DOI:
10.1039/D1RA09112C
(Paper)
RSC Adv., 2022,
12, 6336-6346
Controlled supramolecular interaction to enhance the bioavailability of hesperetin to targeted cancer cells through graphyne: a comprehensive in silico study†
Received
16th December 2021
, Accepted 24th January 2022
First published on 23rd February 2022
Abstract
In the current study, the drug carrier efficiency of graphyne (GRP) for the transfer of the hesperetin (HPT) drug is evaluated for the first time. The GRP efficacy as a carrier is investigated using the density functional theory (DFT) technique to calculate various physiochemical characteristics such as dipole moment, bandgap, and chemical reactivity-descriptors for HPT, GRP and HPT@GRP complex. The non-covalent-interaction (NCI) plot indicated that GRP and HPT have weak interaction force, which is fundamental for the drug's noticeable offloading from the GRP carrier at its target location. According to frontier molecular orbital analysis, the highest occupied molecular orbital (HOMO) of HPT distributes the charge to the GRP, the lowest unoccupied molecular orbital (LUMO) during excitation. Charge transfer is further supported by charge-decomposition-analysis, which interprets the extensive overlap between HPT and GRP orbitals. In the case of the gas phase, the λmax of the HPT@GRP-complex is redshifted by 9 nm from GRP. In the solvent phase, the λmax value is also redshifted. These theoretically calculated spectra also match experimentally observed spectra rather well. The PET (photoinduced electron-transfer) method and electron–hole theory were used for the graphical explication of distinct excited states. The photoinduced electron transfer (PET) mechanism interprets fluorescence dimming because of interaction. Furthermore, GRP with cationic (+1) and anionic (−1) charge states (GRP+1/−1) showed minor structural disfigurement and formed stable HPT complexes. In the current study, HRP is loading and unloading on GRP very effectively, that can potentially be used in the oncology field. Due to this theoretical study, researchers will be interested in looking at other 2D nanomaterials for drug delivery applications.
Introduction
Cancer is a deadly disease with more than 10 million new cases each year1 due to low specificity, poor solubility, poor bioavailability, multi-drug resistance, low therapeutic results, and high cytotoxicity of conventional chemotherapeutic medication.2 Prostate, lungs,3 stomach,4 and colorectal cancer5 are different and the most common types of cancer. Indeed, careful investigation of biologically related features of living systems has always been an important subject of several works to provide insightful information in such cases of immunotherapy,6,7 chemotherapy,8,9 radiotherapy,10 and surgery.11 However, this treatment still exhibits unavoidable side effects by killing healthy cells and severe toxicity.12 For metastatic cancers, the most efficient treatment is chemother.13 In chemotherapy treatment, chemotherapeutic agents, “medicine” is utilized to kill the cancer cells. Still, this treatment method has severe cytotoxic side effects in various tissues and organs, recurrence of cancer because the medicine is not released to a target site. Some significant obstacles in chemotherapy are the resistance of tumor cells to medicine, reduced water-solubility of drugs, and low concentrations of drugs in cancer sites. Researchers and oncologists were trying to minimize these problems.14,15 Therefore, a highly efficient therapeutics treatment would be required, such as “magic bullets” that can overcome the biological hurdles, distinguish between benign and malignant cells, specifically target the cancerous cell and desirably release the drug on the target tissue.16 The application of nanotechnology (nanomedicine) is predicted to help us move toward the purpose mentioned above. The development of various nano-drug-delivery methods performed an essential role in disease diagnosis, detection, and therapy. These nano-drug-delivery systems safely transfer the drugs towards the cancer tissues in a controlled concentration, avoiding the interrelation with the reticuloendothelial system.17 Nanocarriers significantly impact a nano-drug-delivery system for cancer therapy because of the similarity of size with biological structures; these nanocarriers can easily penetrate the cell membrane and have a prolonged circulation time.18–20 Due to rapid and defective angiogenesis (synthesis of new blood vessels from old ones), the permeability of blood vessels in tumours increases, allowing nanocarrier entry. Furthermore, the poor lymphatic drainage in tumours trapped the nanocarrier and allowed them to offload the drug into the vicinity of cancerous cells. These pharmacokinetics modifications provide a better result by explicitly targeting the cancerous site and retaining the increased concentration of the therapeutic agent at its specific defective site for the duration of the activity. This targeted chemotherapeutic agent uses apoptosis and narcosis to kill cancerous cells.21–23 An emerging generation of nanocarrier is 2D nanomaterials, such as tungsten diselenide24 (WSe2), silicene,25 germanene,26 molybdenum disulfide27 (MoS2), bismuth selenide28 (Bi2Se3), manganese dioxide,29 transition metal di-chalcogenides (TMDs), hexagonal boron nitride30 (h-BN), and GRP are some important nanocarriers due to their specified and unique physicochemical properties.31–34 GRP created a honeycomb 2D crystal lattice structure in which all carbon atoms are sp2 hybridized, leading to incredible mechanical and electrical performance is commonly used in optoelectronic devices, in a solar cell as photoconductive material, in drug delivery, and medical imaging due to the presence of good surface reactivity and free π electrons.35 The free surface π electrons are efficient for π–π interaction, electrostatic or hydrophobic interaction with poor soluble drug, and noncovalent interaction in drug delivery system.36 GRP interaction with the biomolecules, tissues and different types of cells is significant for its biomedical application, toxicity, and biocompatibility.37 GRP as a nanocarrier can quickly enter the cell by endocytosis and successfully release the drug in the cytosol in response to stimuli.38 The weight ratio of loaded drug to the carrier is 200% in the case of GRP, which makes GRP a more efficient and preferred nanocarrier over others.39 The drug-carrying capacity of GRP for quercetin, 5-fluorouracil, and daunorubicin has been studied for cancer treatment.40 The interaction between drugs and GRP was investigated by DFT calculation41,42 and the molecular-dynamics simulation. HPT (3′,5,7-trihydroxy-4-methoxy flavanone) and its metabolites are bioactive flavonoids used as an antioxidant, antidiabetic, anticancer, estrogenic, anti-inflammatory, and cardio-neuroprotective.43 This polyhydroxy flavone is common in vegetables, young citrus fruits, tomatoes, apples, and flowers.44 HPT is hydrophobic (poor water solubility) inadequate stability in the digestive tract, resulting in deficient oral absorption.45 Many research groups are working to improve the oral bioavailability and activity of HPT by a nano-drug delivery system such as nano-formulation, cyclodextrin, and phospholipids complex formation, which increase its medicative properties.46 The anticancer effect and possible singling mechanism of HPT on the glioblastoma cells and malignant hematolymphoid cells have been reported a significant inhibitory effect on these cells. The PCT (photo-induced-charge transfer) and PET (photoelectron charge transfer) processes are critical in the biological system because both processes determine the phosphorescence and fluorescence properties of the complex.47 The charge or electron transfer from a chelator to a fluorophore origin. The fluorescence detecting technique is critical for the intended distribution of the HPT drug to the target spot to increase the drug's therapeutic efficiency.
Several drugs are frequently used to treat cancer, along with side effects and toxicities. For example, HPT also causes severe harmful effects on human health, such as bioavailability, absorption levels, anticoagulants, blood pressure drugs, and calcium channel blockers. Therefore, many researchers to improve therapeutic index and decrease its harmful effects, focus on finding nano carrier such as suitable for drug, non-reactive, it does not affect the geometry of medicine, bonded with weak forces due to loading and unloading efficiently. Therefore, we find 1st-time new combination in which carrier is effectively suitable for HTP. It is conformed to density-functional-theory (DFT) and TD-DFT calculations; the drug delivery potential of GRP was assessed for HPT drug. HPT, a chemotherapy medicine used in the treatment of breast cancer, lung cancer, and ovarian cancer.48
Experimental section
Computational details
All inorganic metal salts, organic ligands and solvents are analytically pure reagent grade. Infrared spectroscopy (IR) was obtained by Thermo IS5 FT-IR Fourier infrared spectrometer with KBr tablet in the range of 4000–400 cm−1. The measurement of UV-vis was performed by TU-1950.
When using the Gaussian 09 software package, the density functional theory (DFT) is applied in all calculations.49 The DFT technique provides molecule orientation, structure, charge transport, and adsorption energy. B3LYP50 functional with Lanl2DZ51 basis set was used to improve the geometry.52,53 The HPT model structure is used for geometry optimization. By examining different potential orientations, the most stable system of HPT on GRP was found. The excited state properties of HPT-loaded GRP complex have been investigated using Time-Dependent Density Functional Theory (TDDFT), and extension of DFT. The computed output file of the Gaussian 09 software, further, corresponding potential energy distributions (PEDs) were computed with Vida software and it is reliable assessment for infrared spectroscopy (IR).54 The adsorption energy is a relevant and vital parameter for determining the interactions between the various species involved in the complex creation process, for the adsorption of HPT drug on the surface of GRP, the adsorption energy of complex computed, which is represented by “Ead” in eqn (1).55
| | |
Ead = EHPT@GRP − (EGRP + EHPT)
| (1) |
EHPT@GRP represents the HPT@GRP complex free energy produced between the GRP and the HPT medication, and
EGRP and
EHPT are the GRP and HPT molecules energies, respectively. For
Ead calculations, we used single point energies. A polarizable-continuum-model with integral equation formalism
56 (IEF-PCM was used to examine the impact of solvent). Because water plays such an essential part in the biomolecular system of humans, it is vital to investigate its influence on the HPT@GRP complex. Furthermore, the following formula is used to determine the solvation energy of the system:
| | |
Esolvation = Esol − Egas
| (2) |
Egas and
Esol show the total energies of the system in the gas phase and solvent phases, respectively. The solvation energy of a complex molecule is referred to as solvation. Optimized geometry, characteristics such as global electrophilicity (
x), chemical potential (
l), hardness (
g), softness (
s), and dipole moment were calculated.
56 Various studies, such as natural bond orbital (NBO), PET, charge decomposition analysis (CDA), noncovalent interaction (NCI), and ELF, have been carried out. The frequency calculations of infrared spectroscopy using the DFT technique
57,58 B3LYP/Lanl2DZ were used to validate those as mentioned earlier optimized molecular frameworks. The interaction between fragments of a complex such as GRP and HPT is determined by NCI (noncovalent interaction). Disintegration analysis is used to determine the orbital interaction of the charge donor and acceptor components of the complex charge, indicating how these fragments combine to produce the complex. The delocalization of charges and intramolecular transitions were investigated between molecules' bonds, NBO analysis is utilized.
59 The electron concentration before and after complex formation was examined using the ELF (electron localization function), which occasionally revealed minute differences. HPT, GRP, and HPT@GRP complex UV-visible absorption spectrum in the aqueous and gas phases were estimated using TD-DFT calculations. At B3LYP/Lanl2DZ, charge transfer (PCT) processes, as well as photoinduced electron transfer (PET) and NCI analysis, were investigated. This research also looked at the impact of surface charges (+1 and −1) on the architecture of the examined molecules. The distribution of charges for complex molecules was also investigated on the base of the atomic dipole-moment corrected-Hirshfeld (ADCH) population. This approach was chosen for the analysis because of its well-known ability to handle Hirshfeld-charge dipole moment repeatability complications.
60,61 The charge values acquired by ADCH charge analysis are more credible.
Results and discussion
Fig. 1 represents the optimized geometric structure of the carrier GRP and the HPT@GRP complex. The optimized system of GRP and HPT@GRP-complex are described in ESI Fig. S1.† The optimized HPT@GRP complex clarifies that C–H noncovalent interaction was established between the HPT and GRP, in which GRP utilized the carbon and hydrogen from HPT during the HPT@GRP complex formation. In the HPT@GRP complex, the medicine HPT acts as a donor, donating hydrogen to an acceptor GRP carrier. During complex formation, HPT is adsorbed to the surface of GRP by two C–H interactions. The bond distance of these two C–H interactions is 2.54 and 3.68 Å. This hydrogen bonding (C–H⋯C–H) between HPT@GRP-complex is shown in Fig. 2. The optimized carrier GRP dipole-moment is negligible. After the complex (HPT@GRP) formed, the dipole-moment raised to 2.02 Debye. In polar solvents, this elevated dipole-moment causes an increase in the hydrophilicity of the complex, which facilitates the drug's-controlled delivery throughout biological systems. For instance, in the case of water, the solvent serves as a vital symbol of living organisms while being used and answerable for the optimal circumstances of critical biochemical, biological functions. To investigate the impact of water on the interaction of HPT with GRP, the solubility and chemical stability of the HPT@GRP complex was conducted. Compared to the GRP phase, the solvent phase enhanced the dipole moment of complex formation more, as shown in ESI Table S1.†
 |
| | Fig. 1 The optimized structures of GRP and the HPT@GRP-complex. | |
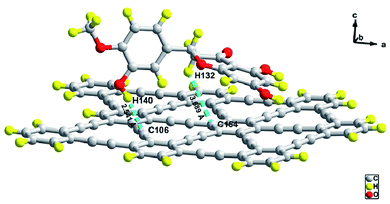 |
| | Fig. 2 Hydrogen bonding between HPT@GRP-complex. | |
Frontier molecular orbital (FMO) analysis
The energy of frontier molecular orbital's (HOMO and LUMO) and the measurement of their energy gap are fundamental factors for determining molecular stability, polarizability, chemical hardness, and chemical reactivity of any molecule. For example, a molecule with a low HOMO–LUMO energy gap is soft, has high chemical reactivity, has more polarizability, and shows low kinetic stability.62 Due to charge transfer from HOMO to LUMO, this energy gap is essential for the biomedical application of the molecule. Fig. S2† represents the EHOMO and ELUMO of GRP, HPT, and HPT@GRP complex, whereby green and red colour distributions represent the positive and negative phases of FMOs wave function, respectively. In HPT ((2S)-5,7-dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-2,3-dihydro-4H-1-benzopyran-4-one), the HOMO is primarily dispersed across the whole molecule, except for the methoxy phenol area. At the same time, the LUMO is generally scattered throughout the molecule, except the dihydroxy benzene region. While in the case of the HPT@GRP-complex, the LUMO is predominantly present on the carrier GRP while the HOMO is distributed on the HPT with total densities separation. As a result, the charge from HPT to the GRP carrier is transferred. The HOMO–LUMO energies and the energy gap between HOMO and LUMO, along with electronic parameters and absorption wavelengths (λmax), are represented in Table 1. HPT has an energy gap of 4.53, having HOMO and LUMO energies of −5.56 and −1.03 eV, respectively. GRP has a band gap of 2.57 eV, having HOMO and LUMO energies of −5.24 and −2.67 eV, respectively. At the same time, the energy gap of the HPT@GRP complex (2.37 eV) is much lower than those of GRP and HPT, which indicates that the complex needs a low amount of energy to move from the ground state to the excited state. The EHOMO of the HPT@GRP complex matches with the EHOMO of HPT.
Table 1 HOMO–LUMO energy, bandgap (Eg), λmax (maximum-adsorption-wavelength), dipole moment (D), chemical potential (μ), hardness (η), global softness (α), electrophilicity index (ω) of GRP, HPT, and HPT-GRP-complex in the solvent phase
| Parameters |
HPT |
GRP |
HPT@GRP |
| EHOMO (eV) |
−5.56 |
−5.24 |
−5.13 |
| ELUMO (eV) |
−1.03 |
−2.67 |
−2.76 |
| ΔE (eV) |
4.53 |
2.57 |
2.37 |
| VIP (eV) |
5.56 |
5.24 |
5.13 |
| VEA (eV) |
1.03 |
2.67 |
2.76 |
| η (eV) |
2.265 |
1.285 |
1.185 |
| μ (eV) |
−3.295 |
−3.955 |
−3.955 |
| X (eV) |
3.295 |
3.95 |
3.945 |
| σ |
1.1325 |
0.6425 |
0.59 |
| ω (eV) |
2.39 |
6.08 |
6.56 |
| Dipole moment |
2.83D |
0.402D |
2.02D |
| λmax |
119.9 |
516.78 |
525.85 |
In contrast, the ELUMO matches with the ELUMO of GRP, indicating that HPT is an electron donor while GRP is an electron acceptor species in a complex molecule. In the UV-vis spectra, the carrier GRP showed λmax at 516 nm while HPT@GRP-complex shows the λmax at 525, redshifted by 9 nm from the carrier molecule. While in this case, the GRP showed λmax at 520 nm, while HPT@GRP-complex shows the λmax at 525, which is redshirted by 5 nm from GRP. The HOMO–LUMO diagrams of HPT, GRP, and HPT@GRP-complex in the solution phase as represented in ESI Fig. S3.† The adsorption energy (Ead) of the HPT@GRP complex in the gas phase and liquid phase is −0.205 eV and −0.0877 eV respectively, which is determined using eqn (1). Because the adsorption energy values are smaller than 1, the it will be easy to unload the HPT from GRP after adsorption. The negative signs with adsorption energy, the HPT@GRP complex are anticipated to be stable. The study is unique in that no one has ever computed the adsorption energy of HPT with GRP previously.
Molecular electrostatic potential (MEP)
The MEP (molecular electrostatic potential) is a valuable descriptor related to the electronic density and evaluates reactivity to nucleophilic and electrophilic attacks.63,64 MEP is useful for studying biological recognition processes, H-bonding interaction, and polarity of the molecule. MEP provides information about the net electrostatic effect around the investigated molecule produced at that point by electron (−ve charge) and proton (+ve charge) distribution. The shape, size, reactive sites, and charge density are predicted from electron density and electrostatic potential iso-surface mapped. The hydrogen donor and hydrogen acceptor ability of molecule for hydrogen bond formation can be easily predicted from a positive and negative region on the MEP map. MEP surfaces have a different color pattern for different electrostatic potential values. Red, orange, and yellow represent negative electrostatics potential regions related to electrophilic reactivity, blue for positive electrostatic potential region ones to nucleophilic reactivity, and green for zero potential region. The region with negative V(r) usually represents the lone pair of the highly electronegative atom in the investigated molecule. The pattern for increasing electrostatic potential is, red < orange < yellow < green < blue. The electrostatic potential map given in Fig. 3 illustrated the 3D charge distribution of GRP, HPT, and HPT@GRP complex molecules.
 |
| | Fig. 3 Molecular electrostatic potential (MEP) map of HPT@GRP complex. | |
Hostile areas in the HPT molecule are mainly located around the oxygen atoms of the hydroxyl group with red color intensity due to lone pair of electrons on the oxygen atom. Therefore, a suitable site for upcoming nucleophiles, as seen in the MEP map; nevertheless, the highest positive regions are centered on the hydrogen of –OH groups, which can be considered suitable spots for the nucleophilic approach. The MEP map of GRP reveals that the potential harmful sites are around carbon atoms, based on these computed results. These sites provide information about the area of the molecule where intermolecular interactions can occur. As a result, Fig. 3 shows that the intermolecular C–H hydrogen bond exists between GRP and HPT in a complex molecule. The MEP diagram of HPT@GRP-complex in the aqueous phase is represented in ESI Fig. S4.†
Infrared spectroscopy studies
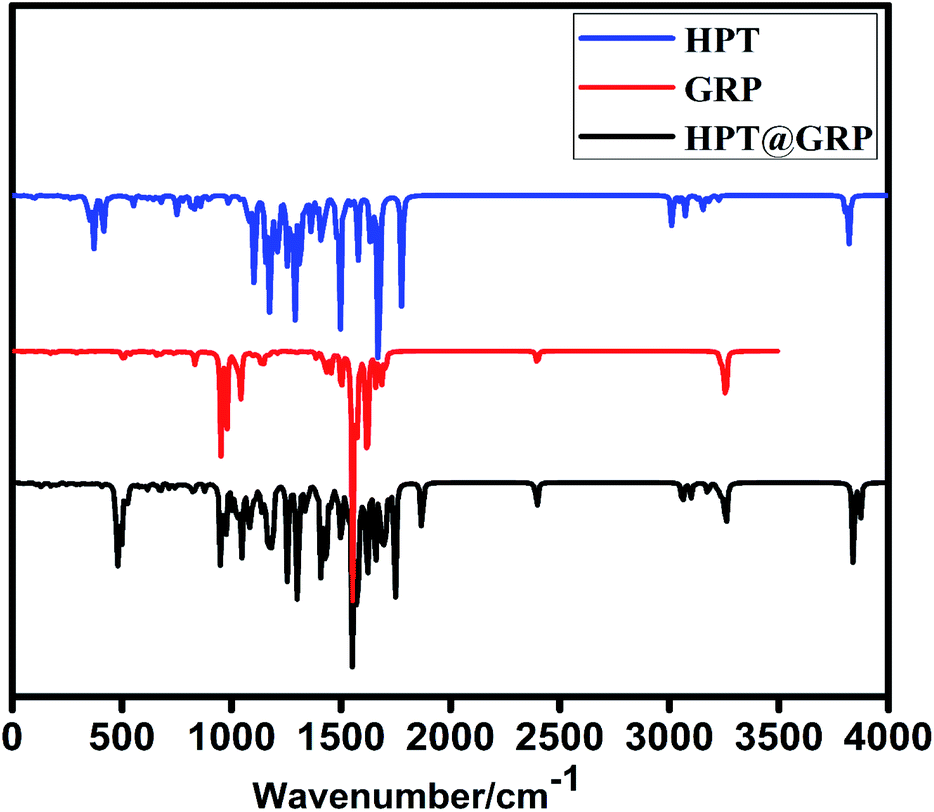
The vibration frequency values of HPT, GRP molecule, and HPT@GRP-complex are given in Table S2.† The adsorption behaviour of the HPT molecule on GRP compounds was investigated by taking into consideration variation in the most prominent peaks in the infrared spectra of HPT@GRP-complex. When looking at the HPT, GRP molecule, and GRP@HPT-complex stretching frequencies, the C–O, C–C, and O–H groups of GRP and HPT show prominent variation in the complex. The vibration frequencies of the free HPT molecule are reported as νsC–O: 1778, νsO–H: 3821, while the νsC–C is registered as 2401.65 IR frequency values were calculated for optimized66 GRP-HPT-complex, and the characteristic vibration peaks obtained are νsO–H: 3870 (cm−1), νsC–C: 2397 (cm−1), and νC–O: 1748 (cm−1). After adsorption (“loading” is better than “absorption”) of HPT on GRP, O–H vibration is redshirted while C–C and C–O stretching frequencies of the complex were blue-shifted according to free HPT and GRP (see Fig. 4). Especially in complex, the significant increase in O–H frequency and decrease in C–C frequency shifts explains the high.
 |
| | Fig. 4 IR spectra of GRP, HPT, and HPT@GRP-complex. | |
Dipole moment and chemical stability descriptor (μ, ω, η, α)
The direction of electron flow in a molecule is described by the chemical potential (μ), while the electron gaining the ability of the molecule is found out by electrophilicity index (ω). A good electrophile has high μ, ω values, whereas in the case of nucleophile vice versa, the value of μ and ω. A softer molecule shows easy charge distribution while the hardness (η) of the molecule shows the symmetrical charge distribution.67 Chemical potential, electrophilicity index, softness and hardness, and dipole moment of the medication HPT, carrier, and complex are summarized in Table 1 as chemical stability descriptors. The potential chemical value of the HPT@GRP complex (μ = −3.94 eV) is lower than the drug (μ = 3.29 eV) and is like that of the GRP carrier (μ = −3.95 eV). Therefore, the complex is more stable than the drug. The hardness value of the HPT@GRP complex is 1.18 eV which is lower than GRP and HPT that have 1.28 eV and 2.26 eV, respectively. Whereas the softness value of the HPT@GRP complex is 0.59 eV and that of drug and carrier is 1.13 eV and 0.64 eV, respectively. The additional charge stability after interaction with the surrounding by gaining or losing of charge compound is determined by the electrophilicity index (ω) value. The dipole moment of the HPT@GRP complex in gas-phase and solvent-phase is 2.02 and 3.55 debye, respectively. The increase in dipole-moment of HPT@GRP complex in solvent phase compared to HPT drug (4.13D) and GRP carrier (0.47D) shows that the HPT drug will show facile movements in the biochemical system after the HPT@GRP complex formation. All stability-descriptors show that drug delivery systems based on HPT@GRP-complex can be used as an effective system for cancer treatment. The values of dipole moment and chemical stability descriptors in an aqueous phase are represented in ESI Table S1.†
CDA and DOS
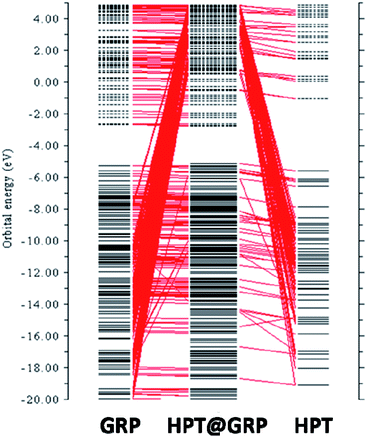
The complex's intermolecular charge transfer between HPT and GRP depends on the HOMO–LUMO energy gap.31 The addition of molecular energy levels in the HPT@GRP-complex MOs by the fragment orbital of HPT and GRP lower the energy gap and facilitate the charge transfer in fragments of the HPT@GRP complex. The orbital contributions from HPT and GRP for the complex formation were represented by plotting a CDA diagram of a complex molecule, as illustrated in Fig. 5. Consequently, the HOMO and LUMO energy is altered by −5.13 eV and −2.76 eV, respectively, resulting in new energy levels. The difference in energy between homo and LUMO has narrowed. As shown in Fig. 6, the peak-maximum of the DOS (density of state) diagram indicates that HPT is appropriately adsorbed on GRP (MOs of GRP and HPT are mixed) and develops new MOs of HPT@GRP complex, resulting in the reduction of the energy gap (2.37 eV) of HPT@GRP process. Furthermore, the DOS diagram reveals that the HPT@GRP complex molecule has gained additional molecular energy levels due to this addition. In this case, the DOS diagram is very consistent with the CDA diagram, showing that the HPT drug was successfully linked to the carrier GRP.
 |
| | Fig. 5 Charge-decomposition analysis of HPT@GRP complex. | |
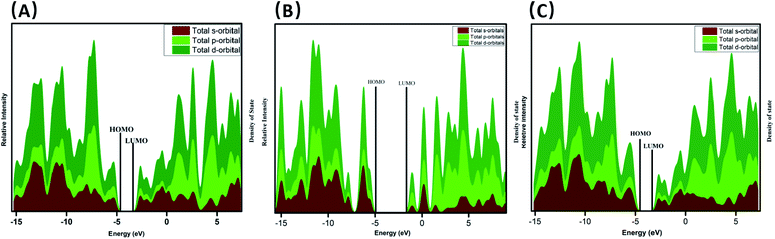 |
| | Fig. 6 DOS spectrum of HPT (A), GRP (B), and HPT@GRP complex (C). | |
Noncovalent interaction (NCI) analysis
The difference between weak-inter-molecular-interaction and strong localized inter-atomic attraction can be clearly distinguished from the NCI plot.68 The NCI graph observed the stiletto-heel shape, reducing density gradient(s) and electron density (r).
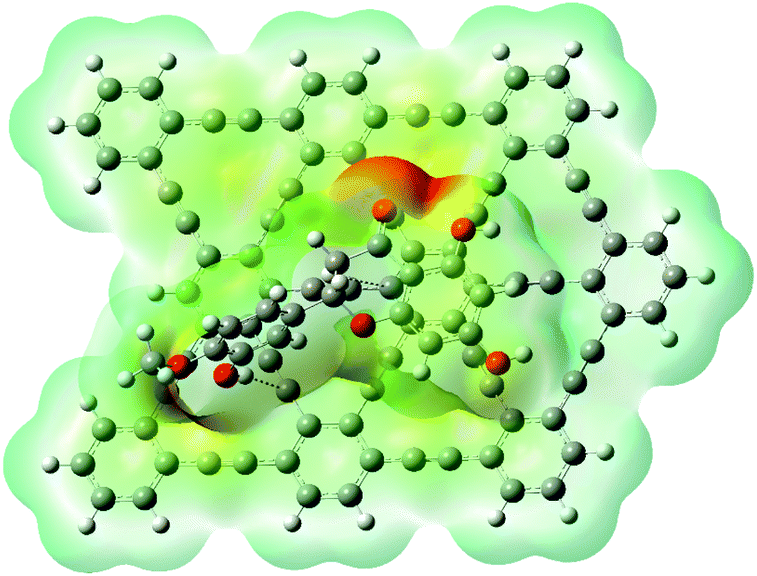
The RDG (s) are obtained via DFT calculation, which is the function of r and s. Different kinds of interactions on the r and s are explained as noncovalent, covalent, and non-interaction. Hydrogen bonding and repulsive force of the interaction is the function of λ2ρ. its values explained in the 2D NCI plot. The 3D colour-filled iso-surface image shows that the red colour is for steric hindrance, blue for hydrogen bonding, and the green represents attractive forces among the HPT and GRP. For an efficient drug transfer system, non covalent interactions between HPT and GRP are essential factors for offloading the drug at the target place. Geometric optimization analysis revealed weak interaction between GRP and HPT. These weak interactions are beneficial because they can quickly unload from the carrier when they approach the targeted site. A 2D noncovalent interaction scatter-graph of RDG against sign (λ2) ρ is plotted for HPT@GRP-complex give stiletto-heel-shape-graph as shown in Fig. 7. This graph explains that sign (λ2) ρ = 0, so there are weak van der Waals forces of interaction, sign (λ2) ρ 0.025 > 0 which indicates the steric-hindrance, and sign (λ2) ρ − 0.02 < 0 indicates that there is week hydrogen-bonding stuck between fragments of the complex, GRP and HPT. These noncovalent interactions are further explored in a 3D colour-filled iso-surface image, as shown in Fig. S5.† The green region among the fragments of the HPT@GRP complex. Represents weak intermolecular forces called van der Waals interactions. The blue colour represents the weak hydrogen bonding among the H-atom of the drug and the C-atom of GRP. The optimization-based geometric study also indicated some weak interaction between GRP and HPT. Due to these weak interactions and H-bonding, the drug molecule can be released quickly from GRP to the cancer site. The orbitals of GRP and HPT overlie in the HPT@GRP-complex, representing the capacity exchange that has appeared.
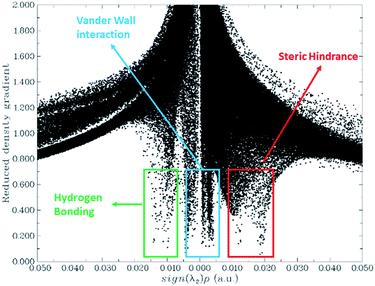 |
| | Fig. 7 Noncovalent interaction (NCI) analysis of HPT@GRP complex. | |
Photophysical properties
In the UV-visible range, the absorption wavelengths of HPT, GRP, and the HPT@GRP complex system measured in aqueous and gas phase is shown in the spectrum given in Fig. 8. The HPT@GRP-complex's maximum absorption λmax of the HPT@GRP-complex is 525 nm, substantially more significant than that of HPT, which is at 119 nm, resulting in the broad absorption pattern in the UV-visible range. The λmax at 525 nm corresponds to 2.37 eV of energy, which is compatible with the HOMO and LUMO energy gaps of the HPT@GRP complex. Absorption at this wavelength drives electron excitation from HOMO to LUMO. The lower the energy gap, the longer the wavelength of light absorbed (for example, Eg = hc/λ). The effect of the aqueous medium on UV-visible calculations was also deemed since the presence of a solvent in the system may induce differences in the excited state energies.
 |
| | Fig. 8 UV-visible absorption spectra of HPT drug, GRP, and HPT@GRP complex in gas-phase (A) and solvent phase (B). | |
Gas and solvent phase
The theoretical UV-visible absorption spectra of HPT, GRP, and HPT@GRP-complex in the solvent phase were computed to explore the effect of the solvent. The wavelengths of HPT, GRP, and its complex in the solvent phase are 200 nm, 520 nm, and 525 nm, respectively. In the UV-vis region, GRP has a maximum at 520 nm, whereas the HPT@GRP complex has a maximum of 525 nm (e.g., it is redshifted by 5 nm in the solvent phase). Excitation was observed at 525 nm, indicating that the charge delocalized from HPT to GRP. All of the changes were observable in the electromagnetic spectrum, revealing that this material might be a promising option for colourimetric and fluorescence monitoring, both of which help identify the particular target site. The ELF map, which shows the regions in molecular space where the electron-density (same spin as neighbour-electron) is superior, was created using covalent connections.69 ELF is denoted as τ(r) and is dependent on the kinetic energy density. The value of τ(r) varies between 0.0 and 1. Delocalization and localization of electrons may both be detected using this value. The areas with enormous ELF-values 0.5–1.0 have localized electrons in and out of bonds. A covalent link, nucleus-shell, or a lone pair in these locations may be responsible for the electron localization. The low ELF value (0.0–0.5) represents the weak van der Waals interactions and spaces between electron shells where delocalization occurs. The delocalization of the π-system in the HPT@GRP-complex molecule is interpreted using ELF analysis. Fig. S6(A and B)† show ELF electron-density pictures of GRP and the GRP-drug combination, respectively. Before establishing the complex, the ELF displays minor variations in electron density. After developing the HPT@GRP complex, the electron density of the GRP is modified. NBO analysis can help with intramolecular and intermolecular interactions by providing relevant information about exchanges within filled orbitals and virtual orbits. In the NBO study of the complexes, these ELF pictures help understand the transition from the drug to GRP and from GRP to medicine. The electron density of carbon atoms in GRP varies due to these transitions. Table S2† shows the changes in energy for internal shifts in GRP before and after the development of the complex.
Surface charge state
Whether neutral, harmful, or positive, charges on the surface play an essential role in regulating how biological substances (in the same way that proteins and plasma membranes exist) interact structurally. The effects of +1 and −1 charges on the bond distances of GRP (carrier) and its HPT@GRP complex are investigated. In the ESI,† geometrical structures with optimal parameters of GRP with +1 and −1 charge states (GRP+1/1) and their related complexes (HPT@GRP+1 and HPT@GRP−1 complex) are shown in Fig. 9 as ESI.† The number of bonds and their length established between the C–H and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C are about 1.06 and 1.21 based on bond distances (measurements) for GRP+1. While the C
C are about 1.06 and 1.21 based on bond distances (measurements) for GRP+1. While the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds sizes ranging from 1.39 to 1.43. Likewise, the bond-length study for GRP−1 revealed that the length of the bonds existing between the C–H and CC is around 1.08 and 1.22, respectively. While the CC bonds sizes ranging from 1.39 to 1.45.
C bonds sizes ranging from 1.39 to 1.43. Likewise, the bond-length study for GRP−1 revealed that the length of the bonds existing between the C–H and CC is around 1.08 and 1.22, respectively. While the CC bonds sizes ranging from 1.39 to 1.45.
 |
| | Fig. 9 Calculated bond length of (a) GRP+1 (b) HPT@GRP+1 complex (c) GRP−1 (d) and HPT@GRP−1 complex. | |
Following HPT adsorption on the surface of GRP−1/+1, the resulting complex of HPT@GRP−1/+1 had a small bond length dispersion w.r.t. GRP−1/+1 carrier, as shown in Fig. 9 shows the calculated bond lengths for GRP−1, HPT@GRP−1, GRP+1, and HPT@GRP+1 hydrogen bonding interaction between H–O and C shows the bond length range from 2.42–2.45 are shown by the optimized structure of theHPT@GRP+1-complex. The bond lengths (distances) between the CC field from 1.4 to 1.46. C–H, O–H, and CC have bond lengths of 1.09, 0.9, and 1.21, respectively. The C–C bonds are separated by 1.4 to 1.56 Å. The bond-lengths of OC are shown by the numbers that range from 1.37 to 1.45.
The hydrogen bonding interaction between H–O and C shows a bond length of 2.55 is shown by the optimized structure of the HPT@GRP−1-complex. The bond distances between C and C range from 1.39 to 1.54. C–H has bond distances ranging from 0.8 to 1.1. The lengths of CC bonds range from 1.39 to 1.5. The bond lengths of OC are 1.36 to 1.45. While the CC has a bond length of 1.21. The type of the bonds and the locations of the significant changes are revealed by charge distribution on the structure. As a result, the charge distribution assists us in identifying the active areas in the network for further reactions. The ADCH charges (atomic dipole-moment corrected-Hirshfeld charges) for HPT@GRP−1/+1 complexes were calculated and presented in Fig. S7.†
A positive charge occurs on the C and H atoms inHPT@GRP−1/+1-complexes, whereas the O atoms have a negative control. In HPT, the O atoms of the OH. group have negative charge values (approximately 0.36 a.u.). If the HPT@GRP−1/+1 complex is used, the extent of ADCH-charge values for other particles practically stays the same. In addition, the estimated adsorption energies for theHPT@GRP+1 complex and theHPT@GRP−1 complex are 1.57 eV and 1.62 eV, respectively. The adsorption energy estimates demonstrate that HPT adsorption on GRP in the +1 and −1 charge states results in stable complexes. The optimized structure of the HPT@GRP+1 complex and the HPT@GRP−1 complex are represented in ESI Fig. S8.†
Photoinduced electron transfer analysis (PET)
The biological activity of a drug carrier can be obtained by evaluating its electronic excitation. The molecular-orbital theory is the basis of the electron–hole theory.70 This electron–hole theory is helpful in determining the electron transfer mechanism and the contributions of the hole and electron orbitals in the singlet-excited state. Fig. 10 shows the electron and hole orbitals of the HPT@GRP complex for the first five excited states, with the data listed in Table 2. According to the findings, LUMO is still present on GRP, and HOMO is present on HPT medication. The HPT@GRP compound has a λmax at 625.3 nm with 0.0009 F in the aqueous phase.
 |
| | Fig. 10 Electron–hole orbitals and PET analysis for n-states = 1–5 of HPT@GRP-complex. | |
Table 2 Electron–hole analysis of 1–5 excited states of the carrier-drug complex
| Excited-state of the carrier-drug complex |
1 |
2 |
3 |
4 |
5 |
| λmax |
625 |
615 |
614 |
599 |
596 |
| ΔE (eV) |
1.983 |
2.014 |
2.019 |
2.067 |
2.079 |
| F |
0.0009 |
0.0067 |
0.0172 |
0.000 |
0.0121 |
| D (Å) |
5.702 |
0.803 |
0.361 |
4.263 |
0.759 |
| S |
0.07667 |
0.80703 |
0.80350 |
0.06179 |
0.76237 |
| Transition-mode |
CT |
CT |
CT |
CT |
CT |
Longer charge transfer times are caused by larger distances (D) between electron–hole orbitals, but shorter overlap integral (S) values between electron–hole orbitals result in faster charge transfer times. For the first five excited-states, in terms of electron–hole orbital distance, D bigger, i.e., 5.702 Å, 0.803 Å, 0.361 Å, 4.263 Å, and 0.759 Å, indicating a long charge transfer length. The orbitals overlap integral (S) is lower for the first five excited states (0.076, 0.807, 0.803, 0.061, and 0.762, respectively), suggesting a rapid charge transfer rate. Because the hole and electron orbitals are well separated, the PET (photoinduced electron transfer) phenomena occur between complicated components. In the excited state at 517 nm, three electrons move from the ground state to LUMO+1. For the third excited state, a transfer of two electrons from the ground state to LUMO+1 takes place at a wavelength of 508 nm. For the 4th and 5th excited states, there is just between the ground state and LUMO+0, one electronic transition, with maximum wavelengths of 470 nm and 456 nm. Fluorescence reduction in the emission spectrum is caused by the PCT (photoinduced charge transfer) and PET processes. This quenching of the biological system reveals useful information.
Because the charge transferring process happens between the pieces of complex HPT@GRP-complex and charge is transferred from HPT (chelator) to carrier GRP, suppression between orbitals occurs in the fluorescence-emission spectrum, according to this research (fluorophore). The molecule and position of the drug in the biological system may be determined using the repressed fluorescence intensity.
Conclusion
In this theoretical work, the adsorption behaviour of HPT on the functionalized GRP is investigated using the DFT method. For this purpose, topological features of electron density, the structural properties, and adsorption energies for the interaction of HPT with the GRP are evaluated in gas and aqueous phases. NCI calculations show that the stability of the complex is usually governed by the development of the intermolecular hydrogen bonds and the electrostatic interactions between GRP and hydroxyl groups of HPT. HPT drugs at the targeted area will quickly offload as weak intermolecular forces exist between HPT and GRP. The adsorption energy value of the HPT@GRP complexes has −0.205 eV and −0.0877 eV in the gas and solvent phases, respectively. The negative values of Ead predicted that adsorption of HPT on GRP is favourable and HPT@GRP complex is stable. The dipole-moment of the HPT@GRP complex is increased as compared to GRP, and HPT specifies the increase in the HPT@GRP-complex hydrophilicity in water (polar solvents), which is helpful in the easy movement of the drug in the biological systems. Frontier molecular orbital analysis explains the charge transfer from HPT to GRP during the complex construction process. The CDA and DOS provide even more assistance with the charge transfer method.
Furthermore, the λmax of the HPT@GRP-complex is redshifted by 9 nm from GRP in the gas phase. The λmax value is also redshifted in the aqueous phase. The PET procedure and its impacts on the HPT@GRP complex's fluorescence phenomena were analyzed using electron–hole theory. The GRP+1/−1 structure is insignificant, and stable complexes with HPT are formed when the two molecules are combined. The results showed that GRP as a medication carrier for HPT in cancer treatment has excellent chemotherapeutic potential. It is hoped that this theoretical work may pique researchers' interest in exploring the potential of alternative 2D nanomaterials for medication delivery. Overall, theoretical data such as enhanced solubility, good complex stability, reduced bandgap, weak van der Waals forces between drug and carrier, and charge transfer mechanism indicate that the HPT@GRP complex is the ideal drug delivery strategy for cancer treatment. In conclusion, GRP has tremendous therapeutic promise as a vehicle or carrier for HPT medication delivery in cancer treatment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21471017).
Notes and references
- H. Ebrahimi, Z. Aryan, S. S. Moghaddam, C. Bisignano, S. Rezaei, F. Pishgar, L. M. Force, H. Abolhassani, E. Abu-Gharbieh and S. M. Advani, Lancet Respir. Med., 2021, 9, 1030–1049 CrossRef.
- E. L. Etter, K.-C. Mei and J. Nguyen, Adv. Drug Delivery Rev., 2021, 113994 CrossRef CAS PubMed.
- M. A. Khan, S. Rubab, A. Kashif, M. I. Sharif, N. Muhammad, J. H. Shah, Y.-D. Zhang and S. C. Satapathy, Pattern Recognit. Lett., 2020, 129, 77–85 CrossRef.
- V. Juvekar, C. S. Lim, D. J. Lee, D. H. Song, C.-K. Noh, H. Kang, S. J. Shin and H. M. Kim, ACS Appl. Bio Mater., 2021, 4, 2135–2141 CrossRef CAS PubMed.
- L. H. Biller and D. Schrag, JAMA, J. Am. Med. Assoc., 2021, 325, 669–685 CrossRef CAS PubMed.
- Y. Zhai, J. Wang, T. Lang, Y. Kong, R. Rong, Y. Cai, W. Ran, F. Xiong, C. Zheng and Y. Wang, Nat. Nanotechnol., 2021, 1–10 CAS.
- Z. Zhang, F. Cui, C. Cao, Q. Wang and Q. Zou, Comput. Biol. Med., 2022, 140, 105092 CrossRef CAS PubMed.
- G. E. Wood, H. Hockings, D. M. Hilton and S. Kermorgant, Oncogene, 2021, 40, 1927–1941 CrossRef CAS PubMed.
- S. Yan, J. Yan, D. Liu, X. Li, Q. Kang, W. You, J. Zhang, L. Wang, Z. Tian and W. Lu, Theranostics, 2021, 11, 6833 CrossRef CAS PubMed.
- M. H. Valstar, B. S. de Bakker, R. J. Steenbakkers, K. H. de Jong, L. A. Smit, T. J. K. Nulent, R. J. van Es, I. Hofland, B. de Keizer and B. Jasperse, Radiother. Oncol., 2021, 154, 292–298 CrossRef CAS PubMed.
- L. Wyld, R. A. Audisio and G. J. Poston, Nat. Rev. Clin. Oncol., 2015, 12, 115–124 CrossRef PubMed.
- Y. Xiong, C. Xiao, Z. Li and X. Yang, Chem. Soc. Rev., 2021, 50, 6013–6041 RSC.
- Y. Yang, M. Xu, Z. Wang, Y. Yang, J. Liu, Q. Hu, L. Li and W. Huang, J. Mater. Chem. B, 2021, 9, 2613–2622 RSC.
- M. A. Essawy, R. M. Abohadida, W. M. Abd-Elkader, H. M. Fathy and H. M. Hassab, Complement Ther. Med., 2021, 102730 CrossRef PubMed.
- C. Grant, B. Loman, M. Bailey and L. Pyter, Brain, Behav., Immun., 2021, 95, 401–412 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nano-Enabled Medical Applications, 2020, pp. 61–91 Search PubMed.
- I. Gaurav, A. Thakur, A. Iyaswamy, X. Wang, X. Chen and Z. Yang, Molecules, 2021, 26, 1544 CrossRef CAS PubMed.
- S. Chen, Z. Song and R. Feng, Anti-Cancer Agents Med. Chem., 2020, 20, 2169–2189 CrossRef CAS PubMed.
- K. Vinothini and M. Rajan, in Characterization and Biology of Nanomaterials for Drug Delivery, Elsevier, 2019, pp. 219–263 Search PubMed.
- M. I. Alam, S. Beg, A. Samad, S. Baboota, K. Kohli, J. Ali, A. Ahuja and M. Akbar, Eur. J. Pharm. Sci., 2010, 40, 385–403 CrossRef CAS PubMed.
- D. J. Prince, D. Patel and S. C. Kachlany, Sci. Rep., 2021, 11, 1–14 CrossRef PubMed.
- M. Mirzaei and H. Nazemi, Biointerface Res. Appl. Chem., 2022, 12, 3752–3761 Search PubMed.
- A. Kakaei and M. Mirzaei, Lab-in-Silico, 2021, 2, 9–14 Search PubMed.
- C. M. Weekley, J. B. Aitken, I. F. Musgrave and H. H. Harris, Biochemistry, 2012, 51, 736–738 CrossRef CAS PubMed.
- R. Hu, Z. Chen, C. Dai, X. Guo, W. Feng, Z. Liu, H. Lin, Y. Chen and R. Wu, Biomaterials, 2021, 269, 120455 CrossRef CAS PubMed.
- J. Ouyang, C. Feng, X. Ji, L. Li, H. K. Gutti, N. Y. Kim, D. Artzi, A. Xie, N. Kong and Y. N. Liu, Angew. Chem., 2019, 131, 13539–13544 CrossRef.
- C. Sweet, A. Pramanik, S. Jones and P. C. Ray, ACS Omega, 2017, 2, 1826–1835 CrossRef CAS PubMed.
- Y. Cheng and H. Zhang, Chem.–Eur. J., 2018, 24, 17405–17418 CrossRef CAS PubMed.
- X. Liu, Y. Zhou, W. Xie, S. Liu, Q. Zhao and W. Huang, Small Methods, 2020, 4, 2000566 CrossRef CAS.
- M. Emanet, Ö. Şen and M. Çulha, Nanomedicine, 2017, 12, 797–810 CrossRef CAS PubMed.
- I. Munir, M. Perveen, S. Nazir, R. A. Khera, A. R. Ayub, K. Ayub and J. Iqbal, J. Mol. Liq., 2021, 336, 116327 CrossRef CAS.
- M. Mirzaei and M. Yousefi, Superlattices Microstruct., 2012, 52, 612–617 CrossRef CAS.
- E. Moezi and M. Mirzaei, Lab-in-Silico, 2021, 2, 25–29 Search PubMed.
- A. Mokhtari, K. Harismah and M. Mirzaei, Superlattices Microstruct., 2015, 88, 56–61 CrossRef CAS.
- R. Majidi and M. Nadafan, Phys. Lett. A, 2020, 384, 126036 CrossRef CAS.
- M. Shahabi and H. Raissi, J. Mol. Liq., 2020, 319, 114334 CrossRef CAS.
- S. Madhumitha, V. Nagarajan and R. Chandiramouli, Comput. Theor. Chem., 2019, 1163, 112514 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS PubMed.
- H. Hashemzadeh and H. Raissi, Appl. Surf. Sci., 2020, 500, 144220 CrossRef CAS.
- K. A. Spandana, M. Bhaskaran, V. R. Karri and J. Natarajan, J. Drug Delivery Sci. Technol., 2020, 57, 101628 CrossRef.
- M. Sherafati, A. S. Rad, M. Ardjmand, A. Heydarinasab, M. Peyravi and M. Mirzaei, Curr. Appl. Phys., 2018, 18, 1059–1065 CrossRef.
- H. Zandi and K. Harismah, Lab-in-Silico, 2021, 2, 50–54 Search PubMed.
- S. U. Tekale, S. S. Kauthale, R. D. Ingle, S. B. Ubale, S. U. Deshmukh, K. Ameta and R. P. Pawar, Natural Heterocycles: Extraction and Biological Activity, Nova Science Publishers, Inc., 2015, p. 165, ISBN: 978-1-63463-424-3 Search PubMed.
- U. N. Wanasundara and F. Shahidi, Food Chem., 1994, 50, 393–396 CrossRef CAS.
- S. Tommasini, M. Calabro, R. Stancanelli, P. Donato, C. Costa, S. Catania, V. Villari, P. Ficarra and R. Ficarra, J. Pharm. Biomed. Anal., 2005, 39, 572–580 CrossRef CAS PubMed.
- N. V. Shah, A. K. Seth, R. Balaraman, C. J. Aundhia, R. A. Maheshwari and G. R. Parmar, J. Adv. Res., 2016, 7, 423–434 CrossRef CAS PubMed.
- R. Sundararaman, K. Letchworth-Weaver, K. A. Schwarz, D. Gunceler, Y. Ozhabes and T. Arias, SoftwareX, 2017, 6, 278–284 CrossRef PubMed.
- I. Erlund, Nutr. Res., 2004, 24, 851–874 CrossRef CAS.
- C. F. Matta, Struct. Chem., 2017, 28, 1591–1597 CrossRef CAS.
- E. Torres and G. A. DiLabio, J. Phys. Chem. Lett., 2012, 3, 1738–1744 CrossRef CAS PubMed.
- Y. Yang and H. Gao, Spectrochim. Acta, Part A, 2012, 85, 303–309 CrossRef CAS PubMed.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem. A, 2001, 105, 8111–8116 CrossRef CAS.
- S. Ghammamy, M. Shahsavary and A. Lashgari, Curr. Res. Chem., 2014, 6, 16–21 CrossRef CAS.
- J. Wang, S. Zhang, J. Zhou, R. Liu, R. Du, H. Xu, Z. Liu, J. Zhang and Z. Liu, Phys. Chem. Chem. Phys., 2014, 16, 11303–11309 RSC.
- M. Shamim, M. Perveen, S. Nazir, M. Hussnain, R. Mehmood, M. I. Khan and J. Iqbal, J. Mol. Liq., 2021, 331, 115607 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- M. Mirzaei, O. Gülseren and N. Hadipour, Comput. Theor. Chem., 2016, 1090, 67–73 CrossRef CAS.
- A. Kouchaki, O. Gülseren, N. Hadipour and M. Mirzaei, Phys. Lett. A, 2016, 380, 2160–2166 CrossRef CAS.
- R. Yankova, S. Genieva, N. Halachev and G. Dimitrova, J. Mol. Struct., 2016, 1106, 82–88 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- E. J. Braga, B. T. Corpe, M. M. Marinho and E. S. Marinho, Int. J. Appl. Sci. Eng. Res., 2016, 7, 315–319 Search PubMed.
- S. Moro, M. Bacilieri, B. Cacciari and G. Spalluto, J. Med. Chem., 2005, 48, 5698–5704 CrossRef CAS PubMed.
- Y. Xie, H. Cui, H. Wu, R. B. Lin, W. Zhou and B. Chen, Angew. Chem., 2021, 133, 9690–9695 CrossRef.
- S. Barusrux, Appl. Sci., 2020, 10(11), 3766 CrossRef.
- A. S. Rad, M. Ardjmand, M. R. Esfahani and B. Khodashenas, Spectrochim. Acta, Part A, 2021, 247, 119082 CrossRef CAS PubMed.
- M. Kamel and K. Mohammadifard, Chem. Rev. Lett., 2021, 4, 54–65 CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
- M. Lin, D. Wang, S. Liu, D. Zhou, H. Zhang, C. Liu and H. Sun, Acta Polym. Sin., 2015, 2, 133–146 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural information, spectra (infrared spectra, UV-visible), HOMO–LUMO diagram, MEP diagram of HPT@GRP-complex, iso-surface image of HPT@GRP complex, electron-density plots, ADCH charges for HPT@GRP−1/+1 complexes, vertical ionization energy, vertical electron affinity and, dipole moment in the aqueous phase, electron-localization function. See DOI: 10.1039/d1ra09112c |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Mubashar Ilyasb,
Ali Raza Ayuba,
Yanhong Zhua and
Hui Li
b,
Mubashar Ilyasb,
Ali Raza Ayuba,
Yanhong Zhua and
Hui Li