
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarvicidal and histopathological efficacy of cinnamic acid analogues: a novel strategy to reduce the dengue vector competence
P. Naresh a,
R. Rajesh Kumarb,
H. N. Vishwasc,
Gopalan Rajagopald,
T. Prabhae and
S. Jubie*a
a,
R. Rajesh Kumarb,
H. N. Vishwasc,
Gopalan Rajagopald,
T. Prabhae and
S. Jubie*a
aDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Ooty, Tamilnadu, India. E-mail: jubie@jssuni.edu.in; Web: https://www.jssuni.edu.in/
bDepartment of Pharmaceutical Biotechnology, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Ooty, Tamilnadu, India
cDepartment of Pharmacy Practice, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Ooty, Tamilnadu, India
dPostgraduate and Research Department of Zoology, Ayya Nadar Janaki Ammal College, Sivakasi, Tamilnadu, India
eDepartment of Pharmaceutical Chemistry, Nandha College of Pharmacy, Affiliated to The Tamilnadu Dr MGR Medical University-Chennai, Erode, Tamilnadu, India
First published on 29th March 2022
Abstract
Background: A novel strategy such as conjugation of amino, Schiff's bases, and thiadiazole moieties to the cinnamic acid nucleus has been adopted in this study to discover new molecules that target the dengue envelope protein (DENVE). Aim: Among the different domains of dengue virus envelope protein (PDB ID 1OKE), we have selected a ligand-binding domain for our structure-based drug design. The designed compounds have also been docked against DENVE protein. Methodology: Based on the in silico results and synthetic feasibility, three different schemes were used to synthesize twenty-three novel cinnamic acid derivatives. Sci-finder ascertained their novelty. The synthesized derivatives were consistent with their assigned spectra. The compounds were further evaluated for their larvicidal activity and histopathological analysis. Multiple linear regression analysis was performed to derive the QSAR model, which was further evaluated internally and externally for the prediction of activity. Results and discussion: Four compounds, namely CA 2, CA 14, ACA 4, and CATD 2, effectively showed larvicidal activity after 24, 48, and 72 h exposure; particularly, compound CA2 showed potent larvicidal activity with LC50 of 82.15 μg ml−1, 65.34 μg ml−1, and 38.68 μg ml−1, respectively, whereas intermittent stages, causes of abscess in the gut, and siphon regions were observed through histopathological studies. Conclusion: Our study identified some novel chemical scaffolds as effective DENVE inhibitors with efficacious anticipated pharmacokinetic profiles, which can be modified further.
1. Introduction
The primary carrier of viruses that cause dengue fever, Aedes aegypti, is found in vast areas of the tropics and subtropics. There are currently no known therapies for dengue fever. However, standard fever treatment is available (nursing care, fluid balance, electrolytes, and blood clotting parameters). As a result, the only way to reduce the prevalence of this disease is by mosquito management, which involves interrupting the disease propagation cycle by attacking mosquito larvae at breeding sites. Besides vector control, a recently authorized dengue vaccine immunizes against all four serotypes. Dengvaxia is Sanofi Pasteur's (Paris, France) chimeric yellow fever-dengue-tetravalent dengue vaccine (CYD-TDV), the first and only of its type, and has been registered for use in 19 countries but is only available in 10.2 of them. In addition, several phase II and phase III trials have resulted in specific vaccine restrictions, such as CYD TDV being delivered exclusively to those who have previously been infected with dengue and live in endemic regions.13–16Presently around the world, dengue is endemic in 112 countries.1,2 Mostly in tropical and subtropical areas, each year, 50–100 million individuals are infected with DENV, resulting in nearly 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 severe life-threatening illnesses and 25000 deaths.3,4 One attractive approach could be the interruption of the virus replication at an early stage of attachment.5 DENV enters the cell by receptor-mediated endocytosis followed by viral E protein-mediated membrane fusion. Membrane fusion is a major molecular event during viral entry into the host cell.6 E (envelope) protein is a significant component of the virion surface, plays an essential role in binding to the host receptor, and assists virus fusion.7 Among the three domains present in the E protein, the hinge region movement of domains I and II facilitates the fusion process.8 Upon lowering the pH, the E protein undergoes significant conformational changes in the hinge region, springing upwards to bring the fusion peptide closer to the host membrane for fusion to occur. Small molecules' rearrangement and/or conformational changes in the hinge region may interrupt the fusion process.9 A small detergent molecule, n-octyl-β-D-glucoside (βOG), occupies the hydrophobic pocket, which is located in the hinge region and plays a significant role in the rearrangement. It has been reported that mutations within this binding pocket lead to the alterations of the pH threshold for fusion. In addition to this, the protonation of histidine residues present in the hydrophobic domain would also impart the conformational change of the E protein. The previously reported fusion inhibitors such as peptidic antivirals suffer from poor absorption from the gastrointestinal tract, necessitating intravenous delivery and high manufacturing costs.10 We, therefore, wanted to develop nonpeptidic small molecules to inhibit dengue virus entry.
000 severe life-threatening illnesses and 25000 deaths.3,4 One attractive approach could be the interruption of the virus replication at an early stage of attachment.5 DENV enters the cell by receptor-mediated endocytosis followed by viral E protein-mediated membrane fusion. Membrane fusion is a major molecular event during viral entry into the host cell.6 E (envelope) protein is a significant component of the virion surface, plays an essential role in binding to the host receptor, and assists virus fusion.7 Among the three domains present in the E protein, the hinge region movement of domains I and II facilitates the fusion process.8 Upon lowering the pH, the E protein undergoes significant conformational changes in the hinge region, springing upwards to bring the fusion peptide closer to the host membrane for fusion to occur. Small molecules' rearrangement and/or conformational changes in the hinge region may interrupt the fusion process.9 A small detergent molecule, n-octyl-β-D-glucoside (βOG), occupies the hydrophobic pocket, which is located in the hinge region and plays a significant role in the rearrangement. It has been reported that mutations within this binding pocket lead to the alterations of the pH threshold for fusion. In addition to this, the protonation of histidine residues present in the hydrophobic domain would also impart the conformational change of the E protein. The previously reported fusion inhibitors such as peptidic antivirals suffer from poor absorption from the gastrointestinal tract, necessitating intravenous delivery and high manufacturing costs.10 We, therefore, wanted to develop nonpeptidic small molecules to inhibit dengue virus entry.
While searching for small molecules, we came across an exciting scaffold, zosteric acid, isolated from marine eelgrass, Zostera marina. It has been reported that zosteric acid, chemically p-sulphoxycinnamic acid, and its analogs act as entry inhibitors by inhibiting the fusion process. Based on the reported antiviral potency of zosteric acid, we moved on to a similar readily available scaffold cinnamic acid, the naturally occurring precursor of flavonoids. It has excellent potential for a therapeutic role, showing antimicrobial, antifungal, and antitumor activities.11 A compound derived from trans-cinnamic acid, p-sulphoxy-cinnamic acid, showed antiviral activity against the dengue virus, suggesting a possible antiviral potential of trans-cinnamic acid.12 Besides, the modification of the –COOH group of cinnamic acid analogs leads to decreased IC50 values against DENV (Fig. 1). Keeping all facts in mind, the present work is aimed to identify novel bioactive molecules bearing cinnamic acid as a key signature of DENV entry.
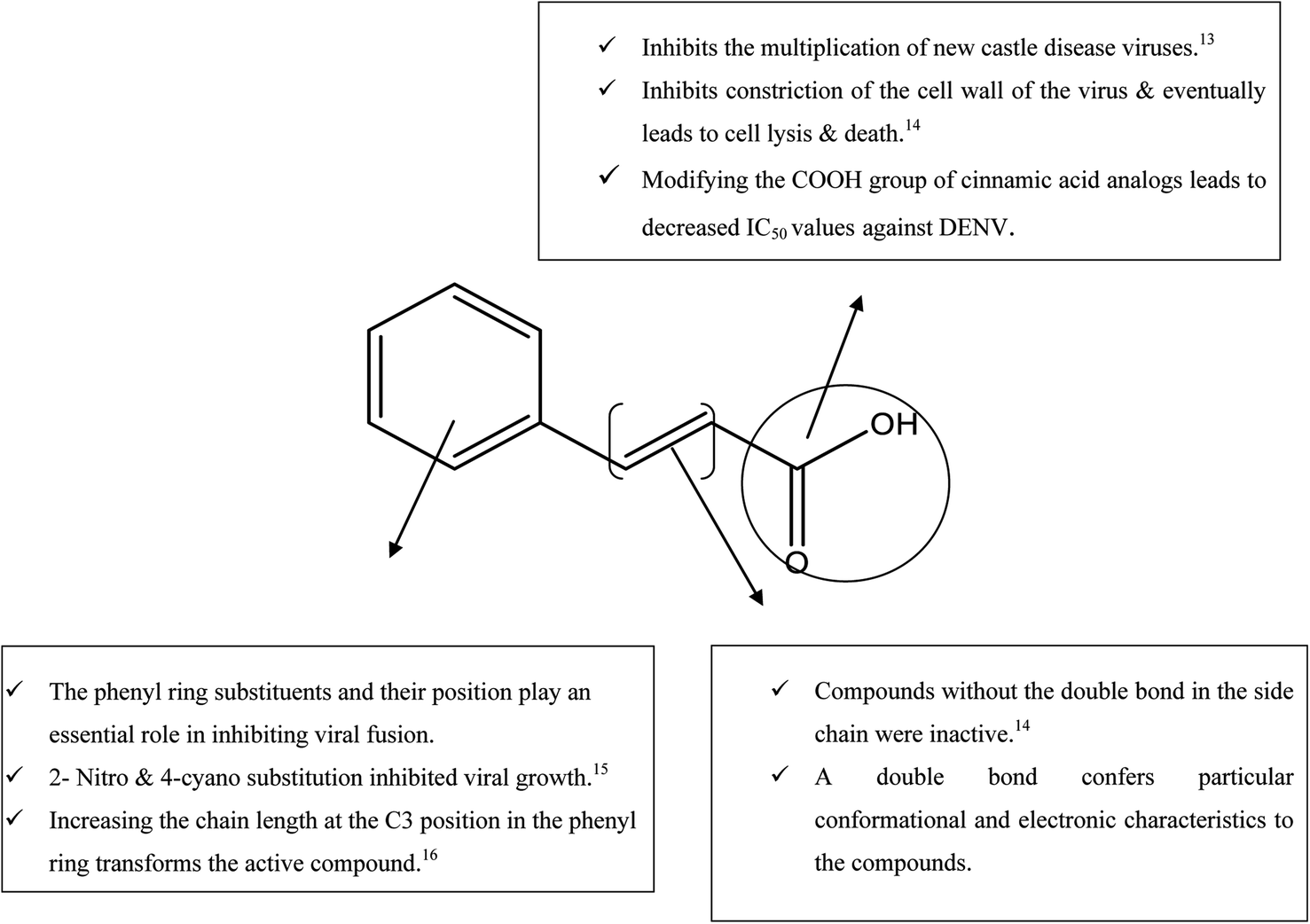 | ||
| Fig. 1 SAR of cinnamic acid. | ||
2. Materials and methods
Sigma Aldrich, SD Fine, and Carbanio supplied all the chemicals used which were of analytical grade. The solvents were purified and dried by industry standards. The 1H and 13C NMR data were analyzed using a Bruker 400 MHZ spectrometer. The internal standard was tetramethyl silane (TMS), and the solvent was DMSO-d6. Chemical shifts were reported downfield from TMS in parts per million (ppm). For analytical thin-layer chromatography, the plates used were MERCK aluminium back pre-coated silica gel 60-F 254 (0.5 mm) plates. An iodine chamber or UV light was used to identify the spots on TLC plates visually. Shimadzu recorded mass spectra.2.1. In silico design
The ADME, molecular docking, and molecular dynamics studies were conducted using Discovery Studio (D.S.) 4.1 and PyRx 0.8. The MMGBSA assay was performed using Schrodinger 2017.2 and the 3D QSAR studies were performed using MOE 2009.10 software.17 The preparation of protein (PDB ID 1OKE) and a library of seventy-two novel ligands was carried out by the standard method.18–20 The CDOCKER module was used for calculating ligand binding affinity and energies.21 The protein binding site was identified using existing ligands in the PDB database, and the sphere concept identified the location.22 All other remaining procedure was followed and maintained by the reported methods.23–25 The protein–ligand interactions were visualized and evaluated with DS 4.1.PyRx 0.8 was used to perform the docking research of designed cinnamic acid analogues against FK506-binding protein (FKBP) from Aedes aegypti (PDB ID 3UQI). PyRx is a Python-based programming language that runs on almost any modern machine, from personal computers to supercomputers. PyRx has been used to determine the binding affinity of a ligand for a protein to facilitate molecular docking. PyRx, a structure-based docking program, was used to screen cinnamic acid analogues for Aedes aegypti (PDB ID 3UQI) at a resolution of 1.30 Å. Additionally, ligands for energy reduction interact in good ways. The MMFF94 force field performed the minimization in 200 steps with an RMS gradient of 0.1. Following the devaluation, the ligands were transferred to the PDBQT format. First, we chose the macromolecule that will define the produced protein's binding site. Next, the active docking site was constructed utilizing bound ligand binding locations. Then, molecular docking was performed on a molecular window, with all produced ligands interacting with the specified active site.34,35 First, all ligands were categorized according to their binding affinity as determined by the PyRx score. Following that, the ligands were classified according to their binding energy levels.
2.2. Chemistry
Twenty-three acid analogues were selected from a library of seventy-two cinnamic acid derivatives that showed significant in silico results and were synthetically feasible.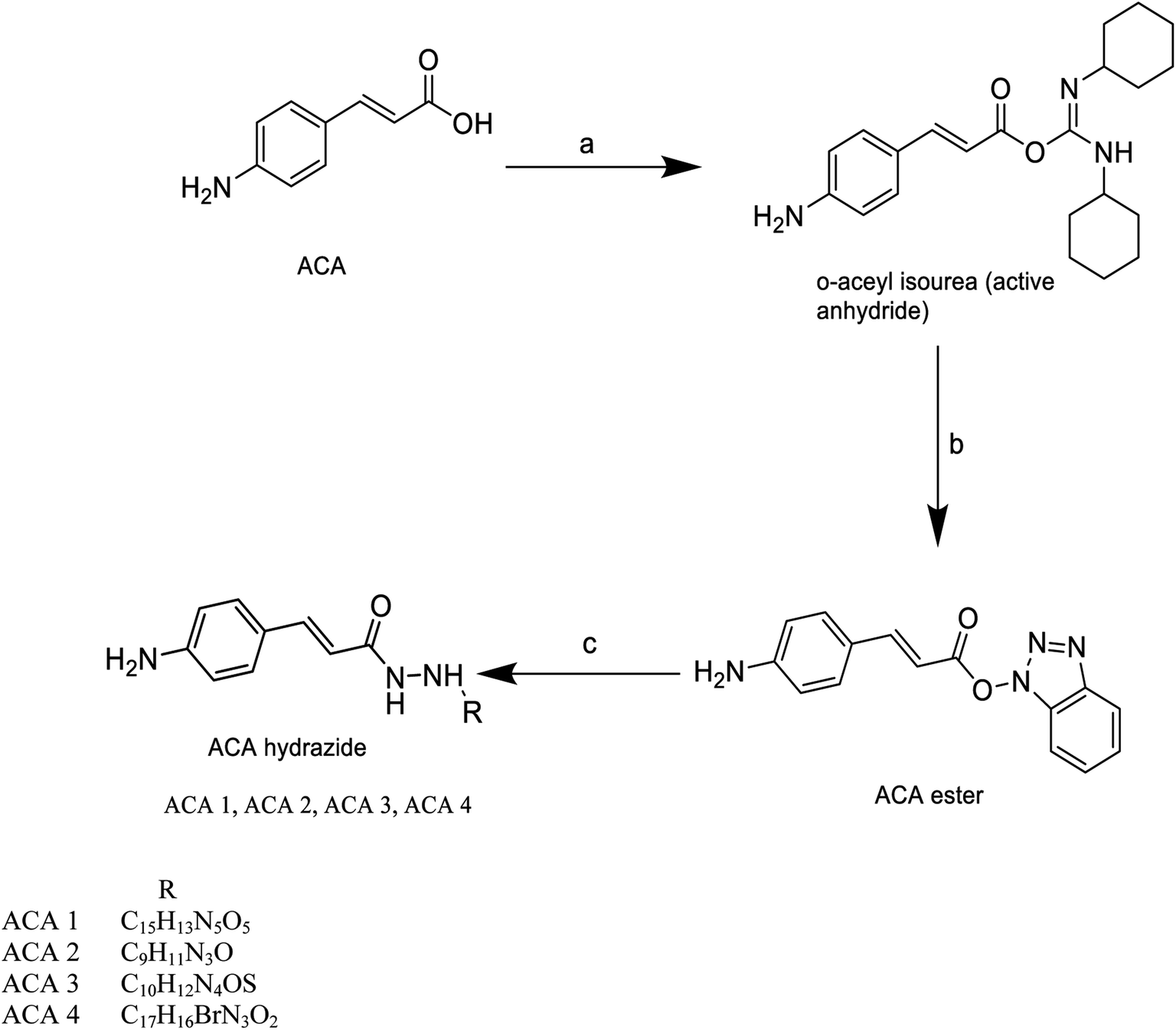 | ||
| Scheme 1 Synthesis of amino cinnamic acid hydrazide derivatives. Reagents and conditions, (a) catalytic DCC (dicyclohexyl carbodiimide). (b) HOBt (1-hydroxy benzotriazole), CH3CN (acetonitrile), stir at room temperature overnight. (c) Different hydrazines, CH3CN, stir overnight at room temperature. | ||
2.2.1.1. (2E)-3-(4-Aminophenyl)-N′-(2,4-dinitrophenyl)prop-2-enehydrazide (ACA 1). Green solid; Rf value = 0.52 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 85%. Mp. 268–270 °C. M.F. C15H13N5O5; MW: 343.25 FTIR (KBr, cm−1) 3423.12 (NH2 str), 3297.48 (C–NH str), 3282.35 (C–NH str), 2892.72 (CH str Ar), 2853.58 (CH str Ali), 1656.05 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1264.54 (C–N str). 1H NMR (300 MHz, DMSO) δ 5.84 (s, 2H, NH2), 7.35–7.62 (m, 4H, Ar H), 7.30–7.45 (m, 3H, Ar H), 9.32 (t, 2H, NH), 9.90 (d, J = 6.3 Hz, 2H, NH2), 5.91 (d, J = 6.3 Hz, 2H, CH), 6.96 (d, J = 6.3 Hz, 2H, CH). 13C NMR (300 MHz, DMSO) δ 114.25, 114.86, 115.00, 121.03, 124.81, 129.40, 129.65, 130.21, 134.52, 139.60, 141.25, 147.46, 152.8, 162.49, 165.80 (aryl carbons), 115.46, 140.15 (CHCH), 163.21 (C–NH). MS (M–H): 343.20; 146.16 (C9H8NO m/z), 155.01 (C5H3N2O4 m/z +1), (C7H6BrN2O m/z).
