
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUpcycling of semicrystalline polymers by compatibilization: mechanism and location of compatibilizers†
Xiaomin Tang a,
Changhao Liua,
Jong Keumbc,
Jihua Chenb,
Brent E. Diala,
Yangyang Wangb,
Wan-Yu Tsaia,
Wim Brasa,
Tomonori Saitoa,
Christopher C. Bowlanda and
X. Chelsea Chen*a
a,
Changhao Liua,
Jong Keumbc,
Jihua Chenb,
Brent E. Diala,
Yangyang Wangb,
Wan-Yu Tsaia,
Wim Brasa,
Tomonori Saitoa,
Christopher C. Bowlanda and
X. Chelsea Chen*a
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: chenx@ornl.gov
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
cNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA
First published on 7th April 2022
Abstract
With the continuous increase of global plastics production, there is a demand to develop energy efficient processes to transform mixed plastic wastes into new products with enhanced utility – a concept that is often referred to as upcycling. Compatibilization is one of the most promising strategies to upcycle communal waste plastics. In this work, poly(ethylene terephthalate) (PET) and high-density polyethylene (HDPE), both widely used semicrystalline packaging polymers, are used as the target polymer blend. We systematically evaluate and compare three commercial ethylene copolymer based compatibilizers, ELVALOY™ AC 2016 Acrylate Copolymer (EAA), ELVALOY™ PTW Copolymer (PTW), and SURLYN™ 1802 Ionomer (Surlyn). They represent different compatibilization mechanisms. Furthermore, this work tackles a challenging question: where the compatibilizers are located in the blend. We discover that the location of the compatibilizer molecules can be predicted by comparing the crystallinity change of PET and HDPE in binary and ternary systems. Gaining this knowledge will facilitate root cause analysis of an ineffective compatibilizer and guide the design strategy to upcycle commingled waste plastics.
Introduction
Plastics are ubiquitous in modern life. Global plastics production is projected to grow to 700 million metric tons (MTs) in 2030.1,2 Among these plastics, about 42% is used for packaging applications that will end up in mixed municipal waste streams.3–5 It is very challenging to recover pure components from commingled wastes, both technically and economically. As a result, most of the packing plastics are single use products, which will be buried in landfills or worse even, leaked out into oceans at the end of life, causing detrimental impact to global environment. Currently there is a demand to develop energy efficient processes to transform mixed plastic wastes to new products with enhanced utility, while retaining their original chemical complexity – referred to as upcycling.6,7Mixed plastic wastes generally consist of polymers that are immiscible with each other. Immiscible polymers phase separates into domains with brittle interfaces, causing diminished properties and thus preventing their upcycling to useful products.8,9 One effective strategy of upcycling immiscible polymer blends is to use compatibilizers with multi-functionality which allows them to establish interactions with different polymers to strengthen the interface.10–12 Three major compatibilization strategies are often used. The first strategy is physical entanglement. For this strategy, a copolymer consisting of two types of repeating units, each selectively mixing with one of the polymers in the blend, creates an entangled interface with improved interfacial adhesion. Recent groundbreaking work on the synthesis of multiblock compatibilizers is very effective at compatibilizing polymer blends such as poly(ethylene terephthalate) (PET)/polyethylene (PE)13 and PE/polypropylene (PP).14,15 The addition of 0.5 wt% of a multiblock copolymer consisting of PET and PE blocks developed by Nomura et al. can convert the PET/PE (4/1 w/w) blend from brittle and weak with elongation at break of 9.8% and a tensile strength of 21.1 MPa to tough and strong with elongation at break of 333% and a tensile strength of 41.9 MPa.13 The second strategy is reactive compatibilization where a functional group on the compatibilizer reacts with one of the polymers in the blend, creating covalent bonding at the interface. Common reactive functional groups include anhydride, epoxy, and isocyanate.16–22 For example, an ethylene glycidyl methacrylate copolymer was used as a reactive compatibilizer in a blend of recycled PET/PE (75/25 w/w). The addition of 5% of the compatibilizer increased the elongation at break from around 10% to greater than 300%.17 The third strategy is to use intermolecular interactions such as hydrogen bonding23–27 and ionic interactions28–30 to strengthen the interface. A study by Hirai et al. has shown a blend of polyamide 11 (PA11) and poly(vinylcatechol) (PVCa) exhibits excellent engineering properties due to the hydrogen bonding between PA11 and PVCa.24
Effective compatibilization will result in the reduction of interfacial tension, stabilization against droplet coalescence, and most importantly, enhancement of adhesion between immiscible phases, leading to improved mechanical properties with very low compatibilizer loadings (<2 wt%).14,21,31,32 When it comes to the evaluation of compatibilizer efficacy, the extent of mechanical property enhancement is often the sole criterion to judge how good a compatibilizer is. A key question that is often omitted is: if a compatibilizer is not good, why is it not good? If a compatibilized blend didn't show greatly enhanced mechanical properties, it may be because the compatibilizer failed to migrate to the interface, or it may be because the compatibilizer successfully migrated to the interface but didn't effectively strengthen it. Where are the compatibilizer molecules in a polymer blend? This question remains largely unanswered because of technique barriers. Previous efforts to investigate the interface activity of additives include the use of pendant drop method to measure the interfacial tension change,33 and transmission electron microscopy (TEM) to directly observe the location of an additive.34 Both methods are very selective on what bulk polymers and copolymer additives to use. A sample for pendant drop test is prepared in a solution state. In order to be visible in TEM, the additive molecule has to be chemically modified so it can be stained by OsO4.34
In this study, we use PET and high-density polyethylene (HDPE) as a model polymer blend for compatibilization. Both PET and HDPE are widely used packing plastics (46% of which are PET and PE3). PET and HDPE are highly immiscible, and they both are semicrystalline polymers. Three commercial ethylene copolymer based compatibilizers, ELVALOY™ AC 2016 Acrylate Copolymer (EAA), ELVALOY™ PTW Copolymer (PTW), and SURLYN™ 1802 Ionomer (Surlyn) are systematically evaluated and compared. Their chemical structures are shown in Scheme 1. Each compatibilizer may represent a different compatibilization mechanism, according to literature reports.17,18 We systematically compare the effect of each compatibilizer on the morphology and physical, thermal, and mechanical properties of PET/HDPE blend. The compatibilization mechanisms of these compatibilizers are thoroughly investigated by Fourier-transform infrared (FTIR) spectroscopy. Further, we report that the location of the compatibilizers can be predicted by probing the effect of compatibilizers on the crystallinity change of PET and PE. This finding offers a facile and effective way to answer the very first important question we must consider for a compatibilization process: where is the compatibilizer and how much is at the interface versus distributed in the bulk. These results will guide effective use of a compatibilizer in immiscible plastic blends and pave ways to improved upcyclability of municipal plastic wastes.
