
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield one-pot synthesis of polyrotaxanes with tunable well-defined threading ratios over a wide range†
Takako Noritomi‡
a,
Lan Jiang‡ a,
Hideaki Yokoyamaa,
Koichi Mayumi*ab and
Kohzo Ito*a
a,
Hideaki Yokoyamaa,
Koichi Mayumi*ab and
Kohzo Ito*a
aMaterial Innovation Research Center (MIRC) and Department of Advanced Materials Science, Graduate School of Frontier Sciences, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8561, Japan. E-mail: kohzo@edu.k.u-tokyo.ac.jp
bThe Institute for Solid State Physics, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8581, Japan. E-mail: kmayumi@g.ecc.u-tokyo.ac.jp
First published on 30th January 2022
Abstract
In this work, we report a high-yield one-pot synthesis of polyrotaxane (PR), composed of (2-hydroxypropyl)-α-cyclodextrin (hpCD) and polyethylene glycol (PEG), with well-defined hpCD threading ratios controllable across a wide range from 0.64% to 10%. In hpCD/PEG aqueous solutions, hpCDs are well dispersed and threaded spontaneously into hpCDs to form a pseudo-PR (pPR) structure. The homogeneous dispersion of hpCDs results in a well-defined threading ratio of hpCDs on PEG, which is suggested by the fact that the dispersity of the molecular weight distribution of PR is almost the same as that of pure PEG. The well-defined hpCD threading ratio of the PRs can be controlled over a wide range by tuning the hpCD concentration in the pPR solutions.
1. Introduction
Cyclodextrins (CDs) spontaneously include various guest molecules to form host–guest supramolecular complexes.1 For small guest molecules, the host–guest ratios are well defined by the molecular recognition of the CDs. Harada et al. reported that α-CDs form pseudo-polyrotaxanes (pPRs) with polyethylene glycol (PEG) in water.2 By end-capping the PEG of the pPRs, they successfully synthesised polyrotaxane (PR–CD),3 in which α-CDs are threaded on PEG. PR–CDs have attracted considerable attention4–6 because the sliding dynamics of the rings along the polymer axis facilitate various unique applications, such as molecular shuttles,7,8 drug delivery systems,9 and tough polymeric materials with slidable cross-links.10–14 A key parameter for the sliding behaviour of PRs is the threading ratio of the rings on the polymer chain.14,15 Hence, controlling the threading ratio of PRs is necessary for further progress in their applications. The average CD threading ratio of the PR–CDs can be changed by the end-modification of PEG,16 extension of PEG chain length,17 and tuning of CD/PEG concentration, solvent, and temperature during pPR formation.18–20 Recently, Jiang et al. reported a high-yield one-pot synthesis method for PR with a low CD threading ratio of 2% from hydroxypropylated CDs (hpCDs) and PEG, PR–hpCD.14In contrast to the uniform host–guest ratios of the supramolecular complexes with small molecules, the threading ratio of α-CD on a PEG chain in PR–CD is not homogeneous, but has a distribution, because there are many inclusion sites on PEG. The inhomogeneous threading ratio of PR–CD is suggested by the wider GPC molecular weight distribution of PR–CD than that of pure PEG.16,17,21 Precise control of the threading ratio for PR with crown ethers and the other rings has been achieved by cyclic redox-driven processes22 and polymerisation of pseudo rotaxanes composed of a ring and short axis.23,24 However, the synthesis procedure is multistep and complicated for large-scale and low-cost production of PR.