O), 1264.54 (C–N str). 1H NMR (300 MHz, DMSO) δ 5.84 (s, 2H, NH2), 7.35–7.62 (m, 4H, Ar H), 7.30–7.45 (m, 3H, Ar H), 9.32 (t, 2H, NH), 9.90 (d, J = 6.3 Hz, 2H, NH2), 5.91 (d, J = 6.3 Hz, 2H, CH), 6.96 (d, J = 6.3 Hz, 2H, CH). 13C NMR (300 MHz, DMSO) δ 114.25, 114.86, 115.00, 121.03, 124.81, 129.40, 129.65, 130.21, 134.52, 139.60, 141.25, 147.46, 152.8, 162.49, 165.80 (aryl carbons), 115.46, 140.15 (CHCH), 163.21 (C–NH). MS (M–H): 343.20; 146.16 (C9H8NO m/z), 155.01 (C5H3N2O4 m/z +1), (C7H6BrN2O m/z).
2.2.1.2. 2-[(2E)-3-(4-Aminophenyl)prop-2-enoyl]hydrazine-1-carbothioamide (ACA 3). Ash solid; Rf value = 0.64 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 81%. Mp. 264–266 °C. M.F. C10H12N4O5; MW: 236.15 FTIR (KBr, cm−1) 3330.23 (NH2 str), 3218.32 (C–NH str), 3033.16 (C–NH str), 2929.97 (CH str Ar), 2854.63 (CH str Ali), 1623.90 (CO), 1783.21 (CN), 1242.24 (C–N str), 1173.72 (CS str). 1H NMR (300 MHz, DMSO) δ 5.23 (s, 2H, NH2), 6.31–7.62 (m, 4H, Ar H), 9.15 (d, J = 6.3 Hz, 1H, NH), 8.44 (d, 1H, NH), 6.12 (s, H, NH2), 6.67 (d, J = 6.5 Hz, 2H, CH), 6.44 (d, J = 6.5 Hz, 2H, CH). 13C NMR (300 MHz, DMSO) δ 113.27, 114.52, 123.50, 127.29, 129.60, 134.62 (aryl carbons), 120.84, 142.12 (CHCH), 178.45 (CS), 162.42 (CO NH). MS (M–H): 236.28; 115.89 (C8H8N m/z), 90.12 (CH4N3S m/z +5), 118.21 (C2H4N3OS m/z +4).
2.2.1.3. (2E)-3-(4-Aminophenyl)-N′-(2,4-dinitrophenyl)prop-2-enehydrazide (ACA 4). Brown solid; Rf value = 0.58 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 88%. Mp. 278–280 °C. M.F. C16H14BrN3O2; MW: 360.20 FTIR (KBr, cm−1) 3472.52 (NH2 str), 3372.65 (C–NH str), 2931.90 (CH str Ar), 2851.85 (CH str Ali), 1681.02 (CO), 1342.52 (C–N str), 523.34 (C–Br str). 1H NMR (300 MHz, DMSO) δ 4.84 (s, 2H, NH2), 7.4 (m, 4H, Ar H), 7.2 (m, 4H, Ar H), 7.9 (d, J = 7.5 Hz, H, NH), 7.6 (d, J = 7.5 Hz, H, NH), 6.65 (d, J = 7.5 Hz 2H, CH), 6.5 (d, J = 7.5 Hz 2H, CH). 13C NMR (300 MHz, DMSO) δ 112.36, 113.29, 118.47, 121.07, 122.41, 125.39, 127.37, 128.34, 128.94, 129.73, 133.30, and 140.56 (aryl carbons), 110.24, 135.15 (CHCH), 163.19, 164.47 (CO NH). MS (M–H): 360.20; 115.89 (C8H8N m/z), 182.01 (C7H4BrO m/z −1), (C7H6BrN2O m/z).
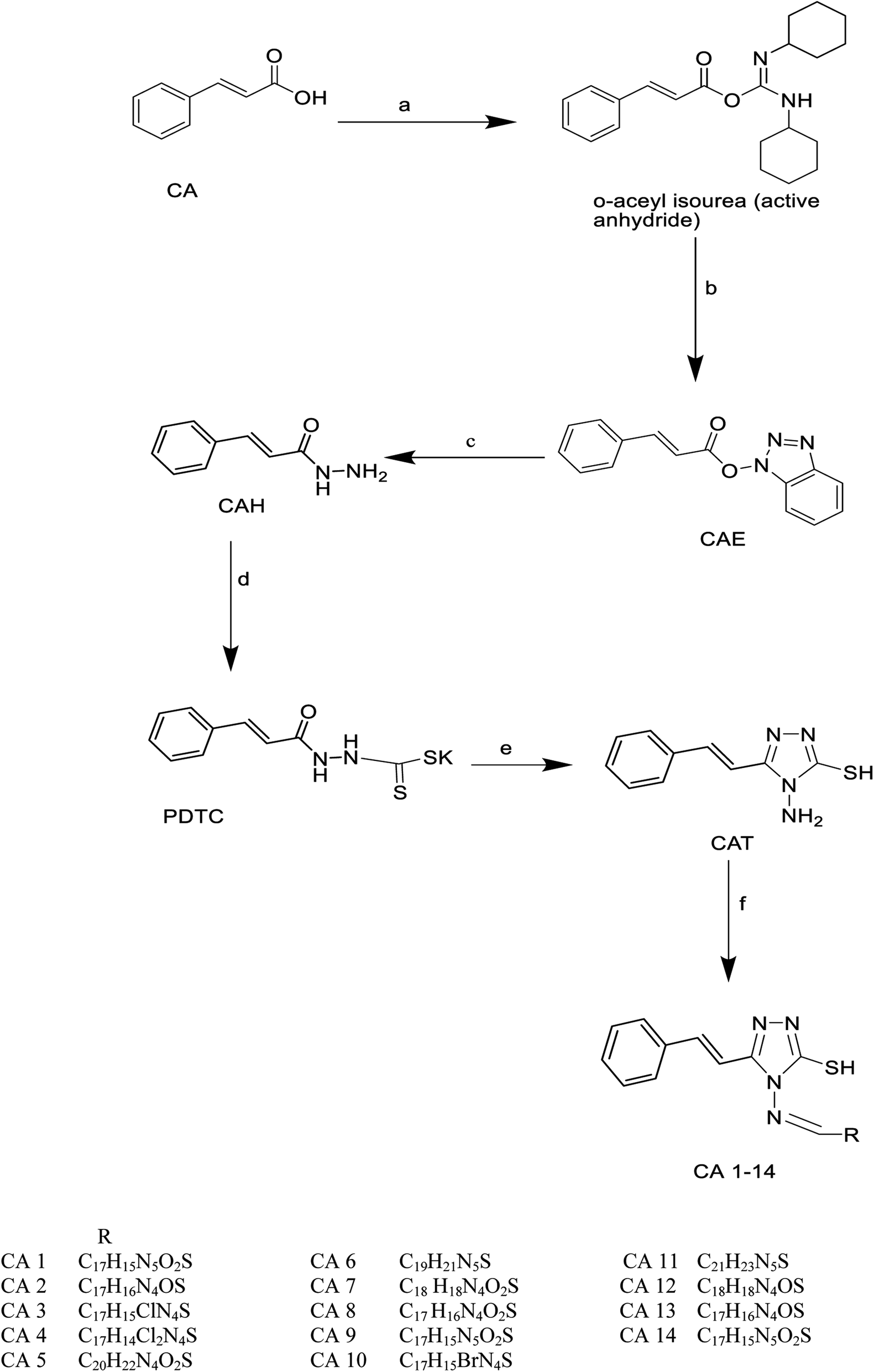 | ||
| Scheme 2 Synthesis of cinnamic acid derivatives. Reagent conditions, (a) catalytic DCC (dicyclohexyl carbodiimide). (b) HOBt (1-hydroxy benzotriazole), CH3CN (acetonitrile), overnight at room temperature, stir. (c) NH2NH2H2O (hydrazine hydrate), CH3CN (acetonitrile), overnight at room temperature, stir. (d) KOH (potassium hydroxide), CS2 (carbon disulfide), ethanol, stir overnight at room temperature. (e) NH2NH2H2O (hydrazine hydrate), H2O, reflux for ten h. (f) Benzaldehyde, ethanol, conc. H2SO4, reflux for 3 h. | ||
2.2.3.1. 2-{[(4-Chlorophenyl) methylidene] amino}-1-[(E)-2-phenylethenyl]-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 1). Yellow solid; Rf value = 0.62 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 83%. Mp. 240–242 °C. M.F. C17H15N5O2S; MW: 352.39 FTIR (KBr, cm−1) 3087.17 (CH Ar), 2927.08 (CH Ali), 2284.25 (C–S str), 1700.31 (CN str), 1268.24 (C–N str) 970.23 (N–C–S str), 806.27 (Ar CH), 370.34 (C–Cl). 1H NMR (300 MHz, DMSO) δ 8.34–8.35 (m, 5H, Ar H), 8.32–8.33 (m, 4H, Ar H), 8.72 (s, NCH), 6.9 (d, J = 6.2 Hz, 2H, CH), 6.2 (d, J = 6.2 Hz, 2H, CH), 3.40 (s, 2H, CH2), 1.51 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 112.25, 116.48, 121.32, 123.25, 125.79, 128.81, 129.93, 134.17, 135.49, 137.25, 140.41, 141.15 (aryl carbons), 64.35 (CH2), 116.23, 132.58 (CHCH), 148.69 (NC), 160.38 (CN, imine). MS (M–H): 352.39 (m/z +2); 103.24 (C8H7 m/z), 203.15 (C10H10N3S m/z +1), 149.32 (C7H5N2O2 m/z).
2.2.3.2. 4-[({2-[(E)-2-Phenylethenyl]-5-sulfanyl-2,3-dihydro-1H-1,2,4-triazol-1-yl}imino)methyl] phenol (CA 2). Milky solid; Rf value = 0.36 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 80%. Mp. 268–270 °C. M.F. C17H16N4OS; MW: 322.38 FTIR (KBr, cm−1) 3177.58 (OH str), 3019.66 (CH str Ar), 2957.94 (CH str Ali), 2363.84 (C–S), 1600.97 (CN str), 1240.27 (C–N str), 973.93 (N–C–S str), 816.88 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 10.63 (s, H, OH), 7.32–7.35 (m, 5H, Ar H), 7.56–7.59 (m, 4H, Ar H), 8.72 (s, H, NCH), 6.9 (d, J = 6.2 Hz, 2H, CH), 6.2 (d, J = 6.2 Hz, 2H, CH), 3.50 (s, 2H, CH2), 1.41 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 112.21, 118.24, 120.21, 122.38, 124.25, 125.26, 127.60, 128.35, 130.12, 133.35, 133.59, 142.40 (aryl carbons), 70.15 (CH2), 116.54, 130.87 (CHCH), 141.15 (NC), 159.16 (CN). MS (M–H): 324.40 (m/z +2); 103.14 (C8H7 m/z), 203.11 (C10H10N3S m/z +1), 125.21 (C7H6NO m/z).
2.2.3.3. 2-{[(4-Chlorophenyl) methylidene] amino}-1-[(E)-2-phenylethenyl]-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 3). White solid; Rf value = 0.42 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 80%. Mp. 221–223 °C. M.F. C17H15ClN4S; MW: 340.82 FTIR (KBr, cm−1) 3173.01 (CH str Ar), 2991.69 (CH str), 2917.43 (CH str Ali), 2349.38 (C–S str), 1700.97 (CN str), 1274.03 (C–N str), 947.08 (N–C–S), 826.88 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 7.85–7.89 (m, 5H, Ar H), 7.47–7.49 (m, 4H, Ar H), 8.5 (s, H, NCH), 6.90 (d, J = 6.3 Hz, H, CH), 6.20 (d, J = 6.3 Hz, H, CH), 3.22 (s, 2H, CH2), 1.50 (s, H, CH). 13C NMR (300 MHz, DMSO) δ 113.52, 114.25, 115.13, 117.48, 120.21, 121.89, 122.10, 123.35, 124.80, 125.50, 127.28, 128.52 (aryl carbons) 72.58 (CH2), 120.14, 138.45 (CHCH), 142.90 (NC), 172.23 (CN). MS (M–H): 342.84 (m/z +1); 144.28 (C7H5ClN m/z −1), 206.77 (C10H10N3S m/z −2).
2.2.3.4. 1-[(E)-2-Phenylethenyl]-2-{[(3,4,5-trimethoxy phenyl) methylidene] amino}-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 5). Pinky solid; Rf value = 0.47 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 91%. Mp. 220–222 °C. M.F. C20H22N4O3S; MW: 396.46 FTIR (KBr, cm−1) 3063.06 (CH str Ar), 2987.89 (CH str), 2917.43 (CH str Ali), 2118.87 (C–S str), 1692.59 (CN str), 1232.55 (C–N str), 948.04 (N–C–S str), 846.88 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 8.42–8.57 (m, 5H, Ar H), 7.08–7.26 (m, 4H, Ar H), 3.93 (s, 9H, CH3), 6.20 (d, J = 6.6 Hz, H, CH), 5.08 (d, J = 6.6 Hz, H, CH), 1.53 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 110.52, 111.22, 112.23, 114.05, 115.15, 116.26, 117.72, 119.28, 120.38, 128.18, 129.45, 131.27 (aryl carbons), 71.28 (CH2), 60.91, 56.32, 56.11 (O–CH3), 115.24, 139.43 (CHCH), 140.82 (NC), 161.51 (CN, imine). MS (M–H): 398.47 (m/z +2); 101.13 (C2H3N3S m/z −2); 106.38 (C8H7 m/z −3); 189.57 (C10H12NO3 m/z +2).
