
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the conformational stability of coiled-coil peptides with a single stereogenic center of a peripheral β-amino acid residue†
Monika Szefczyk *a,
Katarzyna Ożgaa,
Magda Drewniak-Świtalskaa,
Ewa Rudzińska-Szostaka,
Rafał Hołubowiczb,
Andrzej Ożyharb and
Łukasz Berlicki*a
*a,
Katarzyna Ożgaa,
Magda Drewniak-Świtalskaa,
Ewa Rudzińska-Szostaka,
Rafał Hołubowiczb,
Andrzej Ożyharb and
Łukasz Berlicki*a
aDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wroclaw, Poland. E-mail: monika.szefczyk@pwr.edu.pl; lukasz.berlicki@pwr.edu.pl
bDepartment of Biochemistry, Molecular Biology and Biotechnology, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wroclaw, Poland
First published on 7th February 2022
Abstract
The key issue in the research on foldamers remains the understanding of the relationship between the monomers structure and conformational properties at the oligomer level. In peptidomimetic foldamers, the main goal of which is to mimic the structure of proteins, a main challenge is still better understanding of the folding of peptides and the factors that influence their conformational stability. We probed the impact of the modification of the peptide periphery with trans- and cis-2-aminocyclopentanecarboxylic acid (ACPC) on the structure and stability of the model coiled-coil using circular dichroism (CD), analytical ultracentrifugation (AUC) and two-dimensional nuclear magnetic resonance spectroscopy (2D NMR). Although, trans-ACPC and cis-ACPC-containing mutants differ by only one peripheral stereogenic center, their conformational stability is strikingly different.
Introduction
The main inspiration for the development of foldamers, artificial oligomers with a strong tendency to adopt a well-defined thermodynamically stable conformation in solution, were proteins. It was assumed that it might be possible to design similar but unnatural oligomers that fold into compact and specific conformations leading to new types of molecules with interesting properties and useful applications.1 Much research has been undertaken on artificial scaffolds capable of mimicking naturally occurring structural motifs, i.e. coiled-coils.2,3 The “bottom-up” approaches use de novo designed foldameric helices to obtain higher-order structures, usually by the segregation of hydrophilic and lipophilic side chains in the helical conformation.4 In the “top-down” methods, a previously characterized α-amino acid sequence of known secondary/tertiary/quaternary structure is applied directly in the design of peptides with nonnatural backbones intended to form helix bundles.5–7 The crucial step in the design is the identification of new backbones with well-defined structural preferences. Oligo(β-amino acids), named β-peptides, were first proved to meet this criterion and have been widely studied i.e. in terms of folding properties.8–10 In addition, β-peptides show improved stability in enzymatic degradation and have various biological activities, e.g. antimicrobial.11 It was soon proved that a very useful approach for the development of foldamers is the introduction of unnatural amino acids into α-peptides in the process called foldamerization.12,13 In terms of peptidomimetics, apart from widely studied foldamers based on higher homologues of α-amino acids,14 there have been also studied the sequences containing α-aminoisobutyric acid (Aib),15–17 oligoureas,18 azapeptides,19 α-hydrazido-peptides,20 polyamides,21 and others.22–24 Foldamerization of sequences using β-amino acids shows two main advantages, namely, it improves both the conformational and proteolytic stability of peptides.25–27 Importantly, retaining activity28 and binding affinity29–31 of the modified peptides after α → β replacement is usually possible. A wide range of possible applications of the reported α,β-peptides prompted extensive studies on the influence of constrained residues on secondary structure formation, mainly in terms of improving their conformational stability and chosen properties.32–34The influence of constrained β-amino acid residues on secondary structure formation could also potentially have a destructive effect. Analysis of the analogues with conformational stability significantly lower than that of the original oligomer could provide knowledge of the folding propensities of the studied oligomers as well as indicate properties of individual monomers. It is worth noting that the reason for the conformational instability upon substitution could be at least of two types: (a) steric impairment of the side chain with the rest of the structure, particularly the hydrophobic core, or (b) propensity of the individual amino acid to induce the requested conformation. Examples of the first mentioned case have already been published for coiled-coils35,36 and other tertiary structures.37 The second case we endeavor to discuss in this paper. Therefore, we report the analysis of the coiled-coils formed by the incorporation of cyclopentane-based β-amino acid in the peripheral parts of peptide. Single and double substitution with trans- and cis-2-aminocyclopentanecarboxylic acid (ACPC) of the coiled-coil model peptide was applied at positions that do not interfere with its hydrophobic core. Obtained peptides are evaluated using circular dichroism (CD), analytical ultracentrifugation (AUC), and two-dimensional nuclear magnetic resonance spectroscopy (2D NMR).