 | ||
| Scheme 1 Chemical structure of two semicrystalline polymers as a model plastic blend: PET and HDPE, and three commercial ethylene copolymer based compatibilizers used in the paper: ELVALOY™ AC 2016 Acrylate Copolymer (EAA), ELVALOY™ PTW Copolymer (PTW), and SURLYN™ 1802 Ionomer (Surlyn). | ||
Materials and methods
Materials
Poly(ethylene terephthalate) (PET, Laser+® C60A) was provided by DAK Americas. The intrinsic viscosity of the PET was 0.83 ± 0.02 dL g−1. High density polyethylene (HDPE, DOWLEX™ 2050B) was obtained from Dow Chemical. The melt mass-flow rate of HDPE is 0.95 g 10 min−1. Compatibilizers used in the paper were purchased from DOW Chemical. They were ELVALOY™ AC 2016 Acrylate Copolymer (EAA), ELVALOY™ PTW Copolymer (PTW), and SURLYN™ 1802 Ionomer (Surlyn). The melt indexes for those additives are 6 g 10 min−1, 12 g 10 min−1, and 8 g 10 min−1, respectively.Preparation of polymer blends
PET pellets were dried in a vacuum oven at 80 °C for 16 h prior to use. HDPE and compatibilizers were dried in a vacuum oven at 60 °C for 12 h. Pellets of PET and HDPE were fed into a twin-screw batch mixer (Xplore micro compounder (MC5), Xplore Instruments BV, The Netherlands) with and without compatibilizers and blended for 5 min at 255 °C and 120 rpm with dry air as the purge gas. The products were extruded from a 5 mm-diameter circular die and cooled in air.The extruded blends were compression-moulded at 270 °C for 3 min with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi pressure by a Genesis hydraulic press and cooled in air and washed with methanol.
000 psi pressure by a Genesis hydraulic press and cooled in air and washed with methanol.
The thermal stability of each component under high process temperature was evaluated through thermogravimetric analysis (TGA) and the data was provided in ESI (Fig. S1†). The TGA was carried out under air flow from 20 °C to 600 °C at 10 °C min−1 heating rate using TA Q500.
Fourier-transform infrared (FTIR) spectroscopy
A Bruker INVENIOR infrared spectrometer was used for IR measurement. All measurements were conducted using a germanium crystal ATR accessory. Each spectrum was collected at a resolution of 4 cm−1 by averaging 128 scans. Baseline correction, normalization, and peak deconvolution were carried out in the software OriginPro. No ATR correction was performed for all the spectra.Differential scanning calorimetry (DSC)
Before the test, the samples were annealed at 90 °C for four days. DSC were performed on a TA Instruments Q2000 DSC with heating and cooling rates of 10 °C min−1 using nitrogen as a purge gas. The data used in the paper was from the first heating cycle. The degree of crystallinity of each polymer component was calculated by taking the ratio of the integrated area under a melting endotherm (heat of fusion) and that of the 100% crystalline polymer (136 J g−1 for PET35 and 293 J g−1 for HDPE36). Three to five specimens were run for each sample.Small and wide-angle X-ray (SAXS/WAXS)
Small and wide-angle X-ray scattering (SAXS and WAXS) measurements were carried out on a Xenocs Xeuss 3.0 instrument equipped with D2+ MetalJet X-ray source (Ga Kα, 9.2 keV, λ = 1.341 Å). The films were aligned perpendicular to the direction of the incident X-ray beam (transmission mode) and the scattered beam was recorded on a Dectris Eiger 2R 4 M hybrid photon counting detector with a pixel dimension of 75 × 75 μm2, where the X-ray exposure time for each measurement was 60 seconds. The collected 2-dimensional (2D) SAXS images were circularly averaged and expressed as absolute intensity versus q, where q = (4πsinθ)/λ after transmission correction and subtraction of background scattering. In order to calibrate the measured intensities to absolute scale intensities, we used direct beam intensity. The sample-to-detector distances for SAXS and WAXS measurements were 1750 and 55 mm, respectively.
Microscopy
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) were performed on a Hitachi TM3030Plus tabletop instrument with a 15 kV acceleration voltage.Thin sections with thickness of approximately 100 nm were obtained by microtoming a piece of bulk film at room temperature using a Leica EM FC7 cryomicrotome. The thin sections were placed onto a lacey carbon supported copper grid (Electron Microscopy Sciences). The samples were not stained. TEM experiments were performed on an aberration-corrected FEI Titan S 80-300 TEM/STEM microscope, using 300 keV acceleration voltage. The flattened cross-section of the bulk films was used for atomic force microscopy (AFM) measurements.
The atomic force microscopy (AFM) experiments were performed on a Cypher (Asylum Research), and the imaging was performed in tapping mode with a scan rate of 0.5 Hz. The AFM probe used in this study is ElectriMulti75-G from BudgetSensor, with a nominal force constant of 3 N m−1.
Mechanical tensile test
Tensile tests were performed by using an Instron 3343 with a 1 kN load cell and analyzed with Bluehill 3 software (Norwood, MA, USA). The samples used for tests had dimensions of l × w × t = 15 × 5 × 0.3 mm. Polymer films were uniaxially strained at a rate of 15 mm min−1. At least three specimens of each sample were tested.Results and discussion
Compatibilization mechanism
The results and discussion section are organized as follows: first a systematic comparison between EAA, PTW and Surlyn is established. Then we use PTW as an example to demonstrate how the location of compatibilizer molecules within a polymer blend can be predicted.We chose three copolymers for systematic investigation. EAA is an ethylene acrylate copolymer consisting of ethylene units copolymerized with up to 35% butyl acrylate units. The olefinic ethylene blocks are expected to interact with PE and the carbonyl groups in the butyl acrylate blocks to interact with PET, which also contains carbonyl groups. With EAA, physical entanglement is the proposed compatibilization mechanism.17,21 PTW is an ethylene acrylate copolymer grafted with epoxide functional groups. With PTW, reactive compatibilization between the epoxide group in PTW and the hydroxyl (–OH) or carboxylic acid (–COOH) end group on PET are expected.21,37,38 Surlyn is a copolymer of ethylene and methacrylate acid partially neutralized with Na. With Surlyn, hydrogen bonding between the –COOH group in Surlyn and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups on PET has been proposed.18 The reaction between –COOH group in Surlyn and PET's-OH end group is also possible. The composition of the polymer blend was fixed to be 1:1 w/w between PET and HDPE throughout the study. The compatibilizer loading was fixed at 10 wt%, unless otherwise specified. The ternary samples examined in this work is described in Table 1.
O groups on PET has been proposed.18 The reaction between –COOH group in Surlyn and PET's-OH end group is also possible. The composition of the polymer blend was fixed to be 1:1 w/w between PET and HDPE throughout the study. The compatibilizer loading was fixed at 10 wt%, unless otherwise specified. The ternary samples examined in this work is described in Table 1.
The morphology of PET/PE blends was investigated by atomic force microscopy (AFM) and scanning electron microscopy (SEM), shown in Fig. 1 and S2.† Near spherical shaped droplets are observed in both the uncompatibilized and the compatibilized blends (Fig. 1a–d and S2a–d†). Elemental mapping shows that the droplets are oxygen rich (Fig. 1e). Since PET contains oxygen and PE does not contain oxygen, it is evident that PET formed droplets in the PE matrix in all the samples. The average diameter of the droplets in each sample, Dcontrol, D+EAA, D+PTW, and D+Surlyn, was obtained by image analysis using ImageJ software. Details of the analysis are shown in Fig. S3 and Table S1.† In the uncompatibilized blend, Dcontrol was 1.1 μm (Fig. 1f). Compared to Dcontrol, D+EAA and D+Surlyn increased by 50% and 38%, to 2.2 and 1.8 μm. D+PTW remained similar to Dcontrol with an average diameter of 1.2 μm. One of the effects of compatibilizers is reducing the interfacial tension between immiscible polymers, leading to decreased droplet sizes. Nomura et al. reported 7 times reduction in droplet size by adding 2 wt% of a multiblock copolymer compatibilizer into a model PET/PE blend.13 Since the addition of EAA, PTW, and Surlyn did not decrease the average droplet size, one likely explanation is that the interfacial tension between PET and HDPE was not significantly modified in these samples.
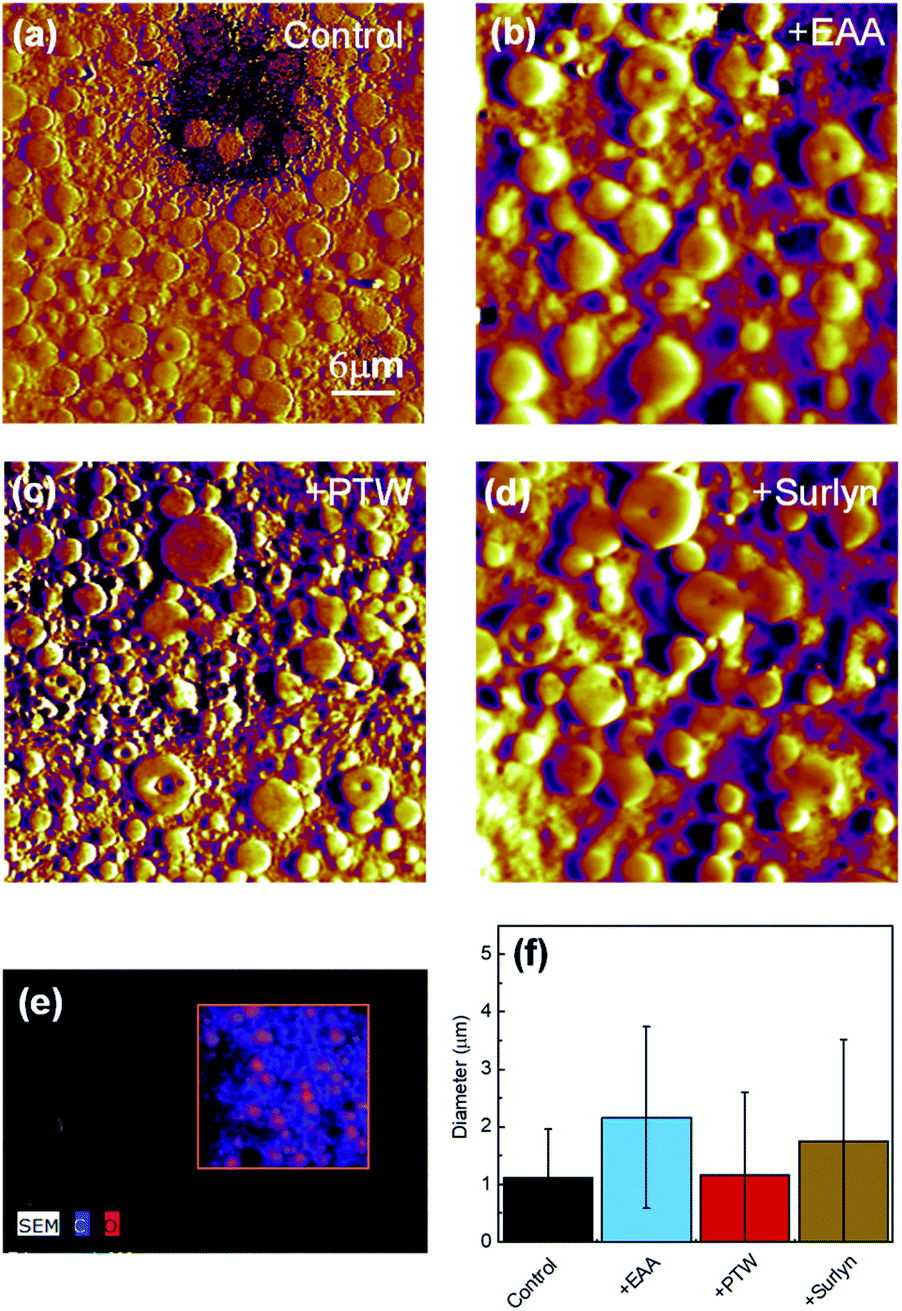 | ||
| Fig. 1 Morphology of PET/PE blends. (a–d) AFM images of control (a), +EAA (b), +PTW (c) and +Surlyn (d). The scale bar in (a) applies to (b), (c) and (d). (e) A representative EDS map of the control sample showing that the droplets are oxygen rich, confirming that PET formed droplets and PE was the matrix; (f) average droplet size in each blend. | ||
The effect of compatibilization on the mechanical properties of HDPE/PET blends was examined by uniaxial tensile tests. The stress–strain curves are shown in Fig. 2 and key parameters are summarized in Table 2. The error bar was based on three repeated tests. Pure PET exhibits high strength (57.4 MPa) and low ductility (elongation at break of 30%); in contrast, pure HDPE has high ductility (elongation at break of 510%) but low strength (19.8 MPa). PET and HDPE are highly incompatible with a large Flory Huggins parameter of 0.4,13 and the 50:50 blend of HDPE/PET (the control sample) is very brittle, resulting in a very low elongation at break of only 3%.
 | ||
| Fig. 2 Representative stress–strain curves of (a), full region of PET/PE blends as well as pure PET and pure HDPE polymers; inset, zoomed-in view between 0 and 30% strain. (b) Magnified view of compatibilized PET/PE blends. | ||
| Sample | Young's Modulus (GPa) | Tensile Strength at Break (MPa) | Elongation at Break (%) | Toughness (MJ m−3) |
|---|---|---|---|---|
| PET | 2.5 ± 0.1 | 57.4 ± 3.1 | 30.3 ± 5.0 | 19.7 ± 1.6 |
| HDPE | 1.3 ± 1.0 | 19.8 ± 1.4 | 510.0 ± 7.3 | 92.0 ± 3.4 |
| Control | 1.2 ± 1.1 | 21.0 ± 0.3 | 3.1 ± 0.1 | 0.4 ± 0.01 |
| +EAA | 1 ± 0.2 | 17.4 ± 0.8 | 5.2 ± 1.1 | 0.6 ± 0.1 |
| +PTW | 0.9 ± 0.5 | 22.6 ± 0.4 | 13.6 ± 1.2 | 2.4 ± 0.07 |
| +Surlyn | 0.9 ± 0.5 | 21.1 ± 0.3 | 13.4 ± 0.9 | 2.5 ± 0.3 |
The addition of EAA into HDPE/PET (sample +EAA) did not show significantly improved mechanical properties. It is not surprising that a random copolymer like EAA does not strengthen the interface. It's been shown that random copolymers create fewer entanglements at the interface compared to block copolymers.39–41 The addition of PTW and Surlyn into HDPE/PET (samples +PTW and +Surlyn) led to improved mechanical properties – approximately 4-fold increase in elongation at break and 6-fold increase in toughness. The presence of the epoxide group on PTW and the carboxylic acid group on Surlyn enhanced the interaction between the compatibilizer and PET, leading to enhanced ductility.
Besides the 10 wt% loading shown in Fig. 2, a series of PTW loadings from 1 wt% to 20 wt% was evaluated and the results are shown in Fig. S4 and Table S2.† Increasing compatibilizer loading from 1 wt% to 10 wt% resulted in further increase in ductility for PTW compatibilized blends. Increasing to 20 wt% led to an increase in elongation at break at the expense of tensile strength for the blend.
We examined the interfaces of control, +PTW and +Surlyn samples, using bright field TEM, shown in Fig. 3. The TEM samples were prepared by microtoming the blend films into thin slices. The TEM images were obtained without staining. PET and HDPE phases are indicated on each image (Fig. 3a–c). They were easily distinguished because PET formed droplets dispersed in the HDPE phase. More zoomed out images are shown in Fig. S5.† In Fig. 3d–f, the bright field images were converted to mass-thickness contrast images. The HDPE/PET interface in the control sample is very sharp, as indicated by the abrupt color change at the interface in Fig. 3d. It can also be observed by the abrupt change in intensity of the line plot in Fig. 3g. Comparatively, both +PTW and +Surlyn samples presented relatively smoother mass-thickness transitions at the interface. These results are not an indication of interfacial thickness, which is on the order of a few nanometers to 10 s of nanometers range and can only be observed under unique circumstances from bilayer samples with staining.42 In our case, the TEM results are an indication of the interface morphology at a much larger scale (hundreds of nms) after being sheared by a diamond knife. The resolution of these images is tens of nms. An abrupt change to low contrast at the control sample's HDPE/PET interface means that the density at the interface is very low. This suggests that after being sheared by a diamond knife, polymer chains pulled away from the HDPE/PET interface, an indication of poor adhesion between PET and HDPE. In contrast, the interfaces of +PTW and +Surlyn samples see smoother transitions from PET phase to HDPE phase. This suggests that with the addition of PTW and Surlyn, the interface of HDPE/PET was less subject to chain pull-out, indicating improved adhesion between the two phases.
 | ||
| Fig. 3 TEM results on PET/PE interfaces. (a–c) Bright field TEM images of control (a), +PTW (b), and +Surlyn (c), (d–f) corresponding mass-thickness contrast images of (a–c), (g) mass-thickness contrast across the interface of PET/PE, indicated by the arrows in (a–c). | ||
We used FTIR spectroscopy to investigate possible compatibilization mechanism for each compatibilizer, shown in Fig. 4. The entire spectra of control, +EAA, +PTW and +Surlyn are shown in Fig. 4a. The control sample showed a band at 1716 cm−1, which is attributed to the CO stretching of the ester group in PET (Fig. 4b). This band is the same to that of pure PET (Fig. S6†), indicating minimal interactions between HDPE and the PET ester group. Pure EAA has a peak at 1735 cm−1, which represents the CO stretching of the ester groups in EAA. Both PET and EAA's ester peaks are clearly observed in +EAA. In fact, examining the whole spectrum, +EAA is a simple combination of control and pure EAA. This suggests that EAA physically mixed into HDPE/PET blend without chemical reactions.
 | ||
| Fig. 4 FTIR spectra revealing compatibilization mechanisms. (a) Whole IR spectra of control, +EAA, +PTW and +Surlyn. Spectra are vertically shifted for clarity; (b) comparison of control, +EAA, and pure EAA in the CO stretching region between 1700 to 1760 cm−1; (c and d) comparison of control, +PTW and pure PTW in the CO stretching region between 1700 to 1760 cm−1; and C–H stretching of the oxirane ring region around 3085 cm−1 and/C–O of the oxirane group stretching region around 942 cm−1 (d); (e) comparison of control, +Surlyn, and pure Surlyn in the CO stretching region between 1700 to 1760 cm−1; (f) peak fitting of +Surlyn with pure Surlyn overlayed for comparison. | ||
Similar comparison was performed between control, pure PTW and +PTW, shown in Fig. 4c. The CO stretching region of +PTW shows two peaks at 1716 and 1733 cm−1, representing PET and PTW's ester groups, respectively. We noticed that two signature peaks that belong to the epoxide group in PTW disappeared after PTW was mixed into HDPE/PET (Fig. 4d). One peak at 942 cm−1 is attributed to the asymmetric C–O stretching and the other peak at 3085 cm−1 is the methylene group of the epoxy ring.43 The lack of these two bands in +PTW in comparison to pure PTW confirms that PTW reacted during the blending process.
Pure Surlyn has two peaks of interest, one peak at 1698 cm−1 is attributed to the CO stretching of the –COOH group; the other band at 1549 cm−1 is attributed to the CO stretching of the Na neutralized carboxylic acid groups (Fig. 4e).44,45 After being blended into PET and PE, the band at 1549 cm−1 can no longer be observed (Fig. 4e, +Surlyn). This may be due to the thermal decomposition of the sodium carboxylate salts.46 Compared with control's CO stretching region, the CO stretching band of +Surlyn is clearly broader. Further analysis of this band reveals that it is a convolution of three peaks (Fig. 4f), representing carboxylic acid groups in Surlyn (1698 cm−1), ester groups in PET (1716 cm−1) and a new peak at 1735 cm−1. This suggests that some of the neutralized and un-neutralized carboxylic acid groups reacted, and the reaction product is reflected by the new peak at 1735 cm−1. Since a portion of the –COOH groups remain unreacted, this portion would be available to form hydrogen bonding. However, the CO stretching region of +Surlyn is a convolution of three peaks. It is difficult to definitively prove the presence of hydrogen bonding.
To this end, the efficacy and mechanism of the three ethylene acrylate copolymer compatibilizers are elucidated. EAA as a random copolymer does not form significant interfacial interactions to compatibilize PET and HDPE. PTW strengthens the HDPE/PET interface through physical interaction with PE and chemical reaction with PET. Surlyn's compatibilization mechanism includes physical interaction with HDPE, and chemical reaction and possible hydrogen bonding with PET. Overall, PTW and Surlyn exhibited better efficacy at enhancing the mechanical properties of PET/HDPE blends. From this systematic comparison, it is evident that when physical interaction is not enough to compatibilize the interface, additional forces such as covalent bonding and hydrogen bonding can help.
Location of compatibilizer
From an upcycling point of view, the three commercial compatibilizers evaluated herein, i.e., EAA, PTW and Surlyn, did not achieve or surpass the original mechanical properties of pure PET or HDPE with a compatibilized blend. Therefore the 50:50 PET/HDPE blend could not be upcycled by adding these compatibilizers. The ineffectiveness of these compatibilizers naturally begs the following questions: why are these compatibilizers not effective? Where are the compatibilizer molecules located in the PET/HDPE blend? For the rest of the results and discussion section, we intend to answer these questions.In literature, many have reported the effect of adding a compatibilizer on the crystallinity of polymer blends. The general trend is that effective compatibilization will result in decreased crystallinity of the polymers.47,48 Most of these literature reports assumed that the compatibilizer molecules are at the interface. However, if the compatibilizer did not locate at the interface, but in the bulk of each individual polymer, crystallinity may also be affected. Therefore, a key piece of missing information in the literature is how a compatibilizer impacts crystallinity when it's located at the interface versus when it's located in the bulk. We use PTW compatibilized HDPE/PET as an example to show that crystallinity may be utilized to estimate the location of the compatibilizer molecules.
In order to compare the effects of a compatibilizer at the interface versus in the bulk, we conducted differential scanning calorimetry (DSC) on a series of binary and ternary blends containing PTW. Since the crystallization kinetics may be different from sample to sample, we annealed all the samples at 90 °C for 4 days prior to the DSC measurements. This annealing temperature is above the glass transition temperature (Tg) of HDPE (−125 °C) and PET (79 °C) but below their melting temperatures. No statistical change in the degree of crystallinity of HDPE or PET was observed when the samples were annealed for 1, 2, 3 and 4 days. This gave us confidence that the degree of crystallinity of annealed samples was near equilibrium. Annealing at temperatures higher than 150 °C for such long periods of time caused the samples to change color, so we did not attempt to anneal at higher temperatures.
The degree of crystallinity of HDPE, Xc,HDPE and that of PET, Xc,PET, were calculated by obtaining the heat of fusion of PE and PET from the first heating scan of the DSC measurements and normalizing them by that of 100% crystalline PE (293 J g−1 for HDPE) and 100% crystalline PET (136 J g−1 for PET). The results are shown in Fig. 5. The DSC thermograms are shown in Fig. S7,† with key parameters listed in Tables S3–S5.†
 | ||
| Fig. 5 Crystallinity of PET and HDPE in PTW binary and ternary blends. (a) Degree of crystallinity of HDPE as a function of PTW weight fraction in HDPE/PTW. (b) Degree of crystallinity of PET as a function of PTW weight fraction in PET/PTW. (c and d) Degree of crystallinity of HDPE (c) and PET (d) in ternary blend HDPE/PET/PTW as a function of PTW weight fraction. Lines are guide to the eye. | ||
For the binary blend of HDPE/PTW, a dramatic decrease in Xc,HDPE was observed with the addition of 5 wt% PTW (Fig. 5a and Table S3†). Xc,HDPE decreased from 67% to 57%. Interestingly, as PTW's concentration increases, Xc,HDPE started to increase back up. At 20 wt% PTW, Xc,HDPE was 67%, back to neat HDPE's value. The results on the binary blends suggest that when PTW's concentration is relatively low (<5 wt%), individual PTW chains are dispersed in HDPE. The physical interaction between the olefinic parts of PTW and PE chains hinders the crystallization of HDPE. As the concentration increases, PTW molecules start to form aggregates in HDPE.49 The aggregates disrupt the crystallization process less, hence the blend regained crystallinity. Based on our results, PTW's critical concentration for aggregates in HDPE is around 5 wt%.
For the binary blend of PET/PTW, Xc,PET remains almost the same at the various of PTW loadings (Fig. 5b and Table S4†), suggesting limited effects of PTW on the crystallinity of PET.
In the ternary blend of HDPE/PET/PTW (the weight ratio of HDPE and PET is 1:1), if PTW located solely at the interface and there was no PTW molecules in the bulk of HDPE, due to the physical entanglement between PTW and HDPE at the interface, one would expect Xc,HDPE to decrease monotonically and eventually reach a plateau as the interface saturates with PTW molecules. Our data indicates that this is not the case. Xc,HDPE as a function of PTW concentration in the ternary blend shows the same trend to the binary HDPE/PTW blend (Fig. 5c, d and Table S5†). Xc,HDPE first decreased and then with increasing PTW concentration it recovered to near the original value in the HDPE/PET blend with no PTW. The lowest Xc,HDPE is seen at approximately 4 wt% PTW. Xc,PET in the ternary blend was not significantly affected by PTW concentration.
The crystalline structures of ternary samples with 10 wt% various compatibilizers were investigated by small and wide-angle X-ray scattering (SAXS/WAXS), shown in Fig. S8.† No significant change in lamellar spacing or crystalline structure was observed.
Combining the crystallinity data with the mechanical and morphological properties discussed earlier, a molecular picture of the HDPE/PET/PTW is emerging: most of the PTW molecules must be located in the PE phase. Only a small amount of PTW chains migrated to the interface. When PTW's concentration is low (<4 wt%), PTW chains are in the form of dispersed chains in HDPE (Fig. 6a). The dispersed PTW chains decreased the crystallinity of HDPE. A small amount of PTW chains that migrated to the interface improved the mechanical properties of the ternary blend and may also contribute to the decreased crystallinity. As PTW's concentration increases (>4 wt%), more PTW chains are added into the HDPE phase and PTW starts to form aggregates, which causes Xc,HDPE to increase back up (Fig. 6b). We hypothesize the aggregates resemble micelles, since macro-scale phase separation from PTW was not observed by the TEM or SEM. At the same time, more PTW chains are also added onto the interface, judging by the enhanced mechanical properties with increasing PTW concentration, but the overall concentration at the interface must be very small, because Xc,HDPE at high PTW concentration was almost the same as that without any PTW. This means that at high PTW concentration, most PTW chains are the in the form of aggregates (micelles) in HDPE. There might also be small amounts of PTW in the PET phase, but the lack of crystallinity change makes it hard to predict whether there is PTW in PET. According to FTIR, nearly all of the epoxy groups reacted. This means that while the PTW molecules at the PET/HDPE interface (and in the PET phase, if any) reacted with PET chains, the PTW molecules in the PE phase must have reacted with itself.
 | ||
| Fig. 6 Schematic of the location and morphology of PTW molecules in HDPE/PET/PTW blends. (a) ≤4 wt% PTW and (b) >4 wt% PTW. The crystallinity of the components is not reflected in the schematic. | ||
Conclusions
In this work, we first investigated the efficacy and compatibilization mechanisms of three commercial ethylene acrylate copolymers, EAA, PTW and Surlyn with 45:45 w/w HDPE/PET model blend at 10 wt% compatibilizer loading. EAA is not an effective compatibilizer for HDPE/PET as it did not decrease the droplet size of the blend, nor did it significantly enhance the mechanical properties. FTIR confirmed that the interaction between EAA and HDPE/PET is purely physical. The addition of PTW and Surlyn into HDPE/PET led to 4-fold increase in elongation at break and 6-fold increase in toughness. The mechanical property enhancement was corroborated by enhanced interfacial adhesion (TEM). FTIR showed that PTW strengthens the HDPE/PET interface through physical interaction with PE and chemical reaction with PET. Surlyn's compatibilization mechanism includes physical interaction with HDPE, and chemical reaction and possible hydrogen bonding with PET.We then demonstrated that the location and morphology of the compatibilizer molecules can be predicted by probing the crystallinity change of binary and ternary blends as a function of compatibilizer concentration, focusing on PTW containing blends. For every PTW concentration investigated, the majority of PTW molecules are in HDPE phase. At concentrations lower than 4 wt%, most of PTW chains are dispersed as individual chains in the bulk PE, disrupting the crystallization process of HDPE. As PTW concentration further increases, micelle formation occurs, and the crystallinity increases back up. Very small amounts of PTW chains are located at the interface, which leads to slightly improved mechanical properties.
This work addresses a key question that is largely unanswered: where are the compatibilizer molecules in a polymer blend? This study demonstrates that DSC may serve as a facile method to predict location and morphology of the compatibilizer molecules in a semicrystalline blend by probing crystallinity change. Knowing the location of a compatibilizer will facilitate root cause analysis of an ineffective compatibilizer and guide design strategy to upcycle commingled waste plastics. One limitation of this method is that it only applies to semicrystalline polymers, as amorphous polymers do not have crystallinity. Nevertheless, due to the wide adoption of semicrystalline polymers in industry and commercial use, this method of predicting the location of an additive may be applied to various semicrystalline systems such as polymer–polymer blends and polymer composites to help more in-depth understanding of fundamentals and further improve materials properties for applications including healthcare, energy, transportation, and aerospace.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research at Oak Ridge National Laboratory, managed by UT Battelle, LLC, for the U.S. Department of Energy (DOE), under contract DE-AC05-00OR22725, was sponsored by the Laboratory Directed Research and Development Program (LDRD) of ORNL. Part of the characterization work was conducted at Oak Ridge National Laboratory's Center for Nanophase Materials Sciences, which a DOE Office of Science User Facility. We thank Professor Chris Ellison from University of Minnesota for helpful discussions.References
- M. R. Pravettoni, Global plastic production and future trends, 2018, http://www.grida.no/resources/6923 Search PubMed.
- UN, The projection of the population of the world, https://www.un.org/sustainabledevelopment/blog/2015/07/un-projects-world-population-to-reach-8-5-billion-by-2030-driven-by-growth-in-developing-countries/.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- C. Giacovelli, Journal, 2018 Single-use plastics: a roadmap for sustainability, United Nations Environment Program, 2018, https://www.unenvironment.org/resources/report/single-useplastics-roadmap-sustainability Search PubMed.
- United States Environmental Protection Agency, Advancing Sustainable Materials Management: 2015 Fact Sheet, EPA530-F-18-004, 2018 Search PubMed.
- A. Rahimi and J. M. Garcia, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- Ellen MacArthur Foundation, The new plastics economy: rethinking the future of plastics & catalyzing action, 2017, https://www.ellenmacarthurfoundation.org/assets/downloads/publications/NPEC-Hybrid_English_22-11-17_Digital.pdf Search PubMed.
- N. F. A. Zainal and C. H. Chan, in Compatibilization of Polymer Blends, ed. A. R. Ajitha and S. Thomas, Elsevier, 2020, pp. 391–433 Search PubMed.
- P. J. Flory, Principles of polymer chemistry, 1953 Search PubMed.
- C. Koning, M. van Duin, C. Pagnoulle and R. Jerome, Prog. Polym. Sci., 1998, 23, 707–757 CrossRef CAS.
- K. Hamad, M. Kaseem and F. Deri, Polym. Degrad. Stab., 2013, 98, 2801–2812 CrossRef CAS.
- A. V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- K. Nomura, X. Y. Peng, H. Kim, K. L. Jin, H. J. Kim, A. F. Bratton, C. R. Bond, A. E. Broman, K. M. Miller and C. J. Ellison, ACS Appl. Mater. Interfaces, 2020, 12, 9726–9735 CrossRef CAS PubMed.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. LaPointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- J. Xu, J. M. Eagan, S.-S. Kim, S. Pan, B. Lee, K. Klimovica, K. Jin, T.-W. Lin, M. J. Howard and C. J. Ellison, Macromolecules, 2018, 51, 8585–8596 CrossRef CAS.
- I. De Schrijver, F. Desplentere, L. Delva, E. De Tandt and K. Ragaert, International Conference on Polymers and Moulds Innovations-PMI, University of Minho, Portugal, 2018 Search PubMed.
- M. Pracella, L. Rolla, D. Chionna and A. Galeski, Macromol. Chem. Phys., 2002, 203, 1473–1485 CrossRef CAS.
- C. Guerrero, T. Lozano, V. Gonzalez and E. Arroyo, J. Appl. Polym. Sci., 2001, 82, 1382–1390 CrossRef CAS.
- M. G. Anagha and K. Naskar, J. Appl. Polym. Sci., 2020, 137, 48727 CrossRef CAS.
- R. Y. Bao, W. R. Jiang, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, RSC Adv., 2015, 5, 34821–34830 RSC.
- N. C. Liu, H. Q. Xie and W. E. Baker, Polymer, 1993, 34, 4680–4687 CrossRef CAS.
- C. R. Chiang and F. C. Chang, Polymer, 1997, 38, 4807–4817 CrossRef CAS.
- N. Yao, H. Wang, L. Zhang, D. Yue and M. Tian, Appl. Surf. Sci., 2020, 530, 147124 CrossRef CAS.
- T. Hirai, J. Kawada, M. Narita, T. Ikawa, H. Takeshima, K. Satoh and M. Kamigaito, Polymer, 2019, 181, 121667 CrossRef CAS.
- J. Kim, H. Zhou, S. T. Nguyen and J. M. Torkelson, Polymer, 2006, 47, 5799–5809 CrossRef CAS.
- X. D. Li and S. H. Goh, Macromol. Chem. Phys., 2002, 203, 2334–2343 CrossRef CAS.
- P. Shokrollahi, H. Mirzadeh, W. T. S. Huck and O. A. Scherman, Polymer, 2010, 51, 6303–6312 CrossRef CAS.
- Y. F. Tian, Y. H. Yang, J. Q. Tan, D. C. Ding, Z. Y. Song, Q. Tao, X. Zheng, T. Hu, X. H. Gong and C. G. Wu, J. Appl. Polym. Sci., 2021, 138, e50127 CrossRef.
- M. Yousfi, S. Livi and J. Duchet-Rumeau, Chem. Eng. J., 2014, 255, 513–524 CrossRef CAS.
- A. Granado, J. I. Eguiazabal and J. Nazabal, Polym. Eng. Sci., 2010, 50, 1512–1519 CrossRef CAS.
- F. Gholami, L. Pakzad and E. Behzadfar, Polymer, 2020, 208, 122950 CrossRef CAS.
- R. J. Spontak and J. J. Ryan, in Compatibilization of Polymer Blends, Elsevier, 2020, pp. 57–102 Search PubMed.
- H. Retsos, I. Margiolaki, A. Messaritaki and S. H. Anastasiadis, Macromolecules, 2001, 34, 5295–5305 CrossRef CAS.
- R. Fayt, R. Jérôme and P. Teyssié, J. Polym. Sci., Polym. Lett. Ed., 1986, 24, 25–28 CrossRef CAS.
- K. H. Yoon, H. W. Lee and O. K. Park, J. Appl. Polym. Sci., 1998, 70, 389–395 CrossRef CAS.
- R. S. Porter, J. Polym. Sci., Polym. Lett. Ed., 1980, 18, 824 CrossRef.
- M. Pluta, Z. Bartczak, A. Pawlak, A. Galeski and M. Pracella, J. Appl. Polym. Sci., 2001, 82, 1423–1436 CrossRef CAS.
- S. Mbarek, M. Jaziri, Y. Chalamet and C. Carrot, J. Appl. Polym. Sci., 2010, 117, 1683–1694 CAS.
- A. Balazs, C. Siemasko and C. Lantman, J. Chem. Phys., 1991, 94, 1653–1663 CrossRef CAS.
- E. A. Eastwood and M. D. Dadmun, Macromolecules, 2002, 35, 5069–5077 CrossRef CAS.
- J. Noolandi, Macromol. Theory Simul., 1992, 1, 295–298 CrossRef CAS.
- K. A. Chaffin, J. S. Knutsen, P. Brant and F. S. Bates, Science, 2000, 288, 2187–2190 CrossRef CAS PubMed.
- T. N. Tran, C. Di Mauro, A. Graillot and A. Mija, Polym. Chem., 2020, 11, 5088–5097 RSC.
- K. Nakamoto, J. Fujita, S. Tanaka and M. Kobayashi, J. Am. Chem. Soc., 1957, 79, 4904–4908 CrossRef CAS.
- L. H. Jones and E. Mclaren, J. Chem. Phys., 1954, 22, 1796–1800 CrossRef CAS.
- M. P. Landoll and M. T. Holtzapple, Biomass Bioenergy, 2012, 45, 195–202 CrossRef CAS.
- S. Moghanlou, M. Khamseh, M. R. Aghjeh and B. Pourabbas, J. Polym. Environ., 2020, 28, 1526–1533 CrossRef CAS.
- J. M. Ferri, D. Garcia-Garcia, E. Rayon, M. D. Samper and R. Balart, Polymers, 2020, 12, 1344 CrossRef CAS PubMed.
- M. D. Whitmore and J. Noolandi, Macromolecules, 1985, 18, 657–665 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09452a |
| This journal is © The Royal Society of Chemistry 2022 |