2.2.3.5. 2-Methoxy-4-[({2-[(E)-2-phenylethenyl]-5-sulfanyl-2,3-dihydro-1H-1,2,4-triazol-1-yl} imino) methyl] phenol (CA 7). Brown solid; Rf value = 0.46 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 85%. Mp. 236–238 °C. M.F. C18H18N4O2S; MW: 354.41 FTIR (KBr, cm−1) 3336.00 (OH str), 3197.23 (CH str Ar), 2973.37 (CH str Ali), 2371.56 (C–S str), 1647.26 (CN str), 1240.27 (C–N str), 977.54 (N–C–S str), 851.60 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 7.33–7.60 (m, 5H, Ar H), 6.24–6.27 (m, 3H, Ar H), 10.23 (s, H, OH), 8.25 (s, H, NCH), 3.00 (S, 2H, CH2), 3.65 (S, 3H, CH3), 1.62 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 112.01, 115.52, 117.13, 119.44, 122.92, 128.53, 130.90, 133.38, 134.24, 135.82, 136.41, 140.27 (aryl carbons), 56.55, 54.12, 54.84 (O–CH3), 20.24, 130.87 (CHCH), 169.64 (CN). MS (M–H): 354.42 (m/z +2); 144.34 (C8H8NO2 m/z +); 204.27 (C10H10N3S m/z +); 251.28 (C10H11N4O2S m/z).
2.2.3.6. 2-{[(4-Nitrophenyl) methylidene] amino}-1-[(E)-2-phenylethenyl]-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 9). Blue solid; Rf value = 0.56 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 87%. Mp. 240–242 °C. M.F. C17H15N5O2S; MW: 353.39 FTIR (KBr, cm−1 3103.57) (CH str Ar), 3044.74 (CH str Ar), 2946.36 (CH str Ali), 2359.98 (C–S str), 1636.65 (CN str), 1264.38 (C–N str), 960.85 (N–C–S str), 853.53 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 8.14–8.28 (m, 5H, ArH), 7.24–7.39 (m, 4H, ArH), 8.44 (s, H, NCH), 6.12 (d, J = 6.8 Hz, H, CH), 5.25 (d, J = 6.8 Hz, H, CH), 3.31 (s, 2H, CH2)1.58 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 116.15, 120.24, 122.31, 124.01, 124.27, 126.14, 128.01, 130.29, 132.57, 134.92, 136.49, 136.48 (aryl carbons), 74.15 (CH2), 119.87, 136.87 (CHCH), 144.68 (NC), 161.10 (CN, imine). MS (M–H): 353.39 (m/z +1); 149.12 (C7H5N2O2 m/z −2); 204.37 (C10H10N3S m/z +1); 250.25 (C9H8N5O2S m/z).
2.2.3.7. 2-{[(4-Bromophenyl) methylidene] amino}-1-[(E)-2-phenylethenyl]-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 10). Brown solid; Rf value = 0.41 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 94%. Mp. 252–254 °C. M.F. C17H15BrN4S; MW: 387.29 FTIR (KBr, cm−3 123.57) (CH str Ar), 2905.31 (CH str Ali), 2315.54 (C–S str), 1636.65 (CN str), 1231.48 (C–N str), 961.47 (N–C–S str), 813.35 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 8.14–8.28 (m, 5H, Ar H), 7.24–7.39 (m, 4H, Ar H), 8.44 (s, H, NCH), 6.10 (d, J = 6.7 Hz, H, CH), 5.17 (d, J = 6.7 Hz H, CH), 2.92 (s, 2H, CH2),1.38 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 116.15, 122.33, 125.31, 128.00, 129.89, 130.16, 131.48, 134.25, 134.54, 136.45, 142.12 (aryl carbons), 69.18 (CH2), 121.24, 135.54 (CHCH),141.19 (NC), 163.41 (CN, imine). MS (M–H): 387.29 (m/z +2); 101.16 (C2H3N3S m/z); 183.02 (C7H5BrN m/z +2); 289.15 (C9H8BrN4S m/z).
2.2.3.8. 2-({(2E)-3-[4-(Dimethylamino)phenyl]prop-2-en-1-ylidene} amino)-1-[(E)-2-phenylethenyl]-2,5-dihydro-1H-1,2,4-triazole-3-thiol (CA 11). Brown solid; Rf value = 0.41 (n-hexane
:ethyl acetate, 6:4, v/v developer, visualization: UV and I2), yield 90%. Mp. 246–248 °C. M.F. C21H23N5S; MW: 377.50 FTIR (KBr, cm−3 2811.34) (CH str Ar), 2361.91 (C–S str), 1636.65 (CN str), 1378.18 (C–N str), 966.38 (N–C–S str), 868.36 (Ar CH str). 1H NMR (300 MHz, DMSO) δ 7.26–7.67 (m, 5H, Ar H), 6.33–7.05 (m, 4H, Ar H), 8.10 (s, H, NCH), 6.02 (d, J = 6.9 Hz, H, CH), 5.35(d, J = 6.9 Hz, H, CH), 2.92 (s, 2H, CH2), 3.21, 3.18 (s, 6H, CH3), 1.58 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 112.25, 116.81, 119.28, 120.12, 123.56, 120.17, 126.49, 128.51, 129.17, 130.26, 130.92, 131.25 (aryl carbons), 65.39 (CH2), 122.42, 138.32 (CHCH), 137.24 (NC), 167.32 (CN, imine). MS (M–H): 377.50 (m/z +2); 173.23 (C11H13N2 m/z −2); 207.42 (C10H10N3S m/z +1).
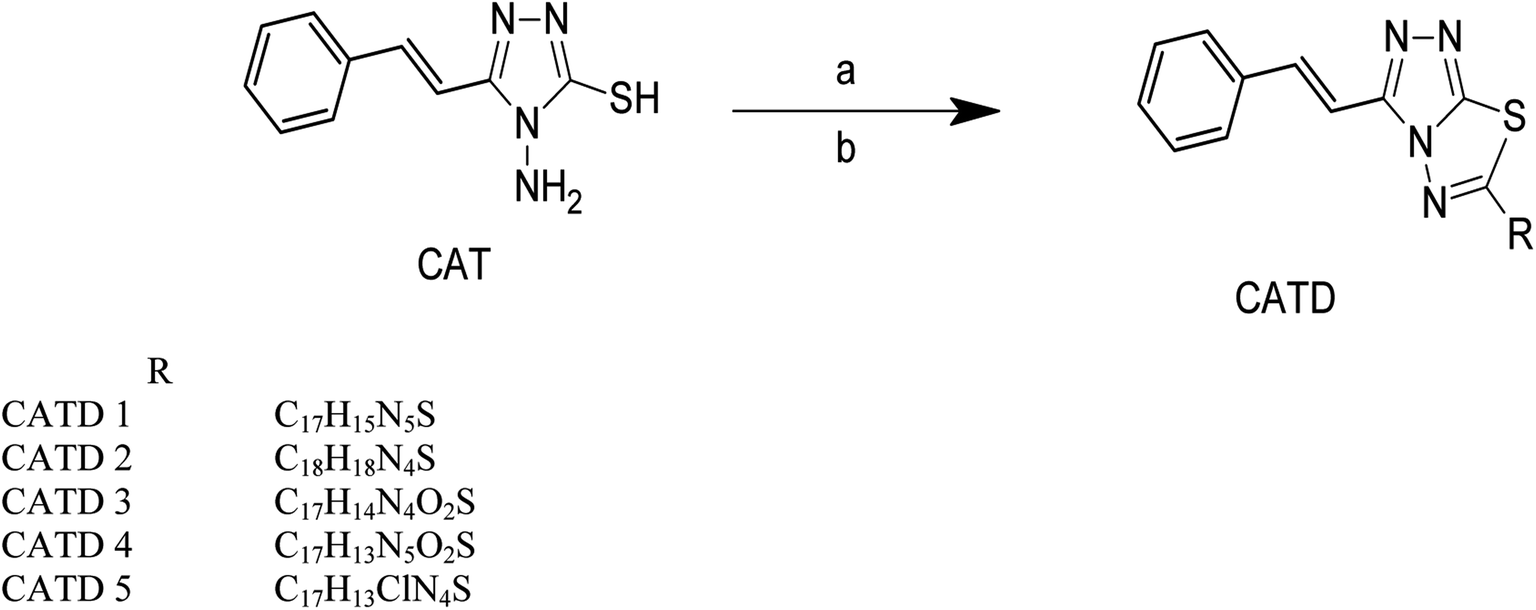 | ||
| Scheme 3 Synthesis of cinnamic acid thiadiazole derivatives. Reagent conditions, (a) 4-amino benzoic acid. (b) POCl3, reflux for three h. | ||
2.2.4.1. 2-(3-Methylphenyl)-5-[(E)-2-phenylethenyl]-5,6-dihydro[1,2,4]triazolo[5,1-b][1,3,4]thiadiazole (CATD 2). White solid; retention factor (Rf) value = 0.68 (ethyl acetate
:n-hexane, 4:6, v/v developer, visualization: UV and I2), yield 90%. Mp. 225–227 °C. M.F. C18H16N4S; MW: 320.41 FTIR (KBr, cm−1) 2923.22 (CH str Ar), 2827.34 (CH str Ali), 1163.11 (C–S str), 1603.86 (CN str), 1247.99 (C–N str), N–C–S (962.54 str), 782.16 (Ar str CH). 1H NMR (300 MHz, DMSO) δ 7.23–7.62 (m, 5H, Ar H), 7.42–7.55 (m, 4H, Ar H), 3.20 (s, H, CH2), 2.42 (T, 3H, CH3), 5.32 (d, J = 6.3 Hz, 2H, CH), 6.45 (d, J = 6.3 Hz, H, CH). 13C NMR (300 MHz, DMSO) δ 124.12, 125.45, 126.20, 127.92, 128.52, 130.12, 131.50, 131.35, 133.56, 136.59, 138.82 (aryl carbons), 71.56 (CH2), 115.82, 131.70 (CHCH), 140.12, 156.70 (CN, imine), 20.81 (CH3). MS (M–H): 320.42 (m/z +2); 103.14 (C8 H7 m/z +1); 229.14 (C11H9N4S m/z +1), 91.13 (C7H7 m/z −1).
2.2.4.2. 4-{5-[(E)-2-Phenylethenyl]-5,6-dihydro[1,2,4]triazolo[5,1-b][1,3,4]thiadiazol-2-yl}benzene-1,2-diol (CATD 3). Brown solid; retention factor (Rf) value = 0.47 (ethyl acetate
:n-hexane, 4:6, v/v developer, visualization: UV and I2), yield 92%. Mp. 245–247 °C. M.F. C17H14N4O2S; MW: 338.38 FTIR (KBr, cm−1) 3086.23 (CH str Ar), 2917.43 (CH str Ali), 2351.65 (C–S str), 1638.58 (CN str), 1237.38 (C–N str), N–C–S (979.87 str), 830.38 (Ar str CH). 1H NMR (300 MHz, DMSO) δ 7.2–7.5 (m, 5H, Ar H), 7.06–7.25 (m, 3H, Ar H), 9.01 (s, H, OH), 9.12 (s, H, OH), 8.34 (s, H, NCH), 6.10 (d, J = 6 Hz, H, CH), 5.52 (d, J = 6 Hz, H, CH), 2.95 (s, 2H, CH2), 1.45 (s, H, SH). 13C NMR (300 MHz, DMSO) δ 110.29, 114.41, 118.52, 120.28, 123.20, 124.61, 126.31, 128.61, 129.54, 130.12, 131.47, 131.87 (aryl carbons), 73.18 (CH2), 124.87, 137.29 (CHCH), 148.08 (C–OH), 171.57 (CN, imine). MS (M–H): 338.38 (m/z +2); 105.31 (C8 H7 m/z −2); 235.44 (C9H7N4O2S m/z +1), 109.31 (C6H5O2 m/z).
2.2.4.3. 2-(3-Methylphenyl)-5-[(E)-2-phenylethenyl]-5,6-dihydro[1,2,4]triazolo[5,1-b][1,3,4]thia diazole (CATD 4). Yellow solid; retention factor (Rf) value = 0.62 (ethyl acetate
:n-hexane, 4:6, v/v developer, visualization: UV and I2), yield 89%. Mp. 262–265 °C. M.F. C17H13N5O2S; MW: 351.38 FTIR (KBr, cm−1) 3096.82 (CH str Ar), 2921.29 (CH str Ali), 1157.94 (C–S str), 1681.02 (CN str), 1277.88 (C–N str), N–C–S (979.87 str), 832.31 (Ar str CH). 1H NMR (300 MHz, DMSO) δ 7.39–7.68 (m, 5H, Ar H), 8.07–8.21 (m, 4H, Ar H), 1.52 (s, H, SH), 3.2 (d, 2H, CH2), 6.41 (d, J = 6.2 Hz, H, CH), 5.37 (d, J = 6.2 Hz H, CH). 13C NMR (300 MHz, DMSO) δ 122.01, 126.52, 127.32, 128.50, 128.87, 130.76, 131.26, 133.58, 134.69, 136.20, 138.51, 14.30 (aryl carbons), 70.54 (CH2), 110.55, 134.42 (CHCH), 143.20, 156.28 (CN, imine). MS (M–H): 351.38 (m/z +2); 105.31 (C8 H7 m/z −2); 235.44 (C9H7N4O2S m/z +1), 109.31 (C6H5O2 m/z).
2.3. Biological activity
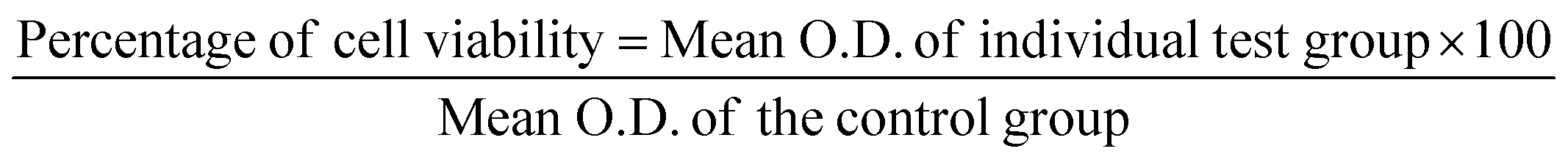
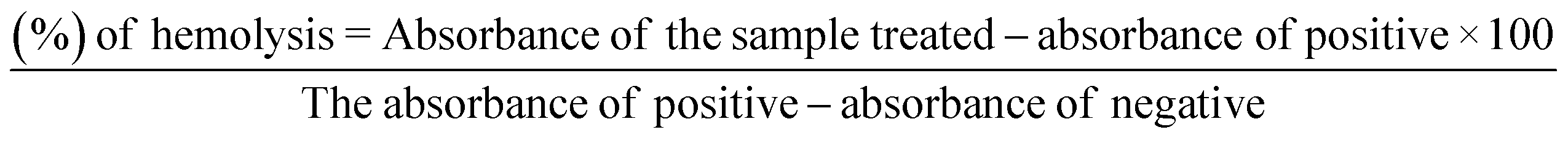
After larval exposure to test compounds after 24 hours and 48 hours, mortality was noted. The LC50 and LC90 values were further determined with the help of probit regression analysis. In addition, the chi-square value, degree of freedom, and p-value were documented. All the analysis was performed using SPSS 16. Any ‘p’-value <0.05 was considered statistically significant. The mortality of the larvae did not exceed 10% in the control group. Hence, the present data did not require any additional statistical analysis.
2.4. QSAR studies
The half-maximal lethal concentration (LC50) measures the effectiveness of a substance in inhibiting a specific biological or biochemical function. The LC50 values were manually converted into pLC50 (predicted LC50) using the formula pLC50 = −log LC50. For performing the QSAR study, a dataset of 24 synthesized compounds was chosen, from which 19 training sets and 5 test set compounds were assigned accordingly. The seven descriptors were chosen to describe the structure of the compounds comprising the series to study viz., dipole moment (AM1_dipole), molar refractivity (M.R.), lowest unoccupied molecular orbital (AM1_LUMO), partition coefficients (logP(o/w)), highest unoccupied molecular orbital (AM1_HOMO), van der Waals energies (EvdW), and topological polar surface area (TPSA).31 The Multiple Linear Regression (MLR) procedures were used to create the QSAR model, which was constructed from the QSAR descriptor panel of the MOE 2009.10 suite,32 and the RMSE and R2 values were derived from the QSAR fit.33 A QSAR correlation plot was generated by plotting the values of pLC50 on the x-axis and the predicted values ($PRED) on the y-axis for all the nineteen compounds (Fig. 9).
