–861 RSC.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- F. Dielmann, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 14071–14073 CrossRef CAS PubMed.
- F. Dielmann, D. M. Andrada, G. Frenking and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 3800–3802 CrossRef CAS PubMed.
- T. Ochiai and S. Inoue, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 624–627 CrossRef CAS.
- T. Ochiai, D. Franz, X. Wu and S. Inoue, Dalton Trans., 2015, 44, 10952–10956 RSC.
- D. Franz, T. Szilvasi, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037–10042 CrossRef CAS PubMed.
- D. Franz, E. Irran and S. Inoue, Dalton Trans., 2014, 43, 4451–4461 RSC.
- D. Franz and S. Inoue, Chem.–Eur. J., 2014, 20, 10645–10649 CrossRef CAS PubMed.
- M. W. Lui, N. R. Paisley, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2016, 22, 2134–2145 CrossRef CAS PubMed.
- M. W. Lui, C. Merten, M. J. Ferguson, R. McDonald, Y. Xu and E. Rivard, Inorg. Chem., 2015, 54, 2040–2049 CrossRef CAS PubMed.
- A. D. K. Todd, W. L. McClennan and J. D. Masuda, RSC Adv., 2016, 6, 69270–69276 RSC.
- H. Liu, M. Khononov, N. Fridman, M. Tamm and M. S. Eisen, Inorg. Chem., 2019, 58, 13426–13439 CrossRef CAS PubMed.
- (a) S. Anga, J. Acharya and V. Chandrasekhar, J. Org. Chem., 2021, 86, 2224 CrossRef CAS PubMed; (b) S. Anga, H. Karmakar, T. K. Panda and V. Chandrasekhar, J. Organomet. Chem., 2021, 954–955, 122091 CrossRef CAS.
- (a) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS; (b) T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC , see also references cited therein..
Footnote |
| † Electronic supplementary information (ESI) available: Text giving experimental details for the catalytic reactions, 1H, 13C{1H} and spectra of guanidines/guanylation products 3a–3v, ligands 1b, 1c and aluminium metal complexes 2a, 2b, 2c. CCDC 2124039–2124043. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ra00242f |
| This journal is © The Royal Society of Chemistry 2022 |