
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of highly substituted poly(furopyrimidine)s via catalyst-free multicomponent polymerizations of diisocyanides, N,N′-dimethylbarbituric acid, and dialdehyde†
Lichao Dong *,
Nan Li,
Hang Yuan and
Meng Wu
*,
Nan Li,
Hang Yuan and
Meng Wu
Aerospace Institute of Advanced Materials & Processing Technology, Beijing, 100074, China. E-mail: 610557405@qq.com
First published on 22nd February 2022
Abstract
In this work, the catalyst-free multicomponent polymerizations of diisocyanides, N,N′-dimethylbarbituric acid, and dialdehyde for highly substituted poly(furopyrimidine)s has been achieved. All the experimental conditions such as polymerization solvents, temperature and time were investigated in detail. Through the systematic optimization of the polymerization conditions, the obtained polymers could have molecular weights of up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1, and excellent yields (up to 84%) can be achieved. All the polymers were well characterized by gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy. The thermal properties of the polymers were investigated and the decomposition temperature (Td, 5%) was 277 °C.
400 g mol−1, and excellent yields (up to 84%) can be achieved. All the polymers were well characterized by gel permeation chromatography (GPC), nuclear magnetic resonance (NMR) and Fourier transform infrared spectroscopy. The thermal properties of the polymers were investigated and the decomposition temperature (Td, 5%) was 277 °C.
Introduction
Multicomponent reactions (MCRs) are defined as reactions in which three or more starting materials are combined in one pot, retaining most of the atoms in the final product.1–3 Thus, MCRs are a very useful way to quickly cover large portions of chemical space and ideal tools for combinatorial chemistry. In recent decades, multicomponent reactions have attracted much attention because of advantages such as high atom economy, simple procedure, and high efficiency. Because of their ability to generate complex structures economically and efficiently, numerous MCRs have been developed and widely studied. And a large and important class of MCRs is the isocyanide multicomponent reactions (IMCRs).Isocyanides is an extraordinary functional group as it displays the reactivity of two extreme resonance forms having either tetravalent or divalent carbon forms. This enables the isocyanide group to undergo both electrophilic and nucleophilic reactions at the divalent carbon atom, which then converts into the tetravalent form in an exothermic reaction.4–6 With the development of organic synthetic chemistry, the isocyanides has attracted considerable attention due to its excellent performance in constructing heterocyclic compounds. The obtained functional products usually possess excellent solubility and stability and have been demonstrated with great application potentials in a variety of fields.
Furopyrimidines represent a very important class of compounds that possess wide range of biological activities such as antibacterial,7 antifungal,8 antitumour,9 antiviral,10 antifolate11,12 and anti-human cytomegalovirus activity.13 Because of the importance of furopyrimidine derivatives, several methodologies for synthesizing them have already been developed.13–19 However, the research on furopyrimidines mainly concentrated in the small molecules at present. There are rarely reports about poly(furopyrimidine)s, which may have new features and functions different from small molecules. Following the daily increasing requirement on molecular functionality, developing new polymerization and providing novel or unprecedented structures has become an important issue.
In 2006, Mohammad Bagher Teimouri et al. reported that the multicomponent cycloaddition of furopyrimidines could be achieved by employing isocyanides, N,N′-dimethylbarbituric acid, and dialdehyde20 (Scheme 1). Inspired by the above-mentioned small molecule's reaction, we have successfully designed and developed a one-pot and catalyst-free polymerization to produce functional poly(furopyrimidine)s in this work (Scheme 2). The reaction conditions were optimized systematically, and poly(furopyrimidine)s with satisfied solubility, thermal stability and molecular weights (up to 16400 g mol−1) were obtained in good yield (up to 84%). Poly(furopyrimidine)s' structures were well characterized by GPC, FT-IR, and NMR. Polymerization mechanism is proposed according small molecule's reaction and the thermal properties were investigated.
 | ||
| Scheme 1 Syntheses of multi-substituted furopyrimidines by one-pot MCRs. | ||
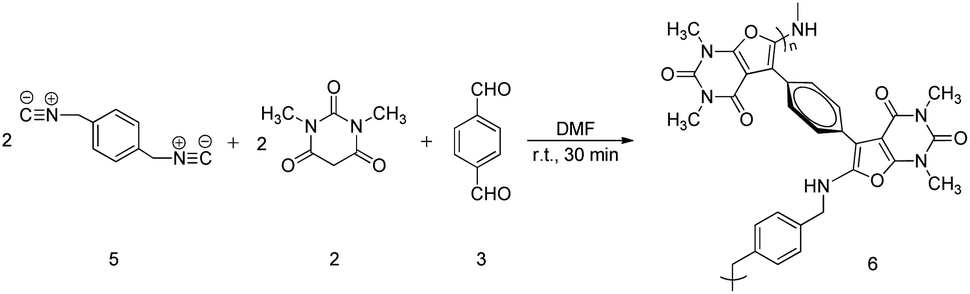 | ||
| Scheme 2 Synthetic route for the poly(furopyrimidine)s. | ||
Experimental
Materials
Unless stated otherwise, all chemicals were obtained from commercial suppliers and used without further purification. The monomers 1,4-bis(isocyanomethyl)benzene 5 were synthesized according to the commonly used synthetic routes (see Scheme 3). N,N′-Dimethylbarbituric acid and terephthalaldehyde were bought from Energy Chemical. 1,4-bis(aminomethyl)benzene and chloroform-d was bought from Innochem. All monomers put into vacuum oven to drying. | ||
| Scheme 3 The synthetic route of monomer diisocyanides. | ||
Instruments
Weight-average molecular weights (Mw) and dispersity indices (Mz/Mw) of the polymers were estimated on a Waters gel permeation chromatography (GPC) system equipped with a Waters 1515 isocratic HPLC pump and Waters 2414 refractive index detector. Polystyrene standards were utilized, and THF was used as the eluent at a flow rate of 1.0 mL min−1. Fourier transform infrared (FT-IR) spectra were measured on a Bruker AV 400 spectrometer. 1H NMR spectra was measured on a Bruker AV 400 spectrometer. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer STA 8000 at a heating rate of 10 °C min−1 under a nitrogen flow.Polymerization
All the polymerization reactions were conducted using standard techniques. A typical polymerization procedure is given below by employing the conditions in Table 2, entry 2 as an example. Into a 10 mL tube equipped with a magnetic stir bar were added 1,4-bis(isocyanomethyl)benzene (0.64 mmol, 100 mg), N,N′-dimethyl-barbituric acid (0.64 mmol, 100 mg), terephthalaldehyde (0.32 mmol, 43 mg) and 3 mL of DMF. The reaction mixture was stirred under air atmosphere at 60 °C for 24 h. Then the solution was added into 200 mL of methanol with vigorous stirring. The precipitate was filtered and washed with hexane and dried in a vacuum at room temperature to a constant weight. Other polymerization reactions also have the same procedure in different conditions.Synthesis of 1,4-bis(isocyanomethyl)benzene
The monomers 1,4-bis(isocyanomethyl)benzene were prepared by the reported procedures, and the synthetic route was shown in Scheme 3. The structural information was characterized by spectroscopic techniques, and the results agreed with its target structures (Fig. S1 and S2†).To a 500 mL round-bottom flask were added 1,4-bis(aminomethyl)benzene (7.8 g, 50 mmol), DCM (20 mL), chloroform (8.044 mL, 100 mmol) and TEBAC (0.2278 g, 1 mmol). A 50% aqueous solution of sodium hydroxide (15 mL) was added. The solution was stirred at 40 °C for 12 h and cooled down to room temperature, the organic layer was combined and washed with DCM and deionized water, respectively. Then the organic phase was dried over anhydrous MgSO4 and concentrated, the crude product obtained was purified by silica gel column chromatography. The 1,4-bis(isocyanomethyl)benzene was obtained as a white solid in 45.0% yield.
Characterization data of 1,4-bis(isocyanomethyl)benzene 5: 1H NMR (400 MHz, CDCl3, δ): 7.39 (s, 4H), 4.66 (m, 4H); 13C NMR (101 MHz, CDCl3, δ): 158.26–158.16, 132.75, 121.31, 45.27–45.12.
Results and discussion
Polymerization
Generally, the optimized conditions for small molecule reactions are not necessarily suitable for polymerizations. Thus, the polymerization conditions were first systematically investigated.Firstly, we investigated the solvent effect on the polymerization; the results are summarized in Table 1. The property of solvent is significant for the specific occurrence of polymerization. Four different solvents including N,N′-dimethylformamide (DMF), ethyl acetate (EA), 1,4-dioxane and toluene were tested. The highest yield (70%), as well as the modest Mw (7600 g mol−1), was achieved in DMF-a suitable solvent for the synthesis of poly(furopyrimidine)s via cycloaddition reaction. Although the polymerization also occurred in other solvents, Mw or yields of the polymer products was lower significantly. Therefore, DMF was used as the polymerization solvent in the following experiments.
| Entries | Solvent | Yield (%) | Mwb | Mzb | Đb |
|---|---|---|---|---|---|
| a Carried out at r.t. for 24 h, concentration: [1] = 0.213 M. [1]:[2]:[3] = 2:2:1.b Determined by GPC in THF using a linear polystyrene as calibration standard; Mz = z − average molecular weight; Đ = Mz/Mw, where Mw = weight − average molecular weight. |
|||||
| 1 | DMF | 70 | 7600 | 10700 |
1.41 |
| 2 | EA | 25 | 5000 | 6700 | 1.34 |
| 3 | 1,4-Dioxane | 10 | 3500 | 4200 | 1.20 |
| 4 | Toluene | — | — | — | — |
Secondly, the effects of temperature on the polymerization are summarized in Table 2. The yield of polymer product increased with the increase of temperature from r.t. to 100 °C. However, the molecular weight almost unchanged at high temperature, which was mainly due to the increased viscosity of the polymerization system and rigidity of polymer structure. This result shown that polymerization is naturally terminated because of the more difficult diffusion of chain segments during the later period of polymerization. Therefore, considering the simplicity of the operation, the polymerization temperature was optimized as 60 °C.
| Entries | Temperature (°C) | Yield (%) | Mw | Mz | Đ |
|---|---|---|---|---|---|
| a Carried out in DMF for 24 h, concentration: [1] = 0.213 M. [1]:[2]:[3] = 2:2:1. |
|||||
| 1 | r.t. | 70 | 7600 | 10700 |
1.41 |
| 2 | 60 | 84 | 16400 |
29700 |
1.81 |
| 3 | 100 | 85 | 16600 |
30800 |
1.86 |
Finally, the effects of time course on the polymerization were evaluated, as shown in Table 3. The yield of the polymer product gradually increased with the polymerization time increased from 2 to 12 h. However, further prolonging the reaction time to 24 h resulted in almost unchanged Mw, suggesting that the polymer chain length growth became difficult after a certain time. One reason is the increased viscosity of the polymerization system and the other is the more difficult diffusion of chain segments during the later period of polymerization due to the large steric hindrance. Based on these results, take the yield into consideration, the reaction time was optimized as 24 h.
| Entries | Time (h) | Yield (%) | Mw | Mz | Đ |
|---|---|---|---|---|---|
| a Carried out in DMF at r.t., concentration: [1] = 0.213 M. [1]:[2]:[3] = 2:2:1. |
|||||
| 1 | 2 | Trace | — | — | — |
| 2 | 6 | 15 | 1900 | 2400 | 1.26 |
| 3 | 12 | 46 | 7600 | 10600 |
1.39 |
| 4 | 24 | 70 | 7600 | 10700 |
1.41 |
To sum up, considering the environmental friendliness, economy and simple experimental operation, we chose N,N′-dimethylformamide (DMF) as solvent, reaction time of 24 h, and polymerization temperature of 60 °C as the optimal polymerization conditions for further investigation.
Structural characterization
As above mentioned, the resultant polymers are all soluble in highly polar organic solvents, which enable us to characterize their structures spectroscopically. Satisfactory analysis data corresponding to their expected molecular structures were obtained from FT-IR and 1H NMR, which could offer detailed information about the polymer structures.Fig. 1 shows the FT-IR spectra of target polymer. The resonance peak at 2114 cm−1 was attributed to the stretching vibration bands of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C in 5 (line D in Fig. 1), which disappeared in the spectra of 6 (lines A in Fig. 1). The peak at 1720 cm−1 in the spectrum of 2 (line C in Fig. 1), associated with the stretching vibration of C
C in 5 (line D in Fig. 1), which disappeared in the spectra of 6 (lines A in Fig. 1). The peak at 1720 cm−1 in the spectrum of 2 (line C in Fig. 1), associated with the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, was disappeared in the spectra of 6 after the formation of the furan ring. And the peak at 1714 cm−1 in the spectrum of 3 (line B in Fig. 1) still exists due to no reaction. Furthermore, the stretching vibrations of N–H and CC were observed at 3428 and 1617 cm−1, respectively, in the spectra of 6, which showed new peaks of amino and furan formations. These results can verify the polymerization occurrence of mixing these monomers together under certain experimental conditions.
O, was disappeared in the spectra of 6 after the formation of the furan ring. And the peak at 1714 cm−1 in the spectrum of 3 (line B in Fig. 1) still exists due to no reaction. Furthermore, the stretching vibrations of N–H and CC were observed at 3428 and 1617 cm−1, respectively, in the spectra of 6, which showed new peaks of amino and furan formations. These results can verify the polymerization occurrence of mixing these monomers together under certain experimental conditions.
 | ||
| Fig. 1 FT-IR spectra of (A) poly(furopyrimidine)s (B) terephthalaldehyde (C) N,N′-dimethylbarbituric acid (D) 1,4-bis(isocyanomethyl)benzene. | ||
In the 1H NMR spectra (Fig. 2), the chemical shift of 1,4-bis(isocyanomethyl)benzene mainly assembled at δ = 3.0 ppm and 7.0–7.5 ppm, which corresponding to the proton resonances of alkyl and benzene ring group. After the polymerization, the chemical shift of benzene ring still resonance at δ = 7.0–7.5 ppm, which was overlapped with the peak of benzene ring. And the proton resonances of alkyl shifted from 3.0 ppm to 1.8 ppm due to the change of chemical environment. In addition, the proton resonances of alkyl (Hd) and aldehyde (He) were disappeared after the formation of the furan ring. Meanwhile, the proton resonances of Hc changed to Hc1 and Hc2. These results suggest that the isonitrile groups, aldehyde and methylene have been converted into furan ring by the polymerization. Moreover, the terminal isonitrile groups and aldehyde has the ability to continue the polymerization according to the small molecule reaction. However, due to the higher viscosity achieved during the later period of polymerization, the diffusion of macromolecular terminal isonitrile groups and aldehyde becomes more difficult, and then, polymerization is naturally terminated.
 | ||
| Fig. 2 1H NMR spectra of (A) 1,4-bis(isocyanomethyl)benzene (B) N,N′-dimethyl-barbituric acid (C) terephthalaldehyde (D) poly(furopyrimidine)s in CDCl3. The solvent peaks are marked with asterisks. | ||
Polymerization mechanism
To study the polymerization mechanism, we referred to the literature studying small molecule's reaction and proposed a tentative mechanism of this polymerization, as shown in Scheme 4. The formation of the fused structure can be rationalized by initial formation of a conjugated electron deficient heterodiene I by Knoevenagel condensation of aldehyde 3 and CH-acid 2, followed by a [1 + 4] cycloaddition reaction or a Michael-type addition reaction (II) with diisocyanides 5 to afford an iminofuran derivative III, which was then isomerized to the target furopyrimidines IV. The IV continue to repeat the whole process from I to IV by the same catalytic mechanism. Ultimately, the poly(furopyrimidine)s are obtained by repeating this process until the polymerization process is over. Due to the higher viscosity achieved during the later period of polymerization, the diffusion of chain segments becomes more difficult, and then, polymerization is naturally terminated. | ||
| Scheme 4 The proposed polymerization process of poly(furopyrimidine)s. | ||
Thermal properties
The thermal stability of polymer was evaluated by thermogravimetric analysis (TGA). As shown in Fig. 3, the polymer lost 5% weight as heated at 277 °C under a nitrogen atmosphere. The TGA indicates that the thermal stability of the obtained polymer is lower, and the degradation starts earlier than the given Td temperature. The above results are mainly due to the different chemical structures of these polymers in their main chains and side chains. Since the main chain is not an overall conjugated structure, there are methylene groups in the chain segments, which will reduce the rigidity of the polymer and the thermal stability; meanwhile, the degradation starts earlier than the given Td temperature was attributed to the removal of methyl groups on the side chains. | ||
| Fig. 3 Thermograms of the poly(furopyrimidine)s. The measurements were performed under N2 atmosphere at the heating rate of 10 °C min−1. | ||
Conclusions
In this paper, the catalyst-free multicomponent polymerizations of diisocyanides, N,N′-dimethylbarbituric acid, and dialdehyde for highly substituted poly(furopyrimidine)s has been achieved. The structure of poly(furopyrimidine)s was well characterized by GPC and NMR and the thermal properties were investigated. Furthermore, the structure of poly(furopyrimidine)s may have potential applications in biopharmaceutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Aerospace Institute of Advanced Materials & Processing Technology.Notes and references
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- A. Dçmling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- A. Dömling and I. Ugi, Angew. Chem., 2000, 112, 3300–3344 CrossRef.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- J. P. Zhu, Eur. J. Org. Chem., 2003, 7, 1133–1144 CrossRef.
- C. G. Dave and R. D. Shah, Molecules, 2002, 7, 554 CrossRef CAS.
- M. M. H. Bhuiyan, K. M. M. Rahman, M. K. Hossain, M. A. Rahim and M. I. Hossain, Croat. Chem. Acta, 2005, 78, 633 CAS.
- A. Gangjee, Y. Zeng, M. Ihnat, L. A. Warnke, D. W. Green, R. L. Kisliuk and F. T. Lin, Bioorg. Med. Chem., 2005, 13, 5475 CrossRef CAS PubMed.
- Z. Janeba, J. Balzarini, G. Andrei, R. Snoeck, E. De Clercq and M. J. Robins, J. Med. Chem., 2005, 48, 4690 CrossRef CAS PubMed.
- A. Gangjee, Y. Zeng, J. J. McGuire and R. L. Kisliuk, J. Med. Chem., 2005, 48, 5329 CrossRef CAS PubMed.
- A. Gangjee, Y. Zeng, J. J. McGuire, F. Mehraein and R. L. Kisliuk, J. Med. Chem., 2004, 47, 6893 CrossRef CAS PubMed.
- M. J. Robins, K. Miranda, V. K. Rajwanshi, M. A. Peterson, G. Andrei, R. Snoeck, E. De Clercq and J. J. Balzarini, J. Med. Chem., 2006, 49, 391 CrossRef CAS PubMed.
- S. Kajikawa, H. Nishino and K. Kurosawa, Heterocycles, 2001, 54, 171 CrossRef CAS.
- B. Mohammad, Z. S. Nader and H. H. Seyyed, Helv. Chim. Acta, 2010, 93, 2189–2193 CrossRef.
- C. M. Li and F. R. Zhang, Tetrahedron Lett., 2017, 58, 1572–1575 CrossRef CAS.
- A. Shaabani, M. B. Teimouria and H. R. Bijanzadehb, Tetrahedron Lett., 2002, 43, 9151–9154 CrossRef CAS.
- L. Dutta, M. Sharma and P. J. Bhuyan, Tetrahedron, 2016, 72, 6654–6660 CrossRef CAS.
- J. I. Pyo, E. J. Hwang, C. S. Chan, S. H. Lee, S. W. Lee, I. T. Kim and S. H. Lee, Synth. Met., 2005, 155, 461–463 CrossRef CAS.
- M. B. Teimouria and R. Bazhrang, Bioorg. Med. Chem. Lett., 2006, 16, 3697–3701 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09336c |
| This journal is © The Royal Society of Chemistry 2022 |