3. Results and discussion
3.1. In silico study of cinnamic acid analogues against DENVE (PDB ID 1OKE)
A compound library of seventy-two new substances substituted with various aldehydes and benzoic acids at the 2nd and 4th positions of cinnamic was designed (Table 1). The proposed compounds were postulated as new molecules by Sci-finder software.| Compounds | Pattern of substitution | Rationale |
|---|---|---|
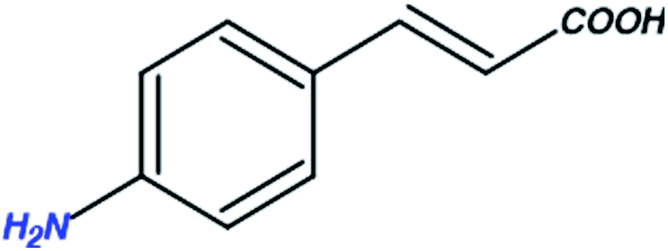 |
NH2 substitution | To inhibit the viral growth |
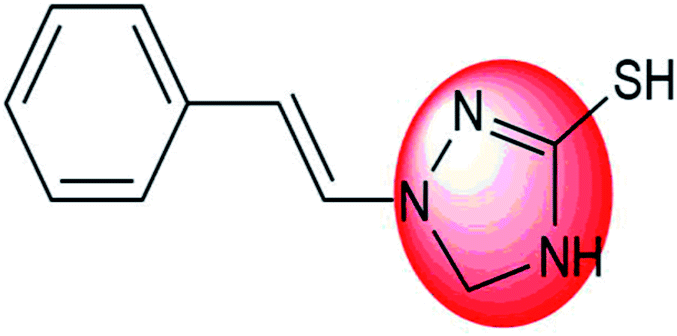 |
Modification of COOH group of 1,2,4-triazole scaffold | To decrease IC50 values against DENV |
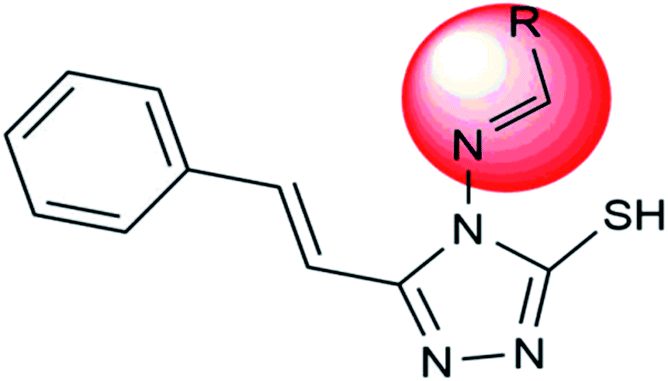 |
Addition of different aromatic aldehydes (2nd & 4th position) (Schiff's base) on the triazole ring | To inhibit the viral growth |
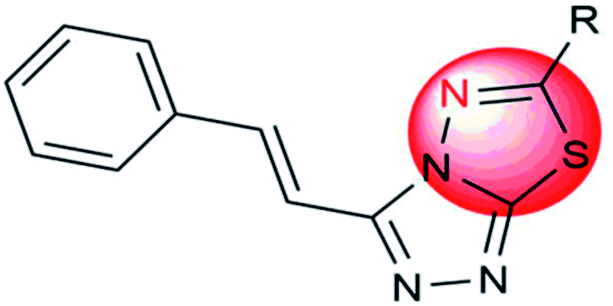 |
Thiadiazole substitution and its position | Play on a significant role in the inhibition of viral fusion |
The most effective ligand binding site was discovered in pocket number 26, which had a volume of 1178.9 Å3 and an area of 901.1 Å2. There are 26 amino acid residues in this pocket, including Thr 48, Glu 49, Ala 50, Lys 51, Gly 271, Leu 275, Thr 280, Lys 120, Ala 205, Lys 198, Val 130, Ala 53, Gly 280, Lys 275, Leu 191, and Arg 57 (Fig. 2). Our study found the same pocket with the above amino acid residues, and the Ramachandran plot further validated the prepared protein (Fig. 3). In addition, the CDOCKER energy of the synthesized compounds was lower than that of the reference inhibitor P02 (−132.553), as shown in Table 2, indicating that these molecules may have a higher binding affinity for DENVE (Fig. 4).
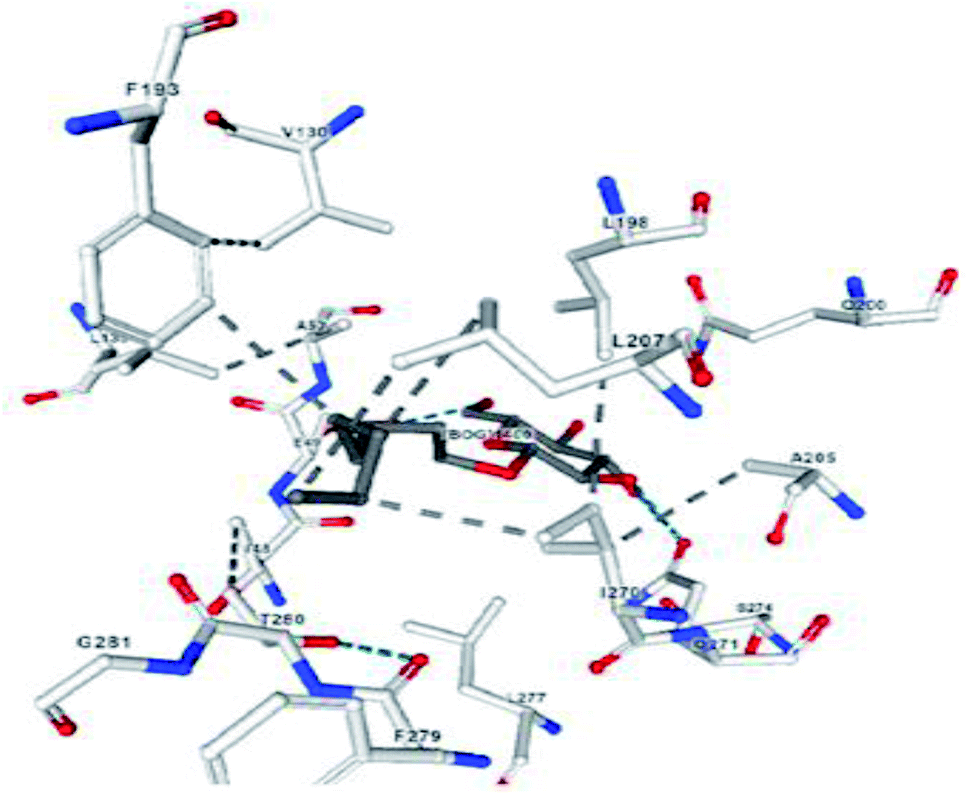 | ||
| Fig. 2 Amino acid residues present in the active site of DENV envelope protein. | ||
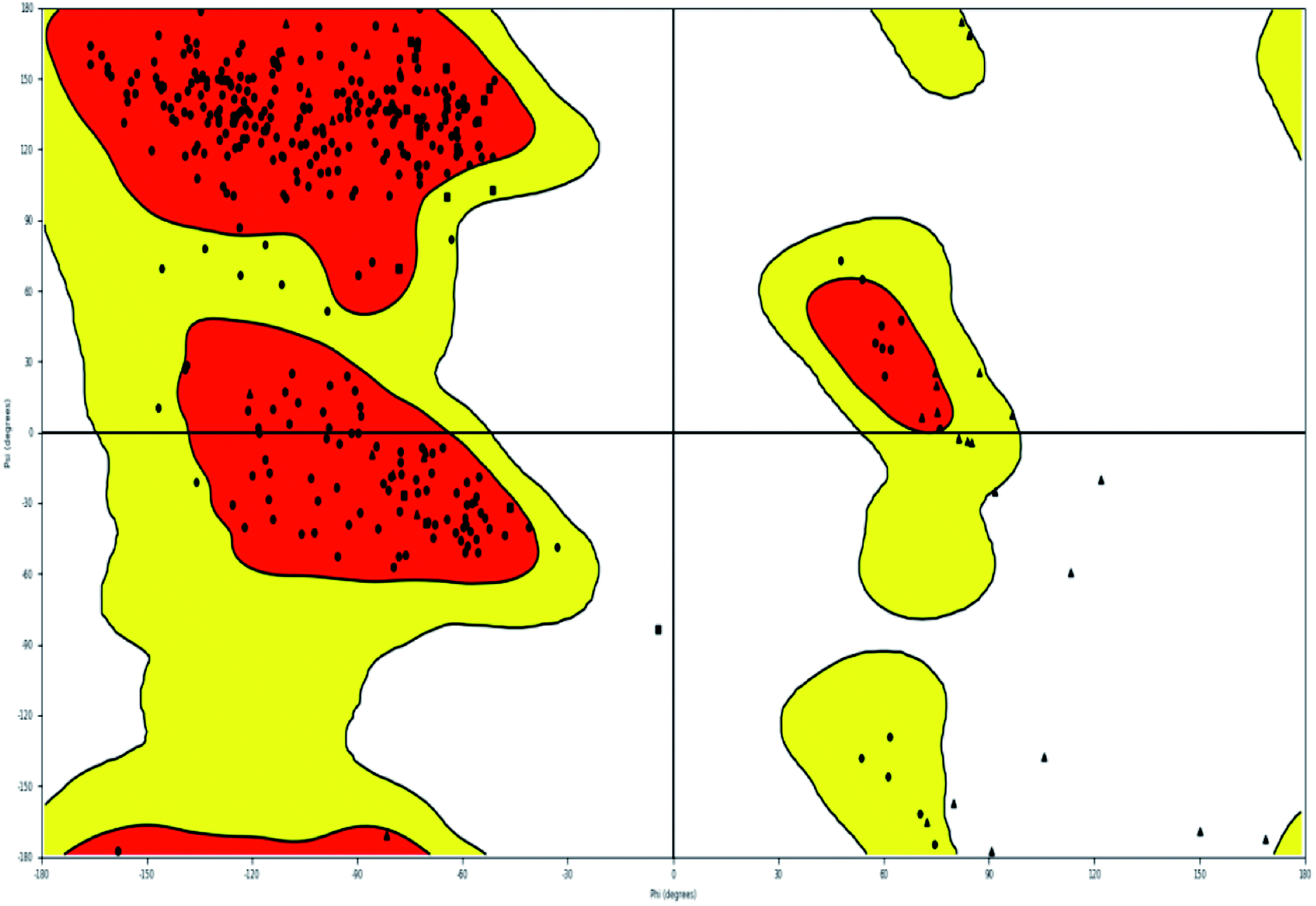 | ||
| Fig. 3 Ramachandran plot of 1OKE. | ||
| S. No. | Comp. name | C docker energy (−kcal mol−1) | C docker interaction energy (−kcal mol−1) |
|---|---|---|---|
| 1 | CA 1 | 31.9484 | 28.2893 |
| 2 | CA 2 | 56.9833 | 45.5343 |
| 3 | CA 3 | 37.9812 | 19.5848 |
| 4 | CA 4 | 42.8537 | 34.7445 |
| 5 | CA 5 | 38.8767 | 30.1751 |
| 6 | CA 6 | 37.5519 | 35.706 |
| 7 | CA 7 | 32.6039 | 35.2159 |
| 8 | CA 8 | 42.3452 | 32.9779 |
| 9 | CA 9 | 31.7342 | 36.3489 |
| 10 | CA 10 | 35.3552 | 30.806 |
| 11 | CA 11 | 30.8311 | 34.2838 |
| 12 | CA 12 | 28.1112 | 36.826 |
| 13 | CA 13 | 33.1193 | 32.1901 |
| 14 | CA 14 | 36.65667 | 24.6668 |
| 15 | ACA 1 | 39.36247 | 31.4295 |
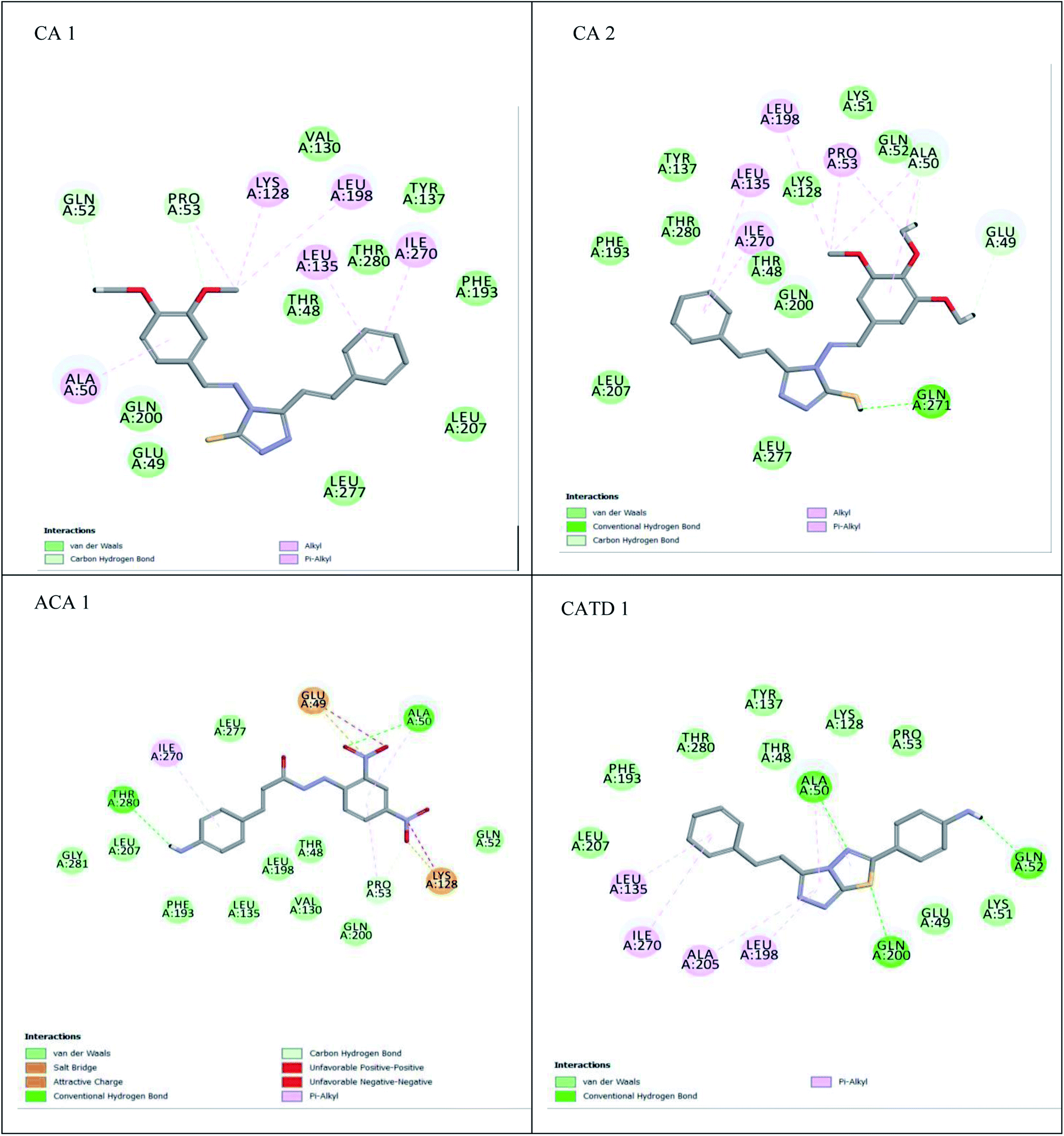 | ||
| Fig. 4 2D interaction of the compounds. | ||
The results of the ADME study showed that all the compounds' logP values were found to be within limits (i.e. less than five). AlogP is a critical parameter that affects drug bioavailability, membrane permeability and drug delivery and clearance paths. This parameter is also important in drug pharmacological and toxicological properties. Plasma protein binding is another parameter to assess the concentration of a medicinal compound. It may be restrictive or permissive and is known to affect drug distribution. A Bayesian score ≥2.226 was observed for the compounds, indicating their likelihood to bind to a carrier protein in the blood. All compounds had a >140 polar surface area (Table 3), indicating the potential for high intestinal absorption (Fig. 5). All of the compounds were found to have high blood barrier levels. All compounds were CYP2D6 non-inhibitors, with none associated with significant drug interaction toxicity.