In this study, we focused on the one-pot synthesis of PR–hpCD in hpCD/PEG aqueous solutions (Fig. 1). We have discovered that the dispersity of the molecular weight distribution of PR–hpCD was almost the same as that of pure PEG, indicating a homogeneous and well-defined threading ratio of hpCDs on PEG. We systematically changed the hpCD/PEG concentration and temperature during pPR formation in homogeneous aqueous solutions and studied the effect of the synthesis conditions on the hpCD threading ratios of the PR–hpCDs. The threading ratio of the PR–hpCDs increased with increasing hpCD concentration but was independent of the PEG concentration and temperature. Based on the synthesis results and X-ray scattering data of the pPR solutions, we discuss the driving force for pPR–hpCD formation and the mechanism of the well-defined threading ratio of PR–hpCD. This study provides a one-pot synthesis method for the precise control of the threading ratios of PRs over a wide range by tuning hpCD concentration in the pPR formation process.
 | ||
| Fig. 1 One-pot synthesis scheme of PR–hpCD. | ||
2. Experimental section
2.1 Materials
All the reagents for the synthesis of PR were used as received. The amino-terminated poly(ethylene glycol) (Mw ∼ 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000680 monomer units), phosphate buffer solution (PBS, 0.1 mol L−1, pH 8.0), (2-hydroxypropyl)-α-cyclodextrin (hpCD, Mw ∼ 1180) and α-cyclodextrin (CD, Mw = 973) were purchased from NOF corporation, Sigma-Aldrich, NIHON SHOKUHIN KAKO Corporation, LTD and Wako, respectively. The 3-hydroxy-1-adamantanecarboxylic acid (OH-Ada-COOH) and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) were purchased from Tokyo Chemical Industry Corporation.
000680 monomer units), phosphate buffer solution (PBS, 0.1 mol L−1, pH 8.0), (2-hydroxypropyl)-α-cyclodextrin (hpCD, Mw ∼ 1180) and α-cyclodextrin (CD, Mw = 973) were purchased from NOF corporation, Sigma-Aldrich, NIHON SHOKUHIN KAKO Corporation, LTD and Wako, respectively. The 3-hydroxy-1-adamantanecarboxylic acid (OH-Ada-COOH) and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) were purchased from Tokyo Chemical Industry Corporation.
2.2 Synthesis of PR
000–14000) for 2 days to remove free hpCD and freeze-dried to obtain the final product, PR–hpCDs. The synthesis scheme is illustrated in Fig. 1.
| Sample name | PEG (×10−3) [mol L−1] | hpCD [mol L−1] | CD [mol L−1] | Threading ratioa [%] | Threading number of CD per PEG NCDb | Mw [×104]c | Mn [×104]d | Mw/Mn | Yield [%] |
|---|---|---|---|---|---|---|---|---|---|
| a Measured by 1H NMR.b Measured by 1H NMR.c Weight average molecular weight determined by GPC.d Number average molecular weight determined by GPC. | |||||||||
| PEG | — | — | — | — | — | 3.18 | 2.99 | 1.06 | — |
| PR–hpCD2 | 4.9 | 0.063 | — | 0.64 | 2.2 | 3.69 | 3.44 | 1.07 | 96.5 |
| PR–hpCD4 | 4.7 | 0.12 | — | 1.09 | 3.7 | 3.85 | 3.63 | 1.06 | 98.5 |
| PR–hpCD7 | 4.3 | 0.2 | — | 2.05 | 7 | 4.08 | 3.82 | 1.07 | 97.6 |
| PR–hpCD11 | 4.5 | 0.3 | — | 3.09 | 10.5 | 4.45 | 4.17 | 1.07 | 92.9 |
| PR–hpCD16 | 4.5 | 0.44 | — | 4.81 | 16.4 | 5.04 | 4.68 | 1.08 | 98.9 |
| PR–hpCD21 | 4.5 | 0.53 | — | 6.03 | 20.6 | 5.69 | 5.2 | 1.09 | 99.6 |
| PR–hpCD34 | 3.8 | 0.65 | — | 10 | 34.2 | 6.88 | 6.28 | 1.09 | 96 |
| PR–CD87 | 1.11 | — | 0.21 | 25.5 | 86.7 | 13.3 | 11.7 | 1.13 | 72.7 |
000–14000) for 2 days to remove free hpCD and freeze-dried to obtain the final product.2.3 Characterization of PR
2.4 Small-angle and wide-angle X-ray scattering (SAXS and WAXS) measurements of pPR solutions
Small-angle and wide-angle X-ray scattering (SAXS and WAXS) measurements of the hpCD, CD, pPR–hpCD and pPR–CD solutions were performed at room temperature to observe the spatial distribution of the hpCD/CD molecules using Nanopix (Rigaku Corporation, Japan). The sample-to-detector distances of SAXS and WAXS were 1300 and 170 mm, respectively, and the two-dimensional scattering patterns were recorded using HyPix-6000. The X-ray wavelength was 1.5 Å. The exposure time was 240–14400 seconds.