Results and discussion
To investigate the influence of peripheral modifications with 2-aminocyclopentanecarboxylic acid (ACPC) isomers on the formation and stability of helices, we have designed a model peptide 1 representing a coiled-coil structure (Fig. 1A). The coiled-coil is a structural proteins motif in which two to seven α-helices are coiled together, usually with a (hpphppp)n repeat pattern, commonly denoted as abcdefg.38 Hydrophobic residues (h) at positions a and d are essential to form the tight knobs into hole packing and control the oligomer state of the coiled-coil, while polar residues (p) at positions e and g are necessary for the formation of salt bridges between the helices. Positions b, c, and f are also responsible for the stability of coiled-coils.3 Following these general principles that govern the design of coiled-coil motifs, we endeavored to construct a trimeric coiled-coil with three heptad repeats to be a model peptide 1 (Fig. 1A). Therefore, isoleucine was incorporated at positions a and d, as it is a hydrophobic residue that ensures trimer formation.39 Lysine and glutamic acid were introduced at positions e and g, respectively, as they can ensure additional stabilization of the structure by the formation of salt bridges. Alanine was chosen to occupy positions b and c, because it is a small, helix-favoring amino acid that prevents undesirable influences of side-chain interactions. Glutamine, a charge-neutral polar amino acid, or tryptophan, a UV chromophore, was incorporated at the most external position f. Furthermore, both ends of the peptide were capped with a glycine residue, and the sequence was appended with a two-residue sequence outside the helical region, in reference to similar peptides previously described.40 Additionally, the N-terminus was acetylated to reduce the net charge. | ||
| Fig. 1 (A) Helical wheel representing the model coiled-coil peptide 1. (B) Sequences of designed peptides, where the red pentagon denotes the trans-aminocyclopentanecarboxylic acid (trans-ACPC) residues and the inverted pentagon denotes the cis-aminocyclopentanecarboxylic acid (cis-ACPC) residues. (C) An example of the application of stereochemical patterning in the design of fragment of peptide 6 based on the ψ][ϕ signs.44 | ||
Subsequently, model peptide 1 was subjected to further modifications. To examine the influence of α → cyclic β residue replacement, peripheral regions of model peptide 1 were substituted with (a) trans-(1S,2S)-aminocyclopentanecarboxylic acid (trans-ACPC) residue, well-known helix-promoting and -stabilizing amino acid41–43 or (b) (1S,2R)-cis-aminocyclopentanecarboxylic acid (cis-ACPC) residue, following the rules of stereochemical patterning. The stereochemical patterning approach is based on the dihedral angle pattern for repeating units and specifies that the signs of the torsions flanking the amide moiety (ψ][ϕ) should be the same sign (–][– or +][+) to promote helix formation (Fig. 1C).44 This strategy has been successfully applied to incorporate cis-ACPC residues into different peptide sequences.45,46 Also, previous studies showed that the most favorable was to employ the S,S enantiomer to α → trans-ACPC substitution, since this replacement occurs in α-helix-forming segments (αR region of ϕ,ψ plot).47,48 Subsequently, the ACPC isomers were introduced: (a) in the position outside the main structure of the coiled-coil (peptides 2 and 5), (b) in the outer position of the core of the coiled-coil (peptides 3 and 6), (c) both in the positions outside and in the outer position of the coiled-coil (peptides 4 and 7) (Fig. 1B).
We used circular dichroism (CD) spectroscopy to elucidate the secondary structure (Fig. 2) and stability (Fig. 3) of the synthesized peptides in phosphate buffer. The model peptide 1 has two minima at λ = 208 and 222 nm with the ratio of R(θ222/θ208) close to 1, which confirms the presence of α-helix structure.49 Introduction of only one trans-β residue (peptides 2 and 3) does not change the position of the minima compared to the model peptide 1 and does not have a significant influence on the R(θ222/θ208) and molar ellipticity values. The α → trans-ACPC residue replacement at both ends of the sequence (peptide 4) resulted in a slight decrease in the molar ellipticity value compared to the peptides mentioned above and the position of two minima was also preserved. In the spectra of peptides 5 and 6, where a single α → cis-ACPC residue replacement was introduced, a more significant decrease in the molar ellipticity values can be observed, compared to peptide 1, and two shallow minima at λ = 208 and 222 nm. The introduction of cis-β residue at both ends (peptide 7) caused total disruption of the helix structure, since only a minimum at 204 nm is present in the CD spectrum, indicating a random coil formation.50
 | ||
| Fig. 2 CD spectra of the studied peptides in phosphate buffer. Cpep = 80 μM, Cbuffer = 0.05 M, pH = 7, T = 20 °C. | ||
 | ||
| Fig. 3 Thermal unfolding curves of the studied peptides followed by CD signal at 222 nm in phosphate buffer with calculated melting temperatures indicated in the legend. Cpep = 80 μM, Cbuffer = 0.05 M, pH = 7. | ||
Conformational stability was determined based on CD measurements in phosphate buffer and was followed in the range of 190–240 nm by a gradual increase in temperature from 4 to 98 °C (Fig. S2†). Thermal unfolding curves followed at 222 nm were used to determine the melting temperatures (Fig. 3). The model coiled-coil peptide 1 is very stable in the phosphate buffer with a cooperative melting transition at temperature 75 °C. A singular α → trans-β residue replacement was shown to have an influence on α,β-peptides stability, since the melting temperatures calculated for peptides 2 and 3 are lower compared to the model peptide 1, however still very high (more than 60 °C). Furthermore, the introduction of two trans-β residues (peptide 4) still preserves a stable helical structure. On the other hand, even a singular peripheral α → cis-β modification has a dramatic influence on the stability of the peptides. The melting temperatures obtained for peptides 5 and 6 are significantly lower compared to other peptides studied. Data for peptide 7 were not obtained as the CD spectrum did not indicate α-helical fold.
In summary, α → trans-β residue replacement does not significantly influence the stability of the peptides in buffer, although α → cis-β residue replacement caused the disruption of the structure.
The peptides modified with trans-ACPC or cis-ACPC were subjected to sedimentation velocity (SV AUC) and sedimentation equilibrium (SE AUC) analytical ultracentrifugation to determine their oligomerization states and the effect of unnatural amino acids incorporation on the stability of the oligomeric forms. Peptides 1, 2, 3, and 4 existed in solution as trimers with sedimentation coefficients 0.90–0.93 S, as determined by the SV AUC (Table 1, Fig. S3†). In addition to trimers, the sedimentation coefficient distributions (c(s)) indicate that low molecular weight components (0.25–0.30 S, 1200–1400 Da) were also present. This could be elongated monomers, which due to their shape would have higher frictional ratios and underestimated molecular weights.51 The apparent molecular weight (MWapp) of 4 was much higher than the MWms of a putative trimer. We conclude that this discrepancy was caused by a tendency of 4 to aggregate. Notably, this made SE AUC analysis impossible, as the peptide aggregated before equilibrium could be attained at any speed. For the other peptides, the trimeric state was confirmed using SE AUC. The data obtained using this technique were fitted to models of single species, monomer-dimer, monomer-trimer, or monomer-tetramer equilibria. The best fits with MWapp compliant with MWms were obtained for the monomer-trimer model for all peptides (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, SE AUC was carried out at 20 °C at 20000, 35000 and 48000 rpma
000 rpm, SE AUC was carried out at 20 °C at 20000, 35000 and 48000 rpma
| Peptide | MWms [Da] | ![[v with combining macron]](https://www.rsc.org/images/entities/i_char_0076_0304.gif) [ml g−1] [ml g−1] |
SV AUC | SE AUC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s [S] | s20.w [S] | MWapp [Da] | n |
|
f/f0 | Oligomer | logKa |
MWapp [Da] | n | |||
a MWms – Molecular Weight determined using mass spectrometry. MWapp – apparent molecular weight. n – quotient of MWapp and MWms. – partial specific volume. s – sedimentation coefficient. s20,w – sedimentation coefficient corrected for water, 20 °C.  – weight averaged s20.w. f/f0 – frictional ratio. Ka – association constant. – weight averaged s20.w. f/f0 – frictional ratio. Ka – association constant. |
||||||||||||
| 1 | 2713.13 | 0.76619 | 0.914 | 0.950 | 8182 | 3.02 | 0.904 | 1.314 | Trimer | 10.0 | 2814 | 1.04 |
| 2 | 2767.22 | 0.77225 | 0.913 | 0.950 | 8301 | 3.00 | 0.902 | 1.289 | Trimer | 9.95 | 2923 | 1.06 |
| 3 | 2696.14 | 0.77439 | 0.926 | 0.964 | 7995 | 2.97 | 0.936 | 1.227 | Trimer | 10.4 | 2941 | 1.09 |
| 4 | 2821.31 | 0.77807 | 0.905 | 0.943 | 9395 | 3.33 | 0.984 | 1.373 | — | |||
| 5 | 2767.22 | 0.77225 | 0.650 | 0.677 | 5768 | 2.08 | 0.677 | 1.420 | Trimer | 8.81 | 2335 | 0.85 |
| 6 | 2696.14 | 0.77439 | 0.680 | 0.708 | 6254 | 2.32 | 0.708 | 1.419 | Trimer | 8.57 | 2655 | 0.98 |
| 7 | 2821.31 | 0.77807 | 0.422 | 0.440 | 3283 | 1.16 | 0.481 | 1.459 | Trimer | 6.91 | 3172 | 1.12 |
| 0.846 | 0.881 | 9315 | 3.30 | |||||||||
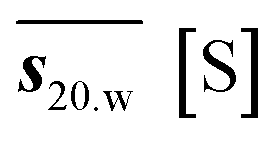
The incorporation of trans-ACPC did not significantly affect the self-affinity of peptide 1 − logKa of the monomer-trimer equilibria was around 10 for all variants tested. In contrast, substitution with cis-ACPC significantly decreased the stability of the obtained peptides compared to model peptide 1. The sedimentation coefficient distributions (c(s)) obtained using SV AUC transitioned from more or less regular distributions indicating the presence of trimers to wide irregular distributions indicating the presence of a mixture of loosely bound monomers, dimers, and trimers in the case of 5 and 6, or to a mixture of monomers and trimers dominated by monomers in the case of 7 (Table 1). The frictional ratio for cis-peptides increased from 1.22–1.37 to around 1.45. This indicates that the hydrodynamic drag increased, supposedly as a result of the more elongated conformation of the peptides. SE AUC analyses indicated that the monomer-trimer model is still valid for the cis-peptides. The self-affinity was smaller compared to the trans-peptides as indicated by the decrease in logKa from 10 to 8.81 (5) 8.57 (6), and 6.91 (7).
In summary, the AUC results showed that foldamerization did not influence the oligomeric state of the studied peptides. However, substitution with cis-ACPC significantly decreased the stability of the obtained peptides compared to model peptide 1, as was also shown by the CD results.
NMR studies
The peptides with trans-ACPC substitution (2 and 3) were chosen for detailed NMR analysis. The unambiguous assignment of nearly all resonances (Tables S2 and S3†) was possible based on the analysis of 1H NMR (Fig. S4 and S7† for 2 and 3 respectively), TOCSY (Fig. S5 and S8†), and 2D NOESY (Fig. S6 and S9†) spectra measured in water solution (pH = 7). In both cases, a significant number of non-sequential inter-proton contacts were identified. Most of the contacts detected were of type i–i + 3 or i–i + 4, suggesting the presence of a α-helical structure consistent with other experiments. Contacts, which were not possible within a helix, were considered to be inter-helical. Based on NMR-derived restraints, the three-dimensional structures of peptides 2 and 3 were calculated (Fig. 4, Table S4†). | ||
| Fig. 4 The averaged structures of peptides 2 and 3 calculated on the basis of restraints derived from 2D NOESY: side view (A and C) and top view (B and D) for 2 and 3, respectively. | ||
As expected, peptides 2 and 3 adopt a well-defined coiled-coil conformation in water solution (Fig. S13†). Positioning of the isoleucine residue side chains forms a hydrophobic core, while the rest of the side chains are exposed to solvent. Although the trans-ACPC residue is placed at the end of the helices, its position is well-defined. Cyclopentane incorporated in trans-ACPC does not contribute to the formation of a hydrophobic core as it is directed toward the solvent.
1H NMR spectra of cis-ACPC mono-substituted peptides 5 and 6 (Fig. S10 and S11†) showed a significant decrease in the dispersion of chemical shifts of proton signals and broadening of resonance lines if compared to their trans-ACPC-substituted analogues 2 and 3. Two-dimensional TOCSY and NOESY spectra were also recorded for these peptides. However, signal broadening and overlapping disable the precise spectral analysis, suggesting the absence of a well-defined structure, which is clearly visible in the comparison of the representative fragments of 2D NOESY spectra recorded for cis-ACPC (6) and trans-ACPC-containing peptides (3) (Fig. S12†). The picture given by NMR measurement is consistent with the CD data and indicates only partial folding of peptides containing the cis-ACPC residue.
Moreover, we have modelled the lowest energy conformations of peptides 2, 3, 5 and 6 in order to analyze network of hydrogen bond around β-amino acid residues (Fig. 5). Trans-ACPC residue present in the structures of peptides 2 and 3 forms hydrogen bonds analogous to those observed in α-helix, thus it is stabilizing the helical structure (Fig. 5A and C, respectively). However, the set of hydrogen bonds formed by cis-ACPC present in peptides 5 and 6 is different (Fig. 5B and D, respectively). Intra-residue hydrogen bond is observed in both cases, and the network of inter-residue hydrogen bonds is affected. Moreover, the β-amino acid residue, due to its constrained conformation, directs neighboring residues (Ac of peptide 5 and Gly–Tyr of peptide 6) outside the helix.
 | ||
| Fig. 5 Zoomed-in fragments of minimized models of peptides containing trans-ACPC (sequences 2 and 3, panels A and C, respectively) and cis-ACPC (sequences 5 and 6, panels B and D, respectively). ACPC residues are shown in green, and hydrogen bonds are shown as green dotted lines. | ||
Conclusions
Although there are several studies on the effect of substitution of β-amino acids on conformational stability of secondary, tertiary or quaternary structures,35,48,52 the comparison of analogues containing units of various stereochemistry are rare, and concerns only single α-helix.53 Here, we applied well-understood coiled-coil structure as a model for better understanding of effects of trans-ACPC and cis-ACPC substitutions on its conformational stability. However, both trans-ACPC and cis-ACPC can be used as building blocks for construction of various helical structures,7,45,54 their effect on α-helix-based coiled-coil is significantly different, as shown in the combined picture derived from all the applied experimental techniques. In particular, CD analysis indicate large difference in conformational stability expressed by Tm values ranging from approximately 40 °C to 60 °C for cis-ACPC and trans-ACPC substituted peptides respectively. The difference in conformational stability was also observed in NMR measurements, as only spectra of trans-ACPC-based peptides showed numerous non-sequential inter-proton contacts allowing building of consistent model. Moreover, AUC measurements confirmed the trimeric structure of all peptides. Importantly, these detailed structural studies indicate that introduced β-amino acid residues do not interfere with hydrophobic core of the coiled-coil or form any direct inter-helical interactions. Therefore, the striking difference of conformational stability between the corresponding trans-ACPC and cis-ACPC mutants is a direct result of their ability to promote helical conformation or not. The insight in conformational preferences of these arrangements was achieved by further molecular modelling studies that revealed that cis-ACPC intra-residue hydrogen bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N) degrades the network of hydrogen bonds stabilizing the helix, while trans-ACPC adjusts well to α-helix structure.
O⋯H–N) degrades the network of hydrogen bonds stabilizing the helix, while trans-ACPC adjusts well to α-helix structure.
It is worth to underline that pairs of peptides 2 and 5, as well as 3 and 6 differ only by one stereogenic center, while their conformational stability are significantly different. Moreover, the discussed changes are introduced at the peripheries of the helices; thus long-range propagation of this effect is observed. Therefore, the small change related to the modulation of the folding propensity of a single amino acid residue has dramatically changed the folding of the coiled-coil structure. In summary, the concept of studying analogues with conformational stability significantly lower than that of the original oligomer can be considered to the be complementary to widely studied foldamers and provide a better understanding of the folding properties of various oligomers.
Experimental section
Peptide synthesis
All commercially available reagents and solvents were purchased from Lipopharm.pl, Sigma-Aldrich, or Merck and used without further purification. Fmoc-(1S, 2R)-2-aminocyclopentanecarboxylic acid (Fmoc-cis-ACPC) was synthesized according to the known protocol.55 Fmoc-(1S, 2S)-2-aminocyclopentanecarboxylic acid (Fmoc-trans-ACPC) was purchased from Synnovator, Inc. The peptides described were obtained with an automated solid-phase peptide synthesizer (Biotage® Initiator + AlastraTM) using rink amide AM resin (loading: 0.59 mmol g−1). Fmoc deprotection was performed using 20% piperidine in DMF for 3 + 10 min. A double coupling procedure was achieved with 0.5 M DIC solution and 0.5 M OXYMA solution (1:1) in DMF, for α-amino acids 2 × 15 min and for β-amino acids 30 min, at 75 °C. The acetylation reaction was carried out using a mixture of NMP/DIPEA/acetic anhydride (80:15:5). The cleavage of the peptides from the resin was accomplished with the mixture of TFA/TIS/H2O (95:2.5:2.5) after 3 h of shaking. The crude peptide was precipitated with ice-cold Et2O and centrifuged (10000 rpm, 15 min, 4 °C). The peptides were purified using preparative HPLC (Knauer Prep) with a C18 column (Thermo Scientific, Hypersil Gold 12 μ, 250 mm × 20 mm) with a water/acetonitrile (0.05% TFA) eluent system.
Analytical high-performance liquid chromatography (HPLC)
Analytical HPLC was performed using a Kinetex 5μ EVO C18 100A 150 × 4.6 mm column. Program (eluent A: 0.05% TFA in H2O, eluent B: 0.05% TFA in acetonitrile, flow 0.5 mL min−1): A: t = 0 min, 90% A; t = 30 min, 10% A.Mass spectrometry (MS)
Mass spectra were recorded using a WATERS LCT Premier XE high-resolution mass spectrometer with an electrospray ionization and time-of-flight (TOF) detector.Circular dichroism (CD)
CD spectra were recorded using a JASCO J-815 spectropolarimeter at 20 °C between 250 and 190 nm in water with the following parameters: 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 50 nm min−1 scanning speed, 5 accumulations, 0.02 cm cuvette path length. The CD spectra of the buffer alone were recorded and subtracted from the raw data. Typically, samples were prepared by dilution of a peptide stock solution at a concentration of 1.3 mM. CD intensity is given as the mean residue molar ellipticity (θ [deg × cm2 × dmol−1]). The melting temperatures of the studied peptides were determined by a method described previously.56Temperature denaturation measurements using CD
To examine the thermal unfolding of the peptide, stock solutions were diluted to 80 μM and two types of measurements were performed: (1) in the range of 190–240 nm and (2) at 222 nm in phosphate buffer pH = 7, 0.05 M. The temperature increased from 4 to 98 °C in increments of 2 °C. Ellipticity measurements were recorded with a 1 mm path length cuvette and other parameters remained unchanged.Analytical ultracentrifugation (AUC)
The weighed portions of the peptides were dissolved in PBS (7.74 mM Na2HPO4, 2.26 mM NaH2PO4, 137 mM NaCl, 2.7 mM NaCl, pH 7.4) at room temperature for at least 15 minutes in an Eppendorf Thermomixer at 500 rpm and subsequently centrifuged at 14000g for 5 minutes at room temperature. The supernatants were collected and the concentrations of the peptides were calculated from absorbance values measured at 280 nm (A280) and molar extinction coefficients of 6990 M−1 cm−1. Sedimentation velocity (SV AUC) and sedimentation equilibrium (SE AUC) analytical ultracentrifugation experiments were conducted at 20 °C using a Beckman Coulter ProteomeLab XLI analytical ultracentrifuge and an An-60Ti rotor. Partial specific volumes of the peptides were estimated using SEDNTERP57 (available at http://www.jphilo.mailway.com/download.htm, 6th April 2020) and corrected for the presence of ACPC, N-terminal acetyl and C-terminal amine moieties using published molar increment values of chemical groups.58 Density and viscosity of PBS were estimated using SEDNTERP. SV AUC was conducted in cells containing charcoal-filled Epon centerpieces with 2 sector-shaped channels for 400 μL of 100 μM peptides in PBS. The samples were centrifuged overnight (approximately 20 h) at 50000 rpm with continuous collection of A280 with 0.003 cm resolution. The time-corrected data59 were fitted to a continuous sedimentation coefficient distribution (c(s)) model60 using SEDFIT software (available at https://sedfitsedphat.nibib.nih.gov/software/default.aspx, 6th April 2020). The fits of 150 sedimentation coefficient values between 0 and 3 S were calculated alternately using simplex and Marquardt–Levenberg algorithms. The quality of the fits was estimated using RMSD values. A p-value for regularization was set at 0.95. SE AUC was conducted at 20000, 35000 and 48000 rpm in cells containing charcoal-filled Epon centerpieces with 6 square channels for 100 μL of approximately 50 μM peptides. For peptide 7, an additional experiment was conducted for 50, 75 and 100 μM peptide samples at 38000, 41000, and 45000 rpm to better describe its properties. The scans of intensities of light at 280 nm were collected separately for reference and sample channels in step mode with 0.001 cm resolution and 10 replicates per scan. Scans representing the peptide solutions at equilibrium were processed using GUSSI (available at http://biophysics.swmed.edu/MBR/software.html, 6th April 2020)61 and analyzed using SEDPHAT (available at https://sedfitsedphat.nibib.nih.gov/software/default.aspx, 6th April 2020) using single species, monomer-dimer, monomer-trimer and monomer-tetramer equilibrium models. The fits were calculated alternately using simplex and Marquardt–Levenberg algorithms. The final fit was performed using a simulated annealing algorithm with default settings. The accuracy of the fits was followed using global RMSD values.
Nuclear magnetic resonance (NMR)
000 steps) under the canonical ensemble (NVT) and 100 ps (50000 steps) under the NPT ensemble.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to gratefully acknowledge National Science Centre, Poland for their financial support through grant no. 2017/26/D/ST5/00341. The Wrocław Center for Networking and Supercomputing (WCSS) is kindly acknowledged for the use of software (GROMACS) and hardware resources (computing grant of Ł. B.).References
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- A. N. Lupas and J. Bassler, Trends Biochem. Sci., 2017, 42, 130–140 CrossRef CAS PubMed.
- E. H. C. Bromley and K. J. Channon, Prog. Mol. Biol. Transl. Sci., 2011, 103, 231–275 CAS.
- D. N. Woolfson, Adv. Protein Chem., 2005, 70, 79–112 CrossRef CAS PubMed.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, JACS, 2007, 129, 4178–4180 CrossRef CAS PubMed.
- W. S. Horne, M. D. Boersma, M. A. Windsor and S. H. Gellman, Angew. Chem., Int. Ed., 2008, 47, 2853–2856 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756 CrossRef CAS PubMed.
- L. K. A. Pilsl and O. Reiser, Amino Acids, 2011, 41, 709–718 CrossRef CAS PubMed.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed.
- T. A. Martinek and F. Fulop, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–362 CrossRef CAS PubMed.
- P. G. Vasudev, S. Chatterjee, N. Shamala and P. Balaram, Chem. Rev., 2011, 111, 657–687 CrossRef CAS PubMed.
- Y.-H. Shin and S. H. Gellman, JACS, 2018, 140, 1394–1400 CrossRef CAS PubMed.
- S. Rinaldi, Molecules, 2020, 25, 3276 CrossRef CAS PubMed.
- J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680–6691 CrossRef CAS PubMed.
- L. Byrne, J. Sola and J. Clayden, Chem. Commun., 2015, 51, 10965–10968 RSC.
- M. De Poli, W. Zawodny, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–878 CrossRef CAS PubMed.
- R. Chingle, C. Proulx and W. D. Lubell, Acc. Chem. Res., 2017, 50, 1541–1556 CrossRef CAS PubMed.
- P. Amabili, M. Calvaresi, G. Martelli, M. Orena, S. Rinaldi and F. Sgolastra, Eur. J. Org. Chem., 2019, 2019, 907–917 CrossRef CAS.
- A. Lobo-Ruiz and J. Tulla-Puche, 2 – Synthetic approaches of naturally and rationally designed peptides and peptidomimetics, Peptide Applications in Biomedicine, Biotechnology and Bioengineering, ed. S. Koutsopoulos, Woodhead Publishing, 2018, pp. 23–49 Search PubMed.
- J. Vagner, H. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292–296 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- G. Olajos, A. Hetenyi, E. Weber, T. Szogi, L. Fulop and T. A. Martinek, Org. Biomol. Chem., 2018, 16, 5492–5499 RSC.
- D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed.
- A. I. Webb, M. A. Dunstone, N. A. Williamson, J. D. Price, A. de Kauwe, W. Chen, A. Oakley, P. Perlmutter, J. McCluskey, M.-I. Aguilar, J. Rossjohn and A. W. Purcell, J. Immunol., 2005, 175, 3810–3818 CrossRef CAS PubMed.
- P. Zubrzak, H. Williams, G. M. Coast, R. E. Isaac, G. Reyes-Rangel, E. Juaristi, J. Zabrocki and R. J. Nachman, Biopolymers, 2007, 88, 76–82 CrossRef CAS PubMed.
- S. Reinelt, M. Marti, S. Dedier, T. Reitinger, G. Folkers, J. A. Lopez de Castro and D. Rognan, J. Biol. Chem., 2001, 276, 24525–24530 CrossRef CAS PubMed.
- J. D. Sadowsky, J. K. Murray, Y. Tomita and S. H. Gellman, ChemBioChem, 2007, 8, 903–916 CrossRef CAS PubMed.
- L. Berlicki, M. Kaske, R. Gutierrez-Abad, G. Bernhardt, O. Illa, R. M. Ortuno, C. Cabrele, A. Buschauer and O. Reiser, Med. Chem., 2013, 56, 8422–8431 CrossRef CAS PubMed.
- U. Strijowski and N. Sewald, Org. Biomol. Chem., 2004, 2, 1105–1109 RSC.
- E. Vass, U. Strijowski, K. Wollschlager, I. M. Mandity, G. Szilvagyi, M. Jewginski, K. Gaus, S. Royo, Z. Majer, N. Sewald and M. Hollosi, J. Pept. Sci., 2010, 16, 613–620 CrossRef CAS PubMed.
- D. Seebach, A. Lukaszuk, K. Patora-Komisarska, D. Podwysocka, J. Gardiner, M.-O. Ebert, J. C. Reubi, R. Cescato, B. Waser, P. Gmeiner, H. Hubner and C. Rougeot, Chem. Biodiversity, 2011, 8, 711–739 CrossRef CAS PubMed.
- M. Szefczyk, N. Szulc, M. Gasior-Głogowska, A. Modrak-Wojcik, A. Bzowska, W. Majstrzyk, M. Taube, M. Kozak, T. Gotszalk, E. Rudzinska-Szostak and L. Berlicki, Nanoscale, 2021, 13, 4000–4015 RSC.
- M. Oba, C. Ito and M. Tanaka, Bioorg. Med. Chem. Lett., 2018, 28, 875–877 CrossRef CAS PubMed.
- M. Drewniak-Switalska, B. Barycza, E. Rudzinska-Szostak, P. Morawiak and L. Berlicki, Org. Biomol. Chem., 2021, 19, 4272–4278 RSC.
- F. Lapenta, J. Aupic, Z. Strmsek and R. Jerala, Chem. Soc. Rev., 2018, 47, 3530–3542 RSC.
- P. B. Harbury, T. Zhang, P. S. Kim and T. A. Alber, Science, 1993, 262, 1401–1407 CrossRef CAS PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9151–9156 CrossRef CAS PubMed.
- J. L. Price, E. B. Hadley, J. . D. Steinkruger and S. H. Gellman, Angew. Chem., Int. Ed., 2010, 49, 368–371 CrossRef CAS PubMed.
- B. F. Fisher, S. H. Hong and S. H. Gellman, JACS, 2018, 140, 9396–9399 CrossRef CAS PubMed.
- I. M. Mandity, E. Weber, T. A. Martinek, G. Olajos, G. K. Toth, E. Vass and F. Fulop, Angew. Chem., Int. Ed., 2009, 48, 2171–2175 CrossRef CAS PubMed.
- L. Berlicki, L. Pilsl, E. Weber, I. M. Mandity, C. Cabrele, T. A. Martinek, F. Fulop and O. Reiser, Angew. Chem., Int. Ed., 2012, 51, 2208–2212 CrossRef CAS PubMed.
- M. Szefczyk, E. Weglarz-Tomczak, P. Fortuna, A. Krzyszton, E. Rudzinska-Szostak and L. Berlicki, Angew. Chem., Int. Ed., 2017, 56, 2087–2091 CrossRef CAS PubMed.
- J. L. Price, W. S. Horne and S. H. Gellman, JACS, 2010, 132, 12378–12387 CrossRef CAS PubMed.
- D. E. Mortenson, D. F. Kreitler, N. C. Thomas, I. A. Guzei, S. H. Gellman and K. T. Forest, ChemBioChem, 2018, 19, 604–612 CrossRef CAS PubMed.
- C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, JACS, 1996, 118, 2744–2745 CrossRef CAS.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- P. Schuck, H. Zhao, C. A. Brautigam and R. Ghirlando, Basic Principles of Analytical Ultracentrifugation, Boca Raton, FL, CRC Press, 2015 Search PubMed.
- M. Drewniak, E. Weglarz-Tomczak, K. Ozga, E. Rudzinska-Szostak, K. Macegoniuk, J. Tomczak, M. Bejger, W. Rypniewski and L. Berlicki, Bioorg. Chem., 2018, 81, 356–361 CrossRef CAS PubMed.
- P. Fortuna, A. Twarda-Clapa, Ł. Skalniak, K. Ożga, T. A. Holak and Ł. Berlicki, Eur. J. Med. Chem., 2020, 208, 112814 CrossRef CAS PubMed.
- T. A. Martinek, A. Hetenyi, L. Fulop, I. M. Mandity, G. K. Toth, I. Dekany and F. Fulop, Angew. Chem., Int. Ed., 2006, 45, 2396–2400 CrossRef CAS PubMed.
- S. G. Davies, O. Ichihara, I. Lenoir and I. A. S. Walters, J. Chem. Soc., Perkin Trans. 1, 1994, 11, 1411–1415 RSC.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, JACS, 2016, 138, 6498–6505 CrossRef CAS PubMed.
- T. M. Laue, B. D. Shah, T. M. Ridgeway and S. L. Pelletier, Analytical Ultracentrifugation in Biochemistry and Polymer Science, Royal Society of Chemistry, ed. S. Harding and A. Rowe, 1992, pp. 90–125 Search PubMed.
- H. Durchschlag and P. Zipper, J. Appl. Crystallogr., 1997, 30, 803–807 CrossRef CAS.
- H. Zhao, R. Ghirlando, G. Piszczek, U. Curth, C. A. Brautigam and P. Schuck, Anal. Biochem., 2013, 437, 104–108 CrossRef CAS PubMed.
- P. Schuck, Biophys. J., 2000, 78, 1606–1619 CrossRef CAS PubMed.
- C. A. Brautigam, Methods Enzymol., 2015, 562, 109–133 CAS.
- T. D. Goddard and D. G. Kneller, Sparky, 3rd edn, University of California, San Francisco, CA, USA, 2001 Search PubMed.
- C. D. Schwieters, J. J. Kuszewski and G. M. Clore, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 47–62 CrossRef CAS.
- T. A. Martinek, G. K. Toth, E. Vass, M. Hollosi and F. Fulop, Angew. Chem., Int. Ed., 2002, 41, 1718–1721 CrossRef CAS PubMed.
- L. J. Németh, Z. Hegedüs and T. A. Martinek, J. Chem. Inf. Model., 2014, 54, 2776–2783 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00111j |
| This journal is © The Royal Society of Chemistry 2022 |