| Comp. | Solubilitya | BBBb | CYP2D6c | Hepatoxicityd | PPBe | Absorptionf | AlogP9g | PSAh |
|---|---|---|---|---|---|---|---|---|
| a Solubility level – 0 extremely low; 1 no very low but possible; 2 yes low; 3 yes good; 4 yes optimal; 5 no too soluble.b BBB – blood-brain barrier level – 0 very high; 1 high; 2 medium; 3 low; 4 undefined.c CYP2D13 – cytochrome 450 inhibition.d Hepatotoxicity.e PPB – plasma-protein binding.f Absorption – 1 very poor, 2 poor; 3 medium; 4 good.g AlogP98 – partition coefficient of octanol/water system,h PSA – polar surface area. | ||||||||
| CA 1 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 4.845 | 152.427 |
| CA 2 | 2 | 1 | FALSE | FALSE | TRUE | 2 | 3.335 | 116.194 |
| CA 3 | 2 | 1 | FALSE | FALSE | TRUE | 2 | 3.135 | 136.491 |
| CA 4 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.308 | 166.139 |
| CA 5 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.118 | 142.319 |
| CA 6 | 2 | 1 | FALSE | TRUE | TRUE | 2 | 3.214 | 132.178 |
| CA 7 | 3 | 1 | FALSE | TRUE | TRUE | 3 | 4.105 | 153.234 |
| CA 8 | 2 | 1 | FALSE | TRUE | TRUE | 2 | 3.214 | 150.178 |
| CA 9 | 3 | 1 | FALSE | TRUE | TRUE | 2 | 3.578 | 146.714 |
| CA 10 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.468 | 150.546 |
| CA 11 | 2 | 1 | FALSE | TRUE | TRUE | 2 | 3.752 | 162.482 |
| CA 12 | 3 | 1 | FALSE | TRUE | TRUE | 3 | 3.312 | 138.814 |
| CA 13 | 2 | 1 | FALSE | TRUE | TRUE | 2 | 3.562 | 157.178 |
| CA 14 | 2 | 1 | FALSE | TRUE | TRUE | 2 | 3.421 | 142.278 |
| ACA 1 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.308 | 126.139 |
| ACA 2 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.178 | 143.116 |
| ACA 3 | 3 | 1 | FALSE | TRUE | TRUE | 3 | 3.751 | 148.142 |
| ACA 4 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 3.892 | 156.178 |
| CATD 1 | 2 | 1 | FALSE | TRUE | TRUE | 3 | 2.932 | 143.911 |
| CATD 2 | 1 | 1 | FALSE | FALSE | TRUE | 4 | 4.513 | 137.954 |
| CATD 3 | 2 | 1 | FALSE | FALSE | TRUE | 3 | 4.113 | 147.954 |
| CATD 4 | 4 | 1 | FALSE | FALSE | TRUE | 4 | 2.625 | 150.491 |
| CATD 5 | 1 | 1 | FALSE | FALSE | TRUE | 4 | 4.357 | 157.954 |
| FMCV | 2 | 1 | FALSE | TRUE | TRUE | 3 | 2.549 | 150.055 |
| P02 | 2 | 2 | FALSE | TRUE | TRUE | 3 | 4.951 | 150.89 |
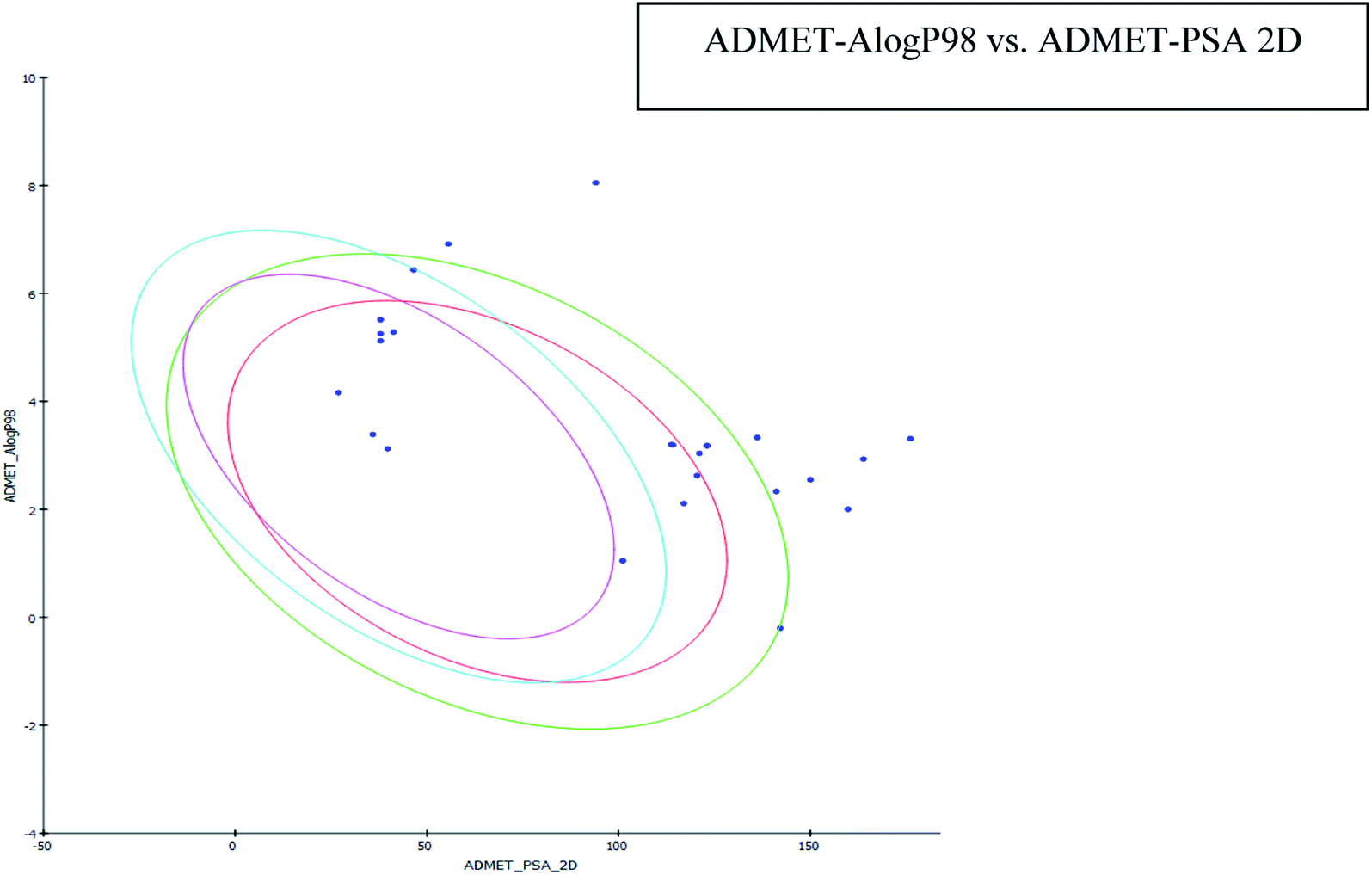 | ||
| Fig. 5 Plot of PSA versus logP with 95% and 99% confidence limit ellipses. | ||
The MM-GBSA approach for binding free energy calculation revealed that van der Waals and coulombic terms were the most relevant positive contributors. A preferential trend towards the van der Waals component was observed, as indicated by the van der Waals and Coulomb energy interactions between −17.19 and −57.5 kcal mol−1, and −1.36 and −41.97 kcal mol−1, respectively (Table 4). CA2 bounds best with DG (−75.87 kcal mol−1), with a slightly higher van der Waals contribution (−43.1 kcal mol−1), favourable lipophilic energy (25.67 kcal mol−1), and a favourable coulomb energy term (−41.97 kcal mol−1). van der Waals was deemed the driving force behind ligand binding.
| S. No. | Comp. code | ΔG bind (kcal mol−1) | ΔG Coulomb | ΔG vdW | ΔG H-bond | ΔG covalent | ΔG lipophilic |
|---|---|---|---|---|---|---|---|
| 1 | CA 1 | −70.87 | −41.97 | −63.1 | −6.46 | 10.44 | −45.67 |
| 2 | CA 2 | −75.71 | −64.22 | −80.41 | −2.14 | −16.89 | −61.27 |
| 3 | CA 3 | −69.31 | 23.56 | −57.5 | −2.5 | −0.01 | −46.19 |
| 4 | CA 4 | −68.22 | −41.21 | −63.24 | −6.53 | 8.44 | −38.42 |
| 5 | CA 5 | −51.47 | −19.36 | −75.55 | −0.18 | 1.72 | −37.68 |
| 6 | CA 6 | −51.3 | −11.27 | −69.4 | −2.86 | 13.24 | −30.19 |
| 7 | CA 7 | −62.49 | −15.11 | −47.19 | −7.4 | −8.27 | −36.96 |
| 8 | CA 8 | −60.78 | −21.56 | −48.43 | −5.69 | 14.76 | −32.12 |
| 9 | CA 9 | −56.286 | −23.041 | −56.205 | −3.13 | 15.602 | −44.468 |
| 10 | CA 10 | −54.268 | −21.465 | −60.112 | −1.684 | 11.377 | −43.595 |
| 11 | CA 11 | −60.243 | −46.524 | −55.803 | 2.073 | 5.477 | −35.024 |
| 12 | CA 12 | −58.177 | −53.035 | −73.522 | 1.433 | 11.726 | −38.407 |
| 13 | CA 13 | −54.562 | −12.145 | −77.397 | −0.208 | 24.994 | −43.145 |
| 14 | CA 14 | −57.471 | −21.185 | −63.714 | 1.699 | 19.058 | −36.654 |
| 15 | ACA 1 | −54.562 | −11.145 | −79.397 | −0.208 | 24.994 | −43.145 |
| 16 | ACA 2 | −57.471 | −21.185 | −63.714 | 1.699 | 19.058 | −36.654 |
| 17 | ACA 3 | −57.177 | −50.035 | −73.522 | 1.433 | 11.726 | −39.407 |
| 18 | ACA 4 | −51.043 | 61.524 | −79.803 | 2.073 | 5.477 | −51.024 |
| 19 | CATD 1 | −56.286 | −23.041 | −56.205 | −3.13 | 15.602 | −44.468 |
| 20 | CATD 2 | −54.268 | −21.465 | −60.112 | −1.684 | 11.377 | −43.595 |
| 21 | CATD 3 | −60.243 | −46.524 | −15.803 | 2.073 | 5.477 | −35.024 |
| 22 | CATD 4 | −58.177 | −53.035 | −73.522 | 1.433 | 11.726 | −38.407 |
| 23 | CATD 5 | −48.770 | −55.237 | −69.722 | 1.521 | 10.614 | −40.701 |
| 24 | Fmcv | −58.177 | −40.015 | −40.221 | 1.278 | 11.425 | −29.728 |
| 25 | PO2 | −63.97 | −23.19 | −41.47 | −4.47 | −4.32 | 0000 |
3.2. In silico study of cinnamic acid analogues against FK506-binding protein (FKBP) from Aedes aegypti (PDB ID 3UQI)
In PyRx, binding affinity parameters were considered for selecting the best “HITS” and compared with the known Co-crystal. PyRx binding energy is the interaction energy between the protein and the ligand. This value strongly indicates the extent of the interaction of proteins and ligands. The synthesized molecules whose binding energy was above the co-crystal FKBP-type peptidyl-prolyl cis–trans isomerase (−4.7) are shown in Table 5, indicating that the compounds were effectively bound to the active site of 3UQI. The hydrogen bonds and Pi–Pi interactions of these compounds were analyzed. The CA 2 showed the highest binding affinity for 3UQI (−7.8) and the 3UQI-6 protein CA 2 complex made one conventional-hydrogen bonding (Arg 43), four hydrophobic interactions (Glu 55, Ile 57, Phe 100 and Trp 60), and two pi–alkyl interactions (Val 56, and Tyr 57).| Ligand | Binding energy (kcal mol−1) | 2D interactions |
|---|---|---|
| CA 2 | −7.8 | 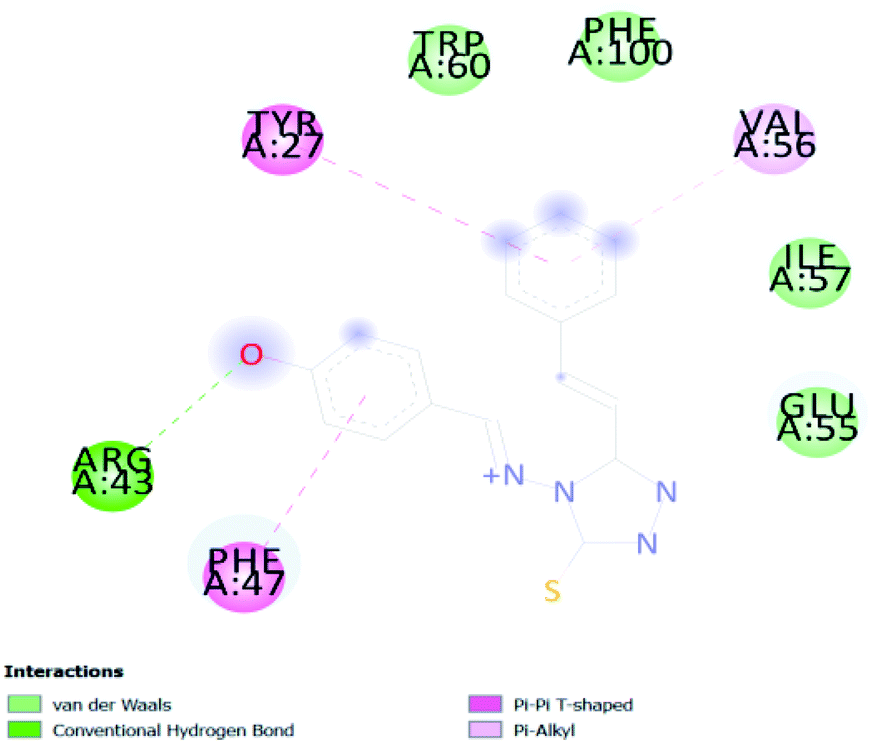 |
| CATD 4 | −7.3 | 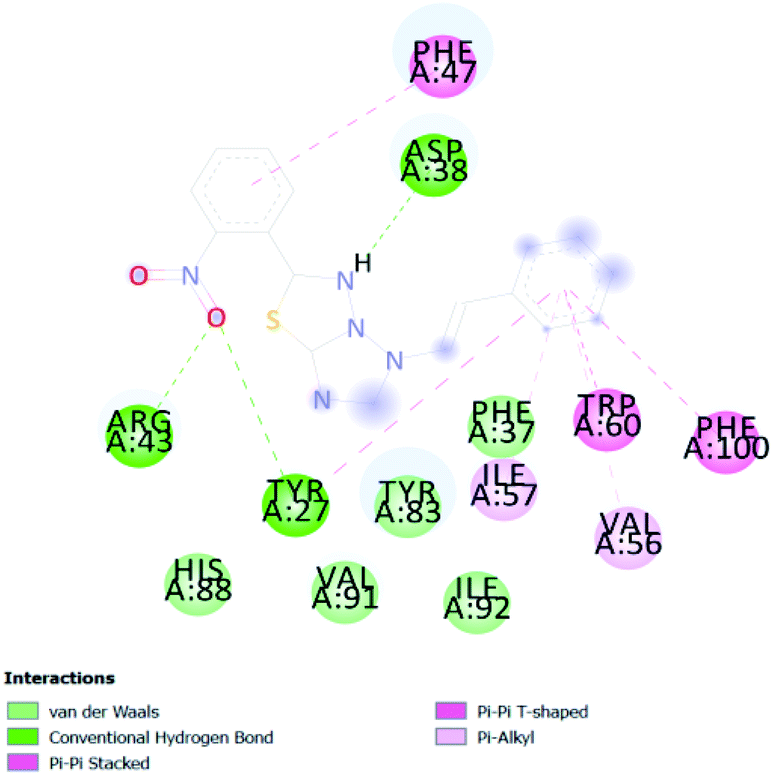 |
| CATD 3 | −7 | 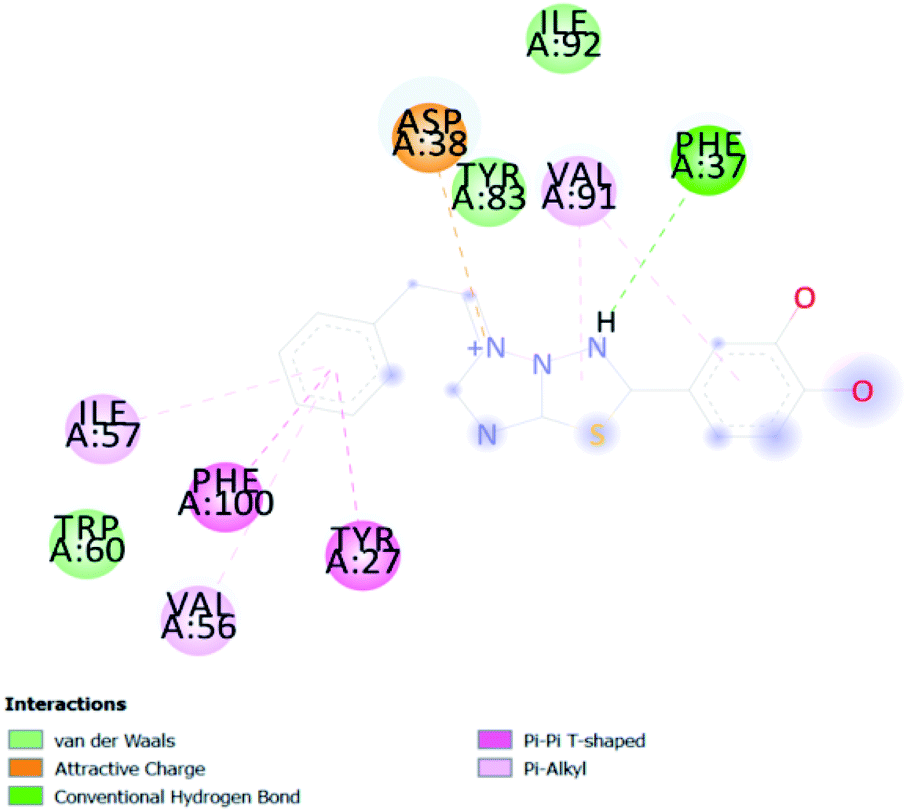 |
| ACA 1 | −6.9 | 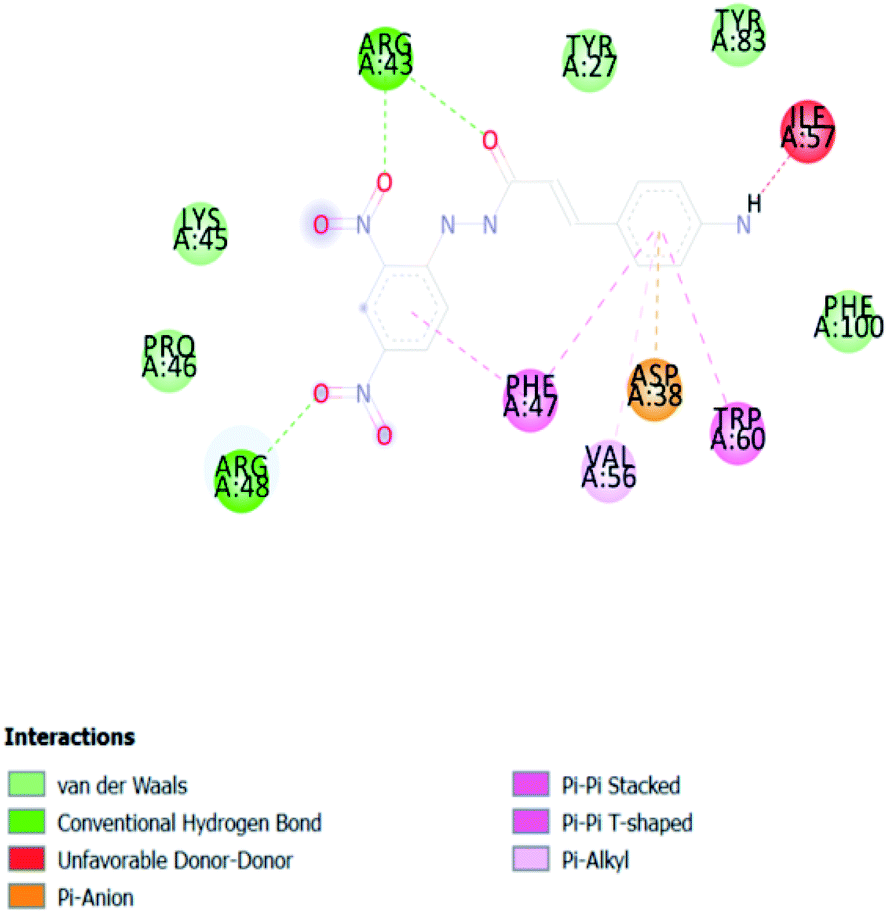 |
| CA 4 | −6.8 | 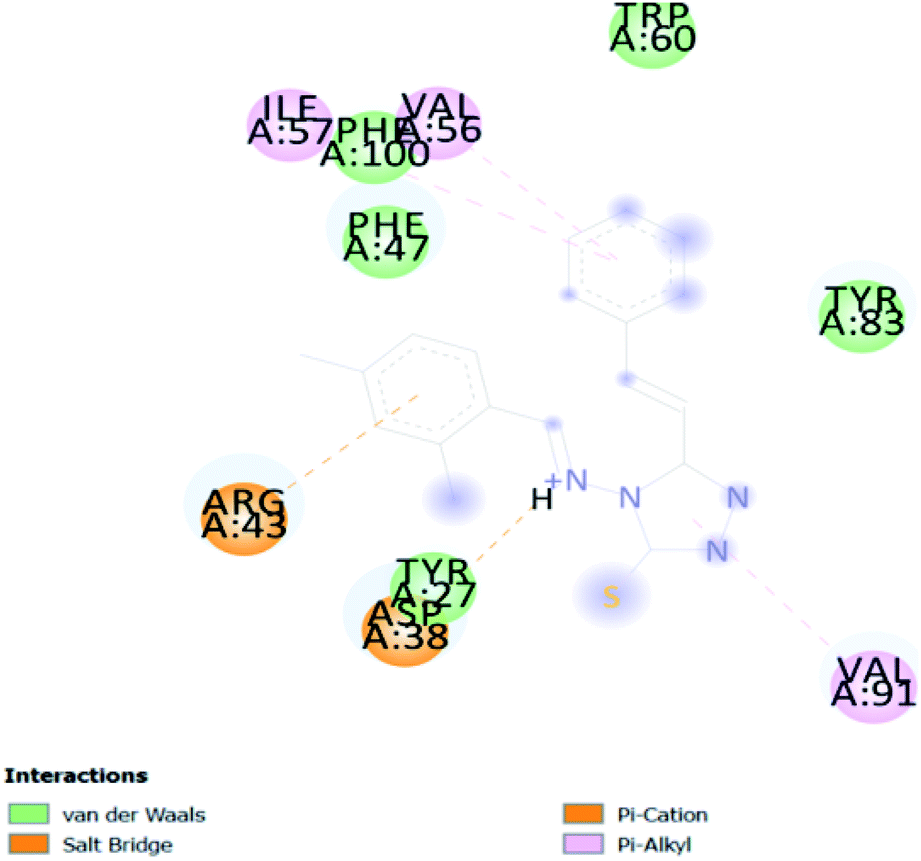 |
| CA 14 | −6.7 | 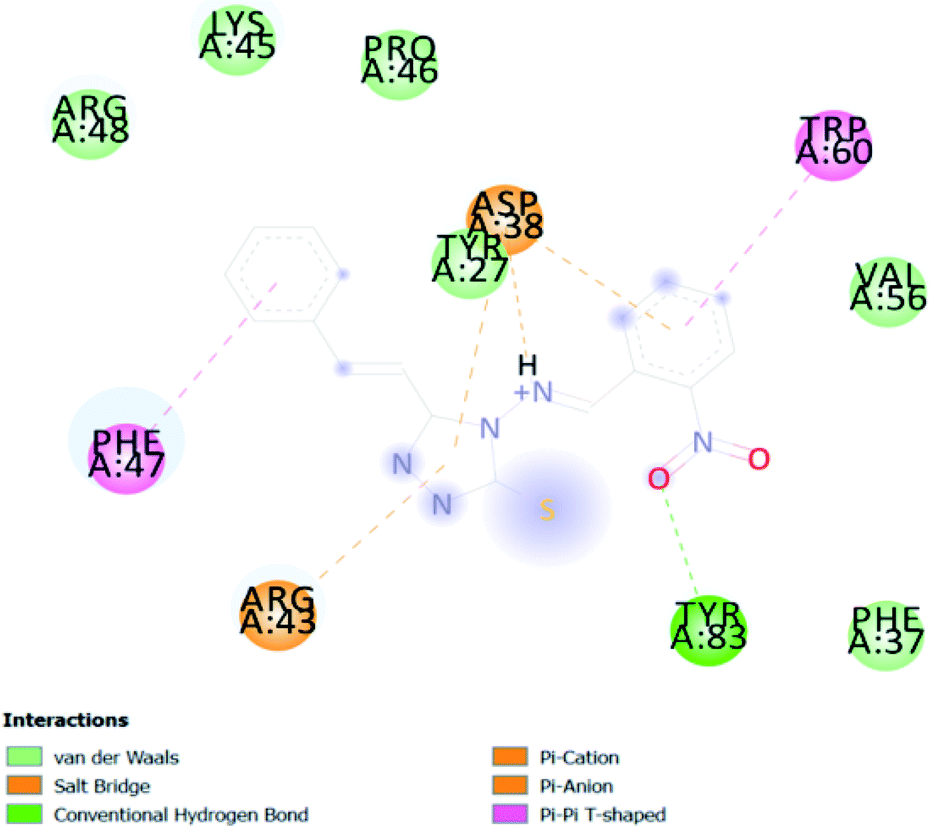 |
| CA 6 | −6.5 | 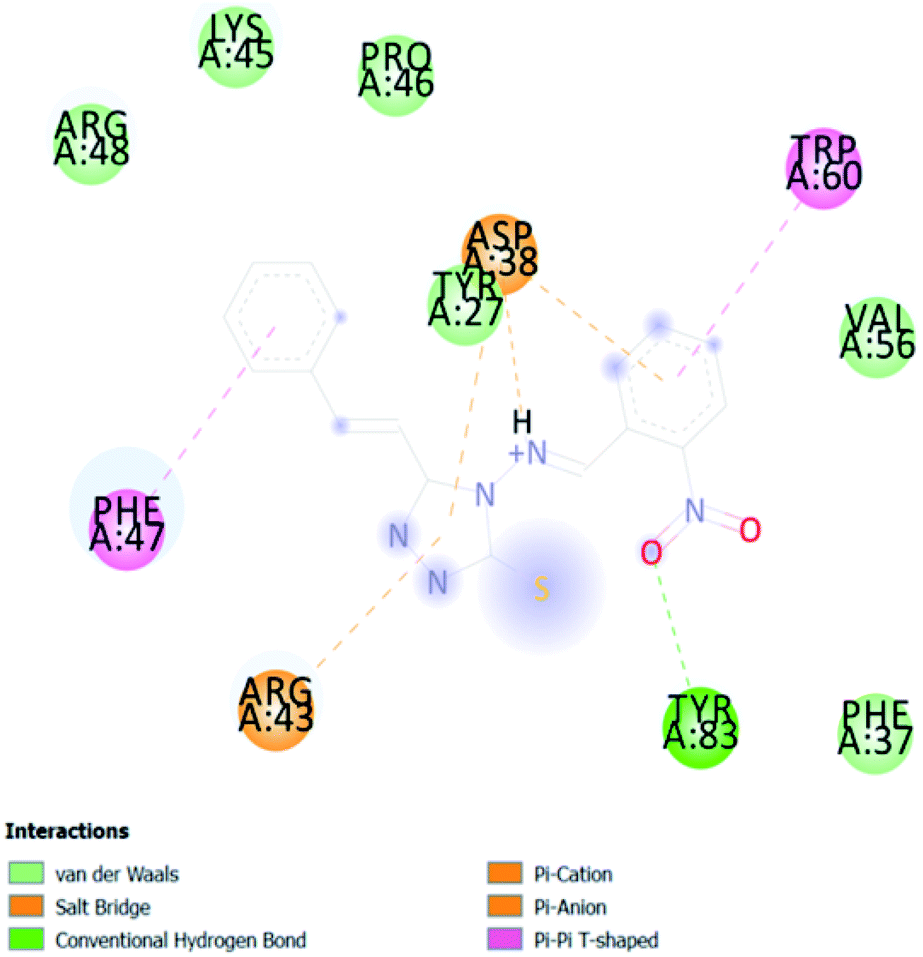 |
| ACA 3 | −6.5 | 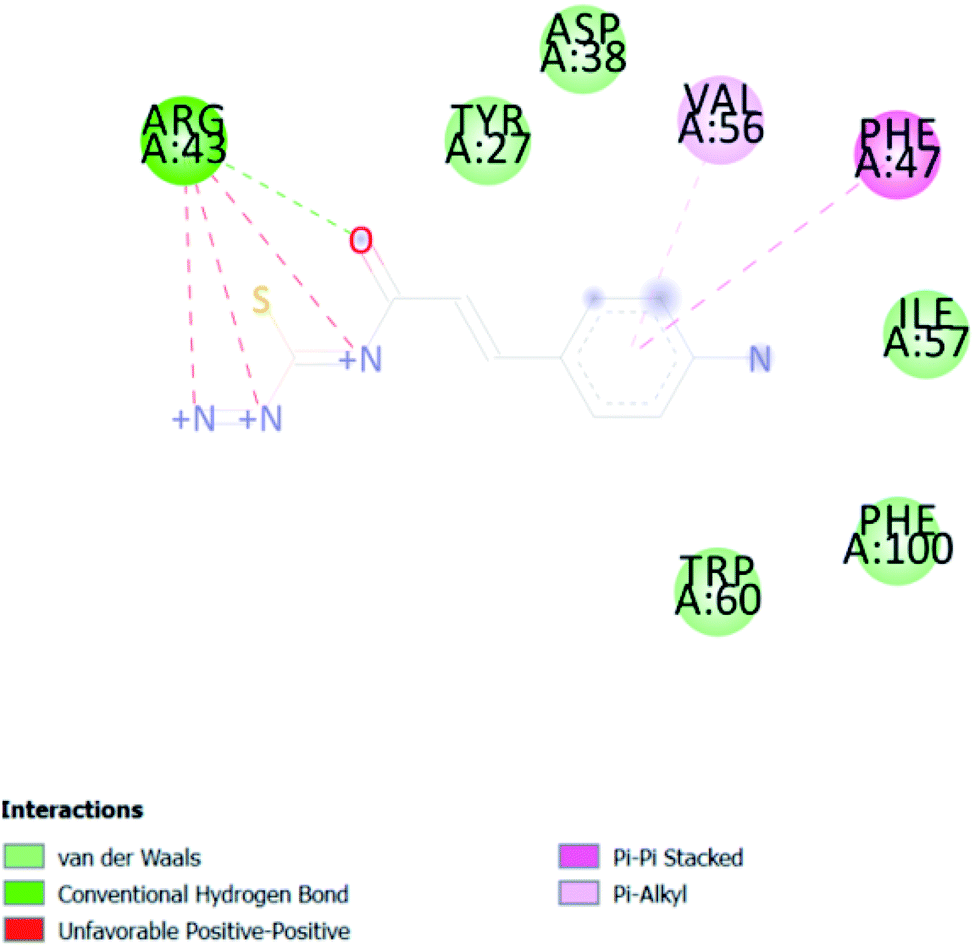 |
| CA 14 | −6.4 | 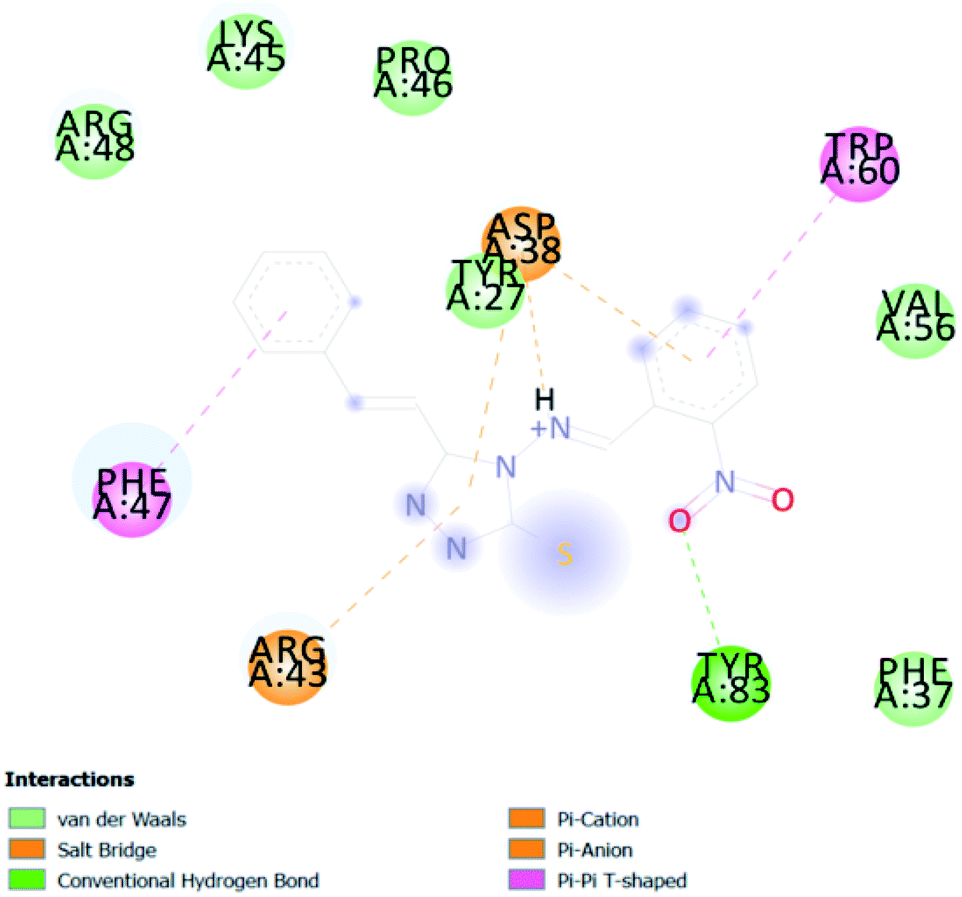 |
| Co-crystal | −4.7 |  |
3.3. Prediction of insecticide likeness
Insecticides are likely to have significantly different bioavailability related property profiles compared with drugs. Therefore, we evaluated the insecticide likeness of our cinnamic acid analogues by the radar (Fig. 6) (the pink area represents the optimal range for each property) and histograms at first glance or according to the insecticide likeness scores (the higher the score, the better the pesticide likeness of our compounds). Our compounds have shown properties which are similar to those of the standard insecticide flupyradifurone, which are given in Table 6. | ||
| Fig. 6 Correlation plot (fitness graph between observed and predicted binding affinity for training): (a) ACA-2 and (b) standard flupyradifurone. | ||
| S. No. | Compound code | MW (g mol−1) | AlogP | HBA | HBD | nRotB | nAromB | RDL | QEI | GAU |
|---|---|---|---|---|---|---|---|---|---|---|
| a MW: molecular weight, AlogP: lipophilicity, HBA: hydrogen bond acceptors, HBD: hydrogen bond donors, nRotB: number of rotatable bonds (flexibility), nAromB: number of aromatic bonds, RDL: relative drug likelihood, QED: quantitative estimate of drug-likeness, Gau.: gaussian scoring function. | ||||||||||
| 1 | CA2 | 322.089 | 2.76 | 4 | 1 | 4 | 17 | 0.807 | 0.567 | 5.021 |
| 2 | CA8 | 384.004 | 3.783 | 4 | 0 | 4 | 17 | 0.945 | 0.574 | 4.466 |
| 3 | CA10 | 385.012 | 1.954 | 4 | 1 | 4 | 12 | 0.9 | 0.65 | 4.979 |
| 4 | CA12 | 337.112 | 1.002 | 4 | 1 | 5 | 12 | 1.045 | 0.697 | 5.369 |
| 5 | CA13 | 324.104 | 1.226 | 4 | 1 | 4 | 12 | 0.942 | 0.678 | 5.077 |
| 6 | CA14 | 351.079 | 3.543 | 4 | 0 | 5 | 17 | 1.097 | 0.637 | 5.142 |
| 7 | ACA4 | 359.027 | 1.475 | 4 | 3 | 4 | 13 | 0.818 | 0.483 | 6.023 |
| 8 | ACA2 | 177.09 | −0.858 | 3 | 3 | 3 | 7 | 1.326 | 0.284 | 4.417 |
| 9 | CATD2 | 320.11 | 0.729 | 4 | 0 | 3 | 13 | 1.184 | 0.707 | 4.255 |
| 10 | Flupyra difurone (standard) | 289.056 | 0.418 | 4 | 1 | 5 | 0 | 1.368 | 0.714 | 5.913 |
| 11 | Limits (as per) | 60–500 | 0.52–4.5 | 0–5 | 0–2 | 0–9 | 0–14 | |||
3.4. Chemistry
Cinnamic acid derivatives were prepared as depicted in Schemes 1–3. The selected cinnamic acids were substituted using hydrazine hydrate at positions 2, 4, and 7 of the phenyl ring with a nitrogen linker. To increase the lipophilicity, permeability, and receptor binding affinity of the cinnamic acid scaffold, 4-amino cinnamic acid (ACA) was esterified using DCC/HOBT to get amino cinnamic acid ester (Scheme 1). The hydrazides were then subjected to conjugation with ACE to yield the cinnamic acid conjugates viz., ACA-1, ACA-2, ACA-3, and ACA-4. Cinnamic acid triazole (8) was used to synthesize compounds viz., CA-1, CA-2, CA-3, CA-4, CA-5, CA-6, CA-7, CA-8, CA-9, CA-10, CA-11, CA-12, CA-13, and CA-14 by Schiff reaction using the same reaction conditions (Scheme 2). The cinnamic acid thiadiazole derivatives viz., CATD-1, CATD-2, CATD-3, CATD-4, and CATD-5 have been synthesized by reactions between the CAT and different benzoic acids (Scheme 3). All the conjugates were purified by column chromatography (ethyl acetate and n-hexane) and characterized by spectroscopic analysis like 1H NMR, 13C NMR, and mass spectra.3.5. Biological studies
| S. No. | Compound | Vero (IC50 μg ml−1) |
|---|---|---|
| 1 | Famciclovir | 270 ± 0.20 |
| 2 | Glabranin | 252 ± 0.20 |
| 3 | CA 1 | 309 ± 0.21 |
| 4 | CA 2 | 321 ± 0.12 |
| 5 | CA 3 | 314 ± 0.12 |
| 6 | CA 4 | 210 ± 0.03 |
| 7 | CA 5 | 223 ± 0.05 |
| 9 | CA 6 | 312 ± 0.05 |
| 10 | CA 7 | 295 ± 0.04 |
| 11 | CA 8 | 318 ± 0.02 |
| 12 | CA 9 | 235 ± 0.05 |
| 13 | CA 10 | 241 ± 0.01 |
| 14 | CA 11 | 229 ± 0.02 |
| 15 | CA 12 | 252 ± 0.04 |
| 16 | CA 13 | 238 ± 0.01 |
| 17 | CA 14 | 240 ± 0.03 |
| 18 | ACA 1 | 285 ± 0.05 |
| 19 | ACA 2 | 242 ± 0.02 |
| 20 | ACA 3 | 254 ± 0.03 |
| 21 | ACA 4 | 328 ± 0.02 |
| 22 | CATD 1 | 211 ± 0.01 |
| 23 | CATD 2 | 218 ± 0.02 |
| 24 | CATD 3 | 270 ± 0.20 |
| 25 | CATD 4 | 252 ± 0.20 |
| 26 | CATD 5 | 314 ± 0.21 |
| Compound | Conc. (PPM) | Absorbance | Hemolytic (%) |
|---|---|---|---|
| FMVC | 100 | 3.244 | 8.901 |
| 50 | 3.065 | 6.448 | |
| 25 | 2.823 | 4.802 | |
| CATD-2 | 100 | 3.188 | 5.759 |
| 50 | 3.033 | 5.367 | |
| 25 | 2.732 | 4.605 | |
| CA-2 | 100 | 2.208 | 3.278 |
| 50 | 1.977 | 2.003 | |
| 25 | 1.783 | 1.202 | |
| CA-14 | 100 | 2.66 | 4.422 |
| 50 | 2.62 | 4.326 | |
| 25 | 1.367 | 1.149 | |
| CA-13 | 100 | 3.121 | 5.589 |
| 50 | 2.994 | 5.268 | |
| 25 | 2.623 | 4.329 | |
| ACA-4 | 100 | 3.228 | 3.460 |
| 50 | 2.739 | 2.122 | |
| 25 | 2.389 | 1.736 | |
| CA-12 | 100 | 3.178 | 5.734 |
| 50 | 3.037 | 5.377 | |
| 25 | 2.55 | 4.144 | |
| Glabranin | 100 | 2.974 | 5.217 |
| 50 | 2.775 | 4.713 | |
| 25 | 1.437 | 2.326 | |
| DMSO | 0.851 | 0 |
| S. No. | Compound code | Molecular formula | Predicted pLC50 (μM) |
|---|---|---|---|
| 1 | CA 5 | C20H22N4O3S | 6.8780 |
| 2 | CA 9 | C17H15N5O2S | 6.7218 |
| 3 | ACA 1 | C15H13N5O5 | 6.6914 |
| 4 | CATD 1 | C17H15N5S | 6.8921 |
| 5 | CATD 5 | C17H13ClN4S | 6.8987 |
Different concentrations (50, 100, 150, 200 and 250 μg ml−1) were used to determine the mortality after 24, 48 and 72 h of subjection. The mortality rate is affected by concentration and subjection period. However, observing CA 2 treatment at a minimum concentration for 24 and 48 hours resulted in the highest mortality range. ACA 4 showed slow mortality in the 24, 48, and 72 h exposure times, but they inhibited larval development at the early pupal stage. At the cellular level, some damage may occur (Fig. 7 and Table 10)
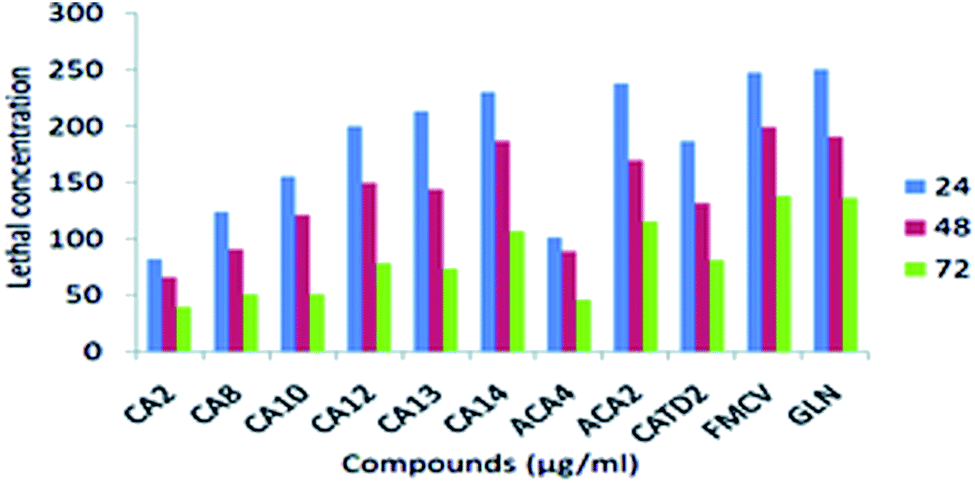 | ||
| Fig. 7 Larvicidal activity of different synthesized compounds against Aedes aegypti. | ||
| Sample | Time (h) | LC50 (ppm) | 95% LCL to UCL (ppm) | LC90 (ppm) | 95% LCL to UCL (ppm) | Intercept | Slope | χ2 value |
|---|---|---|---|---|---|---|---|---|
| a LCL: lethal concentration limit, UCL: upper concentration limit, LC50: lethal concentration 50%, LC90: lethal concentration 90%, estimated air concentration of a substance administered via inhalation route, and DF: degree of freedom. | ||||||||
| CA2 | 24 | 82.150 | 060.708–124.814 | 109.721 | −2.821 | 0.013 | 0.379 | 0.944 |
| 48 | 65.342 | 097.638–186.016 | 96.144 | −1.496 | 0.010 | 0.269 | 0.966 | |
| 72 | 38.685 | 010.970–118.460 | 79.422 | −1.014 | 0.012 | 0.694 | 0.875 | |
| CA8 | 24 | 123.885 | 054.531–167.921 | 265.404 | −1.122 | 0.009 | 1.217 | 0.749 |
| 48 | 90.793 | 048.979–118.498 | 165.954 | −1.548 | 0.017 | 0.833 | 0.842 | |
| 72 | 50.137 | 013.314–090.657 | 122.039 | −1.516 | 0.023 | 0.763 | 0.858 | |
| CA10 | 24 | 155.581 | 094.019–162.201 | 210.414 | −2.643 | 0.014 | 0.802 | 0.849 |
| 48 | 121.420 | 081.828–140.880 | 172.096 | −1.396 | 0.011 | 0.357 | 0.949 | |
| 72 | 50.285 | 031.654–107.105 | 122.913 | −1.129 | 0.015 | 0.901 | 0.825 | |
| CA12 | 24 | 200.037 | 167.480–258.244 | 295.409 | −2.957 | 0.013 | 1.939 | 0.585 |
| 48 | 149.114 | 126.982–224.856 | 229.123 | −1.667 | 0.010 | 0.236 | 0.972 | |
| 72 | 78.125 | 054.652–130.490 | 166.552 | −1.278 | 0.013 | 0.453 | 0.929 | |
| CA13 | 24 | 213.150 | 190.708–244.8 14 | 309.721 | −2.829 | 0.013 | 0.379 | 0.944 |
| 48 | 143.342 | 097.638–186.016 | 266.144 | −1.496 | 0.010 | 0.269 | 0.966 | |
| 72 | 73.685 | 050.970–108.460 | 189.422 | −1.014 | 0.012 | 0.694 | 0.875 | |
| CA14 | 24 | 230.111 | 215.757–615.686 | 389.073 | −2.491 | 0.009 | 0.748 | 0.862 |
| 48 | 186.546 | 175.129–259.690 | 280.065 | −2.830 | 0.014 | 0.381 | 0.944 | |
| 72 | 106.898 | 084.119–180.863 | 237.905 | −1.339 | 0.010 | 0.330 | 0.954 | |
| CA15 | 24 | 227.608 | 213.318–466.134 | 353.117 | −2.858 | 0.011 | 1.463 | 0.691 |
| 48 | 209.434 | 169.671–300.198 | 308.221 | −2.084 | 0.010 | 1.144 | 0.766 | |
| 72 | 125.295 | 118.817–194.307 | 260.714 | −1.888 | 0.012 | 0.409 | 0.938 | |
| ACA4 | 24 | 101.176 | 89.885–276.910 | 358.428 | −2.636 | 0.011 | 1.446 | 0.695 |
| 48 | 88.823 | 62.926–175.039 | 323.217 | −2.103 | 0.010 | 1.723 | 0.632 | |
| 72 | 47.347 | 032.563–93.148 | 240.784 | −1.702 | 0.012 | 1.755 | 0.625 | |
| ACA2 | 24 | 238.252 | 227.451–491.294 | 306.392 | −4.857 | 0.019 | 0.628 | 0.890 |
| 48 | 169.114 | 126.982–224.856 | 259.123 | −1.667 | 0.010 | 0.236 | 0.972 | |
| 72 | 115.394 | 068.626–148.713 | 207.730 | −1.445 | 0.013 | 1.177 | 0.759 | |
| CATD2 | 24 | 187.076 | 168.650–250.990 | 311.453 | −1.928 | 0.010 | 0.483 | 0.923 |
| 48 | 131.513 | 098.030–168.174 | 241.010 | −1.539 | 0.012 | 1.823 | 0.610 | |
| 72 | 086.556 | 068.446–108.142 | 179.532 | −1.193 | 0.014 | 2.145 | 0.543 | |
| FMCV | 24 | 247.578 | 188.650–280.990 | 311.453 | −1.928 | 0.010 | 0.483 | 0.723 |
| 48 | 202.983 | 168.030–230.174 | 241.010 | −1.293 | 0.022 | 1.823 | 0.501 | |
| 72 | 138.785 | 118.446–188.142 | 179.532 | −1.193 | 0.016 | 2.145 | 0.443 | |
| GLA | 24 | 258.231 | 230.650–290.990 | 311.453 | −1.782 | 0.014 | 0.538 | 0.823 |
| 48 | 198.960 | 172.030–268.174 | 241.010 | −1.539 | 0.017 | 1.675 | 0.610 | |
| 72 | 140.249 | 1298.446–180.142 | 179.532 | −1.132 | 0.011 | 2.238 | 0.534 | |
In both larvae, the drug samples CA 2, CA 14, ACA 4, and CATD 2 resulted in severe lesions that primarily affected the midgut epithelium and the caeca to a lesser extent (Fig. 8). Histopathological variations were observed qualitatively based on location along the midgut and quantitatively based on incubation time. After 72 h of infection, anterior midgut cells did not differ morphologically from normal ones, but for mild apical lysis of transparent cells and no lateral junctional complex disturbances. Compared with monitors, transparent cell lysis was accelerated at the brush border, basal membranes, and cytoplasmic organelles before bursting into the gut lumen. A posterior midgut partial lysis was initiated following local detachment in dark cells, with basal membrane dilatation and loss of the peritrophic membrane, with enlargement of cells.
 | ||
| Fig. 8 Histopathological observation of 72 h of exposure to selected compounds CA2, CA14, ACA4, and CATD 2 against A. aegypti. The black-coloured arrow refers to damage in mosquitoes: B.M. – basal membrane; N – nucleus border; GL – gut lumen; PM – peritrophic membrane; CM – cytoplasmic membrane of the larvae compared to the control. | ||
3.6. QSAR studies
The QSAR approach is used to identify the structural parameters for the enhanced bioactivity, which leads to the design of the new scaffold with improved biological effects. A QSAR correlation between larvicidal activities with the series of nineteen synthesized compounds was performed by multiple linear regression analysis. The LC50 (μM) values of the training set were first converted into a negative logarithmic scale (pLC50) to achieve the normal distribution (Table 9). The derived QSAR model shows a good correlation between biological activity and parameters, further validated through test set compounds (Table 11). The descriptors showed a positive correlation among all parameters selected for the QSAR model. The positive coefficients suggest that including such hybrid structures together leads to an increase in biological activity. Further, the regression graph was plotted for pLC50 vs. $PRED. (Fig. 9). The cross-validated correlation coefficient (r2) value is 0.61664, and the root mean square error value is 0.06233.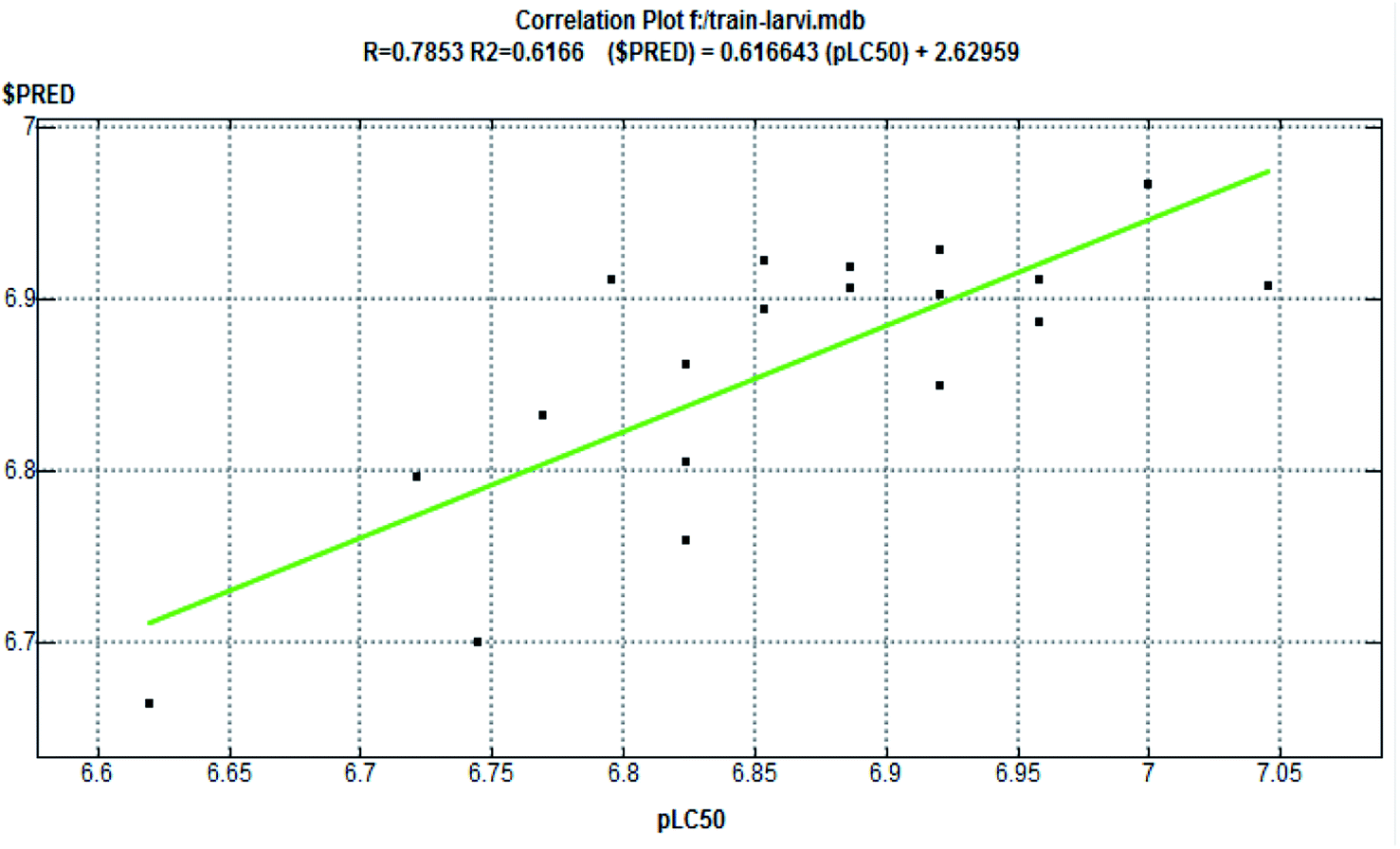 | ||
| Fig. 9 Correlation plot (fitness graph between observed and predicted binding affinity for training). | ||
The formula for the generated QSAR linear model is
| pLC50 = 8.45519–0.02346 × AM1_dipole−0.00034 × TPSA + 0.00052 × E_vdw + 0.08078 × AM1_LUMO+0.16393 × AM1_HOMO−0.01678 × mr + 0.01392 × logP(o/w) |
Thus, the developed QSAR model can be applied to develop potent modified molecules with nitrogen, sulfur, and oxygen atoms, as potential anti-dengue agents.
4. Conclusion
The present research study suggests that our synthesized compounds may be the “LEAD” molecules for early DENV inhibition, which prevents or lessens the chances of patients progressing to dengue hemorrhagic fever/dengue shock syndrome. Cost-effectiveness is a crucial issue for this still poverty-linked disease, and this issue is also figured out by selecting early DENV inhibitors. Overall, this study concludes CA-2 and ACA-4 as potent compounds for biologically controlling Aedes aegypti. The docking studies also support the insecticidal potency of the synthesized compounds. Interestingly, in silico docking findings show that the molecule CA-2 also regulates the 1OKE protein through the β-OG bag. As a result, CA-2 may be used as a lead molecule in anti-viral entry inhibition. The QSAR model developed may be used to design candidates, with nitrogen, sulfur, and oxygen atom substituents, as promising anti-dengue compounds. The compounds CA 2 and ACA 4 may also be considered lead compounds for other mosquito-borne viral diseases.| S. No. | Comp. code | LC50 (μM) | pLC50 (μM) | ||
|---|---|---|---|---|---|
| Observed | Predicted | Residual | |||
| 1 | CA 1 | 0.13 | 6.8861 | 6.9184 | −0.0323 |
| 2 | CA 2 | 0.09 | 7.0458 | 6.9070 | 0.1388 |
| 3 | CA 3 | 0.12 | 6.9208 | 6.9290 | −0.0082 |
| 4 | CA 4 | 0.16 | 6.7959 | 6.9113 | −0.1154 |
| 5 | CA 6 | 0.14 | 6.8539 | 6.8945 | −0.0406 |
| 6 | CA 7 | 0.11 | 6.9586 | 6.8864 | 0.0722 |
| 7 | CA 8 | 0.15 | 6.8239 | 6.8617 | −0.0378 |
| 8 | CA 10 | 0.14 | 6.8539 | 6.9230 | −0.0691 |
| 9 | CA 11 | 0.17 | 6.7696 | 6.8326 | −0.0630 |
| 10 | CA 12 | 0.11 | 6.9586 | 6.9119 | 0.0467 |
| 11 | CA 13 | 0.12 | 6.9208 | 6.9025 | 0.0183 |
| 12 | CA 14 | 0.18 | 6.7447 | 6.7000 | 0.0447 |
| 13 | ACA 2 | 0.15 | 6.8239 | 6.8050 | 0.0189 |
| 14 | ACA 3 | 0.19 | 6.7212 | 6.7963 | −0.0751 |
| 15 | ACA 4 | 0.10 | 7.0000 | 6.9670 | 0.0330 |
| 16 | CATD 2 | 0.13 | 6.8861 | 6.9068 | −0.0207 |
| 17 | CATD 3 | 0.12 | 6.9208 | 6.8501 | 0.0707 |
| 18 | CATD 4 | 0.15 | 6.8239 | 6.7601 | 0.0638 |
| 19 | FMCV | 0.24 | 6.6198 | 6.6645 | −0.0447 |
Funding
The work was supported through the JSS Academy of Higher Education & Research, Mysuru, to provide a research grant for the study (ref. no. REG/DIR(R)/URG/54/2011–12/10887/1, Date: 23.02.2017).Conflicts of interest
The authors declare that there are no conflicts of interest.References
- S. P. Lim, Q. Y. Wang, C. G. Noble, Y. L. Chen, H. Dong, B. Zou, F. Yokokawa, S. Nilar, P. Smith, D. Beer and J. Lescar, Antiviral Res., 2013, 100, 500 CrossRef CAS PubMed.
- C. G. Noble, Y. L. Chen, H. Dong, F. Gu, S. P. Lim, W. Schul, Q. Y. Wang and P. Y. Shi, Antiviral Res., 2010, 85, 450 CrossRef CAS PubMed.
- D. J. Gubler, Trends Microbiol., 2002, 10, 100 CrossRef CAS.
- S. S. Whitehead, J. E. Blaney, A. P. Durbin and B. R. Murphy, Nat. Rev. Microbiol., 2007, 5, 518 CrossRef CAS PubMed.
- R. Fritz, K. Stiasny and F. X. Heinz, J. Cell Biol., 2008, 183, 353 CrossRef CAS PubMed.
- Y. Modis, S. A. Ogata, D. E. Clements and S. C. Harrison, Nature, 2004, 427, 313 CrossRef CAS PubMed.
- K. Stiasny, C. Kossl, J. Lepault, F. A. Rey and F. X. Heinz, PLoS Pathog., 2007, 3, 20 CrossRef PubMed.
- T. Kampmann, D. S. Mueller, A. E. Mark, P. R. Young and B. Kobe, Structure, 2006, 14, 1481 CrossRef CAS PubMed.
- Z. Zhou, M. Khaliq, J. E. Suk, C. Patkar, L. Li, R. J. Kuhn and C. B. Post, ACS Chem. Biol., 2008, 19, 765 CrossRef PubMed.
- Q. Y. Wang, S. J. Patel, E. Vangrevelinghe, H. Y. Xu, R. Rao, D. Jaber, W. Schul, F. Gu, O. Heudi, N. L. Ma, M. K. Poh, W. Y. Phong, T. H. Keller, E. Jacoby and S. G. Vasudevan, Antimicrob. Agents Chemother., 2009, 53, 1823 CrossRef CAS PubMed.
- Y. Qian, H. J. Zhang, H. Zhang, C. Xu, J. Zhao and H. L. Zhu, Bioorg. Med. Chem., 2010, 18, 4991 CrossRef CAS PubMed.
- C. R. Rees, J. M. Costin, R. C. Fink, M. McMichael, K. A. Fontaine, S. Isern and S. F. Michael, Antiviral Res., 2008, 80, 135 CrossRef CAS PubMed.
- R. Yin, Z. Ding, X. Liu, L. Mu, Y. Cong and T. Stoeger, J. Virol. Methods, 2010, 167, 107 CrossRef CAS PubMed.
- D. R. Green and F. Llambi, Cold Spring Harbor Perspect. Biol., 2015, 7, a006080 CrossRef PubMed.
- G. S. Singh, Eur. J. Med. Chem., 2009, 44, 2265 CrossRef CAS PubMed.
- B. Podobnik, J. Stojan, L. Lah, N. Krasevec, M. Seliskar, T. L. Rizner, D. Rozman and R. Komel, J. Med. Chem., 2008, 51, 3480 CrossRef CAS PubMed.
- R. R. Craig, M. C. Joshua, C. F. Ryan, M. M. Matthew, A. F. Krystal, I. Sharon and F. M. Scott, Antiviral Res., 2008, 80, 135 CrossRef PubMed.
- F. M. Scott, I. Sharon and M. C. Joshua, US Pat., US2011/010037/A1, 2011 Search PubMed.
- C. D. Andersson, B. Y. Chen and A. Linusson, Proteins, 2011, 79, 1363 CrossRef CAS.
- K. R. Cousins, Chem draw ultra 12.0, J. Am. Chem. Soc., 2011, 133, 8388 CrossRef CAS PubMed.
- T. Usha, K. G. Arvind, L. Syed, H. P. Prashanth, T. Madhan Mohan, P. Veena and K. M. Sushil, Asian Pac. J. Cancer Prev., 2015, 15, 10345 CrossRef PubMed.
- T. Usha, S. K. Middha and A. K. Goyal, J. Biomed. Res., 2014, 28, 406 CAS.
- V. B. Sulimov, D. C. Kutov and A. V. Sulimov, Curr. Med. Chem., 2019, 26, 7555 CrossRef CAS PubMed.
- K. Kalani, J. Agarwal, S. Alam, F. Khan, A. Pal and S. K. Srivastava, PLoS One, 2013, 8, e74761 CrossRef CAS PubMed.
- S. Sarvagalla, V. K. Singh, Y. Y. Ke, H. Y. Shiao, W. H. Lin, H. P. Hsieh, H. Hsu and M. S. Coumar, J. Comput.-Aided Mol. Des., 2015, 29, 89 CrossRef CAS PubMed.
- T. Geerts and Y. Vander Heyden, Comb. Chem. High Throughput Screening, 2011, 14, 339 CrossRef CAS PubMed.
- M. Kurokawa, A. Wadhwani, H. Kai, M. Hidaka, H. Yoshida, C. Sugita, W. Watanabe, K. Matsuno and A. Hagiwara, Phytother. Res., 2016, 30, 797 CrossRef CAS PubMed.
- R. Gopalan, M. Nanthagopal, S. Mahalingam, G. K. Arumugam, S. K. Samuthirarajan, M. Narayanasamy and I. Sakkanan Ilango, Nano Express, 2021, 2, 010010 CrossRef.
- R. G. Monnerat, A. C. Batista, P. T. de Medeiros, E. S. Martins, V. M. Melatti, L. B. Praça, V. F. Dumas, C. Morinaga, C. Demo and A. C. M. Gomes, Biol. Control, 2007, 41, 291 CrossRef.
- B. Banumathi, T. Vaseeharan, S. Chinnasamy, M. Vijayakumar, N. S. Govindarajan, P. Vijayan, U. Muthukumaran and G. Benelli, J. Cluster Sci., 2017, 28, 2857 CrossRef CAS.
- T. Prabha and T. Sivakumar, Asian J. Pharm. Clin. Res., 2018, 11, 233 Search PubMed.
- S. M. Hipparagi and M. D. Bhanushali, J. Sci. Soc., 2013, 40, 80 CrossRef.
- Molecular Operating Environment (MOE): Chemical Computing Group Inc. 2009; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7.
- L. K. Wolf, Chem. Eng. News, 2009, 87(45), 48 CrossRef.
- Y. C. Chen, Trends Pharmacol. Sci., 2015, 36(2), 78–95 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |