
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the synthesis of thienoindole analogs and their diverse applications
Shubham Pandey†
,
Simran Aggarwal†
,
Ritu Choudhary
and
Satish K. Awasthi
 *
*
Department of Chemistry, Chemical Biology Laboratory, University of Delhi, 110007, Delhi, India. E-mail: satishpna@gmail.com; remarkableshubh@gmail.com; aggsimran22@gmail.com; ritu.choudhary0190@gmail.com
First published on 25th May 2022
Abstract
Thiophene-fused heterocyclic organosulfur systems, especially the thieno[3,2-b]indole moiety have attracted significant attention because they show a wide spectrum of biological activities such as antituberculosis, antitumor, antifungal, antibacterial, and human 5-HT5A receptor binding inhibition. Moreover, they also find applications in material chemistry and chemical engineering. Thus, due to their intriguing properties and applications, researchers are continually attempting to create more effective and environment-friendly methods for their preparation. In this review, we present a complete assessment of the current advances in the field of thieno[3,2-b]indole synthesis.
 Shubham Pandey | Shubham Pandey received his Bachelor's Honours in Chemistry from University of Delhi, India in 2018. Later, he completed his post-graduate studies with specialization in Organic Chemistry from Department of Chemistry, University of Delhi, India in 2020. He qualified for the National Eligibility Test (NET) jointly conducted by CSIR & UGC (New Delhi, Govt of India). His research interest includes various aspects of synthetic organic chemistry. |
 Simran Aggarwal | Simran Aggarwal received her undergraduate degree from University of Delhi, India in 2018. Later, she completed her post-graduate studies with specialization in Organic Chemistry from Department of Chemistry, University of Delhi, India in 2020. She qualified for the National Eligibility Test-Junior Research Fellowship (NET-JRF) jointly conducted by CSIR & UGC (New Delhi, Govt of India). She is currently pursuing her research at Department of Chemistry, University of Delhi under the supervision of Prof. S. K. Awasthi. She has been working on various applications of nitrogen- and sulfur-based organic compounds and bio-organic chemistry. |
 Ritu Choudhary | Ritu Choudhary received her undergraduate degree from Gargi College, University of Delhi, India in 2018 and post-graduate degree from Department of Chemistry, University of Delhi, India in 2020. Her research interests include heterocyclic and medicinal chemistry. |
 Satish K. Awasthi | Dr Satish Kumar Awasthi was awarded a DPhil in 1991 from Allahabad University, UP, India. He received a National Research Associateship from Department of Biotechnology, Government of India to work at the Molecular Biophysics Unit, Indian Institute of Science, Bangalore, India. He is a recipient of several awards including the INSA Visiting Scientist in Germany, ICMR-Biomedical Young Scientist Award at the University of Copenhagen, Denmark, TIT-UNICEF Fellowship Award, Tokyo, Japan, and The Commonwealth Academic Fellowship award at The Royal Veterinary College, University of London, London. Dr Awasthi has been working in diversified areas ranging from peptide chemistry to drug delivery. Specifically, he has been working on the antisense properties of peptide nucleic acids (PNAs), the design and synthesis of small molecules for antibacterial, antifilarial and antimalarial studies and their X-ray crystal structure analysis. |
1 Introduction
Thiophene-fused heterocyclic organosulfur systems have piqued the interest of chemists around the world as they exhibit a diverse set of biological properties and are considered safe compounds for agricultural and pharmaceutical applications.1The thieno[3,2-b]indole moiety is specifically useful in the development of antituberculosis,2 human 5-HT5A receptor binding inhibition, antitumor,3 anti-infective, anti-osteoarthritis,4 antibacterial,5 and antifungal6 drugs and also potent in curing neurological diseases such as senile dementia and Parkinson's disease (Fig. 1).
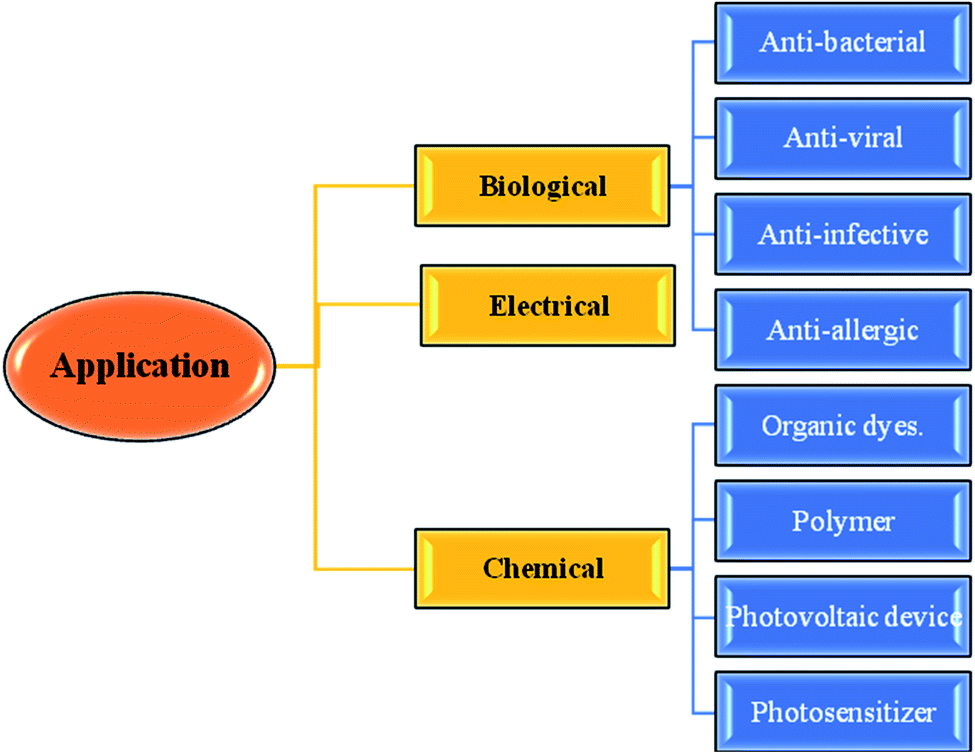 | ||
| Fig. 1 Applications of thienoindoles. | ||
Moreover, it is an important type of π-extended electron-rich system, which can be used for designing molecules for photosensitive and photovoltaic devices. In the past few years, it has also been widely used in designing and engineering fused molecules for organic electronic application, which basically works via the electron push–pull mechanism. This moiety is present in several functionalized organic dyes7–9 such as MKZ-39 and DPP-r-TI, which is an effective photosensitizer for photothermal and photodynamic therapies and polymers10,11 such as PTITBT, PTTICN, PTTIF, and PTTI (Fig. 2).
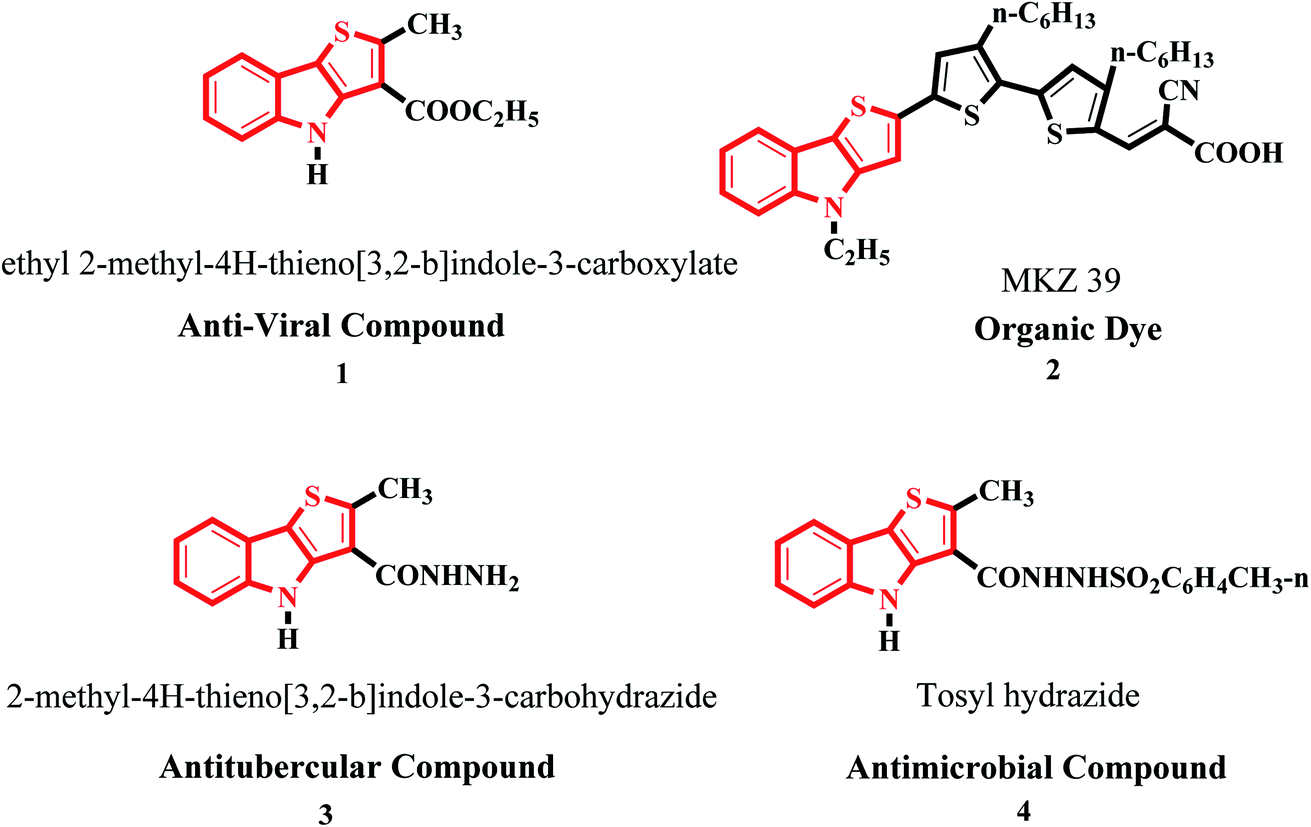 | ||
| Fig. 2 Substituted thieno[3,2-b]indole derivatives and their application. | ||
According to the fusion of the thiophene ring on the indole ring, thienoindole can be categorized into different types. It is a tricyclic heterocyclic compound in which a thiophene ring is fused to an indole ring having one nitrogen atom. The fusion may occur in four different ways, i.e., 8H-thieno[2,3-b]indole 5, 4H-thieno[3,2-b]indole 6, 4H-thieno[3,4-b]indole 7 and 6H-thieno[3,2-e]indole 8, resulting in four important types of thienoindoles (Fig. 3).
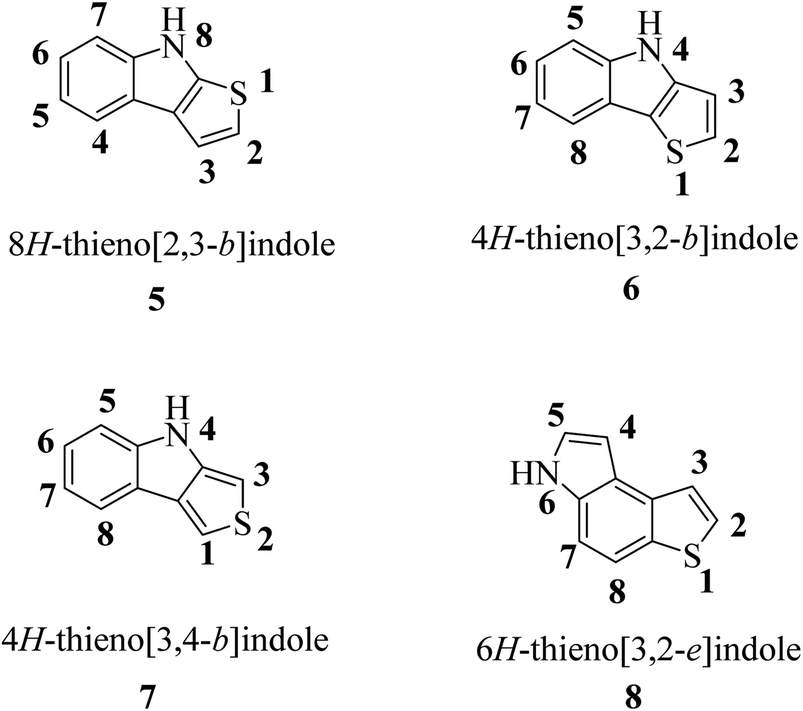 | ||
| Fig. 3 Types of thienoindoles. | ||
2 Synthesis of thienoindoles
In the past few decades, thiophene-fused indoles have gradually been established as a novel class of valuable compounds having intriguing chemical activities and distinct biological activities. Plant growth regulators are critically important for crop production in high yield and enhanced quality, and therefore many plant growth-regulating chemicals have been synthesized to yield good seedlings by promoting root elongation. Nowadays, synthetic chemicals are used at various stages of rice plant development. A streptomycete strain identified as Streptomyces albogriseolus MJ286-76F7 produces a novel active chemical named thienodolin, an alkaloid having a thienoindole skeleton, which exhibits growth-promoting and growth-inhibiting activities in rice seedlings. In 1950, P. A. S. Smith and co-workers reported the first synthesis of 4H-thieno[3,2-b]indole 6 from the diazotization of o-nitroaniline 9 and thiophene 10 via the formation of 2-(2-nitrophenyl)thiophene 11 (Scheme 1a). Later, in 1960, Kobayashi et al. synthesized thieno[2,3-b]indoles 5, starting from 3-(2-oxo-2-phenylethyl)indolin-2-one 12 and phosphorus pentasulfide 13 (Scheme 1b). In the early 90 s, Nakamura et al. structurally elucidated and Kanbe et al. isolated and characterized thienodolin by actively extracting thienodolin from a Streptomyces albogriseolus culture broth using ethyl acetate as the solvent followed by purification via preparative HPLC and silica gel column chromatography. It was found that when rice seedlings were treated with 1.2 × 10−6 to 1.2 × 10−5 M thienodolin, it exhibited growth-promoting activity, whereas 4.0 × 10−5 M thienodolin showed inhibitory activity in rice seedlings.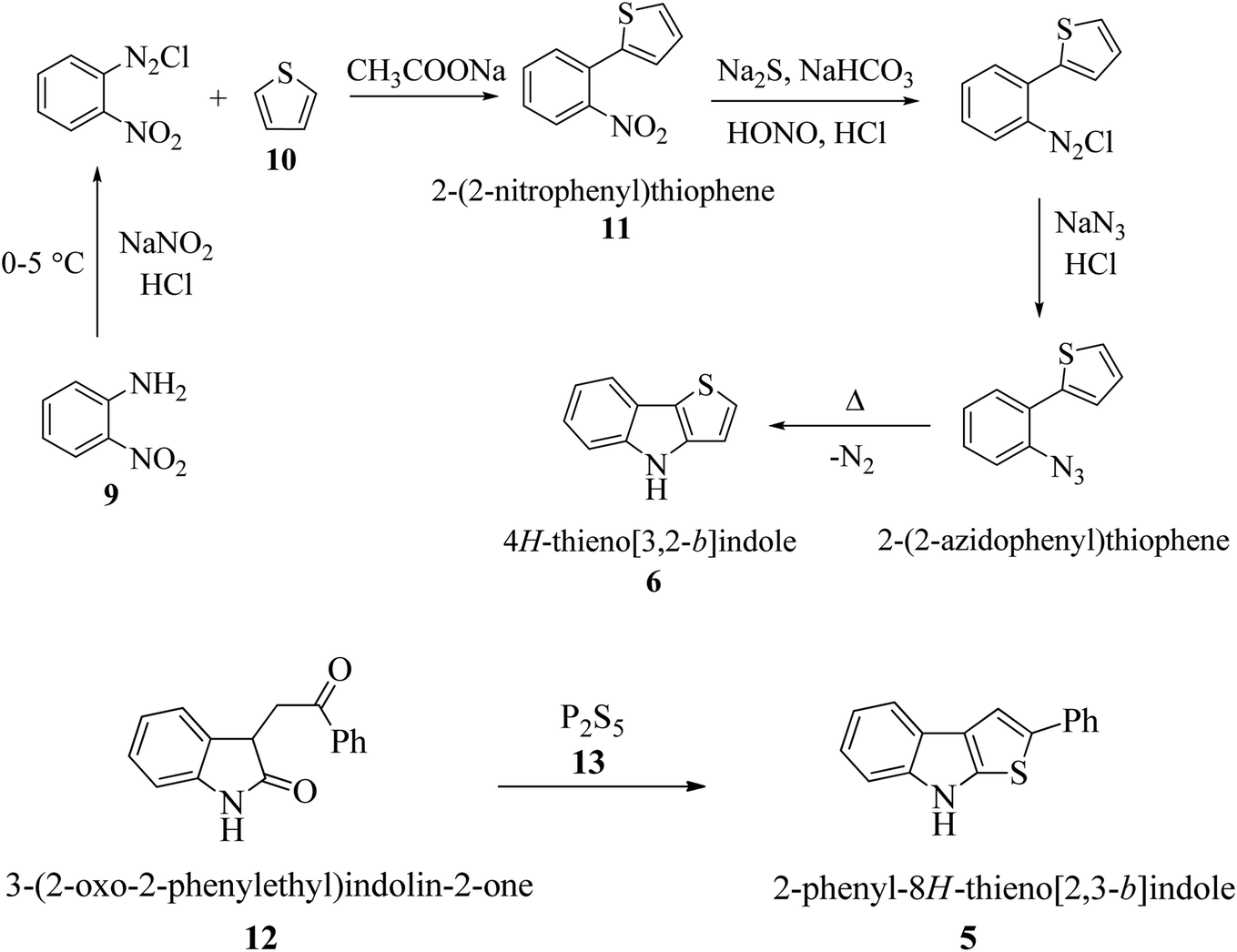 | ||
| Scheme 1 (a) First synthesis of thieno[3,2-b]indole via diazotization of o-nitroaniline and thiophene. (b) First synthesis of thieno[2,3-b]indole from 3-phenacyloxindole and phosphorous pentasulfide. | ||
2.1 Synthesis of thieno[2,3-b]indole by radical cyclization
Singh et al.12 synthesized substituted thieno[2,3-b]indole 15 using the radical cyclization approach in 2011 (Scheme 2). However, although this is an effective approach for the synthesis of thieno[2,3-b]indole in high yield, the substrate, (o-bromoindolyl)acrylonitrile 14, used is synthesized in many steps via the base-induced condensation of (o-bromoindolyl)acrylonitrile with various aryl/heteroaryldithioesters. Also, the 1H NMR spectrum of the substrate showed that the substrate is produced as an inseparable mixture of (E)/(Z) isomers. Moreover, the push–pull nature of the double bond forced the substrate to undergo thermal (Z)/(E) isomerization.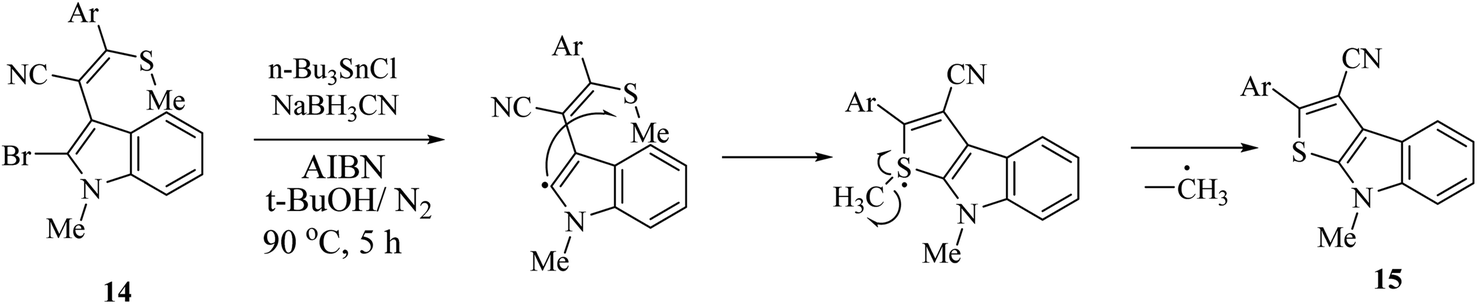 | ||
| Scheme 2 Radical cyclization approach for the synthesis of 15. | ||
Thieno[2,3-b]indole and its derivatives were also prepared via the nitrene-mediated Cadogan cyclization of 3-(o-nitrophenyl)thiophene, AlCl3-induced electrophilic recyclization of 2-(2-furyl)aryl isothiocyanates and oxidative cyclization of indolin-2-thiones. However, most of these methods are afflicted by drawbacks of multistep precursor synthesis, limited scope and generality. Accordingly, considering their applications in pharmaceuticals and materials science, more versatile and effective strategies for the synthesis of thiophene-fused heterocycles are needed.
2.2 Synthesis of 2-substituted thieno[2,3-b]indoles by Lawesson's reagent (LR)
Igrashev and co-workers reported the convenient, short and reliable synthesis of 2-substituted thieno[2,3-b]indoles from readily available reagents involving the two-step reaction-aldol-crotonic condensation of the starting materials and treatment of the intermediate with Lawesson's reagent. The reaction of the intermediate with LR takes place in two steps. Initially, the ethylidene double bond of indolin-2-ones undergoes reduction, and then Paal–Knorr cyclization occurs to give the tricyclic product.When isatins 16 are treated with methyl ketones 17 in mild base, e.g., secondary and tertiary amines, aldol-type adduct 18 is formed, which undergoes dehydration under acidic conditions to form the crotonic condensation product 3-(2-oxo-2-(hetero)arylethylidene)indolin-2-one 19. Compound 19 is more stable than compound 18 given that compound 19 is an unsaturated 1,4-diketone. The carbon–carbon double bond of 19 undergoes reduction in the presence of Na2S2O4,13 H2/Pd(C)14 or Me3P–H2O (ref. 15) (Scheme 3) to give indolin-2-one 20. Compound 20, which bears a 4-oxobutyramide fragment (1,4-dicarbonyl derivatives), undergoes Paal–Knorr reaction in the presence of thionation agents such as P4S10 or Lawesson's reagent and gets cyclized into thieno[2,3-b]indole 21.
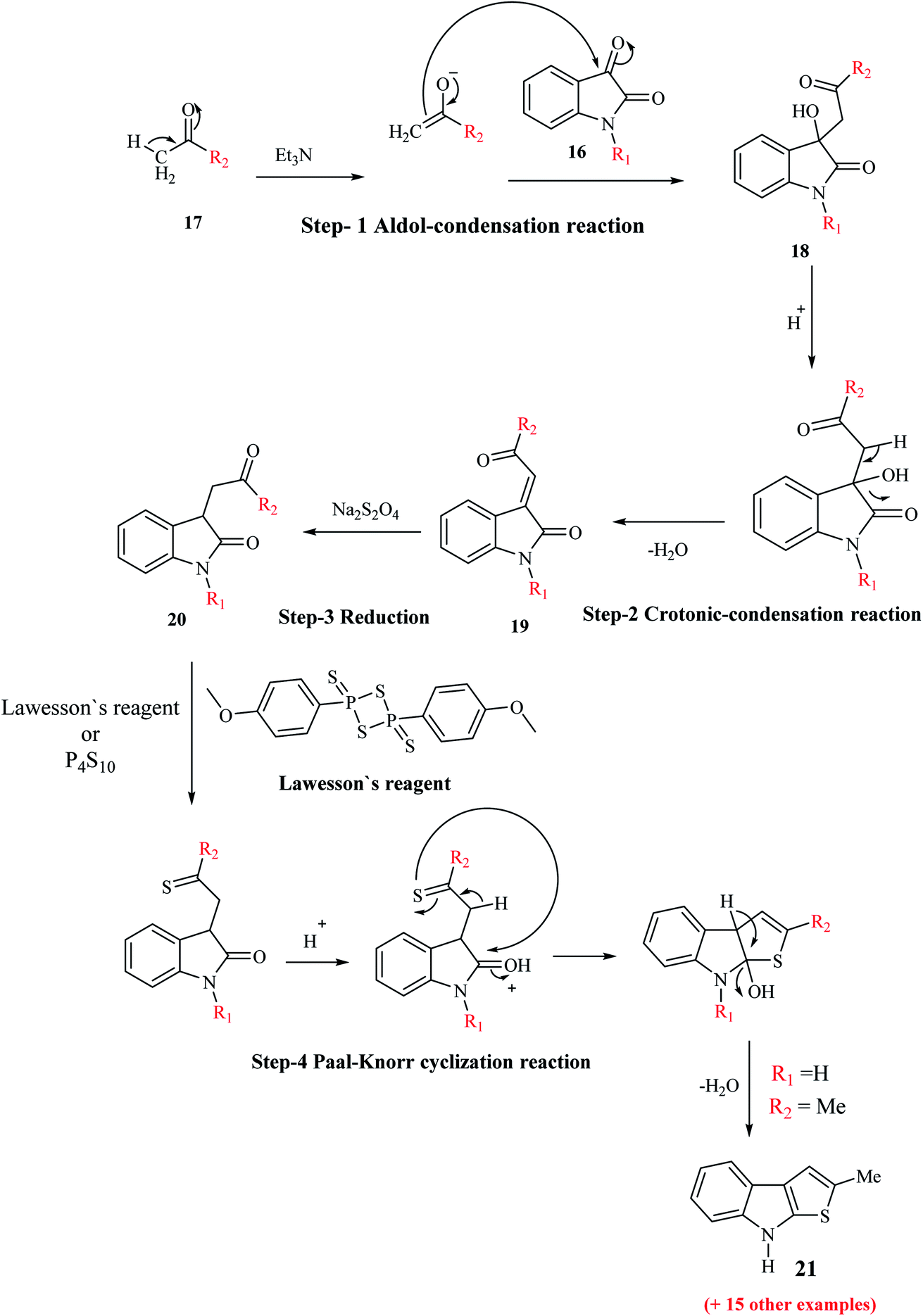 | ||
| Scheme 3 Synthesis of 2-substituted thieno[2,3-b]indole 21 via thionation of indolin-2-ones 19. | ||
Depending on the reaction conditions, the Paal–Knorr reaction produces pyrroles, furans or thiophenes from 1,4-diketones. Thiophene is obtained by using sulfurization agents such as phosphorus pentasulfide and Lawesson's reagent. LR is used as a thiation agent and is a powerful dehydrator, driving the reaction towards completion. This reagent has a four-membered ring of alternating phosphorus and sulfur atoms.
Although this synthetic strategy appears to be appropriate, it has little preparative interest given that thienoindoles are obtained in low yields. For example, 2-methyl-8H-thieno[2,3-b]indole was obtained in 15% yield via this four-step pathway using unsubstituted isatin and acetone.
This procedure was further modified using path A to path D to enhance the overall yield of the desired product. Thieno[2,3-b]indole 21a was prepared using 1-ethyl-isatin 16a and acetophenone 17a in the presence of base and ethanol via path A to path D (Scheme 4).
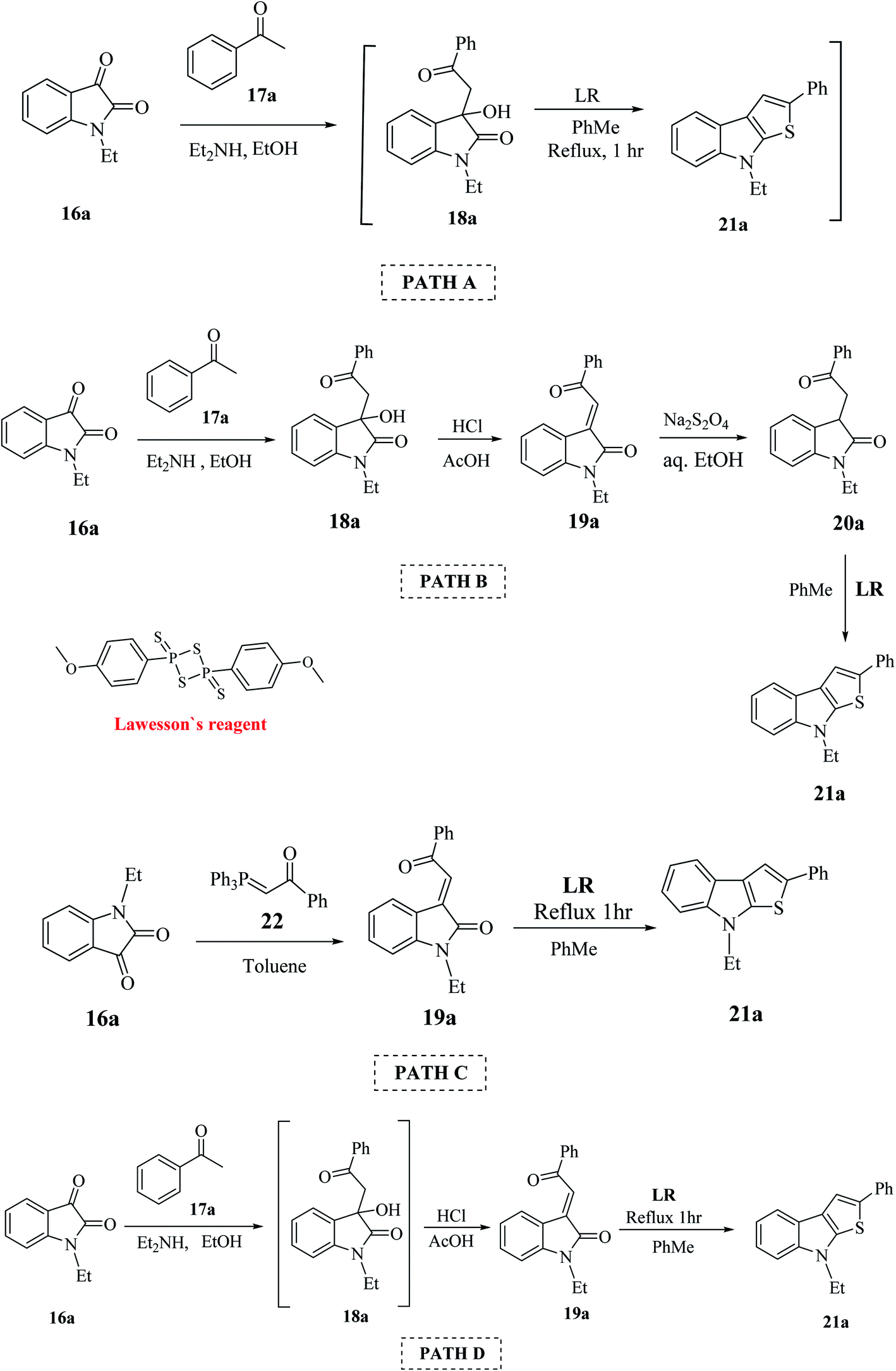 | ||
| Scheme 4 Various paths undertaken for enhancing the yield of thienoindoles. | ||
Lawesson's reagent first acts as the source of H2S to reduce C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in 19a, and then acts as the thiation agent to give 21a via Paal–Knorr reaction. Thus, the four-step procedure is reduced to a two-step procedure, leading to an overall good yield of the product. Hence, path D is the most effective, shortest and most robust method for the synthesis of thienoindoles. In some specific cases, path C is also used as an alternative synthetic route.
C in 19a, and then acts as the thiation agent to give 21a via Paal–Knorr reaction. Thus, the four-step procedure is reduced to a two-step procedure, leading to an overall good yield of the product. Hence, path D is the most effective, shortest and most robust method for the synthesis of thienoindoles. In some specific cases, path C is also used as an alternative synthetic route.
Thieno[2,3-b]indoles containing electron-withdrawing groups such as 4-cyano or 2-nitro-phenyl substituent at the C-2 position were prepared in high yields of 92% and 82%, respectively (Fig. 4).
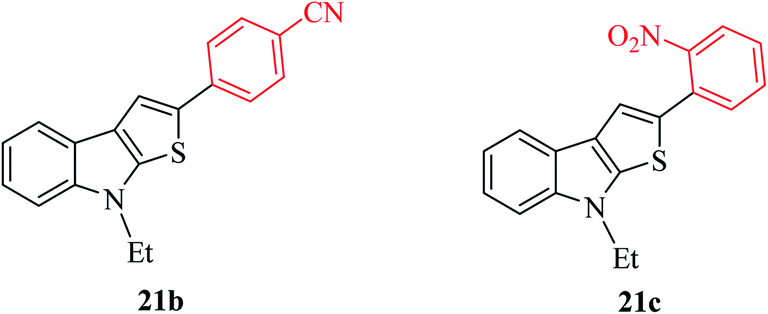 | ||
| Fig. 4 Thieno[2,3-b]indole containing electron-withdrawing groups. | ||
2.3 Synthesis of thienoindoles via Pd-catalyzed cross coupling reaction
2.3.2.1 Role of base and solvent. In most organoboron compounds, the C–B bond is extremely covalent and the complex does not undergo transmetalation easily. Thus, the notable and significant role of the base such as K2CO3, K3PO4, Na2CO3, NaOH and NaHCO3 in the Suzuki–Miyaura reaction is to activate the organoboron derivative by making a hypervalent, anionic boron-“ate” complex, which promptly undergoes transmetalation. In an alternative process, the base displaces the halide in the [PdXR2] complex to form the [Pd(OtBu)R2] complex (Scheme 5).
The activity and selectivity of the Suzuki–Miyaura reaction are influenced by the solvent such as PhMe, DMF, 1,4-dioxane, benzene, THF and CH3CN. Moreover, a mixture of organic solvents and water can be utilized to increase the rate, selectivity and yield of the coupled product. In the current scenario of synthesis, a mixture of toluene and 1,4-dioxane is used as the organic solvent and water as the co-solvent.
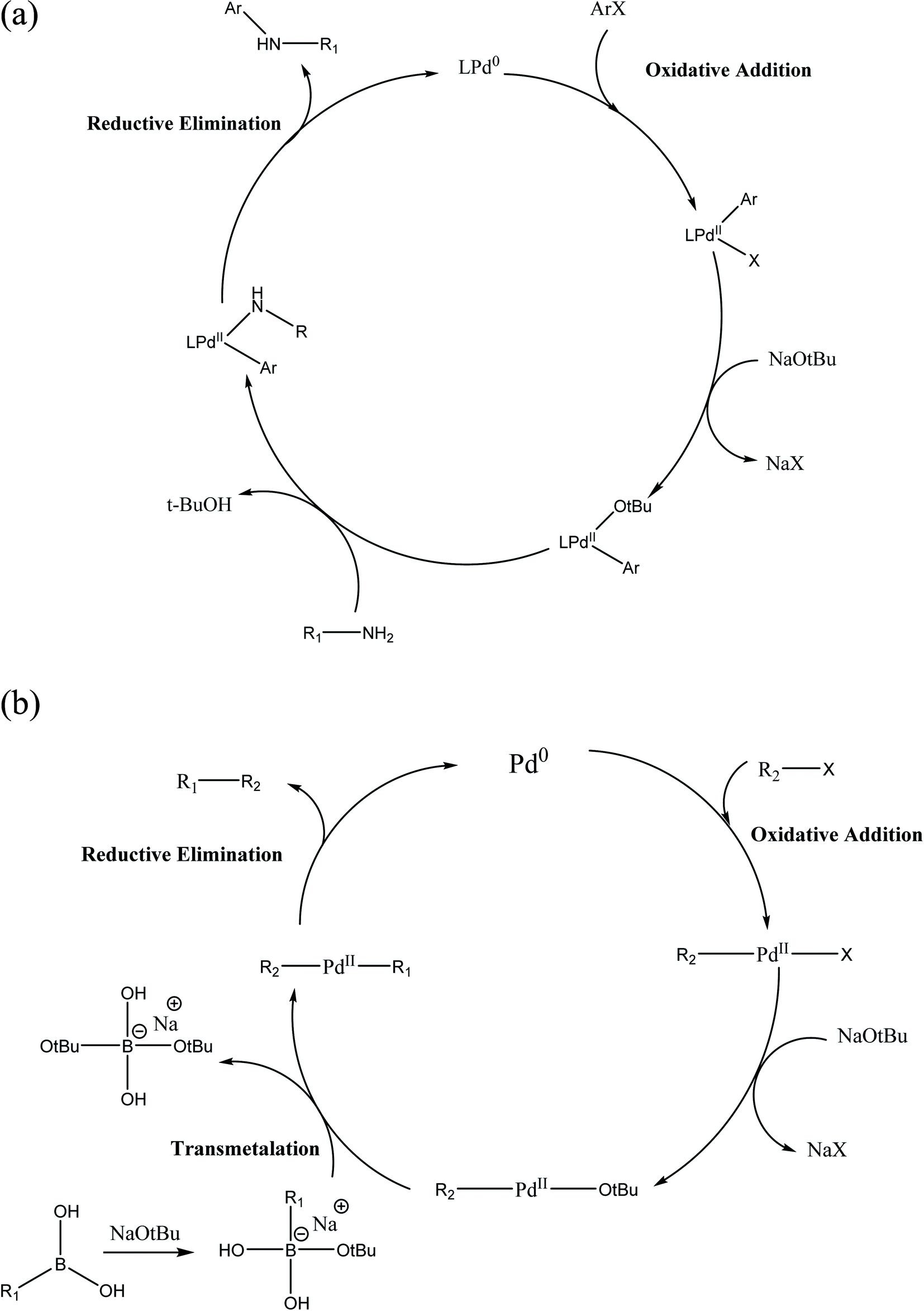 | ||
| Scheme 5 (a) Mechanism of Suzuki–Miyaura cross-coupling reaction; (b) Buchwald–Hartwig coupling reaction. | ||
Toluene and 1,4-dioxane are frequently used as solvents given that they have a high boiling point and can solubilize most organic compounds. Generally, strong bases such as NaOtBu and KOtBu in toluene are used to increase the reaction rate and product yield.
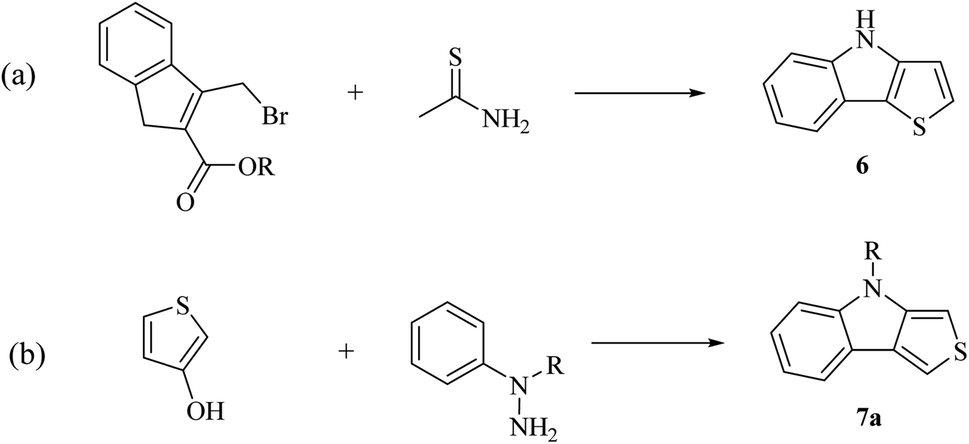 | ||
| Scheme 6 Classical synthetic approaches for synthesizing 6 and 7a. | ||
(a) Reaction of substituted indole with thioamide.16
(b) Reaction of 3-hydroxy thiophene with hydrazine.2
However, these methods were tedious, inflexible and no improvement was reported until 2000. Later, the development of Pd cross-coupling chemistry facilitated the synthesis of thienoindoles and a palladium-catalyzed two step procedure has been developed, which involves a Suzuki reaction to form a C–C bond between benzene and thiophene, followed by ring closure. Ring closure reaction can be of three types, as follows: (a) nitrene insertion,17 (b) oxidative C–N coupling18 and (c) Cadogan cyclization.19
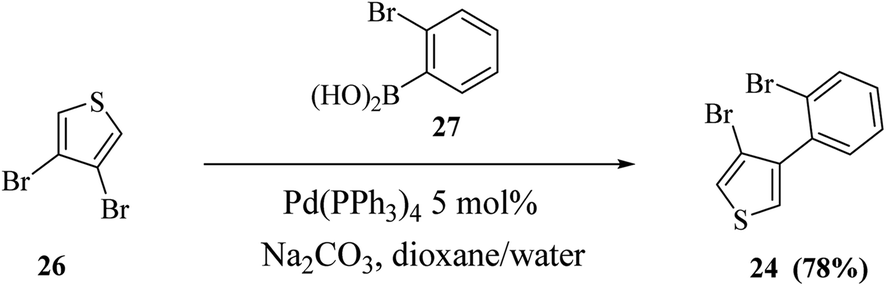
Thieno[3,2-b]indoles were synthesized efficiently via the site-selective Suzuki–Miyaura coupling of 2,3-dibromothiophene with 2-bromophenylboronic acid,20 and subsequent two-fold palladium catalyzed C–N coupling (Buchwald–Hartwig reaction). In the first step, 2,3-dibromothiophene 23 is converted to 3-bromo-2-(2-bromophenyl)thiophene 24 in 82% yield (Scheme 7).
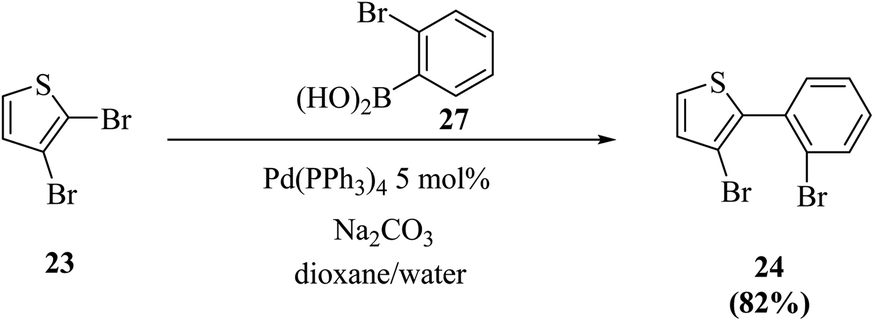 | ||
| Scheme 7 Site-selective Suzuki cross-coupling of 2,3-dibromothiophene. | ||
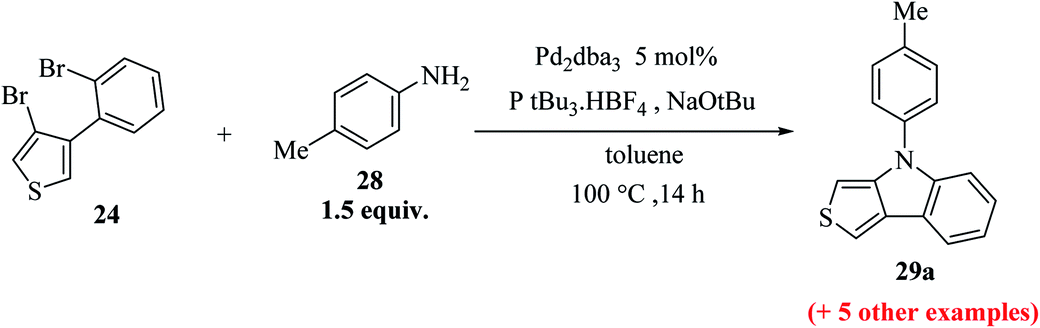
Further, 3-bromo-2-(2-bromophenyl)thiophene 24 undergoes Pd-catalyzed two-fold C–N coupling with p-methyl aniline 28 to give 25a (Scheme 8). Four ligands were screened using NaOtBu in toluene and Pd2(dba)3 to enhance the yield of 25a and it was observed that the use of bidentate ligands dppf and (S)-BINAP gave 25a in 97% and 67% yield, respectively, whereas the bulky monodentate ligands SPhos and P(tBu)3·HBF4 gave 25a in 65% and 86% yield, respectively. However, the bidentate ligand DPPF was found to give the highest yield,21 i.e., 97% (Table 1).
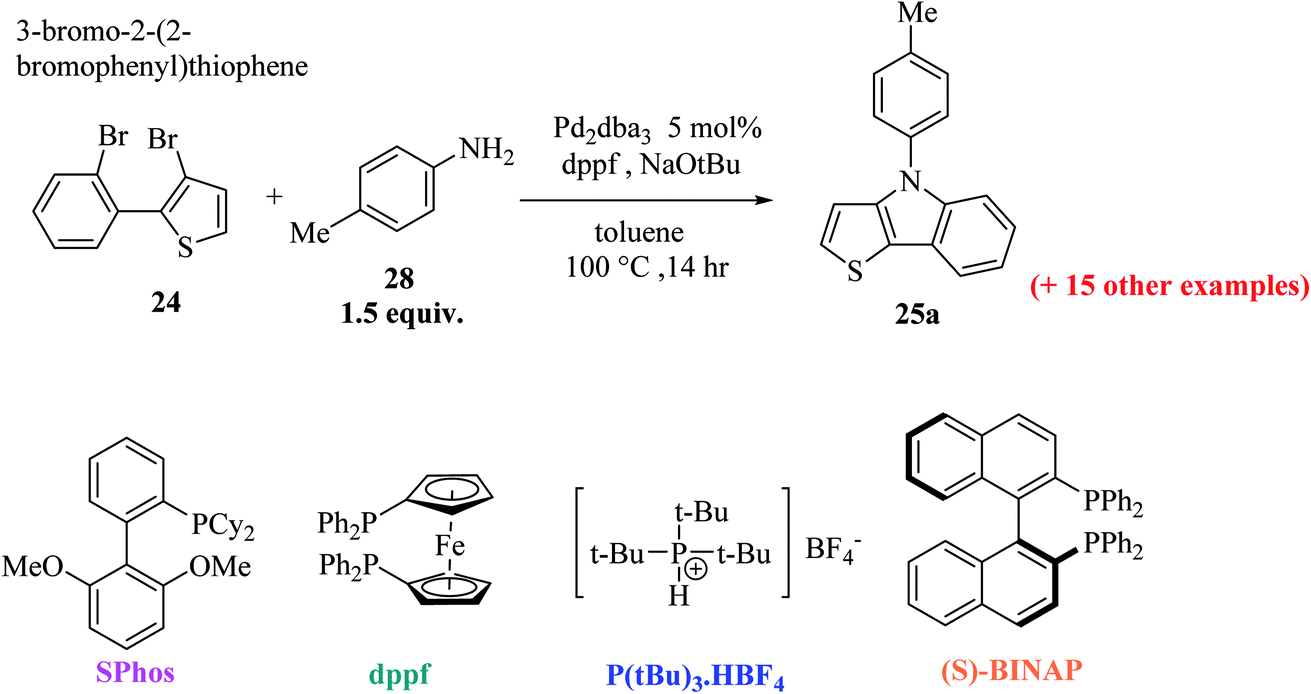 | ||
| Scheme 8 Pd-Catalyzed C–N coupling of 3-bromo-2-(2-bromophenyl)thiophene 24 to give 25a. | ||
| S. No. | Ligand [mol%] | Yield (%) |
|---|---|---|
| 1 | P(tBu)3·HBF4[10%] | 86 |
| 2 | dppf [5%] | 97 |
| 3 | SPhos [10%] | 65 |
| 4 | (S)-BINAP[5%] | 67 |
Using these optimized conditions, compound 24 was treated with electron-rich amines, namely, 4-(methylthio)aniline and p-anisidine, which produced the corresponding thienoindoles 25b and 25c in excellent yields. When electron-poor, benzylic and aliphatic amines were used, 1,1′-bis(diphenylphosphino)ferrocene (dppf) resulted in poor yield of the products. Thus, dppf was replaced by (S)-BINAP, which gave the corresponding thienoindoles 25d and 25e in good yields. Treatment of 1-naphthylamine gave the product 25f in a comparatively low yield (78%), whereas sterically hindered amine gave the product 25g in very good yield. Hence, the maximum yield was observed for electron-rich amines. In total, 20 substituted thieno[3,2-b]indoles were synthesized using this method, some of which are shown in Fig. 5.
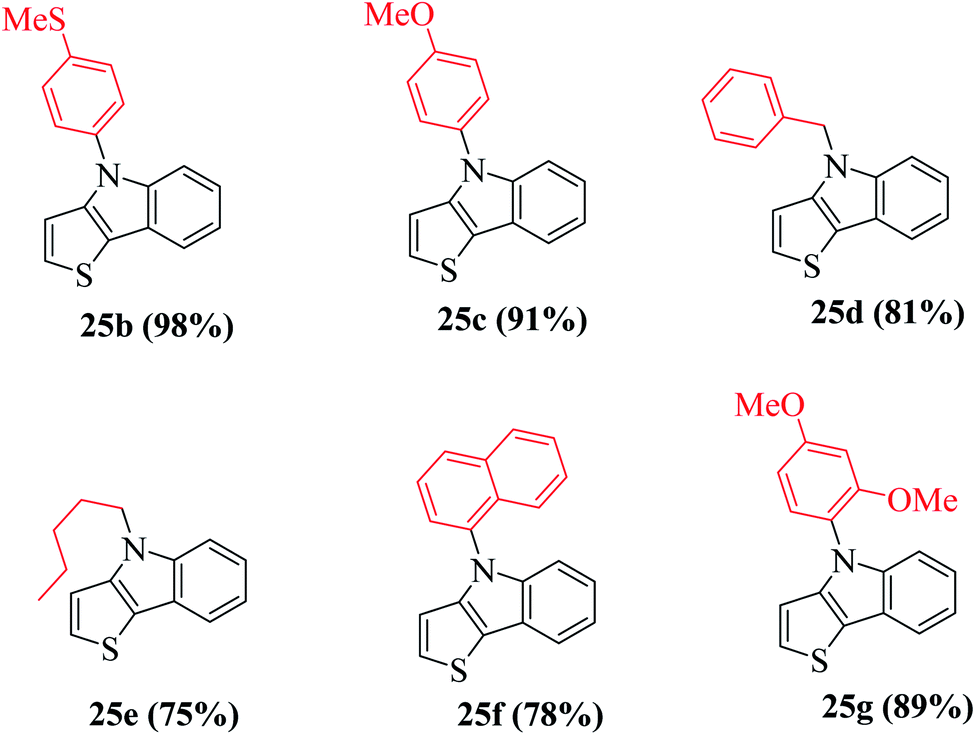 | ||
| Fig. 5 Yield of products having different substituents. | ||
Also, thieno[3,4-b]indoles were synthesized via the site-selective Suzuki cross-coupling reaction of 3,4-dibromothiophene 26 to form 3-bromo-4-(2-bromophenyl)thiophene 24 in 78% yield (Scheme 9).
 | ||
| Scheme 9 Site-selective Suzuki cross-coupling of 3,4-dibromothiophene. | ||
Further, 3-bromo-4-(2-bromophenyl)thiophene 24 undergoes Pd-catalyzed two-fold C–N coupling with p-methyl aniline 28 to give 29a (Scheme 10).
 | ||
| Scheme 10 Two-fold C–N coupling of compound 24. | ||
Upon screening of various ligands, the monodentate ligand P(tBu)3.·HBF4 was found to be the most effective, giving the corresponding thieno[3,4-b]indole 29a in 95% yield (Table 2).