3. Results and discussion
Table 1 summarises the characterisation results of the obtained PR–CD and PR–hpCDs prepared at various hpCD concentrations ranging from 0.063 to 0.65 mol L−1. The threading ratios and average number of hpCD (CD) per PEG, NCD were evaluated by 1H NMR measurements based on the previous work.14 The theoretical maximum number of hpCDs on the PEG chain is 340. The 1H NMR spectra of PR–hpCD and PR–CD were shown in Fig. S1–S8.† The threading ratios calculated from the NMR results were consistent with those estimated using TG-DTA (Fig. S9 and Table S1†). With increasing hpCD concentration in the pPR solutions, the threading ratio of PR–hpCD increased from 0.64% to 10%. The PRs were named PR–hpCD NCD (NCD = 2.2–34). The yields of all PR–hpCDs were over 90%.Fig. 2(a) shows the molecular weight (MW) distributions of PR–hpCD and pure PEG. With increasing the threading ratio and NCD, the MW distribution profile was shifted in parallel to a higher molecular weight regime. The MW distribution of the PR–hpCDs reflects the distribution of the hpCD threading ratio and NCD. Interestingly, as shown in Fig. 2(b), the MW distributions normalised by the average molecular weight Mw almost overlapped with each other for all the PR–hpCDs and pure PEG, indicating that the NCD and hpCD threading ratios of PR–hpCD were almost uniform. In contrast, the MW distribution of PR–CD prepared from native CDs and PEG was wider than that of PEG (Fig. 2(c) and (d)), indicating a broad distribution of the threading ratio and NCD of PR–CD.
 | ||
| Fig. 2 Distribution of (a) molecular weight MW and (b) MW normalized by average molecular weight Mw for PEG, PR–hpCD, and PR–CD. | ||
To determine the main factors influencing the NCD and threading ratio, PR–hpCD was synthesised at different PEG concentrations and temperatures in pPR solutions. The details of the synthesised PR–hpCDs are summarized in Tables S2 and S3.† As shown in Fig. S9 and S10,† the normalised molecular weight distribution, MW/Mw of all the PR–hpCDs almost overlapped with that of the pure PEG, suggesting well-defined threading ratios of the PR–hpCDs. As indicated in Table 1, the threading ratio of PR–hpCD increased with increasing hpCD concentration (Fig. 3(a)), but was independent of the PEG concentration and inclusion temperature (Fig. 3(b) and (c)). These results suggest that the threading ratio of PR–hpCD is dominated by the hpCD concentration during the pPR formation process. As shown in Table S2,† the molar ratio of CD to PEG in the pPR solutions was varied from 98 to 400, which is much larger than the number of hpCDs in a PR–hpCD, NCD = 15–19. Therefore, the threading ratio and NCD were independent of PEG concentration.
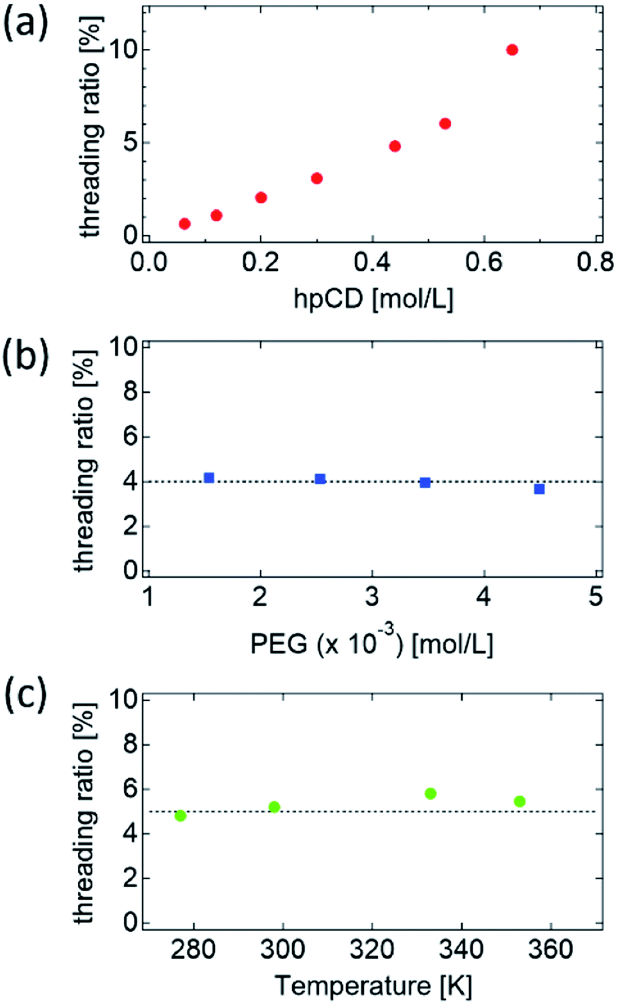 | ||
| Fig. 3 Plot of threading ratio of PR–hpCD against (a) hpCD concentration, (b) PEG concentration, and (c) inclusion temperature in pPR solution. | ||
The temperature-independence of the hpCD threading ratio indicated that the inclusion complexation of hpCD and PEG in water was driven by enthalpic interactions. The spatial distribution of hpCDs in pPR solutions was measured using SAXS and WAXS (Fig. 4(a) and S12–S17†). As shown in Fig. 4(a), the scattering profile of the pPR–hpCD7 solution was close to that of the hpCD solution with the same hpCD concentration. This suggests that the hpCDs in the pPR solution were homogeneously dispersed, similar to the hpCD solution (Fig. 4(b)). From the fact that the spatial distribution of hpCD and PEG conformation were not changed by the pPR formation (Fig. 4(a) and S18†), the entropic penalty by the inclusion complexation of hpCD and PEG can be negligible. In contrast, as shown in Fig. 4(a), the pPR–CD solution exhibited diffraction peaks and an increase in low-q intensity attributed to the crystallisation of native CDs (Fig. 4(c)).2,3 The hydrogen bonding between native CDs in the crystal structure plays an important role in the inclusion complexation of the CD/PEG system.19,25 However, the heterogeneous aggregation of CDs caused an inhomogeneous CD threading ratio of PR–CD (Fig. 4(c)). In the case of the hpCD/PEG systems, the hydroxypropyl group on hpCD weakened the attractive interaction between hpCDs and prevented the crystallisation of hpCD. The homogeneous dispersion of hpCD in the pPR–hpCD solutions resulted in the well-defined threading ratio of PR–hpCD (Fig. 4(b)). The driving force of the pPR–hpCD formation is the enthalpic benefit originated from the molecular interaction between the inside of the hpCD and the PEG axis.
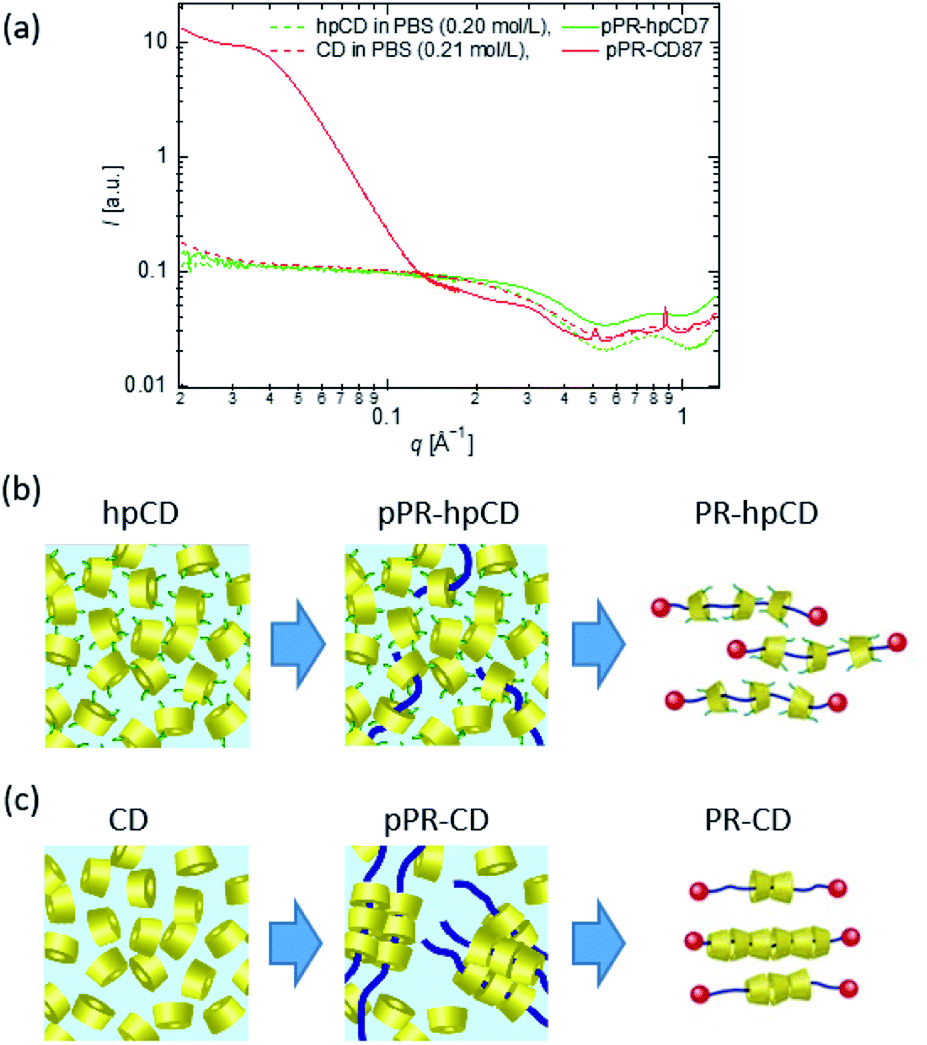 | ||
| Fig. 4 (a) Scattering profiles of CD, hpCD, pPR–CD, and pPR–hpCD7 solutions measured by SAXS and WAXS. Schematic illustrations of pPR and PR prepared from (b) CD/PEG and (c) hpCD/PEG solutions. | ||
4. Conclusion
In conclusion, we established a simple synthesis strategy to control PR threading ratios over a wide range. Native CDs form crystallised domains in pPR–CD solutions, resulting in the inhomogeneous CD threading ratio of PR–CD. Hydroxypropyl modification of CDs prevented hydrogen bonding between the CDs, generating well-dispersed hpCDs in the pPR–hpCD solutions. The pPR formation of hpCD and PEG in water was driven by enthalpic interactions between the inside of the hpCD and PEG. The threading ratio of PR–hpCD increased with hpCD concentration in pPR solutions but was independent of the PEG concentration. Surprisingly, the dispersity of the molecular weight distribution of PR–hpCDs with various hpCD threading ratios was almost the same as that of PEG. This indicates that the threading ratio of PR–hpCDs was well-defined, which was related to the homogeneous dispersion of hpCDs in the pPR solutions. This work enables the high-yield one-pot synthesis of PRs with various well-defined threading ratios by tuning the hpCD concentration, which is expected to facilitate the industrial use of PRs for functional devices and materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JST-Mirai Program Grant Number JPMJMI18A2, JST CREST Grant Number JPMJCR1992, JSPS KAKENHI Grant Number JP20K05627, NEDO JPNP18016m, NEDO JPNP18016, and the Materials Education Program for the future leaders in the Research, Industry, and Technology (MERIT). The SANS experiment was performed with the approval of Institute for Solid State Physics, the University of Tokyo (Proposal No. 21540), at Japan Atomic Energy Agency, Tokai, Japan.References
- W. Sliwa and T. Girek, Cyclodextrins: Properties and Applications, Wiley-VCH, Weinheim, 2017 Search PubMed.
- A. Harada and M. Kamachi, Macromolecules, 1990, 23, 2821–2823 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Nature, 1992, 356, 325–327 CrossRef CAS.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS PubMed.
- G. Wenz, B. H. Han and A. Muller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- A. Harada, A. Hashidzume and Y. Takashima, Adv. Polym. Sci., 2006, 201, 1–43 CrossRef CAS.
- P. L. Anelli, N. Spencer and J. F. A. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed.
- H. Fujita, T. Ooya, M. Kurisawa, H. Mori, M. Terano and N. Yui, Macromol. Rapid Commun., 1996, 17, 509–515 CrossRef CAS.
- T. Ooya, H. Mori, M. Terano and N. Yui, Macromol. Rapid Commun., 1995, 16, 259–263 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–499 CrossRef CAS.
- K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- K. Koyanagi, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2017, 50, 5695–5700 CrossRef CAS.
- S. Choi, T. W. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- L. Jiang, C. Liu, K. Mayumi, K. Kato, H. Yokoyama and K. Ito, Chem. Mater., 2018, 30, 5013–5019 CrossRef CAS.
- K. Mayumi, Polym. J., 2021, 53, 581–586 CrossRef CAS.
- K. Kato, Y. Okabe, Y. Okazumi and K. Ito, Chem. Commun., 2015, 51, 16180–16183 RSC.
- Y. Kobayashi, Y. Nakamitsu, Y. Zheng, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2018, 54, 7066–7069 RSC.
- T. Ooya, M. Eguchi and N. Yui, J. Am. Chem. Soc., 2003, 125, 13016–13017 CrossRef CAS PubMed.
- G. Fleury, C. Brochon, G. Schlatter, G. Bonnet, A. Lapp and G. Hadziioannou, Soft Matter, 2005, 1, 378–385 RSC.
- N. Jarroux, P. Guegan, H. Cheradame and L. Auvray, J. Phys. Chem. B, 2005, 109, 23816–23822 CrossRef CAS PubMed.
- J. Araki, C. Zhao and K. Ito, Macromolecules, 2005, 38, 7524–7527 CrossRef CAS.
- Y. Qiu, B. Song, C. Pezzato, D. Shen, W. Liu, L. Zhang, Y. Feng, Q. H. Guo, K. Cai and J. F. Stoddart, Science, 2020, 368, 1247–1253 CrossRef CAS PubMed.
- K. Nakazono, T. Ishino, T. Takashima, D. Saeki, D. Natsui, N. Kihara and T. Takata, Chem. Commun., 2014, 50, 15341–15344 RSC.
- J. E. Lewis, J. Winn, L. Cera and S. M. Goldup, J. Am. Chem. Soc., 2016, 138, 16329–16336 CrossRef CAS PubMed.
- S. Takahashi, N. L. Yamada, K. Ito and H. Yokoyama, Macromolecules, 2016, 49, 6947–6952 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09475k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |