DOI:
10.1039/D1RA09318E
(Paper)
RSC Adv., 2022,
12, 11974-11991
Novel adamantyl clubbed iminothiazolidinones as promising elastase inhibitors: design, synthesis, molecular docking, ADMET and DFT studies†
Received
29th December 2021
, Accepted 24th March 2022
First published on 19th April 2022
Abstract
Porcine Pancreatic Elastase (PPE) is a serine protease that is homologous to trypsin and chymotrypsin that are involved in various pathologies like inflammatory disease, Chronic Obstructive Pulmonary Disease (COPD), acute respiratory distress syndrome, cystic fibrosis, and atherosclerosis. PPE if remained uninhibited would lead to digestion of important connective tissue. We developed new structurally diverse series of adamantyl-iminothiazolidinone hybrids to divulge elastase inhibition assay. To identify potent derivatives, in silico screening was conducted and in vitro studies disclosed that the compounds 5a, 5f, 5g, and 5h showed excellent binding energies and low IC50 values. In silico studies including molecular docking, DFT studies (using the B3LYP/SVP basis set in the gas phase) drug likeness scores and molecular dynamic simulation studies were conducted to evaluate protein–ligand interactions and to determine the stability of top ranked conformation. In silico studies further supported the results of in vitro experiments and suggest these derivatives as novel inhibitors of elastase enzyme.
Introduction
Elastase is a pancreatic enzyme that is resistant to degradation in the gastrointestinal tract. It is made by pancreatic acinar cells. Its measurement in faeces has been widely used to determine pancreatic exocrine adequacy because to its simplicity; nonetheless, it is insensitive to mild to moderate pancreatic insufficiency.1,2 It is initially synthesized as an inactive zymogen before being activated by trypsin in the duodenum. Reduced faecal elastase concentrations have been seen in various gastrointestinal illnesses (e.g., duodenal enteropathy), in circumstances where elastase is destroyed by bacteria (e.g., inflammatory bowel disease), and as a consequence of diarrhea's dilution effect.3 Because the test looks for human elastase, it does not react with the pancreatic enzyme replacement drug derived from swine that is used to treat cystic fibrosis. Elastin is also present in the skin's bottom layer, alongside collagen, as well as nerves, blood arteries, lymph nodes, and muscles.4 Collagen is a fundamental component of the extracellular matrix (ECM) and has a direct impact on the skin. The preservation of collagen structure is linked to the intrinsic and photoaging processes of the skin.5,6 Elastase are serine proteases that hydrolyze the amides and esters present in many proteins, including elastin. Elastic fibers are composed of elastin, a protein found in the majority of connective tissues and tendons. Because elastase destroys elastic fibers and leads to skin inflammation and other skin-related issues, decreasing elastase activity may be an effective method of avoiding skin ageing.7 Adamantyl moiety is a non-polar cage structure that provides lipophilic character to the molecule and improves the barrier crossing ability of a molecule to the biological system. In most cases, the adamantyl group tunes the hydrophobicity of the molecule by enhancing the penetrating ability and therapeutic activity of several trial compounds through various mechanisms.8–10 The steric factor of the adamantyl group promotes the stability and pharmacologic half-life of compounds. From the last few decades, adamantyl appending compounds as potential pharmacophore, attracted the attention of several synthetic chemists and pharmacists.11 As a result, adamantyl analogs passed from the intensive biological investigation.12 To date, several marketed drugs incorporating with adamantyl scaffold are now available. Adamantyl-containing compounds exhibit a broad spectrum of biological activities such as antiviral, anti-HIV, antifungal, anti-diabetic, antibacterial,13 anti-inflammatory,14 antimicrobial, hypoglycemic activity, treatment of COVID-19,15,16 and treatment for neurological disease.17,18
Acyl thioureas signify a class of pharmaceutically important compounds to develop the architect of a potential drug. These molecules have been explored for many pharmaceutical activities.19–21 and in agriculture as an insecticide and weed killer. In addition, they are the precursor for the synthesis of a number of heterocyclic compounds and metal complexes22 Acyl thiourea have also been applied as ion sensors and for the estimation of metal ions.23–25 These electron-rich molecules act as ligand and organocatalysts in various asymmetric synthesis26–28
Heterocyclic skeletons containing five-member rings with two heteroatoms are promising core structures, commonly encountered in organic and medicinal chemistry. Thiazolidinone motifs are biologically significant five-membered heterocycle that is very common substructures in various pharmacological active molecules and known to exhibit a wide range of biological activities such as anticancer, antifungal, anti-inflammatory, antimicrobial, antidiabetic, inhibiting neuraminidase of influenza virus, and anti-HIV,29 antischistosomal activity. Among these are inhibitors for necroptosis, selective GSK-3β inhibitor, anticancer activity,30 aldose reductase inhibitor,31 potent antiproliferative agent and inhibitor for non-membrane protein tyrosine phosphate. The aforesaid facts inspired us to synthesize novel ethyl 3-adamantanyl-2-((2-methyl benzoyl)imino)-4-oxothiazolidin-5-ylidene acetates (5a–j) for the appraisal of elastase inhibition assay. The structures of some pharmacologically active compounds and designed molecules are shown in Fig. 1.
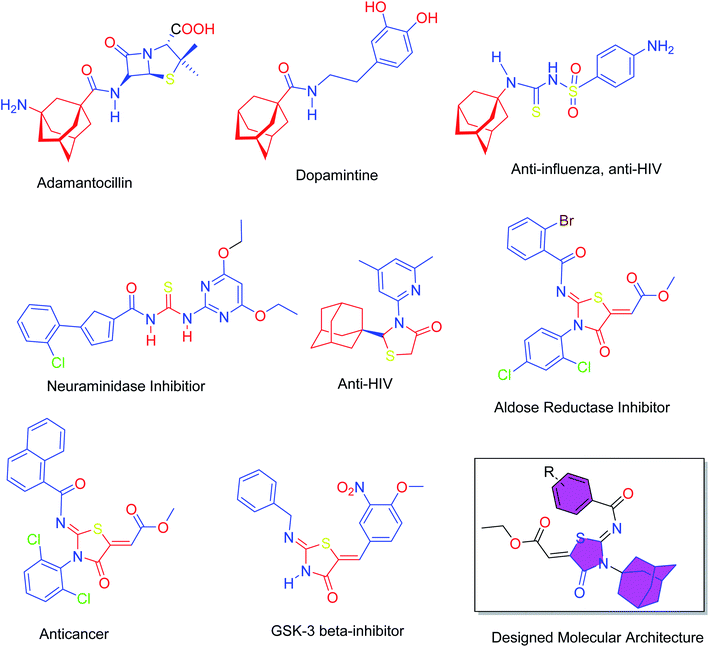 |
| | Fig. 1 Pharmacological active compounds containing adamantyl, thiourea, thiazolidinone moieties and the designed molecules. | |
Results and discussion
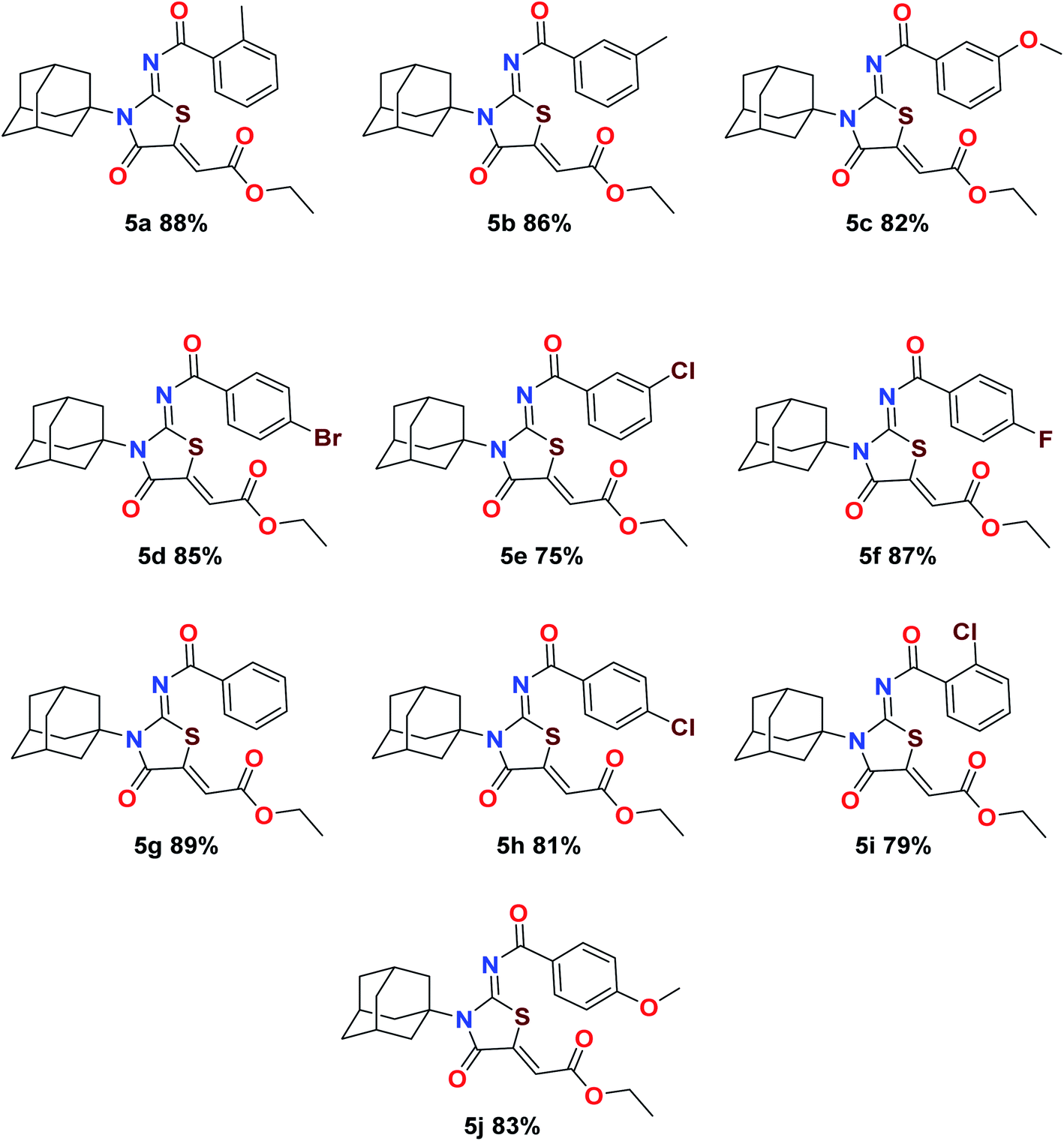
The target compounds were synthesized according to synthetic route described in Scheme 1. Suitably substituted benzoic acids were converted to corresponding acid chlorides (1a–j) using dichloromethane as solvent. The acid chloride was converted to isothiocyanate by adding potassium thiocyanate in acetone. Reaction of the latter in situ with an equimolar amount of amantadine (2) afforded the acyl thioureas (3a–j). Ethyl 4-ethoxypent-4-en-2-ynoate with (3a–j) at room temperature in dry ethanol furnished the desired compounds (5a–j) in excellent yields and high purity. The complete structures of synthesized derivatives are given in (Fig. 2).
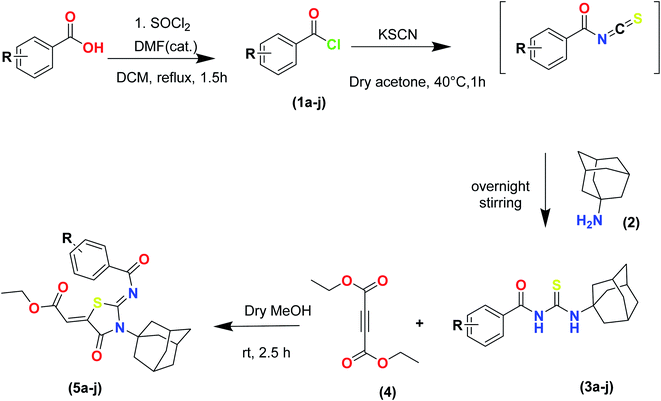 |
| | Scheme 1 Synthesis of ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((2-methylbenzoyl)imino)-4-oxothiazolidin-5-ylidene)acetates (5a–j). | |
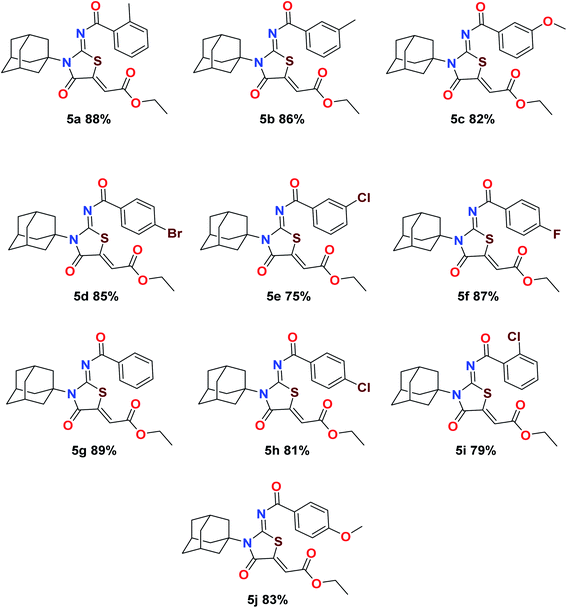 |
| | Fig. 2 Molecular structures and percent yields of final compounds (5a–j). | |
Structure–activity relationship (SAR) of iminothiazolidinones derivatives
The important SAR features can be deduced from this study. The inhibitory potential of the synthesized amantadine derivatives was investigated using elastase from porcine pancreas. The relationships between different substituted and unsubstituted patterns were analyzed on their ability to inhibit elastase. Such type of analysis assists to infer the SAR among the synthesized screened derivatives. Oleanolic acid is a pentacyclic triterpenoid and has been used as stander. In the current study, amantadine thiazolidinone derivatives were synthesized to find out more effective and selective antagonists of elastase. Among the synthesized analogues, compounds 5a, 5f, 5g, and 5h were showed high potency against elastase. The presence of unsubstituted phenyl group in 5g, provide effectiveness to develop high potency against elastase with IC50 of 0.124 ± 0.022 μM (Table 1) which is 50 times more potent as compared to oleanolic acid value of 5.996 ± 0.882 μM. With respect to structural comparison compounds with a methyl 5a, flouro 5f, chloro 5h, and bromo 5d substituted phenyl group exhibit excellent inhibitory activity. This is due to positive mesomeric effect of these substituents. Interestingly the compound 5g doesn't have any substitution on benzoylimino group of a compound, but it showed excellent IC50 value and good binding energy. It can be perceived that absence of mesomeric effect might increase activity against PPE enzyme.
Table 1 Elastase (elastase from porcine pancreas) inhibitory activity of derivatives (5a–j)a
| Compound |
Elastase IC50 + SEM (μM) |
Compound |
Elastase IC50 + SEM (μM) |
| SEM = standard error of the mean; values are expressed in mean ± SEM. |
| 5a |
0.854 ± 0.241 |
5f |
0.384 ± 0.624 |
| 5b |
0.982 ± 0.286 |
5g |
0.124 ± 0.022 |
| 5c |
0.738 ± 0.117 |
5h |
0.221 ± 0.059 |
| 5d |
0.597 ± 0.087 |
5i |
1.791 ± 0.213 |
| 5e |
1.961 ± 0.336 |
5j |
3.257 ± 0.541 |
| Oleanolic acid |
5.996 ± 0.882 |
Kinetic mechanism
An inhibition kinetic study was carried out in order to better understand the mode of action of the synthetic compound against elastase. Based on our IC50 results, we choose our most potent compound 5g for further investigation to determine the type of inhibition and the constant of inhibition. 1/V versus substrate N-succinyl-Ala-Ala-p-nitroanilide 1/[S] in the presence of different inhibitor concentrations was plotted using a Lineweaver–Burk plot in which the results were a series of straight lines; however, the result of a Lineweaver–Burk plot of compound 5g revealed that Vmax remained constant without significantly affecting the slopes. While Km increases with increasing concentration, Vmax remains constant with only a statistically insignificant difference. A competitive inhibition model was used to explain this behavior (Fig. 3A), and a second plot (Fig. 3B) of slope against concentration of 5g revealed the EI dissociation constant. The inhibitor concentration of 5g was plotted against the slope of the graph, and the calculated Ki value was found to be 0.6 μM.
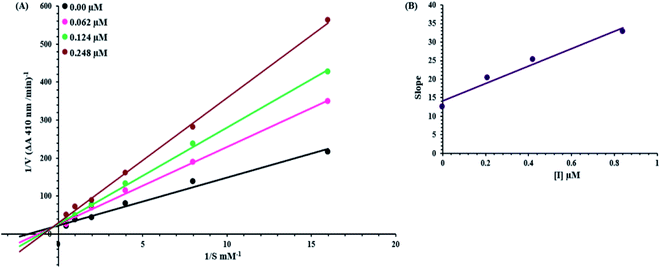 |
| | Fig. 3 Lineweaver–Burk plots for inhibition of elastase from porcine pancreas in the presence of compound 5g (A) concentrations of 5g were 0.00, 0.062, 0.124 and 0.248 μM, substrate N-succinyl-Ala-Ala-Ala-p-nitroanilide concentrations were 2, 1, 0.5, 0.25, 0.125 and 0.0625 mM. (B) The insets represent the plot of the slope. | |
Computational studies
Quantum chemistry through density functional theory calculation. All the selected molecules (5a–j) were optimized firstly, using the B3LYP/SVP basis set in the gas phase, and their values are shown in Table 2.
Table 2 Geometric parameters of the compounds (5a–j)
| S. no. |
Compound |
Gas phase |
| Optimization energy (hartree) |
Polarizability (α) (a.u.) |
Dipole moment (Debye) |
| 1 |
5a |
−1747.578754 |
175.593333 |
2.632463 |
| 2 |
5b |
−1756.398850 |
215.067333 |
2.151600 |
| 3 |
5c |
−1821.412424 |
174.135000 |
1.454307 |
| 4 |
5d |
−4262.687518 |
309.789333 |
1.774701 |
| 5 |
5e |
−2162.996044 |
170.469000 |
1.848393 |
| 6 |
5f |
−1806.453269 |
166.362333 |
1.037827 |
| 7 |
5g |
−1717.551017 |
201.042667 |
1.104426 |
| 8 |
5h |
−2163.003741 |
172.960333 |
2.863819 |
| 9 |
5i |
−2163.010784 |
173.656000 |
2.127731 |
| 10 |
5j |
−1843.417542 |
311.708667 |
4.266993 |
The geometries of selected compounds were optimized to lowest energy gradient and no imaginary frequencies were observed which demonstrate that all structures were true local minima. The optimized structures of selected compounds are shown in Fig. 4.
 |
| | Fig. 4 Optimized structures of the compounds (5a–j). | |
The molecular orbital (MO) analysis is of prime importance in quantum chemistry. It has been broadly defining chemical behavior. The highest occupied molecular orbital (HOMO) and lower unoccupied molecular orbital (LUMO) are the chief molecular orbitals in a compound. They are used to explain chemical properties such as reactivity, stability, and kinetics. The FMO orbitals of synthesized compounds are shown in Fig. 5.
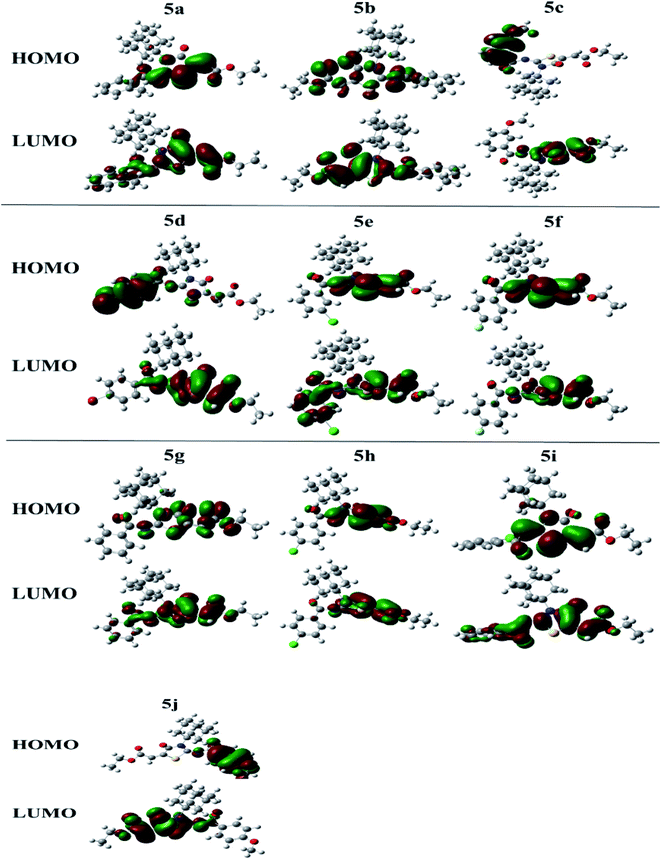 |
| | Fig. 5 FMOs of the compounds (5a–j). | |
The hardness (η) defined the extent to which a molecule is hard or soft. The softer the molecule, the better will be the reactivity.
Electronegativity (X) characterizes the power to attract electrons. All these properties were estimated at. The compound 5c showing smallest HOMO–LUMO energy gap value (0.064 eV) predicts a good chemical reactivity. The compound 5j is showing a small hardness value (0.071), high polarizability thus leading to be the soft molecule among all compounds. The higher the electronegativity value in the case of compound 5d showing (0.173) value exposes that 5d has good electron attracting power and acts as a better electrophile among all compounds. While compounds 5f and 5a also showed good reactivity after 5c, with the energy gap values 0.067 eV and 0.069 eV, respectively. In the same manner, just after 5j the compounds 5b, 5g and 5d showing high polarizability with the values of 0.072, 0.076 and 0.078, respectively. Energetic parameters of compounds (5a–j) are showing in Table 3 below.
Table 3 Energetic parameters of the compounds (5a–j)
| Compound |
EHOMO (eV) |
ELUMO (eV) |
ΔEgap (eV) |
Hardness (η) |
Softness (S) |
Electronegativity (X) |
| 5a |
−0.25326 |
0.18451 |
0.069 |
0.219 |
2.28 |
0.034 |
| 5b |
−0.13569 |
0.00824 |
0.127 |
0.072 |
6.95 |
0.064 |
| 5c |
−0.25591 |
0.19175 |
0.064 |
0.224 |
2.23 |
0.032 |
| 5d |
−0.25186 |
−0.09513 |
0.157 |
0.078 |
6.38 |
0.173 |
| 5e |
−0.26176 |
0.18998 |
0.072 |
0.226 |
2.21 |
0.036 |
| 5f |
−0.25943 |
0.19229 |
0.067 |
0.226 |
2.21 |
0.034 |
| 5g |
−0.13817 |
0.01304 |
0.125 |
0.076 |
6.61 |
0.063 |
| 5h |
−0.26359 |
0.18141 |
0.082 |
0.223 |
2.25 |
0.041 |
| 5i |
−0.26101 |
0.18360 |
0.077 |
0.222 |
2.25 |
0.039 |
| 5j |
−0.23092 |
−0.08884 |
0.142 |
0.071 |
7.04 |
0.160 |
Molecular docking studies. Porcine Pancreatic Elastase (PPE) is a serine protease that is homologous to trypsin and chymotrypsin. It is composed of single polypeptide chain of 240 amino acids that are linked with four disulphide linkages. Moreover, it is complexed with Ca2+ atom which is necessary for its stability.32 PPE consist of four subsites, among these most of inhibitors bind to S1 and S2 active sites. S1 site is consider as a primary specificity active site (Fig. 6). PPE plays pivotal role in onset and progression of inflammatory disease like cancer and COPD.33 PPE if remained uncontrolled would lead to digestion of important connective tissue. Keeping in view the importance of PPE, in silico studies were conducted to support the result of in vitro assays. Crystallographic structure of PPE was downloaded from Protein data bank. (PDB ID: 1BMA; resolution 1.99). The important amino acids residues of catalytic site were selected and docked with potent thiazolidinone derivatives. The important amino acid residues of PPE protein are as follows; THR221, SER203, HIS60, SER222, PHE223, VAL224, SER225, TRP179, CYS199, GLY201, GLY198, THR152, LEU227, CYS229, ALA104 THR152, GLY228, ARG226, VAL103, SER203, HIS60, and GLN200. Synthesized derivatives were selected on the basis of IC50 values obtained during in vitro assays. Based on IC50 value, 5a, 5f, 5g and 5h was selected for in silico studies as these derivatives showed excellent inhibitory potential during in vitro assays.
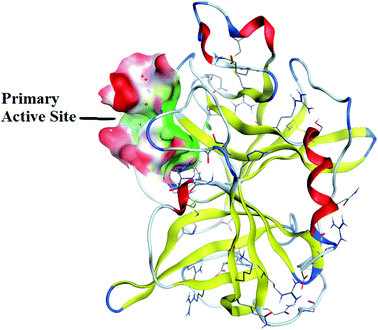 |
| | Fig. 6 Crystallographic structure of porcine pancreatic elastase (PPE). | |
Synthesized derivatives were evaluated for binding affinities and binding scores with amino acid residues of active pocket. Binding affinities were determined on the basis of predicted inhibitory constant value (ki). Most of the compounds exhibited potent binding scores and demonstrated good binding affinities. The best conformation of protein–ligand was selected and analyzed further for bonding and non-bonding interactions. Docking scores of potent compounds with their IC50 values are given in Table 4.
Table 4 Docking score of potent derivatives
| Code |
Docking score (kJ mol−1) |
IC50 + SEM (μM) |
Predicted inhibitory constant (ki) (μM) |
| 5a |
−24.68 |
0.854 ± 0.241 |
2.7 |
| 5f |
−23.61 |
0.384 ± 0.624 |
1.95 |
| 5g |
−23.56 |
0.124 ± 0.022 |
1.94 |
| 5h |
−28.82 |
0.221 ± 0.059 |
1.63 |
| Co-crystal ligand benzyl methyl aminimide |
−28.03 |
— |
5.9 |
The detailed 3D and 2D binding interactions of derivative 5a within active pocket of PPE enzyme is shown in Fig. 7. The amino acid residues involved in bonding and non-bonding interactions with 5a were THR152, LEU227, CYS229, GLY228, SER225, ARG226, TRP179, VAL103, PHE223, VAL224, SER203, HIS60, SER222 and GLN200. Briefly substituted benzoyl imino ring of parent compound was making strong pi-sulfur linkage with C229. Compound 5a showed good binding energy and IC50 value which might be due to substitution of methyl group on benzoylimino ring. It was observed that compound 5a was producing hydrogen bonding of moderate strength with GLN200 with bond length of 3.1 Å. Presence of methyl group induce positive mesomeric effect by donating electrons. Moreover, acetate group of molecule was making alkyl and pi-alkyl interactions with PHE223 and VAL103. Amantadine ring was making van der Waals interactions with VAL222 and SER225.
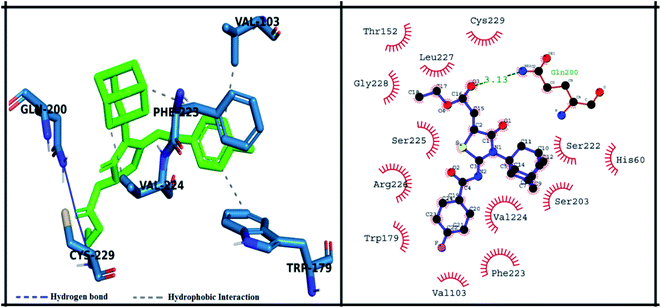 |
| | Fig. 7 Putative 2D and 3D binding mode of compound 5a (green colored) within active pocket of elastase enzyme (amino acid residues in blue color). | |
The detailed 3D and 2D binding interactions of derivative 5f within active pocket of PPE enzyme is shown in Fig. 8. The amino acid residues involved in bonding and non-bonding interactions with 5f TRP179, SER225, VAL224, PHE223, GLN200, CYS199, SER203, SER222 and ARG226. Briefly flouro substituted benzoylimino ring of parent compound was making strong π–π stacked interaction with PHE223. Compound 5f showed good binding energy and IC50 value which might be due to substitution of fluorine on benzoyl imino ring. Fluorine induce positive mesomeric effect by donating electrons. Moreover, amantadine ring was making alkyl interaction with VAL103. In addition, single hydrogen-acceptor bonding was observed with ARG226 with bond length of 2.9 Å. The van der Waals interactions were exhibited by oxothiazolidinylidene ring with TRP179 and CYS199.
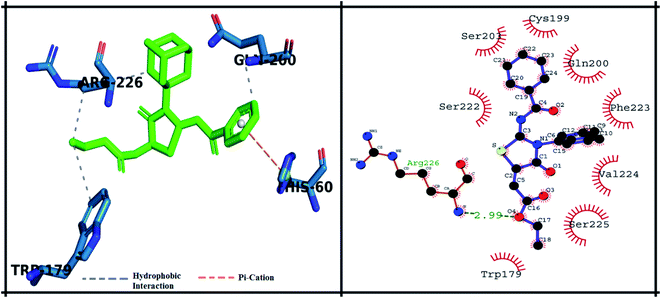 |
| | Fig. 8 Putative 2D and 3D binding mode of compound 5f (green colored) within active pocket of elastase enzyme (amino acid residues in blue color). | |
The detailed 3D and 2D binding interactions of derivative 5g within active pocket of PPE enzyme is shown in Fig. 9. The amino acid residues involved in bonding and non-bonding interactions with 5g were PHE223, ARG226, VAL103, ASP63, THR100, HIS60, ASP202, SER222, SER203, THR221, GLN200, CYS199 and VAL224. Briefly, compound 5g was making two hydrogen bonds with VAL224 and GLN200 with bond length of 2.95 Å and 3.21 Å respectively. Interestingly, compound 5g doesn't bear any substitution on benzoylimino group, but it showed excellent in vitro IC50 value and exhibited good binding energy during molecular docking studies. It can be predicted that presence of stabilizing hydrogen bonds with good cutoff value might have increased the activity against PPE enzyme. Moreover, central amantadine ring was making hydrophobic interaction with SER222. Other amino acids involved in van der Waals interactions were SER203 and SER103.
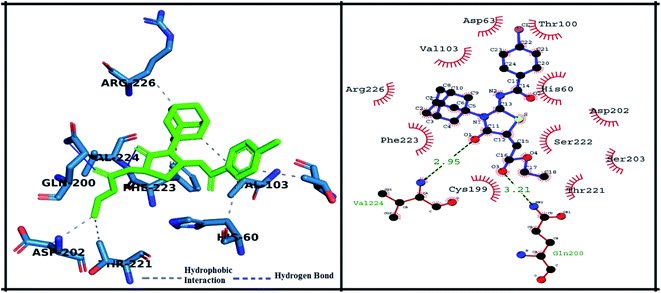 |
| | Fig. 9 Putative 2D and 3D binding mode of compound 5g (green colored) within active pocket of elastase enzyme (amino acid residues in blue color). | |
The detailed 3D and 2D binding interactions of derivative 5h within active pocket of PPE enzyme is shown in Fig. 10. The amino acid residues involved in bonding and non-bonding interactions with 5h were SER225, GLN200, SER203, SER222, HIS60, VAL224, VAL103, PHE223, TRP179 and ARG226. Briefly, 5h compound is appeared to be most potent derivative which showed highest binding energy of −28.82 kJ mol−1 and excellent in vitro activity (IC50 value). It might be due to substitution of chlorine on benzoyl imino ring of compound. Substituted chlorine was making strong π-alkyl interaction with TRP179. Also, benzoyl imino ring was making π–π stacked interaction with PHE223. Moreover, it was observed that amantadine ring was playing pivotal role in making hydrophobic interactions with amino acid residues f active site. Amantadine ring of 5h compound was producing π and π-alkyl interaction with VAL103. Whereas acetate group of 5h compound was involved in π-alkyl interaction with VAL224. Furthermore, compound 5h was producing hydrogen bond with SER225 having bond length of 2.3 Å. The amino acid residues involved in van der Waals interaction with 5h compound were GLN200, SER203, SER222 and HIS60.
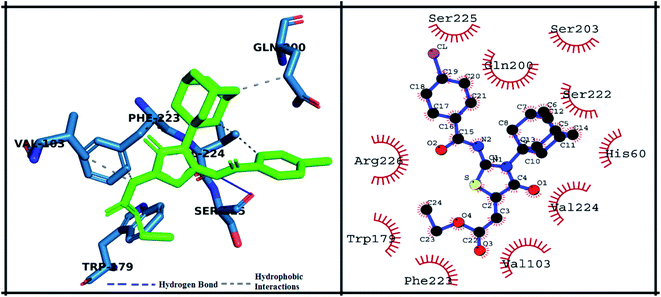 |
| | Fig. 10 Putative 2D and 3D binding mode of compound 5h (green colored) within active pocket of elastase enzyme (amino acid residues in blue color). | |
The co-crystal ligand was re-docked into active pocket of elastase enzyme which was aimed to validate the docking protocol and evaluation of binding score and binding affinity. It was observed that co-crystal ligand was producing important bonding and nonbonding interactions with targeted protein. The amino acid residues which were involved in important stabilizing interactions were THR221, SER203, HIS60, SER222, PHE223, VAL224, SER225, TRP179, GLN200, CYS199, GLY201, GLY198, THR152, LEU227, CYS229, ALA104 and VAL103. Co-crystal ligand was involved in important stabilizing hydrogen bonding with GLN200, VAL224, PHE223 and HIS60. The docking score was found to be −28.03 kJ mol−1. In comparison to synthesized derivatives, it was observed that compound 5h was producing better docking scores than co-crystal ligand. It can be predicted that synthesized derivatives are better inhibitors of PPE enzyme than co-crystal ligand. The 3D interactions of co-crystal ligand with targeted protein is shown in Fig. 11.
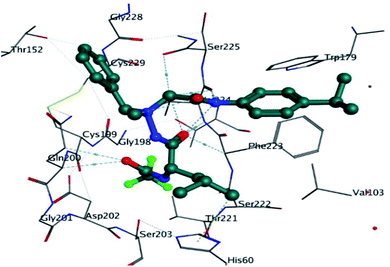 |
| | Fig. 11 Putative binding interactions of the co crystal ligand with in the active pocket of porcine pancreatic elastase enzyme. | |
Molecular dynamic simulations: analysis of RMSD and RMSF. The root mean square deviation (RMSD) is a metric that indicate the average change in displacement of atoms with respect to frame. The value of RMSD can be obtained by analysis of MD trajectory with respect to time. Y-Axis of a graph depicts the evolution of protein RMSD and x-axis indicate the course of time in nanosecond. The RMSD of ligand, targeted protein and protein ligand complex is shown in Fig. 12.
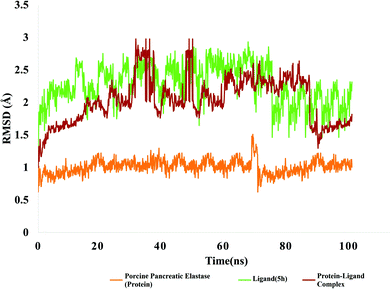 |
| | Fig. 12 RMSD value of protein (porcine pancreatic elastase), compound 5h and protein–ligand complex. | |
The RMSD of ligand is plotted by aligning protein–ligand on the reference protein backbone and RMSD of ligand heavy atoms were calculated. Fig. 12 depicts the evolution of RMSD of protein, ligand and protein–ligand complex. The RMSD value of protein ligand shows that complex attained stability initially up to 30 ns and after that it showed fluctuations in RMSD up to 3 angstrom. After 60 ns of simulation time, complex again stability and remained around 2 angstrom which is perfectly acceptable. After 80 ns, complex RMSD dropped to 1.2 angstrom and remained stable around 1.5 angstrom. In case of targeted protein, RMSD value was quite stable and it attained stability after 10 ns and remained stable around 1 angstrom which is perfect. Green line is indicating evolution of ligand RMSD which is showing fluctuations within 2 angstrom and after 80 ns of simulation time, it got stable at 2 angstrom.
The local changes around protein chain are characterize by root mean square fluctuation (RMSF) values. Fig. 13 depicting the RMSF value of residues index of targeted protein.
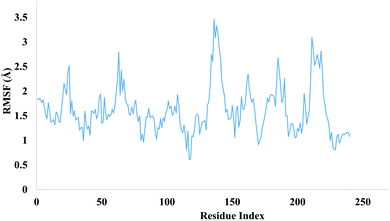 |
| | Fig. 13 Evolution of RMSF value for targeted protein. | |
Peaks sections are showing those residues which fluctuate more during MD simulation. Usually a residue belonging to N and C terminal tends to fluctuate more whereas more compact regions like secondary structures part are more rigids and show less fluctuation. The stability of ligand complexed with protein are indicated by low RMSF values of amino acid residues of binding site. MD simulations demonstrated total 13 hydrogen bonds were produced between ligand and protein. Hydrogen bonds formed during simulations are tabulated in Table 5.
Table 5 Hydrogen observed during MD simulations
| S. no. |
Donor |
Acceptor |
| 1 |
VAL224-main |
5h-side |
| 2 |
5h-side |
GLN200-side |
| 3 |
GLN200-side |
5h-side |
| 4 |
ARG64-side |
5h-side |
| 5 |
5h-side |
LEU227-main |
| 6 |
5h-side |
ARG226-main |
| 7 |
TYR170-side |
5h-side |
| 8 |
ARG36-side |
5h-side |
| 9 |
SER39-side |
5h-side |
| 10 |
GLY38-main |
5h-side |
| 11 |
5h-side |
ASN153-side |
| 12 |
5h-side |
SER225-main |
| 13 |
THR44-side |
5h-side |
ADMET properties of compounds (5a–j). Prediction of ADMET properties is prime step in the process of drug discovery and drug development. In silico ADMET evaluation models were developed to assist the scientist in drug design and lead optimization. Physicochemical properties, absorption, distribution, metabolism, toxicity, excretion, and medicinal properties of adamantyl hybrid iminothiazolidinones derivatives are calculated through the online web server ADMET lab 2.0. It is an integrated online platform for accurate and comprehensive prediction of ADMET properties. ADMET factors that were considered are physicochemical properties, blood–brain barrier (BBB), Caco-2 permeability, volume of distribution (VD), P-glycoprotein (PGP) substrate, P-glycoprotein (PGP) substrate, plasma protein binding, human intestinal absorption (HIA), MDCK permeability, clearance (CL), half-life (T1/2), eye corrosion, eye irritation, respiratory toxicity, AMES toxicity, carcinogenicity, and synthetic accessibility score.Total ten adamantyl hybrid iminothiazolidinones derivative were subjected ADMET study using online web server ADMETlab 2.0. It was observed that physicochemical properties of all compounds were meeting the criteria of drug like rule i.e., Lipinski rule of five (Table 6).
Table 6 Physiochemical properties of the compounds (5a–j)
| Physicochemical properties |
| |
Molecular weight |
Density |
nHA |
nHD |
TPSA |
log S |
log P |
log D |
| 5a |
452.18 |
0.999 |
6 |
0 |
76.04 |
−6.135 |
4.727 |
4.124 |
| 5b |
452.18 |
0.999 |
6 |
0 |
76.04 |
−6.157 |
4.704 |
4.416 |
| 5c |
468.17 |
1.014 |
7 |
0 |
85.27 |
−5.992 |
4.502 |
4.201 |
| 5d |
516.07 |
1.135 |
6 |
0 |
76.04 |
−6.378 |
5.05 |
4.34 |
| 5e |
472.12 |
1.048 |
6 |
0 |
76.04 |
−6.282 |
4.922 |
4.502 |
| 5f |
456.15 |
1.033 |
6 |
0 |
76.04 |
−6.0 |
4.519 |
4.195 |
| 5g |
438.16 |
1.006 |
6 |
0 |
76.04 |
−5.78 |
4.413 |
4.182 |
| 5h |
472.12 |
1.048 |
6 |
0 |
76.04 |
−6.177 |
4.937 |
4.09 |
| 5i |
472.12 |
1.048 |
6 |
0 |
76.04 |
−6.28 |
4.626 |
4.288 |
| 5j |
468.17 |
1.014 |
7 |
0 |
85.27 |
−6.013 |
4.466 |
4.179 |
Moreover, it was observed that all compounds showed a positive value of Caco-2 permeability. Caco-2 permeability is human colon epithelial cell line which corresponds to better human intestinal absorption. A drug having a Caco-2+ value corresponds to high intestinal absorption. The calculated value for human intestinal absorption (HIA) showed that all compounds have probability of good absorption from intestinal membrane. Furthermore, the calculated value for human intestinal absorption was better than standard oleanolic acid for compound 5h and 5i. A compound having positive value of blood–brain barrier have better lipophilicity profile and it can absorb readily from plasma membranes. Calculated values for blood–brain barrier (BBB) and blood placental barriers (BPB) for all compounds were found to be better than standard. It was noticed that the compounds 5a, 5d, 5h and 5i showed better lipophilicity profile and values was observed as BBB++. In terms of Plasma glycoprotein (PGP) substrate and PGP-inhibitor, it was observed that the output value of all compounds had the probability of being PGP-substrate, and PGP-inhibitor. Plasma protein binding (PPB) is an important factor to determine safety of drugs, as drug having high value of PPB (>90%) have narrow therapeutic index whereas drugs having low value of PPB are much safer. In the present study, compounds 5b, 5c, 5f, 5g, and 5j showed low PPB values, which means that these compounds have a broad therapeutic index. In terms of carcinogenicity, compounds 5f, 5h, and 5i were found to be non-carcinogenic whereas all other compounds have the probability of being carcinogenic. AMES toxicity profile depicted that compounds have probability of being toxic. Synthetic accessibility score (SA score) is designed to estimate ease of synthesis of drug-like molecules, and it was observed that all compounds have positive value of SA score. Overall, all compounds showed better ADMET profile, all values are tabulated in Table 7 given below.
Table 7 ADMET profile of the compounds (5a–j)
| Absorption & distribution properties |
| Mode |
Volume of distribution (VD) |
Human intestinal absorption (HIA) |
Caco-2 permeability |
Blood brain barrier (BBB) & blood-placenta barrier (BPB) |
Plasma protein binding (PPB) |
PGP-Inhibitor |
p-Glycoprotein substrate (PGP-substrate) |
MDCK Permeability |
| Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
| 5a |
++ |
0.609 |
+ |
0.023 |
+ |
−4.564 |
++ |
0.596 |
− |
95.90% |
+ |
0.003 |
+ |
0.0 |
+ |
6.3 × 10−5 |
| 5b |
+ |
0.621 |
+ |
0.04 |
+ |
−4.622 |
+ |
0.738 |
+ |
85.93% |
+ |
0.016 |
+ |
0.001 |
+ |
2.7 × 10−5 |
| 5c |
+ |
0.572 |
+ |
0.108 |
+ |
−4.607 |
+ |
0.799 |
+ |
84.80% |
+ |
0.013 |
+ |
0.001 |
+ |
2.4 × 10−5 |
| 5d |
++ |
0.795 |
+ |
0.136 |
+ |
−4.593 |
++ |
0.495 |
− |
95.43% |
+ |
0.237 |
+ |
0.001 |
+ |
2.3 × 10−5 |
| 5e |
+ |
0.707 |
+ |
0.012 |
+ |
−4.571 |
+ |
0.516 |
− |
95.67% |
+ |
0.035 |
+ |
0.001 |
+ |
4.3 × 10−5 |
| 5f |
+ |
0.595 |
+ |
0.026 |
+ |
−4.568 |
+ |
0.741 |
+ |
86.58% |
+ |
0.026 |
+ |
0.001 |
+ |
2.7 × 10−5 |
| 5g |
+ |
0.618 |
+ |
0.036 |
+ |
−4.605 |
+ |
0.799 |
+ |
82.55% |
+ |
0.01 |
+ |
0.001 |
+ |
3.2 × 10−5 |
| 5h |
+ |
0.624 |
+ |
0.01 |
+ |
−4.532 |
++ |
0.313 |
− |
98.30% |
+ |
0.033 |
+ |
0.0 |
+ |
6.4 × 10−5 |
| 5i |
+ |
1.052 |
+ |
0.012 |
+ |
−4.615 |
++ |
0.136 |
− |
99.81% |
+ |
0.611 |
+ |
0.001 |
+ |
2.8 × 10−5 |
| 5j |
+ |
0.58 |
+ |
0.079 |
+ |
−4.599 |
+ |
0.77 |
+ |
85.15% |
+ |
0.011 |
+ |
0.001 |
+ |
2.2 × 10−5 |
| Metabolism |
Excretion |
| |
CYP1A2 Inhibitor |
CYP2C19 Inhibitor |
CYP2C9 Inhibitor |
CYP2D6 Inhibitor |
CYP3A4 Inhibitor |
CL |
T1/2 |
| Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
| 5a |
Inh |
0.173 |
Inh |
0.872 |
Inh |
0.933 |
Inh |
0.167 |
Inh |
0.497 |
Low |
3.831 |
Long |
0.006 |
| 5b |
Inh |
0.167 |
Inh |
0.872 |
Inh |
0.918 |
Inh |
0.07 |
Inh |
0.416 |
Low |
3.263 |
Long |
0.016 |
| 5c |
Inh |
0.215 |
Inh |
0.886 |
Inh |
0.944 |
Inh |
0.056 |
Inh |
0.589 |
Low |
4.483 |
Long |
0.018 |
| 5d |
Inh |
0.216 |
Inh |
0.823 |
Inh |
0.908 |
Inh |
0.063 |
Inh |
0.276 |
Low |
1.328 |
Long |
0.008 |
| 5e |
Inh |
0.265 |
Inh |
0.867 |
Inh |
0.935 |
Inh |
0.157 |
Inh |
0.388 |
Low |
2.767 |
Long |
0.012 |
| 5f |
Inh |
0.213 |
Inh |
0.84 |
Inh |
0.886 |
Inh |
0.054 |
Inh |
0.286 |
Low |
3.103 |
Long |
0.006 |
| 5g |
Inh |
0.247 |
Inh |
0.875 |
Inh |
0.927 |
Inh |
0.089 |
Inh |
0.376 |
Low |
3.146 |
Long |
0.016 |
| 5h |
Inh |
0.253 |
Inh |
0.847 |
Inh |
0.912 |
Inh |
0.278 |
Inh |
0.316 |
Low |
2.461 |
Long |
0.007 |
| 5i |
Inh |
0.211 |
Inh |
0.866 |
Inh |
0.942 |
Inh |
0.154 |
Inh |
0.418 |
Low |
2.311 |
Long |
0.005 |
| 5j |
Inh |
0.174 |
Inh |
0.855 |
Inh |
0.932 |
Inh |
0.025 |
Inh |
0.413 |
Low |
4.229 |
Long |
0.014 |
| Medicinal properties |
Toxicity |
| |
Synthetic accessibility score |
Lipinski rule |
AMES toxicity |
Carcinogenicity |
Eye corrosion |
Eye irritation |
Respiratory toxicity |
| Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
| 5a |
+ |
4.078 |
+ |
Accepted |
+ |
0.017 |
+ |
0.526 |
+ |
0.003 |
+ |
0.031 |
+ |
0.022 |
| 5b |
+ |
4.079 |
+ |
Accepted |
+ |
0.006 |
+ |
0.313 |
+ |
0.003 |
+ |
0.046 |
+ |
0.025 |
| 5c |
+ |
4.061 |
+ |
Accepted |
+ |
0.006 |
+ |
0.209 |
+ |
0.003 |
+ |
0.044 |
+ |
0.024 |
| 5d |
+ |
4.086 |
+ |
Accepted |
+ |
0.007 |
+ |
0.443 |
+ |
0.003 |
+ |
0.049 |
+ |
0.025 |
| 5e |
+ |
4.091 |
+ |
Accepted |
+ |
0.006 |
+ |
0.278 |
+ |
0.003 |
+ |
0.025 |
+ |
0.029 |
| 5f |
+ |
4.049 |
+ |
Accepted |
+ |
0.005 |
− |
0.425 |
+ |
0.003 |
+ |
0.032 |
+ |
0.04 |
| 5g |
+ |
4.009 |
+ |
Accepted |
+ |
0.008 |
+ |
0.292 |
+ |
0.003 |
+ |
0.063 |
+ |
0.03 |
| 5h |
+ |
4.045 |
+ |
Accepted |
+ |
0.007 |
− |
0.459 |
+ |
0.003 |
+ |
0.018 |
+ |
0.025 |
| 5i |
+ |
4.081 |
+ |
Accepted |
+ |
0.016 |
− |
0.935 |
+ |
0.003 |
+ |
0.019 |
+ |
0.101 |
| 5j |
+ |
4.02 |
+ |
Accepted |
+ |
0.007 |
+ |
0.257 |
+ |
0.003 |
+ |
0.038 |
+ |
0.025 |
| TOX21 pathway |
| |
NR-AR |
NR-AR-LBD |
NR-ER |
Antioxidant response element |
| Result |
Probability |
Result |
Probability |
Result |
Probability |
Result |
Probability |
| 5a |
Active |
0.002 |
Active |
0.027 |
Active |
0.117 |
Active |
0.906 |
| 5b |
Active |
0.001 |
Active |
0.308 |
Active |
0.261 |
Active |
0.972 |
| 5c |
Active |
0.001 |
Active |
0.583 |
Active |
0.28 |
Active |
0.932 |
| 5d |
Active |
0.001 |
Active |
0.347 |
Active |
0.285 |
Active |
0.928 |
| 5e |
Active |
0.0 |
Active |
0.355 |
Active |
0.265 |
Active |
0.94 |
| 5f |
Active |
0.0 |
Active |
0.439 |
Active |
0.199 |
Active |
0.921 |
| 5g |
Active |
0.0 |
Active |
0.484 |
Active |
0.269 |
Active |
0.935 |
| 5h |
Active |
0.001 |
Active |
0.024 |
Active |
0.244 |
Active |
0.915 |
| 5i |
Active |
0.001 |
Active |
0.561 |
Active |
0.286 |
Active |
0.913 |
| 5j |
Active |
0.001 |
Active |
0.599 |
Active |
0.129 |
Active |
0.933 |
Experimental
General information
Solvent and reagents were purchased from Sigma-Aldrich and dried before use. The melting point was finding a digital Gallenkamp (SANYO) model MPD BM 3 apparatus and was uncorrected. 1H NMR and 13C NMR spectra were determined in CDCl3 or acetone-d6 solutions at 300 MHz and 75.4 MHz, respectively using a Bruker AM-300 spectrophotometer. FTIR spectra were recorded using an FTS 3000 MX spectrophotometer. The chemical shifts were measured in ppm units and coupling constants (J) in hertz.
Synthesis of the compounds
Substituted aryl carboxylic acid (3.97 mmol, 1.2 mol eq.) was reacted with thionyl chloride (4.30 mmol, 1.3 mol eq.) under inert conditions at reflux in dichloromethane. When the reaction was completed, the dichloromethane was removed under reduced pressure. The resulting acid chloride was converted to isothiocyanate by adding potassium thiocyanate (4.96 mmol, 1.5 mol eq.) in acetone. Amantadine (3.31 mmol, 1 mol eq.) was added to the reaction mixture and heated to 40 °C for 1.5 hours. The reaction mixture was pure in crushed ice the crude product precipitated out and recrystallized in ethanol. The obtained acyl thioureas (1.27 mmol, 1 mol eq.) was further reacted with ethyl 4-ethoxypent-4-en-2-ynoate (2.54 mmol, 2 mol eq.) at room temperature. The final product precipitated out during reaction. The product was filtered, dried, recrystallized in ethanol. Other compounds were prepared using the same method.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((2-methylbenzoyl)imino)-4-oxothiaz olidin-5-ylidene)acetate (5a). White solid: yield: 88%; Rf: 0.6; mp 173–176 °C; IR: 2980, 2907, 2849 (aliphatic C–H stretching), 1715, 1697, 1639 (3C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1540 (NC), 1257 (C–S) cm−1. 1H NMR: (300 MHz, CDCl3, 300 MHz) δ ppm, 8.14 (d, 1H, J = 7.5 Hz, Ar-H), 7.45 (t, 1H, J = 6.9, 7.5 Hz, Ar-H), 7.33–7.29 (m, 2H, J = 8.1 Hz, Ar-H), 6.82 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.78–2.72 (m, 8H, CH3, CH2 adamantyl), 2.28 (s, 3H, CH adamantyl), 1.85–1.72 (m, 6H, CH2), 1.35 (t, 3H, J = 7.2 Hz). 13C NMR: (75 MHz, CDCl3, 300 MHz) δ ppm, 177.8 (CO of benzoyl), 166.3 (CO of ester), 165.6 (CO), 163.4 (CN of imine), 141.9 (α, β CC) 141.5, 133.7, 132.4, 132.1, 131.3 (Ar-C), 125.7 (α, β CC), 118.1 ((Ar-C)), 67.96 (CH2), 61.6, 39.7, 36.1, 30.3 (adamantyl), 22.2 (CH3-Ar), 14.2 (CH3). HRMS (ESI): calculated for C25H28N2O4S+: 453.1770. Found m/z 453.1750 [M + H]+.
O), 1540 (NC), 1257 (C–S) cm−1. 1H NMR: (300 MHz, CDCl3, 300 MHz) δ ppm, 8.14 (d, 1H, J = 7.5 Hz, Ar-H), 7.45 (t, 1H, J = 6.9, 7.5 Hz, Ar-H), 7.33–7.29 (m, 2H, J = 8.1 Hz, Ar-H), 6.82 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.78–2.72 (m, 8H, CH3, CH2 adamantyl), 2.28 (s, 3H, CH adamantyl), 1.85–1.72 (m, 6H, CH2), 1.35 (t, 3H, J = 7.2 Hz). 13C NMR: (75 MHz, CDCl3, 300 MHz) δ ppm, 177.8 (CO of benzoyl), 166.3 (CO of ester), 165.6 (CO), 163.4 (CN of imine), 141.9 (α, β CC) 141.5, 133.7, 132.4, 132.1, 131.3 (Ar-C), 125.7 (α, β CC), 118.1 ((Ar-C)), 67.96 (CH2), 61.6, 39.7, 36.1, 30.3 (adamantyl), 22.2 (CH3-Ar), 14.2 (CH3). HRMS (ESI): calculated for C25H28N2O4S+: 453.1770. Found m/z 453.1750 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((3-methylbenzoyl) imino)-4-oxothiazolidin-5-ylidene)acetate (5b). White solid: yield: 86%; Rf: 0.59; mp 177–179 °C; IR: 2980, 2907, 2847 (aliphatic C–H stretching), 1719, 1693, 1638 (3CO), 1525 (NC), 1272 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.21 (s, 1H, Ar-H), 8.10 (d, 1H, J = 7.8 Ar-H), 7.58 (d, 1H, J = 7.8 Ar-H), 7.45 (t, 1H, J = 7.8 Ar-H), 6.87 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.78–2.72 (m, 8H, CH3, CH2 adamantyl), 2.28 (s, 3H, CH adamantyl), 1.85–1.72 (m, 6H, CH2), 1.35 (t, 3H, J = 7.2 Hz). HRMS (ESI): calculated for C25H28N2O4S+: 453.1770. Found m/z 453.1753 [M + H]+.
Ethyl (Z)-2-((Z)-3-((1s,3s)-adamantan-1-yl)-2-((3-methoxybenzoyl)imino)-4 oxothiazolidin-5-ylidene) acetate (5c). White solid: yield: 82%; Rf: 0.58; mp 235–237 °C; IR: 2906 (aliphatic C–H stretching), 1709, 1638, 1613 (3CO), 1526 (NC), 1276 (C–S) cm−1. 1H NMR: (300 MHz, CDCl3, 300 MHz) δ ppm, 7.81 (d, 1H, J = 7.5 Hz, Ar-H), 7.73 (d, 1H, J = 2.4 Hz, Ar-H), 7.41 (t, 1H, J = 8.1 Hz, Ar-H), 7.16 (dd, 1H, J = 7.8, 2.1 Hz, Ar-H), 6.85 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 3.89 (s, 3H, O– CH3), 2.77 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.80 (q, 6H, J = 12 Hz, CH2 adamantyl), 1.35 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 176.3 (CO of benzoyl), 166.5 (CO of ester), 165.8 (CO), 165.4 (CN of imine), 159.7 (Ar-C), 141.6 (α, β CC), 136.4 (Ar-C), 129.5 (α, β CC), 122.6, 120.2, 118.7, 113.9 (Ar-C), 68.2 (CH2), 61.6, 55.4 (O–CH3), 39.7, 36.1, 30.4, 14.2 (CH3). HRMS (ESI): calculated for C25H28N2O5S+: 469.1719. Found m/z 469.1714 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((4-bromobenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5d). White solid: yield: 85%; Rf: 0.52; mp 181–183 °C; IR: 3057, 2905, 2848 (aliphatic C–H stretching), 1721, 1691, 1650 (3CO), 1509 (NC), 1265 (C–S) cm−1.1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.07 (d, 2H, J = 8.7 Hz, Ar-H), 7.65 (d, 2H, J = 8.7 Hz, Ar-H), 6.86 (s, 1H, CCH), 4.32 (q, 2H, J = 7.2 Hz, CH2), 2.75 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.80 (q, 6H, J = 12.3 Hz, CH2, adamantyl), 1.36 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.6 (CO of benzoyl), 166.9 (CO of ester), 166.4 (CO), 165.4 (CN of imine), 141.4 (α, β CC), 134.1, 131.9, 131.5. HRMS (ESI): calculated for C24H25BrN2O4S+: 517.0718. Found m/z 517.0715 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((3-chlorobenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5e). White solid: yield: 75%; Rf: 0.50; mp 193–194 °C; IR: 2977, 2914, 2848 (aliphatic C–H stretching), 1724, 1692, 1637 (3CO), 1519 (NC), 1287 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.21 (s, 1H, Ar-H) 8.08 (d, 1H, J = 7.8 Ar-H), 7.57 (d, 1H, J = 7.8 Ar-H), 7.45 (t, 1H, J = 7.8 Ar-H), 6.87 (s, 1H, CCH), 4.32 (q, 2H, J = 7.2 Hz, CH2), 2.77 (s, 6H, CH2, adamantyl), 2.28 (s, 3H, CH adamantyl), 1.82–1.80 (q, 6H, J = 12.3 Hz, CH2, adamantyl), 1.36 (t, 3H, J = 7.2 Hz, CH3), 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.1 (CO of benzoyl), 167.4 (CO of ester), 166.4 (CO), 165.4 (CN of imine), 141.3 (α, β CC), 136.9, 134.7, 133.2, 130.2 (Ar-C), 129.9 (α, β CC), 119.2 (Ar-C), 113.9 (Ar-C), 68.5 (CH2), 61.7, 39.7, 36.1, 30.4, 14.2 (CH3). HRMS (ESI): calculated for C24H25ClN2O4S+: 473.1224. Found m/z 473.1219 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((4-fluorobenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5f). White crystalline solid: yield: 87%; Rf: 0.48; mp 231–233 °C; IR: 2980, 2907, 2850 (aliphatic C–H stretching), 1719, 1697, 1639 (3CO), 1535 (NC), 1270 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.25–8.20 (dd, 2H, J = 5.4 Hz, J = 8.7 Hz,Ar-H) 7.21–7.15 (t, 2H, J = 8.7 Hz, Ar-H), 6.85 (s, 1H, CCH), 4.31 (q, 2H, J = 7.2 Hz, CH2), 2.76 (s, 6H, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.86–1.74 (q, 6H, J = 12.3 Hz, CH2, adamantyl), 1.35 (t, 3H, J = 7.2 Hz, CH3), 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.2 (CO of benzoyl), 167.7 (CO of ester), 166.4 (CO), 165.4 (CN of imine), 164.3 (Ar-C) 141.5 (α, β CC),132.6, 132.5, 131.1 (α, β CC), 118.9, 115.9, 115.6 (Ar-C), 68.2 (CH2), 61.7, 39.7, 36.1, 30.3, 14.2 (CH3). HRMS (ESI): calculated for C24H25FN2O4S+: 457.1519. Found m/z 457.1517 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-(benzoylimino)-4-oxothiazolidin-5-ylidene) acetate (5g). White solid: yield: 89%; Rf: 0.61; mp 167–169 °C; IR: 2979, 2913, 2848 (aliphatic C–H stretching), 1719, 1697, 1634 (3CO), 1530 (NC), 1270 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.21 (d, 2H, J = 7.9 Hz, Ar-H), 7.64–7.59 (tt, 1H, J = 7.8 Hz, J = 1.2 Hz Ar-H), 7.51 (t, 2H, J = 7.8 Hz, Ar-H), 6.85 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.77 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.80 (q, 6H, J = 12 Hz, CH2 adamantyl), 1.35 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 176.4 (CO of benzoyl), 166.5 (CO of ester), 165.7 (CO), 165.4 (CN of imine), 141.6 (α, β CC), 135, 133.4, 130 (Ar-C), 128.6 (α, β CC), 118.7 (Ar-C), 68.2 (CH2), 61.7, 39.7, 36.1, 30.4, 14.2 (CH3). HRMS (ESI): calculated for C24H26N2O4S+: 439.1613. Found m/z 439.1610 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((4-chlorobenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5h). White solid: yield: 81%; Rf: 0.49; mp 181–183 °C; IR: 2980, 2908, 2849 (aliphatic C–H stretching), 1721, 1699, 1639 (3CO), 1535 (NC), 1269 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.15 (dd, 2H, J = 6.9 Hz, J = 1.8 Hz Ar-H), 7.49 (dd, 2H, J = 6.6 Hz, J = 1.8 Hz Ar-H), 6.87 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.76 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.80 (q, 6H, J = 12 Hz, CH2 adamantyl), 1.35 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.5 (CO of benzoyl), 166.9 (CO of ester), 166.4 (CO), 165.4 (CN of imine), 141.6 (α, β CC), 139.8, 133.6, 131.3 (Ar-C), 128.9 (α, β CC), 119.1 (Ar-C), 68.3 (CH2), 61.7, 39.7, 36.1, 30.4, 14.2 (CH3). HRMS (ESI): calculated for C24H25ClN2O4S+: 473.1224. Found m/z 473.1220 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((2-chlorobenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5i). White solid: yield: 79%; Rf: 0.63; mp 166–167 °C; IR: 2984, 2906, 2849 (aliphatic C–H stretching), 1724, 1697, 1646 (3CO), 1533 (NC), 1256 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.02 (dd, 1H, J = 7.5 Hz, J = 1.5 Hz Ar-H), 7.51–7.35 (m, 3H, Ar-H), 6.87 (s, 1H, CCH), 4.31 (q, 2H, J = 9 Hz, CH2), 2.71 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.22 (s, 3H, CH adamantyl), 1.75 (q, 6H, J = 12 Hz, CH2 adamantyl), 1.35 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.7 (CO of benzoyl), 166.3 (CO of ester), 165.4 (CO), 165.2 (CN of imine), 141.2 (α, β CC), 134.3, 134, 132.7, 131.8, 131.4 (Ar-C), 126.6 (α, β CC), 118.1 (Ar-C), 68.3 (CH2), 61.7, 39.6, 36, 30.3, 14.2 (CH3). HRMS (ESI): calculated for C24H25ClN2O4S+: 473.1224. Found m/z 473.1222 [M + H]+.
Ethyl (Z)-2-((Z)-3-((3s,5s,7s)-adamantan-1-yl)-2-((4-methoxybenzoyl) imino)-4-oxothiazolidin-5-ylidene) acetate (5j). White solid: yield: 83%; Rf: 0.58; mp 211–213 °C; IR: 2944, 2896, 2842 (aliphatic C–H stretching), 1703, 1638, 1602 (3CO), 1535 (NC), 1247 (C–S) cm−1. 1H NMR (300 MHz, CDCl3, 300 MHz) δ ppm, 8.17 (d, 2H, J = 8.7 Hz, Ar-H), 6.82 (d, 2H, J = 8.7, Ar-H), 6.83 (s, 1H, CCH), 4.30 (q, 2H, J = 9 Hz, CH2), 3.90 (s, 3H, O–CH3), 2.76 (d, 6H, J = 2.4 Hz, CH2, adamantyl), 2.26 (s, 3H, CH adamantyl), 1.80 (q, 6H, J = 12 Hz, CH2 adamantyl), 1.34 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (75 MHz, CDCl3, 300 MHz) δ ppm, 175.7 (CO of benzoyl), 166.5 (CO of ester), 165.4 (CO), 164.8 (CN of imine), 163.9 (Ar-C), 141.9 (α, β CC), 132.3 (Ar-C), 127.8 (α, β CC), 118.4, 113.8 (Ar-C), 68 (CH2), 61.6, 55.5 (O–CH3), 39.7, 36.2, 30.4, 14.2 (CH3). HRMS (ESI): calculated for C25H28N2O5S+: 469.1719. Found m/z 469.1717 [M + H]+.
In vitro methodology
Elastase inhibition assay. The inhibition activity of elastase (elastase from porcine pancreas) was determined by following the methods of ref. 34 and 35 with minor modifications as discussed in our previously reported article.36 By measuring the absorbance of the solution at 410 nm, the amount of released p-nitroaniline, which had been hydrolyzed by the substrate (N-succinyl-Ala-Ala-Ala-p-nitroanilide), could be used to determine the elastase inhibition activity. It was necessary to prepare a 0.8 mM solution of the N-succinyl-Ala-Ala-Ala-p-nitroanilide in a 0.2 M Tris–HCl buffer in order to conduct the experiment (pH 8.0). The buffer solution (130 μL) was added to the test sample (10 μL) in a 96-well microplate, and the mixture was mixed thoroughly. The microplate was pre-incubated for 10 minutes at 25 °C before the addition of an commercially available elastase stock solution (10 mL, 0.0375 unit per mL) was performed. Following the addition of the enzyme, the microplate was maintained at 25 °C for 30 minutes, and the absorbance at 410 nm was measured using a microplate reader (SpectraMax ABS, USA). The IC50 values were determined through the series of triplicate experiments with eight different concentrations as a serial dilution using default protocol in the SoftMax pro software. Using the following formula, it was possible to calculate the elastase inhibition activities:
| Elastase inhibition activity (%) = (ODcontrol − ODsample × 100)/ODcontrol |
where ODcontrol and ODsample represents the optical densities in the absence and presence of sample, respectively. Oleanolic acid was used as the standard inhibitor for elastase. The IC50 of the derivatives was calculated by assaying the eight different concentrations of the tested derivatives (serial dilutions). The observed values were analyzed through non-linear calculations using graph pad prism software 5 as discussed in previously reported article.35
Protocol for kinetics
To determine the mode of inhibition, a kinetic analysis was performed using our previously published method34 in the following manner: the compound 5g was chosen based on its high IC50 value and high potency. It was possible to study the kinetics of compound 5g by varying the concentration of N-succinyl-Ala-Ala-Ala-p-nitroanilide in the presence of various concentrations of compound 5g (0.00, 0.062, 0.124, and 0.248 μM). Briefly, the N-succinyl-Ala-Ala-Ala-p-nitroanilide concentration was changed from 2, 1, 0.5, 0.25, 0.125, and 0.0625 mM for kinetics studies, and the rest of the procedure was the same as described in the elastase inhibition assay protocol. The maximum initial velocities were determined from the initial linear portion of absorbance up to 10 minutes after the addition of the enzyme at a rate of one minute intervals until the enzyme was removed. The type of inhibition on the enzyme was determined by plotting the inverse of velocities (1/V) against the inverse of substrate concentration 1/[S] mM−1 on a Lineweaver–Burk plot. SoftMaxPro was used to process the data after it was collected. An analysis of a secondary plot of 1/V against inhibitor concentration revealed that the EI dissociation constant Ki was observed.
Computational studies
Quantum chemistry through density functional theory calculation. The density functional theory (DFT) is one of widely used method to determine the electron density and energetic parameter of the compounds. It is used to determine the structure of atoms, molecules, crystals, surfaces, and their interactions. These calculations are done by using the Gaussian 09W program on the Linux based workstation.37 B3LYP method was used to determine vibrational wavenumbers with an SVP basis set. It is known that the B3LYP functional assists a good description of harmonic vibrational wavenumbers for small and medium-sized compounds. The optimized structural parameters are further use for calculations. The output check files were analyzed by GuassView 6.0.38
Molecular docking studies. Molecular docking studies were performed using Molecular Operating Environment (MOE) 2015.10 software package.39 The docking studies were conducted to evaluate and investigate binding energies and binding affinities of synthesized derivatives within activation loop of targeted protein. The crystallographic structure of targeted protein porcine pancreatic elastase (PDB ID = 1 BMA; resolution 1.99 angstrom) was obtained from protein data bank (https://www.rcsb.org/).40 All the steps for protein preparation that are pre requisite for docking procedure were performed. Initially co-crystal ligand and het atoms were removed moreover, water molecules were also removed as they interfere with ligand molecules during docking calculations. The addition of polar hydrogen atoms with their standard 3D geometry, addition of partial Gasteiger charges using MMFF94x as a force field was undertaken. Furthermore, fixation of errors in connections, repairing missing residues, 3D protonation, and fixation of potential and energy minimization was also performed using protein preparation wizard of MOE software. The selection of same active site amino acid residues of co crystal inhibitor was carried out using site finder utility of MOE 2015.10. Dummies atoms were created at the site of selected amino acid residues. The structures synthesized thiazolidinone derivatives (5a–j) were generated using Chemdraw ultra 12.0. The generated structures were optimized to steepest energy gradient prior to docking with targeted protein. The ligand database comprising of synthesized thiazolidinone derivatives were docked into selected amino acid residues of active site. Docking protocol was as follows, placement method was set to triangular matcher, scoring methodology was set to London dG, refinement methodology was set to rigid receptor and GBVI/WSA dG was the scoring function for selection of best pose from 100 poses for each compound. After completion of docking protocol, best poses for each compound were evaluated for their fittings into active pocket and best protein–ligand conformations were selected on the basis of docking score for further analysis. The analysis and visualization of best docked pose was carried out using PyMol.41 The putative 2D ligand interactions were created using LigPlot+ 2.2.42 It is run from intuitive java interface which provide detailed 2D interactions of protein–ligand complex. Molecular docking protocol was validated by re-docking co-crystal ligand benzyl methyl aminimide inhibitor with amino acid residues of active site.
ADMET properties. Determination of biochemical processes from drug administration to elimination plays a vital role in lead optimization. An ideal drug candidate should be administered and absorbed into the systemic circulation and must be non-toxic and eliminate without affecting the biological activity. These processes are seeming to be distinct, but they are closely inter-related, so the determination of ADMET properties have prime importance in the drug discovery process. Since traditional methods were time-consuming and much research till 1990 went into vain due to the appearance of undesirable effects in the middle of the drug discovery process. So, the determination of ADMET properties is of prime importance to rolling out undesired effects of a drug candidate at the initial stage of the drug discovery process. Efficient and reliable online prediction models developed for in silico determination of ADMET properties. ADMET properties like absorption, distribution, metabolism, excretion, toxicity and physicochemical properties of eight compounds were done using online in silico prediction model ADMET lab 2.0.43 All compounds were converted to SMI format using ChemDraw Ultra and these SMI structures were loaded into online server ADMET lab 2.0. It also facilitates the manually generating the desired structures using JMSE editor.
Molecular dynamic simulations. The molecular dynamic study of top ranked conformation was performed using Nano scale molecular dynamic (NAMD) software44 on CUDA-accelerated GPU system having 16 core processor and 64 GB RAM memory. Visualization of MD simulations was carried out using VMD software.45 MD simulations were carried out in order to determine binding interactions and stability of protein–ligand complex under accelerated conditions. Top ranked protein–ligand complex was selected and topology files were generated for protein and ligand using CHARMM36 forcefield.46 The system was neutralized by addition of NaCl charges. The system was minimized to steepest-energy gradient to remove any close contacts between atoms. The system was equilibrated in NVT ensemble for 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps followed by equilibration in NPT ensemble for additional 500000 steps. Afterward, simulation was performed for 100 ns under periodic boundaries condition.47 The calculation of binding energy, van der Waals and electrostatic interactions were carried out using PME method.48
000 steps followed by equilibration in NPT ensemble for additional 500000 steps. Afterward, simulation was performed for 100 ns under periodic boundaries condition.47 The calculation of binding energy, van der Waals and electrostatic interactions were carried out using PME method.48
Conclusions
A structurally diverse series of adamantyl-iminothiazolidinone conjugates was synthesized, characterized, and tested for in vitro elastase inhibition assay and subjected to in silico ADMET prediction. The inhibition studies revealed compounds 5a, 5f, 5g, and 5h to show significant activity. Compound 5g with no substitution on benzoyl ring having IC50 value (IC50 0.124 μM) elastase was 50 times more potent as compared to oleanolic acid having IC50 5.996 μM. Kinetic studies showed competitive mode of inhibition and excellent binding energies. Overall, all compounds showed better ADMET profile, indicating their potential to serve as lead compounds in drug discovery. The comprehensive in silico studies including molecular docking studies, drug likeness properties and stability of top ranked protein–ligand complex was conducted to evaluate and support in vitro studies. All these studies suggested potent inhibitory potential of synthesized derivatives.
Conflicts of interest
The authors declare no conflict of interests.
Acknowledgements
The authors gratefully acknowledge the Quaid-i-Azam University for environment conducive for research.
References
- A. Janoff, Annu. Rev. Med., 1985, 36(1), 207–216 CrossRef CAS PubMed.
- P. Guilloteau, R. Zabielski and J. W. Blum, Physiol. Pharmacol., 2009, 60, 37–46 Search PubMed.
- C. Dumont, C. Bourgeois, H. Fessi, P. Y. Dugas and V. Jannin, Int. J. Pharm., 2019, 565, 409–418 CrossRef CAS PubMed.
- D. C. Whitcomb and M. E. Lowe, Dig. Dis. Sci., 2007, 52(1), 1–17 CrossRef CAS PubMed.
- D. M. Goldberg, Clin. Chim. Acta, 2000, 291(2), 201–221 CrossRef CAS.
- N. Arshad, M. Rafiq, R. Ujan, A. Saeed, S. I. Farooqi, F. Perveen, P. A. Channar, S. Ashraf, Q. Abbas, A. Ahmed and T. Hokelek, RSC Adv., 2020, 10(35), 20837–20851 RSC.
- P. A. Henriksen, Curr. Opin. Hematol., 2014, 21(1), 23–28 CrossRef CAS PubMed.
- E. S. Al-Abdullah, H. H. Asiri, S. Lahsasni, E. E. Habib, T. M. Ibrahim and A. A. El-Emam, Drug Des., Dev. Ther., 2014, 8, 505–518 Search PubMed.
- J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, Eur. J. Med. Chem., 2011, 46, 1949–1963 CrossRef CAS PubMed.
- A. Saeed, U. Flörke and M. F. Erben, J. Mol. Struct., 2014, 1065, 150–159 CrossRef.
- L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113(5), 3516–3604 CrossRef CAS PubMed.
- G. Lamoureux and G. Artavia, Curr. Med. Chem., 2010, 17(26), 2967–2978 CrossRef CAS PubMed.
- L. L. Gladkov, S. V. Gaponenko, E. V. Shabunya-Klyachkovskaya, A. N. Shimko, E. S. Al-Abdullah and A. A. El-Emam, Spectrochim. Acta Mol. Biomol. Spectrosc., 2014, 128, 874–879 CrossRef CAS PubMed.
- F. J. Jiménez-Jiménez, H. Alonso-Navarro, E. García-Martín and J. A. G. Agúndez, J. Pers. Med., 2020, 10(4), 217 CrossRef PubMed.
- F. J. Jiménez-Jiménez, H. Alonso-Navarro, E. García-Martín and J. A. G. Agúndez, J. Pers. Med., 2020, 10(4), 217 CrossRef PubMed.
- S. R. Brenner, J. Med. Virol., 2020, 92(11), 2341–2342 CrossRef CAS PubMed.
- R. F. Butterworth, J. Parkinsons Dis., 2020, 7(1), 4 Search PubMed.
- E. Wolf, K. Seppi, R. Katzenschlager, G. Hochschorner, G. Ransmayr, P. Schwingenschuh, E. Ott, I. Kloiber, D. Haubenberger and M. D. Auff, Mov. Disord., 2010, 25(10), 1357–1363 CrossRef PubMed.
- U. Zahra, S. Zaib, A. Saeed, M. ur Rehman, G. Shabir, H. O. Alsaab and I. Khan, Int. J. Biol. Macromol., 2021, 198, 157–167 CrossRef PubMed.
- N. Arshad, A. Saeed, F. Perveen, R. Ujan, S. I. Farooqi, P. A. Channar, G. Shabir, H. R. El-Seedi, A. Javed and M. Yamin, Bioorg. Chem., 2021, 109, 104707 CrossRef CAS PubMed.
- H. Aziz, A. Saeed, M. A. Khan, S. Afridi and F. Jabeen, Mol. Diversity, 2021, 25(2), 763–776 CrossRef CAS PubMed.
- S. Parveen, K. K. Tong, M. Khawar Rauf, M. Kubanik, M. A. Shaheen, T. Söhnel, S. M. Jamieson, M. Hanif and C. G. Hartinger, Chem.–Asian J., 2019, 14(8), 1262–1270 CrossRef CAS PubMed.
- V. Kshtriya, B. Koshti, D. K. Pandey, S. Kharbanda, D. K. Singh, D. Bhatia and N. Gour, R. Soc. Chem. Spec. Publ., 2021, 17(16), 4304–4316 CAS.
- A. Kalaiyarasi, J. Haribabu, D. Gayathri, K. Gomathi, N. Bhuvanesh, R. Karvembu and V. Biju, J. Mol. Struct., 2019, 1185, 450–460 CrossRef CAS.
- R. K. Mohapatra, P. K. Das, M. K. Pradhan, M. M. El-Ajaily, D. Das, H. F. Salem, U. Mahanta, G. Badhei, P. K. Parhi and A. A. Maihub, Comments Mod. Chem., 2019, 39(3), 127–187 CAS.
- H. Nkabyo, G. Bosman, R. Luckay and K. Koch, Inorg. Chim. Acta, 2020, 508, 119644 CrossRef CAS.
- A. M. Plutin, R. Ramos, R. Mocelo, A. Alvarez, E. E. Castellano, M. R. Cominetti, K. M. Oliveira, T. D. de Oliveira, T. E. Silva and R. S. Correa, Polyhedron, 2020, 184, 114543 CrossRef CAS.
- T. Parvin, R. Yadav and L. H. Choudhury, Org. Biomol. Chem., 2020, 18(29), 5513–5532 RSC.
- J. Balzarini, B. Orzeszko-Krzesińska, J. K. Maurin and A. Orzeszko, Eur. J. Med. Chem., 2009, 41(1), 303–311 CrossRef PubMed.
- S. Ashraf, A. Saeed, S. H. Moon, U. Flörke, S. H. Kim, Z. Ashraf, M. Yaseen and M. Latif, J. Med. Chem., 2020, 5(13), 3965–3970 CAS.
- S. Ali, A. Saeed, N. Abbas, M. Shahid, M. Bolte and J. Iqbal, MedChemComm, 2012, 3(11), 1428–1434 RSC.
- M. S. Weiss, S. Panjikar, E. Nowak and P. A. Tucker, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1407–1412 CrossRef PubMed.
- G. Hansen, H. Gielen-Haertwig, P. Reinemer, D. Schomburg, A. Harrenga and K. Niefind, MedChemComm, 2011, 409, 681–691 CAS.
- R. Mathammal, N. Jayamani and N. Geetha, J. Spectrosc., 2013, 171735 Search PubMed.
- A. Saeed, S. A. Ejaz, A. Ul-Hamid, H. R. El-Seedi and J. Iqbal, J. Mol. Struct., 2021, 130821 CrossRef CAS.
- R. A. Costa, E. S. A. Junior, J. d. A. Bezerra, J. M. Mar, E. S. Lima, M. L. B. Pinheiro, D. V. Mendonça, G. B. P. Lopes, A. D. S. Branches and K. M. Oliveira, J. Chem., 2019, 9627404 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian09, R. A. J. I., Gaussian Inc., Wallingford C. T., 2009, 121, pp. 150–166 Search PubMed.
- R. Dennington, T. A. Keith, and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- MOE (Molecular Operating Environment), Version 2015.10, Chemical Computing Group Search PubMed.
- S. K. Burley, H. M. Berman, G. J. Kleywegt, J. L. Markley, H. Nakamura and S. Velankar, Methods Mol. Biol., 2017, 627–641 CrossRef CAS PubMed.
- S. Yuan, H. S. Chan and Z. Hu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 7, e1298 Search PubMed.
- R. A. Laskowski and M. B. Swindells, J. Chem. Info. Model., 2011, 51(10), 2778–2786 CrossRef CAS PubMed.
- ADMETlab 2.0, https://admetmesh.scbdd.com/, accessed 29 September 2021 Search PubMed.
- J. C. Phillips, D. J. Hardy, J. D. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin and W. Jiang, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- S. Kim, J. Lee, S. Jo, C. L. Brooks III, H. S. Lee and W. Im, J. Comput. Chem., 2017, 38, 1879–1886 CrossRef CAS PubMed.
- P. L. Barclay and D. Z. Zhang, J. Comput. Phys., 2021, 435, 110238 CrossRef CAS.
- E. K. Guckel, Large scale simulations of particulate systems using the PME method, University of Illinois at Urbana-Champaign, 1999 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Syeda Abida Ejaz
*a,
Syeda Abida Ejaz