DOI:
10.1039/D2RA00199C
(Paper)
RSC Adv., 2022,
12, 10088-10096
Kinetics and mechanisms of flumequine degradation by sulfate radical based AOP in different water samples containing inorganic anions†
Received
11th January 2022
, Accepted 24th March 2022
First published on 30th March 2022
Abstract
Many studies have reported that hydroxyl radical (HO˙) driven advanced oxidation processes (AOPs) could degrade fluoroquinolones (FQs) antibiotics effectively. Compared with HO˙, sulfate radical (SO4˙−) shows a similar oxidation capacity but a longer half-life. SO4˙− could cause chain reactions and resulted in the generation of halogen radicals and carbonate radicals from the main anions in sea water including Cl−, Br− and HCO3−. However, few studies were focused on the degradation of FQs in marine aquaculture water and seawater, as well as the bioaccumulation of transformation products. As a typical member of FQs, flumequine (FLU) was degraded by UV/peroxodisulfate (PDS) AOPs in synthetic fresh water, marine aquaculture water and seawater. The reaction rate constants in the three water samples were 0.0348 min−1, 0.0179 min−1 and 0.0098 min−1, respectively. The reason was attributed to the inhibition of the anions as they could consume SO4˙− and initiate the quenching reaction of free radicals. When the pH value increased from 5 to 9, the reaction rate decreased from 0.0197 min−1 to 0.0066 min−1. The energy difference between HOMO and LUMO of FLU was calculated to be 8.07 eV indicating that FLU was a stable compound. The atoms on quinolone ring of FLU with high negative charge would be more vulnerable to attack by free radicals through electrophilic reactions. Two possible degradation pathways of FLU were inferred according to the degradation products. Preliminary bioaccumulation analysis of transformation products by the EPI suite software proved that the values of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow and logBCF of the final product P100 were less than those of FLU and the intermediates.
Kow and logBCF of the final product P100 were less than those of FLU and the intermediates.
1. Introduction
Fluoroquinolones (FQs) antibiotics have been widely used in veterinary medicine, animal husbandry and commercial aquaculture due to their potent antibacterial activity.1,2 However, these compounds have been continuously introduced into the environment and caused direct ecological risks impacts.3 As their stability and biological resistance, most antibiotics cannot be efficiently degradated by traditional biological water treatment processes.4,5 It is necessary to develop an efficient process for FQs removal because of the low degradation efficiencies by the traditional waste water treatments plants.
As a member of FQs group, flumequine (FLU) has been frequently detected in various waterbodies at concentrations ranging from ng L−1 to μg L−1.6,7 The degradations of FLU by different methods have been investigated in the past few years. Lou et al. indicated that the flumequine efficiency removal reached 94% by coupling of heterogeneous photocatalysis with ozonation and hydrogen peroxide.8 Harrabi et al. found that the degradation of FLU was improved by the presence of hydroxyl radicals and sulfate radicals.9 A study indicated that the effective removal of flumequine by combined UV/ClO2 advanced oxidation processes (AOPs).10 Solis et al. found that the enhanced elimination of flumequine in the presence of peroxymonosulfate (PMS) and magnetic graphene catalysts.11 Feng et al. pointed out that multiple additions of Fe(VI) improved the degradation efficiency of FLU, and provided greater degradation than a single addition of Fe(VI).12 Zeng et al. indicated that the TiO2/AN sample exhibited high photocatalytic performance under simulated sunlight irradiation, and 100% removal efficiency of FLU was achieved after 4 hours of illumination.13 Ashrafi et al. found that the effective removal of FLU from aqueous solution by laccase from Trametes versicolor, and the removal rate can reach 77.76% under the optimum conditions.14 Many studies have reported that hydroxyl radical (HO˙, E0 = 2.5–3.1 V, <1 μs) driven advanced oxidation processes (AOPs) such as photocatalytic and Fenton processes could degrade FLU effectively.15,16 Compared with HO˙, sulfate radical (SO4˙−) shows a similar oxidation capacity (E0 = 1.9–2.8 V) but a longer half-life (30–40 μs).17–19 Moreover, the radical precursors peroxodisulfate (PDS) and peroxymonosulfate (PMS) are stable and easily activated.20 As a potential alternative for traditional AOPs, SO4˙− based AOP has been proven to be effective for the removal of antibiotics.21,22
SO4˙− is generated through homolysis or heterolysis of the peroxide (O–O) bonds in persulfate anions via electron or energy transfer.23 There are various methods of activating PS/PMS to produce SO4˙−, such as ultraviolet (UV) irradiation, heating, ultrasound, transition metal catalysis, nanostructured carbons,alkali activation and carbon based material activation.24 Among them, ultraviolet (UV) irradiation is environmentally friendly and commonly used as the activation methods (eqn (1) and 2).25,26 At present, UV/PMS has been widely used in degradation of various antibacterial agents in water and wastewater, such as the chloramphenicol,27 CIP and AMO,28 sulfaquinoxaline,29 norfloxacin,30 ofloxacin,31 ciprofloxacin, metronidazole (MNZ),32 sulfamethoxazole (SMX) and sulfamethoxypyridazine (SMP).33 The UV/PDS (PMS) process has been reported to be a possible method for removing antibiotics including FLU. For example, Qi et al. reported that FLU was able to be degraded by UV/PMS in different water matrices, followed the order of tap water > ultrapure water > river water > secondary clarifier effluent, and the presence of HCO3− could inhibited FLU removal, and Cl− concentrations did not significantly affect the reaction. The results showed that UV/PMS is a feasible technology for treating FLU in waters and wastewaters.34 On the contrast, Cl− could inhibited FLU removal in our experiments. Therefore, it is necessary to explore the degradation of FLU in synthetic fresh water, marine aquaculture water and seawater and their constituents.
Some studies suggest that the addition of persulfate to water is not pernicious to humans. For example, the effective removal of 2-methylisoborneol and 2-methylisoborneol-producing algae by sulfate radical-based technology in drinking water sources.35
Considering that FLU has been directly applied as feed additive in marine aquaculture, the process of FLU degradation may be different as the water was a mixture of seawater and fresh water at ratio of 1:2.36 SO4˙− was reported to be more susceptible to halide scavenging than that of HO˙. Consequently, SO4˙− could cause chain reactions and resulted in the generation of halogen radicals and carbonate radicals from the main anions in sea water including Cl−, Br− and HCO3− (eqn (3)–(7)).37,38 Such as HO˙, SO4˙− can relatively oxidize all organic matters without selectivity with near-diffusion-limited rate constants. However, the oxidation abilities of halogen radicals (except for Cl˙) to organic matters are selective, for example, they can react faster with certain organic compounds (e.g., alkenes, aromatics, and organothiols) (k = 106 to 1010 M−1 s−1) than other organic compounds (e.g., aliphatic molecules) (k = 103 to 106 M−1 s−1).39 Therefore, the new second generation radicals may also be responsible for the degradation of FLU due to its complex structure. Our previous studies investigated the degradation of sulfamethoxazole (SMX) and ofloxacin (OFL) by UV/PDS in different water samples.31,33 The results indicated that the reaction rates for both SMX and OFL followed the order of synthetic seawater > synthetic marine aquaculture water > freshwater. Anions in seawater showed different effects on SMX and OFL degradation. Therefore, the effects of main anions in seawater on FLU degradation of SO4˙− based AOP need to be further investigated. Furthermore, the toxicity evaluation of the degraded products is also very necessary for the food safety of seafood.
| |
 | (1) |
| |
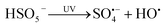 | (2) |
| | |
SO4˙− + Cl− → Cl˙ + SO42− k = 2.7 × 108 M−1 s−1
| (3) |
| | |
Cl˙ + Cl− → Cl2˙− k = 8 × 109 M−1 s−1
| (4) |
| | |
Cl˙ + Cl2˙− → Cl2 + Cl−
| (5) |
| | |
SO4˙− + Br− → Br˙ + SO42− k = 3.5 × 109 M−1 s−1
| (6) |
| | |
Br˙ + Br− → Br2˙− k = 1.2 × 1010 M−1 s−1
| (7) |
| | |
Br˙+Br2˙− → Br2 + Br−
| (8) |
| |
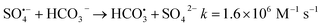 | (9) |
| | |
SO4˙− + HO− → HO˙ + SO42− k = 6.5 × 107 M−1 s−1
| (10) |
| | |
HO˙ + HCO3− → CO3˙− + H2O
| (11) |
This study aimed to investigate the degradation of FLU in UV/PDS system in different water samples including seawater, marine aquaculture water and freshwater. The effects of water quality parameters especially the concentrations of the anions in seawater on FLU degradation were tested. The transformation pathways of FLU and the oxidation products were analyzed by theoretical calculation and LC-MS/MS.40 The corresponding bioaccumulations of the oxidation products were also estimated by EPI-suite.41
2. Materials and methods
2.1. Materials and water samples
FLU (99%) was purchased from Shanghai Yuanye Biological Company. Sodium persulfate (Na2S2O8), sodium chloride (NaCl), sodium sulfate (Na2SO4), sodium bromide (NaBr) and sodium bicarbonate (NaHCO3) were analytical-reagent grade purchased from Chengdu Jinshan Chemical Reagent Company. HPLC grade formic acid, acetonitrile and methanol (CH3OH) were purchased from Thermo Fisher Scientific. Ultrapure water obtained from the Millipore Milli-Q Ultrapure Gradient system was used to prepare all solutions. The essential anions including Cl− (19 g L−1), Br− (65 mg L−1), HCO3− (152 mg L−1) and SO42− (2.74 g L−1) were added into ultrapure water to prepare the synthetic seawater according the components of actual seawater. The synthetic marine aquaculture was prepared by synthetic seawater and ultrapure water in a ratio of 2:1. Therefore, the concentrations of the essential anions were approximately one-third of that in synthetic seawater. The pH of all samples was adjusted to 7 with phosphate buffer.
2.2. Experimental procedures
The UV/PDS photo-reactor was purchased from Shanghai Bilang Instruments and Equipment Company. The diameter and length of the quartz reaction tube was 2.5 cm and 18 cm, respectively. The UV light source is a high-pressure mercury lamp with a light power of 240 W. The initial concentration of FLU was set at 0.05 mM to analyze the reaction rates and products accurately. Different concentration of PDS was added and mixed with FLU. About 1 mL pure methanol was added to terminate the reaction at the set time periodically. After the reaction was quenched, the reaction solutions were collected and followed by filtration and quantitative detection. In the kinetics studies, the effects of water quality, anions and pH on the reaction were studied. When the effect of different inorganic anions on the degradation of FLU was investigated individually, the concentrations of the other three ions did not changed. All kinetic experiments were carried out in a 100 mL reaction tube.31 The degradation kinetic of FLU in UV/PDS system was described by a pseudo first-order kinetic model. All kinetic experiments were performed at least in duplicate.
2.3. Analytical methods
Ultra-high-performance liquid chromatography (UPLC, Agilent, USA) system equipped with a Zorbax RRHD Eclipse plus C18 column (2.1 mm × 100 mm, 1.8 μm), a quaternary gradient pump, a UV diode array detector and a fluorescence detector was used to determine the concentrations of FLU. The mobile phase was a mixture of formic acid (0.1%) and acetonitrile at a ratio of 70:30 (v/v%) with a flow rate of 0.35 mL min−1. FLU was detected at 234 nm.
2.4. Identification of the transformation products
ESI-high resolution mass spectrometry was used to identify the oxidation products. The accurate mass, isotope patterns and MS/MS patterns were obtained from a Thermo Fisher LC-MS/MS (Q-Exactive) system equipped with a Zorbax SB-C18 column (2.4 mm × 150 mm, 5 mm), a diode-array UV/vis detector and a mass spectrometer. The gradient elution was performed with a mixture of acetonitrile and 0.2% formic acid at a flow rate of 0.30 mL min−1. The oxidation products were identified in a positive electrospray ionization mode (ESI+) with a mass scan range from 50 to 1000. The Thermo Xcalibur Qual Browser and ChemSpider databases were used to infer the structures and molecular formula of the unknown products.
3. Results and discussion
3.1. Oxidation of FLU by UV/PDS in different water samples and reaction kinetics
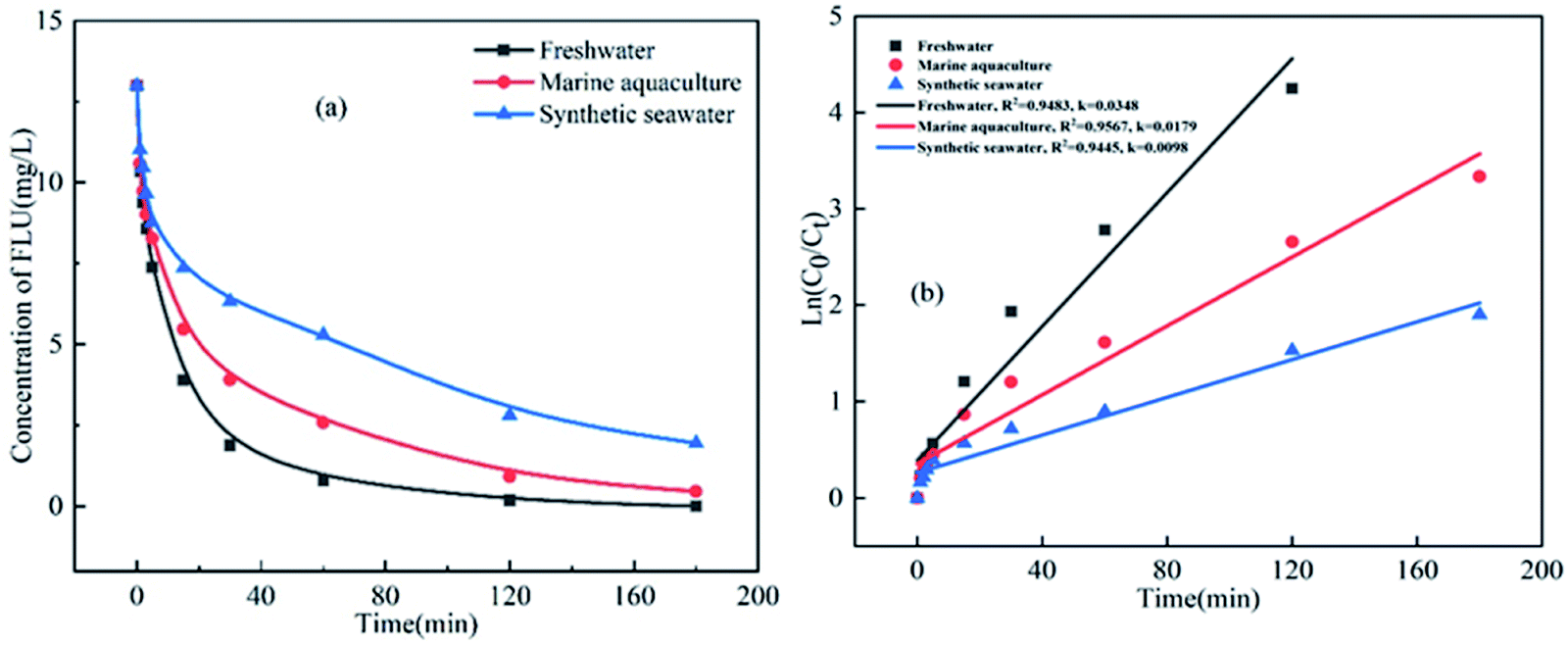
The oxidation kinetics of FLU by UV/PDS in three water samples including fresh water, synthetic marine aquaculture water and synthetic seawater were compared. As shown in Fig. 1, the concentrations of FLU decreased continuously with time at different rates. After 3 hours of reaction, FLU was almost completely degraded in fresh water. While the degradation rates of FLU in synthetic mariculture water and seawater were 96% and 85%, respectively. In the previous study, we found that FLU could not be degraded by NaClO and showed almost no reaction.42 Pseudo-first-order rate constants were calculated by the slopes of the fitted linear plots of ln([FLU]) versus reaction time t (Fig. 1b). It can be seen that the reaction rate constants in the three water samples were 0.0348 min−1, 0.0179 min−1 and 0.0098 min−1, respectively, followed the order of fresh water > synthetic mariculture water > synthetic seawater, which was contrary to the degradation law of OFL and SMX by UV/PDS in our previous research. As the main cations including sodium ions (Na+) and potassium ions (K+) in sea water were proved to have no impact on the degradation of organic compound by UV/PDS.34 The reason may be attributed to the high concentration of anions (e.g., Cl−, Br−, HCO3−) in the mariculture water and seawater, which showed promoting or inhibiting effects on the FLU degradation by the second-generation radicals such as Cl˙, Br˙ and 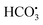 .43–45 Thus, the anions in the water would greatly affect the degradation of FLU.
.43–45 Thus, the anions in the water would greatly affect the degradation of FLU.
 |
| | Fig. 1 Oxidation of FLU by UV/PDS in different water samples (a) and the linear fit of the kinetic data (b) (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, pH = 7, T = 298.15 K). | |
Additionally, organic matter also has an effect on the degradation of FLU in different water samples. Qi et al. investigated the humic acid can affect the NP and TCS removal, as the HA concentration (>1 mg L−1) increased, the inhibition of NP degradation becomes enhanced.46 Peng et al. pointed out that NOMs (e.g., SRHA and PLFA) adversely affected MeP removal by Co/CNTs + PMS system. The reason may be attributed to the organic matters can affect the oxidation reaction based on radicals through the scavenging effect and competing with the target contaminants for the oxidant radicals.47 On the other hand, organic matters might restrict the interaction of PMS with catalytic active sites. Thus, the anions and organic matters in the water would greatly affect the degradation of FLU.
3.2. Effects of coexisting inorganic anions on the oxidation of FLU by UV/PDS
To analyze the effects of coexisting inorganic anions on the oxidation of FLU by UV/PDS, the reaction kinetics were studied in water samples containing different amounts of inorganic anions including Cl−, Br−, HCO3−and SO42−. The concentrations of these inorganic anions were set according to their actual concentrations in various water samples. For example, the Cl− concentrations in fresh water, marine aquaculture water and seawater were 0 g L−1, 6.6 g L−1 and 19 g L−1, respectively. It can be seen from Fig. 2a that when the concentration of Cl− increased from 0 g L−1 to 19 g L−1, the degradation efficiency of FLU decreased by 65%. The reaction rates were 0.0348 min−1, 0.0178 min−1 and 0.0120 min−1. The results indicated that Cl− obviously inhibited the reaction and slowed down the oxidation rate of FLU. The phenomenon was consistent with the degradation of SMX, OFL and chloramphenicol by the UV/PDS system which could be attributed to the transformation of Cl− in the UV/PDS system as shown in eqn (3)–(5).27,31,33 Therefore, the inhibitory effect of Cl− on the degradation efficiency of FLU could be attributed to: Cl− could capture SO4˙− to generate Cl˙ (E0 = 2.40 V) and Cl2˙− (E0 = 2.0 V) with oxidation properties weaker than that of SO4˙−. Moreover, the quenching reaction between Cl˙ and Cl2˙− (eqn (5)) led to the reduction of the amounts of free radicals in the reaction system.
 |
| | Fig. 2 Oxidation of FLU by UV/PDS at different concentrations of Cl− (a) and the linear fit of the kinetic data (b) (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, pH = 7, T = 298.15 K). | |
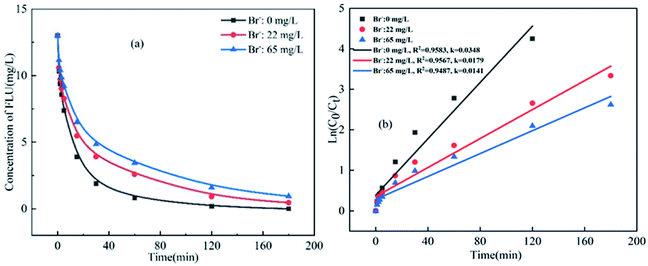 |
| | Fig. 3 Oxidation of FLU by UV/PDS at different concentrations of Br− and the linear fit of the kinetic data (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, pH = 7, T = 298.15 K). | |
The effect of Br− on the degradation of FLU was the same as that of Cl− (Fig. 3). It can be seen that the degradation efficiency of FLU decreased gradually with the increase of Br−. When the concentration of Br− increased from 0 mg L−1 (in fresh water) to 65 mg L−1 (in sea water), the reaction rate decreased from 0.0348 min−1 to 0.0141 min−1. The results showed that Br− inhibited the degradation of FLU by UV/PDS as reported by Luca et al.48 This may be due to the fact that the transformation of Br− and the properties of FLU, the new radical Br˙ (E0 = 2.00 V) and Br2˙− were formed by Br− reacted with SO4˙−, the generated radicals not only quenched each other, but also reacted with organic matters with at a slower rate than of SO4˙−.48
HCO3− is an important component in seawater with a concentration of about 0.142 g L−1 and the concentration in marine aquaculture water is about 0.047 g L−1. As shown in Fig. 4, the degradation efficiency of FLU decreased gradually with the increase of HCO3−. In the study of Wang et al.,49 HCO3− also inhibited the degradation of ciprofloxacin by BiFeO3/PDS. The reason may be due to that HCO3− was considered as a strong free radical scavenger for both SO4˙− and HO˙.50,51 The HCO3− reacted with SO4˙− and HO˙ (eqn (9)–(11)) to form CO3˙− and HCO3˙ with weak oxidability resulting in a decrease of the total amount of free radicals in the reaction system. On the other hand, the pH value of the three water samples were different (6.6, 7.6 and 8.4 for simulated fresh water, marine aquaculture water and seawater, respectively) as HCO3− could be hydrolyzed.
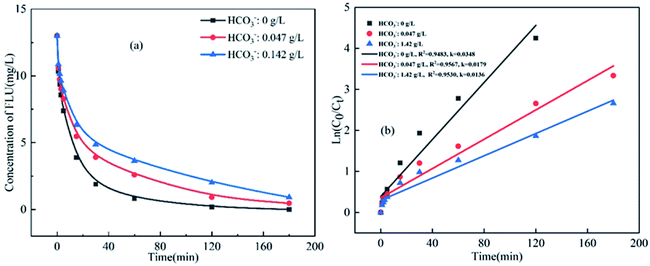 |
| | Fig. 4 Oxidation of FLU by UV/PDS at different concentrations of HCO3− and the linear fit of the kinetic data (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, pH = 7, T = 298.15 K). | |
There is high concentration of SO42− in mariculture water and seawater. Thus, the influence of SO42− on the degradation of FLU in UV/PS was also studied. As shown in Fig. 5, the degradation efficiency of FLU was not significantly affected by SO42−. In the different concentrations of SO42−, the degradation rate of FLU was 96% within 180 min. When the concentration of SO42− increased from 0.0903 g L−1 to 2.71 g L−1, the reaction rates were 0.0179 min−1 and 0.0182 min−1, respectively. The phenomenon was consistent with the degradation of OFL.31 The much slower reaction of SO42− with radicals such as ˙OH and SO4˙− compared to other inorganic anions (Cl−, Br−, HCO3−). Thus, the neglectable effects of SO42− on the degradation of FLU can be attributed to the fact that SO42− neither participated in the UV/PDS reaction nor changed the characteristics of the solution.
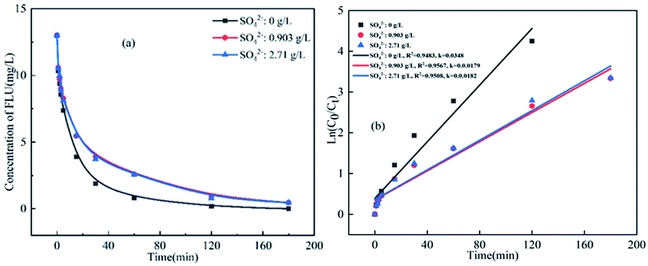 |
| | Fig. 5 Oxidation of FLU by UV/PDS at different concentrations of SO42− and the linear fit of the kinetic data (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, pH = 7, T = 298.15 K). | |
3.3 Effects of pH on the degradation of FLU
The pH value of reaction is a critical parameter influencing the progress of oxidation. Thus, the effects of different pH on the reaction rate of UV/PDS degradation of FLU were studied. Obviously, pH has a important effect on the degradation of FLU by UV/PMS. As shown in Fig. 6, the degradation efficiency of FLU decreased gradually with the increase of pH value. When the pH value increased from 5 to 9, the reaction rate decreased from 0.0197 min−1 to 0.0066 min−1. This phenomenon was consistent with the effect of HCO3− on degradation of FLU.
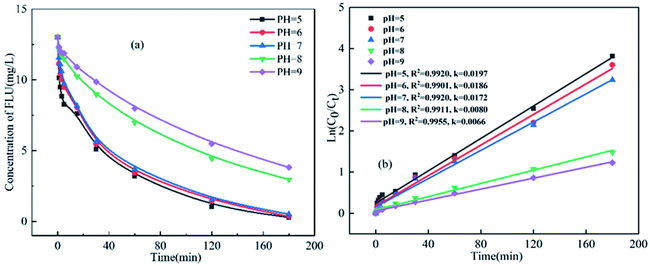 |
| | Fig. 6 Oxidation of FLU by UV/PDS at different pH values and the linear fit of the kinetic data (reaction conditions: [FLU] = 0.05 mM, 50 mL, [PDS] = 20 mM, 1 mL, light power = 240 W, T = 298.15 K). | |
The change of reaction rate with pH value may be explained from the following aspects: (1) in alkaline solution, SO4˙− reacted with OH− to produce HO˙ with a lower redox potential (eqn (10)). In addition, the coexistence of SO4˙− and HO˙ would lead to mutual quenching which reduced the removal rate of FLU. (2) It was reported that the redox potential of SO4˙− under acidic conditions was higher than that under alkaline conditions.52 (3) FLU is an amphoteric antibacterial compound, which contains amino possessing a positive charge and carboxyl groups with a negative charge. The morphology of FLU would change with different pH values of the solution. Under acidic conditions, zwitterionic functional groups would be protonated, resulting in positive charge on the piperazine ring, the electrostatic attraction between positively charged FLU+ and negatively charged SO4˙− made the oxidation of FLU by UV/PDS easier. On the contrary, due to the negative charge of FLU in alkaline solution, the interaction between FLU− and negatively charged SO4˙− increased the repulsive force that resulted in the slowdown of the degradation rate. Thus, more and more FLU in protonated form were formed with the decrease of the pH, which was advantageous to the oxidation of FLU by free radicals, especially by SO4˙−.
To sum up, the increase of pH will not only change the morphology of FLU, but also affect the composition and quantity of free radicals in the solution, thus affecting the degradation rate of FLU by UV/PDS in many ways.
3.4 Product identification and path way analysis
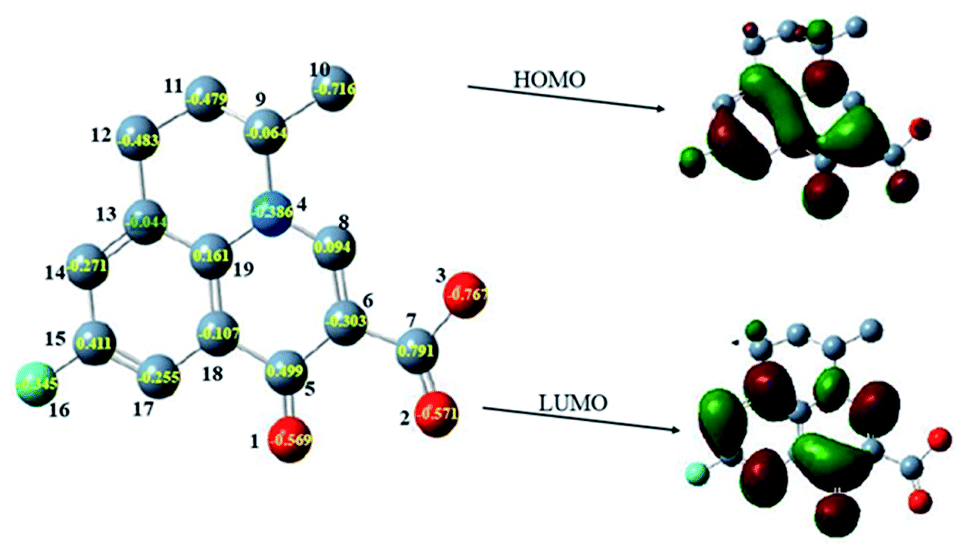
The oxidation products were identified on the basis of the chemical structures and characteristics of parent compound FLU first. The density functional theory (DFT) has been widely used as it can obtain the physicochemical properties of compounds to reveal their changing tendencies.53 A large number of researchers have used DFT to calculate and predict the environmental behavior of organic pollutants, as well as conduct risk assessment on the toxicity of transformation products.54 The quantum chemical parameters including the frontier orbital characteristics and charge distribution of FLU were calculated by Gaussian09 program according to DFT. As it was difficult to extract electrons from a low-lying highest occupied molecular orbital (HOMO) and add electrons to a high-lying lowest unoccupied molecular orbital (LUMO), the degradation potential of FLU was preliminarily analyzed by the difference calculation of quantum chemical parameters between EHOMO and ELUMO (ΔE). A large value of ΔE indicated a high kinetic stability and low chemical reactivity.55 ΔE was calculated to be 8.07 eV which was much higher than that of other fluoroquinolones indicating that FLU is very stable. The main reaction active sites on FLU molecules were determined according to the net charge distribution by natural bond orbital analysis. As shown in Fig. 7, the electrons of HOMO and LUMO were mainly distributed on quinolone group indicating that the quinolone ring would easy to be attacked not only by electrophilic reagent but also nucleophilic reagent. The charge values of the atoms C6, O1, O2, O3 and N4 on the quinolone group were calculated to be −0.386 eV, −0.569 eV, −0.571 eV, −0.767 eV and −0.303 eV, respectively. These atoms with high negative charge would be more vulnerable to attack by free radicals through electrophilic reactions.
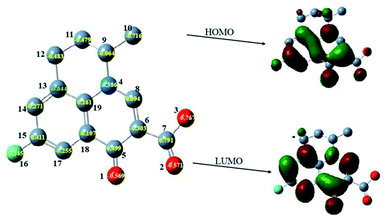 |
| | Fig. 7 The HOMO, LUMO and net charge distribution of FLU. | |
The transformation products were identified by the accurate masses obtained from LC-MS/MS combined with the reaction active sites analysis. One final product and three intermediate products were identified, while no halogenated compound was found in marine aquaculture water and seawater. Based on the above transformation products and reaction sites analysis, the degradation of FLU was inferred including two possible pathways G and H (Fig. 8). Both the two pathways had the same intermediate product p234 (M/Z = 234) which was formed by the oxidation of carboxyl (–COOH) of FLU to hydroxyl group (–OH). Through the pathway G, hydroxyl group (–OH) of p234 was oxidized to form p220-1 (M/Z = 200). In pathway H, the C–N bond of p234 were attacked and broken to form p220-2 (M/Z = 200). And then the quinolone groups on P220-1 and p220-2 were oxidized to form the final product P100 (M/Z = 100). The reaction products and pathways were the same in the three water bodies.
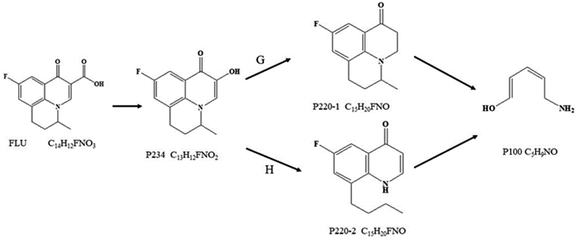 |
| | Fig. 8 Possible degradation pathway of FLU by UV/PDS system in freshwater, marine aquaculture water and seawater. | |
3.5 Preliminary analysis of bioaccumulations
The bioaccumulation characteristics of the degradation products were analyzed by Epi suite software. LogKow and logBCF were selected as the representative indexes. The compound with greater values of LogKow and logBCF indicated that it would be more easily absorbed by cellular organisms and dissolved in nonpolar media. The calculation results were shown in Table 1. The logKow and logBCF of FLU was 1.60 and 0.579, respectively. LogKow of the three intermediates p234, p220-1 and p220-2 was 2.50, 3.28 and 3.51, respectively. LogBCF of the three intermediates was 0.731, 1.248 and 1.401, respectively. LogKow and logBCF of the final degradation product P100 were both less than 0. The results showed that the values of LogKow and logBCF of the three intermediates were greater than those of FLU, indicating that compared with FLU, the three intermediates were easier to accumulate and more toxic in organisms. The values of logKow and logBCF of the final product P100 were less than those of FLU and the intermediates, indicating that P100 exhibited weak bioaccumulation and toxicity. The above results suggested that UV/PDS is suitable for the application of FLU degradation in fresh water, marine aquaculture water, synthetic seawater.
Table 1 Bioaccumulations of FLU and the oxidation products
| Compound |
Structure |
LogKow |
LogBCF |
| FLU C14H12FNO3 |
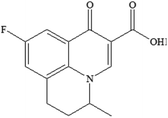 |
1.60 |
0.579 |
| P234 C13H12FNO2 |
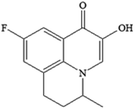 |
2.50 |
0.731 |
| P220-1 C15H20FNO |
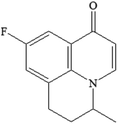 |
3.28 |
1.248 |
| P220-2 C15H20FNO |
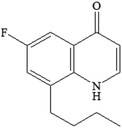 |
3.51 |
1.401 |
| P100 C5H9NO |
 |
<0 |
<0 |
4. Conclusions
As a member of FQs group, FLU was degraded by UV/PDS in synthetic freshwater, marine aquaculture water and seawater with different amounts of inorganic anions. The results showed that the reaction rate constants of FLU in the three water samples followed the order of fresh water > marine aquaculture water > synthetic seawater. The anions with higher concentrations in seawater including Cl−, Br− and HCO3− inhibited the degradation of FLU. The reason should be partly attributed to that the anions could capture SO4˙− to generate new reactive radicals such as Cl˙, Br˙ and CO3˙− with oxidation properties weaker than that of SO4˙−. The degradation efficiency of FLU was neglectable affected by SO42−, which can be attributed to the fact that SO42− neither participated in the UV/PDS reaction nor changed the pH of the solution. The degradation efficiency of FLU decreased gradually with the increase of pH value. The quenching reaction of free radicals, the difference of redox potential of SO4˙− and the morphology of FLU under different pH values should be responsible for the effect of pH on the degradation of FLU. The main reaction active sites on FLU molecules were the atoms C6, O1, O2, O3 and N4 on the quinolone group. Three intermediate products with greater values of logKow and logBCF were identified. The final product P100 exhibited weak bioaccumulation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 51668005), the Open fund of Guangxi Key Laboratory of Clean Pulp & Papermaking and Pollution Control (No. KF201719), the Innovation Project of Guangxi-Graduate Education (No. YCSW2019029), the Natural Science Foundation of Guangxi Province (No. 2020GXNSFAA159135) and the Opening Foundation of Key Laboratory of Environment Change and Resources Use in Beibu Gulf Ministry of Education (Nanning Normal University) (No. NNNU-KLOP-K1937).
References
- Y. J. Gou, P. Chen, L. Yang, S. J. Li, L. Peng, S. X. Song and Y. F. Xu, Chemosphere, 2021, 270, 129481 CrossRef CAS PubMed.
- C. Wang, J. Zhang, J. Y. Du, P. F. Zhang, Z. W. Zhao, W. X. Shi and F. Y. Cui, J. Hazard. Mater., 2021, 416, 125893 CrossRef CAS PubMed.
- G. Y. Ding, G. L. Chen, Y. D. Liu, M. Li and X. Liu, Sci. Total Environ., 2020, 738, 139802 CrossRef CAS PubMed.
- E. Aghdam, Y. Y. Xiang, J. L. Sun, C. Shang, X. Yang and J. Y. Fang, J. Environ. Sci., 2017, 58, 146–154 CrossRef CAS PubMed.
- Q. Q. Cai and J. Y. Hu, Water Res., 2018, 140, 251–260 CrossRef CAS PubMed.
- Z. Q. Ye, H. S. Weinberg and M. T. Meyer, Anal. Chem., 2007, 79, 1135–1144 CrossRef CAS PubMed.
- C. Wang, J. Y. Du, Z. J. Liang, J. L. Liang, Z. W. Zhao, F. Y. Cui and W. X. Shi, J. Hazard. Mater., 2022, 422, 126884 CrossRef CAS PubMed.
- W. R. Lou, A. Kane, D. Wolbert, S. Rtimi and A. A. Assadi, Chem. Eng. Process., 2017, 122, 213–221 CrossRef CAS.
- M. Harrabi, H. B. Ammar, K. Mbarki, I. Naifar, C. Yaiche, F. Aloulou and B. Elleuch, Ultrason. Sonochem., 2018, 48, 517–522 CrossRef CAS PubMed.
- W. K. Ye, F. X. Tian, B. Xu, D. S. Zhao, J. Ye, B. Wang, F. Lai, Y. J. Tan and X. J. Hu, Sep. Purif. Technol., 2022, 280, 119846 CrossRef CAS.
- R. R. Solis, O. Dinc, G. D. Fang, M. N. Nadagouda and D. D. Dionysiou, Environ. Sci.: Nano, 2021, 8, 960–977 RSC.
- M. B. Feng, X. H. Wang, J. Chen, R. J. Qu, Y. X. Sui, L. Cizmas, Z. Y. Wang and V. K. Sharma, Water Res., 2016, 103, 48–57 CrossRef CAS PubMed.
- X. L. Zeng, X. Z. Sun and Y. Wang, Environ. Eng. Res., 2021, 26, 200524 CrossRef.
- S. D. Ashrafi, S. Nasseri, M. Alimohammadi, A. H. Mahvi and M. A. Faramarzi, Global NEST J., 2020, 22, 240–249 CAS.
- C. Rodrigues-Silva, M. G. Maniero, S. Rath and J. R. Guimaraes, Sci. Total Environ., 2013, 445, 337–346 CrossRef PubMed.
- C. Rodrigues-Silva, M. G. Maniero, S. Rath and J. R. Guimaraes, Chem. Eng. J., 2013, 224, 46–52 CrossRef CAS.
- J. Ali, W. L. Lei, A. Shahzad, J. Ifthikar, G. G. Aregay, I. I. Shahib, Z. Elkhlifi, Z. L. Chen and Z. Q. Chen, Water Res., 2020, 181, 115862 CrossRef CAS PubMed.
- J. S. Du, W. Q. Guo, D. Che and N. Q. Ren, Chem. Eng. J., 2018, 351, 532–539 CrossRef CAS.
- A. A. Batoeva, M. R. Sizykh and V. A. Munkoeva, Environ. Earth Sci., 2019, 229, 012016 Search PubMed.
- J. Lee, U. von Gunten and J. H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- S. Z. Wang and J. L. Wang, Chemosphere, 2021, 263, 127964 CrossRef CAS PubMed.
- Y. Liu, R. N. Guo, G. H. Shen, Y. H. Li, Y. Q. Li, J. F. Gou and X. W. Cheng, Chem. Eng. J., 2021, 405, 126990 CrossRef CAS.
- Q. Lin and Y. Deng, Environ. Sci. Technol., 2021, 55, 15010–15012 CrossRef CAS PubMed.
- R. Y. Xiao, Z. H. Luo, Z. S. Wei, S. Luo, R. Spinney, W. C. Yang and D. D. Dionysiou, Curr. Opin. Chem. Eng., 2018, 19, 51–58 CrossRef.
- X. W. Ao and W. J. Liu, Chem. Eng. J., 2017, 313, 629–637 CrossRef CAS.
- Y. Q. Gao, N. Y. Gao, W. H. Chu, Y. F. Zhang, J. Zhang and D. Q. Yin, Chem. Eng. J., 2019, 370, 706–715 CrossRef CAS.
- C. C. Lin, Y. C. Wang and M. S. Wu, J. Taiwan Inst. Chem. Eng., 2020, 111, 155–161 CrossRef CAS.
- M. Pirsaheb, H. Hossaini and H. Janjani, Microchem. J., 2020, 153, 9 CrossRef.
- L. Boudriche, Z. Safaei, D. Ramasamy, M. Sillanpaa and A. Boudjemaa, Water Environ. Res., 2019, 91, 1412–1419 CrossRef CAS PubMed.
- H. H. Xue, S. Y. Gao, N. Zheng, M. Li, X. Wen and X. D. Wei, Water Sci. Technol., 2019, 79, 2387–2394 CrossRef CAS PubMed.
- Y. J. Zhu, M. Wei, Z. H. Pan, L. Y. Li, J. Y. Liang, K. F. Yu and Y. Y. Zhang, Sci. Total Environ., 2020, 705, 135960 CrossRef CAS PubMed.
- A. A. Zarei, P. Tavassoli and E. Bazrafshan, Water Sci. Technol., 2018, 1, 126–133 CrossRef PubMed.
- Y. Y. Zhang, L. Y. Li, Z. H. Pan, Y. J. Zhu, Y. N. Shao, Y. H. Wang and K. F. Yu, Chem. Eng. J., 2020, 379, 122354 CrossRef CAS.
- Y. M. Qi, R. J. Qu, J. Q. Liu, J. Chen, G. Al-Basher, N. Alsultan, Z. Y. Wang and Z. L. Huo, Chemosphere, 2019, 237, 124484 CrossRef CAS PubMed.
- L. Li, W. J. Zhang, Y. F. Zhang, S. Kuppers, L. Jin, Y. N. Zhang, N. Y. Gao and D. Zhang, Chem. Eng. J., 2019, 356, 43–52 CrossRef CAS.
- Y. Y. Zhang, C. Rong, Y. Q. Song, Y. H. Wang, J. Y. Pei, X. Y. Tang, R. J. Zhang and K. F. Yu, Chemosphere, 2017, 182, 245–254 CrossRef CAS PubMed.
- H. V. Lutze, R. Bakkour, N. Kerlin, C. von Sonntag and T. C. Schmidt, Water Res., 2014, 53, 370–377 CrossRef CAS PubMed.
- P. C. Xie, J. Ma, W. Liu, J. Zou, S. Y. C. Yue, X. Li, M. R. Wiesner and J. Y. Fang, Water Res., 2015, 69, 223–233 CrossRef CAS PubMed.
- M. S. Chen, C. A. Rholl, T. C. He, A. Sharma and K. M. Parker, Environ. Sci. Technol., 2021, 55, 9395 CrossRef CAS PubMed.
- I. Kaur, W. L. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
- Y. A. Shao, Z. H. Pan, C. Rong, Y. H. Wang, H. X. Zhu, Y. Y. Zhang and K. F. Yu, Environ. Pollut., 2018, 241, 9–18 CrossRef CAS PubMed.
- Z. H. Pan, Y. J. Zhu, M. Wei, Y. Y. Zhang and K. F. Yu, J. Environ. Sci., 2021, 102, 170–184 CrossRef PubMed.
- H. V. Lutze, N. Kerlin and T. C. Schmidt, Water Res., 2015, 72, 349–360 CrossRef CAS PubMed.
- Y. Yang, X. L. Lu, J. Jiang, J. Ma, G. Q. Liu, Y. Cao, W. L. Liu, J. Li, S. Y. Pang, X. J. Kong and C. W. Luo, Water Res., 2017, 118, 196–207 CrossRef CAS PubMed.
- K. H. Wammer, A. R. Korte, R. A. Lundeen, J. E. Sundberg, K. McNeill and W. A. Arnold, Water Res., 2013, 47, 439–448 CrossRef CAS PubMed.
- Y. M. Qi, J. Y. Wei, R. J. Qu, G. Al-Basher, X. X. Pan, A. A. Dar, A. Shad, D. M. Zhou and Z. Y. Wang, Chem. Eng. J., 2021, 403, 126396 CrossRef CAS.
- J. B. Peng, Z. X. Wang, S. Y. Wang, J. Liu, Y. Z. Zhang, B. J. Wang, Z. M. Gong, M. J. Wang, H. Dong, J. L. Shi, H. J. Liu, G. X. Yan, G. G. Liu, S. X. Gao and Z. G. Cao, Chem. Eng. J., 2021, 409, 128176 CrossRef CAS.
- A. De Luca, X. X. He, D. D. Dionysiou, R. F. Dantas and S. Esplugas, Chem. Eng. J., 2017, 318, 206–213 CrossRef CAS.
- C. C. Wang, S. W. Gao, J. C. Zhu, X. F. Xia, M. X. Wang and Y. N. Xiong, J. Environ. Sci., 2021, 99, 249–259 CrossRef PubMed.
- F. Ghanbari, M. Khatebasreh, M. Mahdavianpour and K. Y. A. Lin, Chemosphere, 2020, 244, 125326 CrossRef CAS PubMed.
- F. Ghanbari, M. Moradi and F. Gohari, J. Water Proc. Eng., 2016, 9, 22–28 CrossRef.
- Y. Su, L. Zhang, F. Wang, Y. Wang, H. Su, P. Chen, W. Lv, K. Yao and G. Liu, Environ. Chem., 2016, 35, 1753–1761 CAS.
- E. J. Vasini, M. V. Mirifico and J. A. Caram, Int. J. Quantum Chem., 2011, 111, 1879–1884 CrossRef CAS.
- S. Y. Zhang, J. W. Chen, X. L. Qiao, L. K. Ge, X. Y. Cai and G. S. Na, Environ. Sci. Technol., 2010, 44, 7484–7490 CrossRef CAS PubMed.
- B. Huang, H. C. Wang, D. Cui, B. Zhang, Z. B. Chen and A. J. Wang, Chem. Eng. J., 2018, 341, 238–247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00199c |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab



 .43–45 Thus, the anions in the water would greatly affect the degradation of FLU.
.43–45 Thus, the anions in the water would greatly affect the degradation of FLU.