| S. No. | Ligand [mol%] | Yield (%) |
|---|---|---|
| 1 | P(tBu)3·HBF4[10%] | 95 |
| 2 | dppf [5%] | 43 |
| 3 | SPhos [10%] | 65 |
| 4 | (S)-BINAP[5%] | 36 |
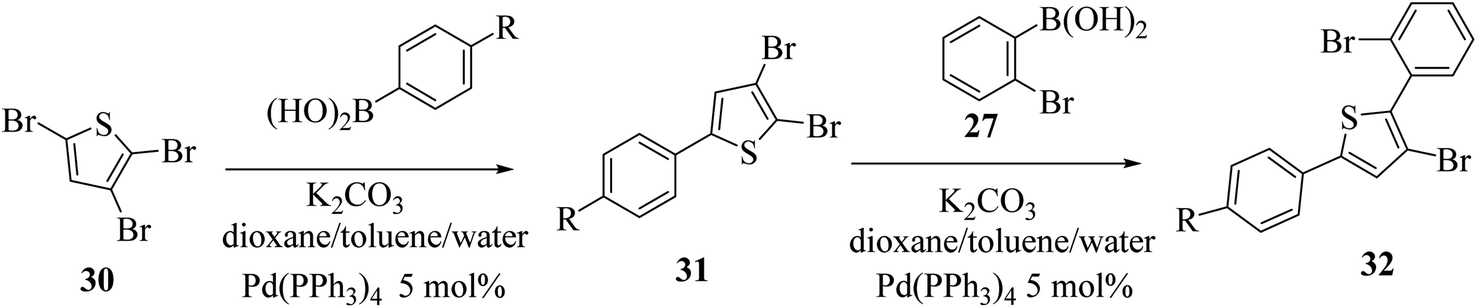
At last, the practicality of the strategy was examined for the preparation of 5-substituted thieno[3,2-b]indoles. The synthesis began with two successive site-selective Suzuki cross-coupling reactions of 2,3,5-tribromothiophene 30 utilising the earlier reported conditions. 2,3,5-Tribromothiophene was initially converted to 2,3-dibromo-5-arylthiophene 31a–d, and later to 3-bromo-2-(2-bromophenyl)-5-arylthiophene 32a–d (Scheme 11), giving product 33 in low yield. This is presumably because of the extra aryl group at position-5, which may affect the overall electronic nature22 of the molecule (Table 3).
 | ||
| Scheme 11 Coupling reaction of 2,3,5-tribromothiophene 30 to give compound 32. | ||
| R | 31 (yield%) | 32 (yield%) |
|---|---|---|
| (a) H | 60 | 43 |
| (b) Cl | 37 | 30 |
| (c) F | 43 | 33 |
| (d) t-Bu | 43 | 26 |
Buchwald's biaryl ligand SPhos gave the final product 33a in the highest yield, i.e., 96% (Scheme 12), whereas ligands such as (t-Bu)3P·HBF4, dppf and xantphos gave compound 33a in 84%, 36% and 43% yield, respectively.
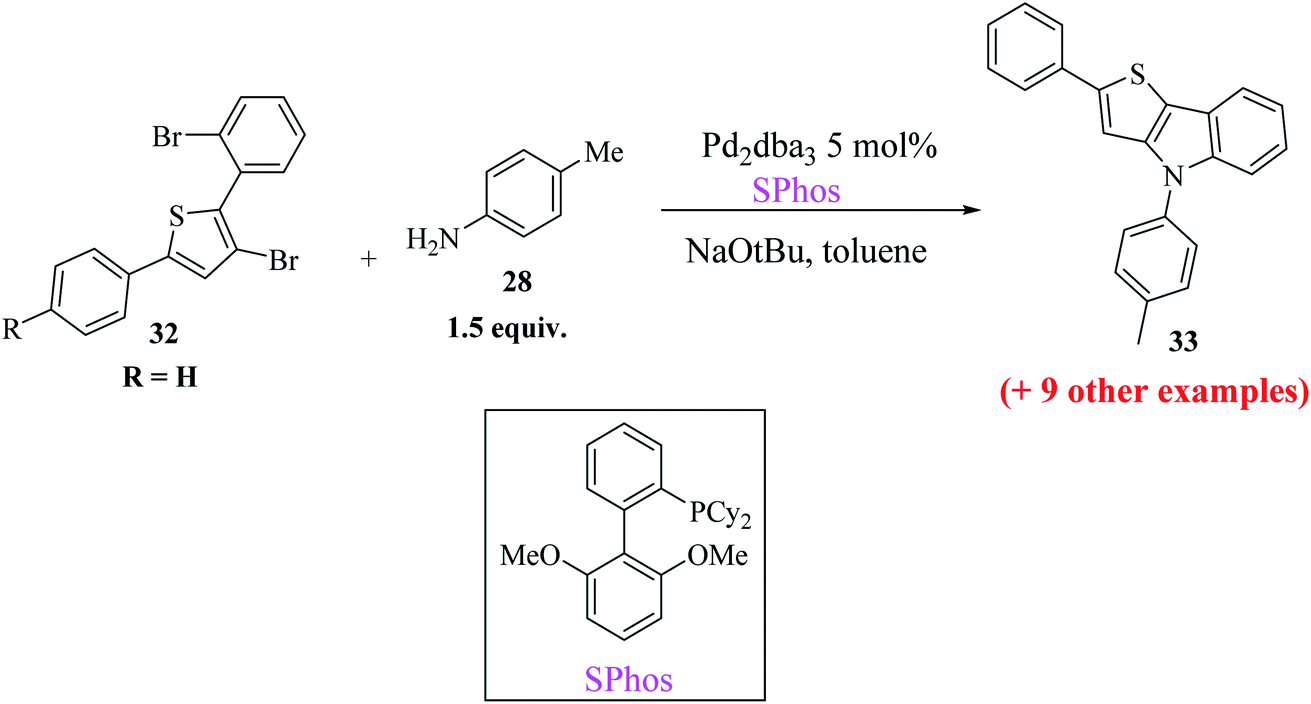 | ||
| Scheme 12 Synthesis of 5-substituted thieno[3,2-b]indole 33. | ||
In conclusion, thieno[3,2-b]indoles and thieno[3,4-b]indoles have been synthesized via a new, more efficient and convenient synthetic methodology, namely, Buchwald–Hartwig cross-coupling. Also, the role of the ligand was found to be more crucial in the second step.
2.4 Synthesis of thieno[3,2-b]indole via Cadogan reductive cyclization
Dehaen et al.19 synthesized thieno[3,2-b]indole in two steps. The first step is the Suzuki–Miyaura coupling reaction between o-nitrophenyl boronic acid 34 and 2-bromothiophene 35. The recent literature revealed that arylboronic acids substituted with electron-withdrawing groups (here, nitro at the ortho position) undergo extensive deboronation under standard Suzuki–Miyaura conditions, which employ aq. Na2CO3 as the base. This premature destruction of the C–B bond causes low yields. Hence, to reduce or protect from proto-deboronation, Suzuki–Miyaura coupling reaction has been performed under microwave-enhanced conditions. The second step is the nitrene-mediated reductive cyclization of 2-(2-nitrophenyl)thiophene 36 under MW irradiation, which leads to a dramatic rate enhancement given that the usual method demands drastic conditions and long reaction time (Scheme 13).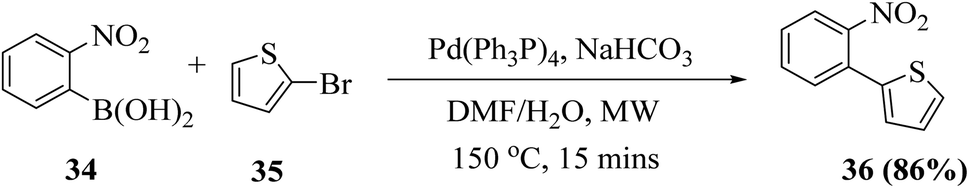 | ||
| Scheme 13 Synthesis of 2-(2-nitrophenyl)thiophene 36 via Suzuki–Miyaura coupling reaction. | ||
Further, a mixture of compound 36 and triethyl phosphite was suspended in a 10 mL sealed glass vial and irradiated with 300 W power at 210 °C. Bunyan and Cadogan demonstrated that aromatic C-nitroso-compounds are promptly deoxygenated by triethyl phosphite. Hence, deoxygenation of 36 led to the formation of a nitroso-compound, which readily underwent deoxygenation, resulting in the formation of an indole ring via a nitrene intermediate.23 The reaction took 15 min to go to completion and acidic work-up removed the phosphate by-products and product 37 was obtained in good yield (Scheme 14).
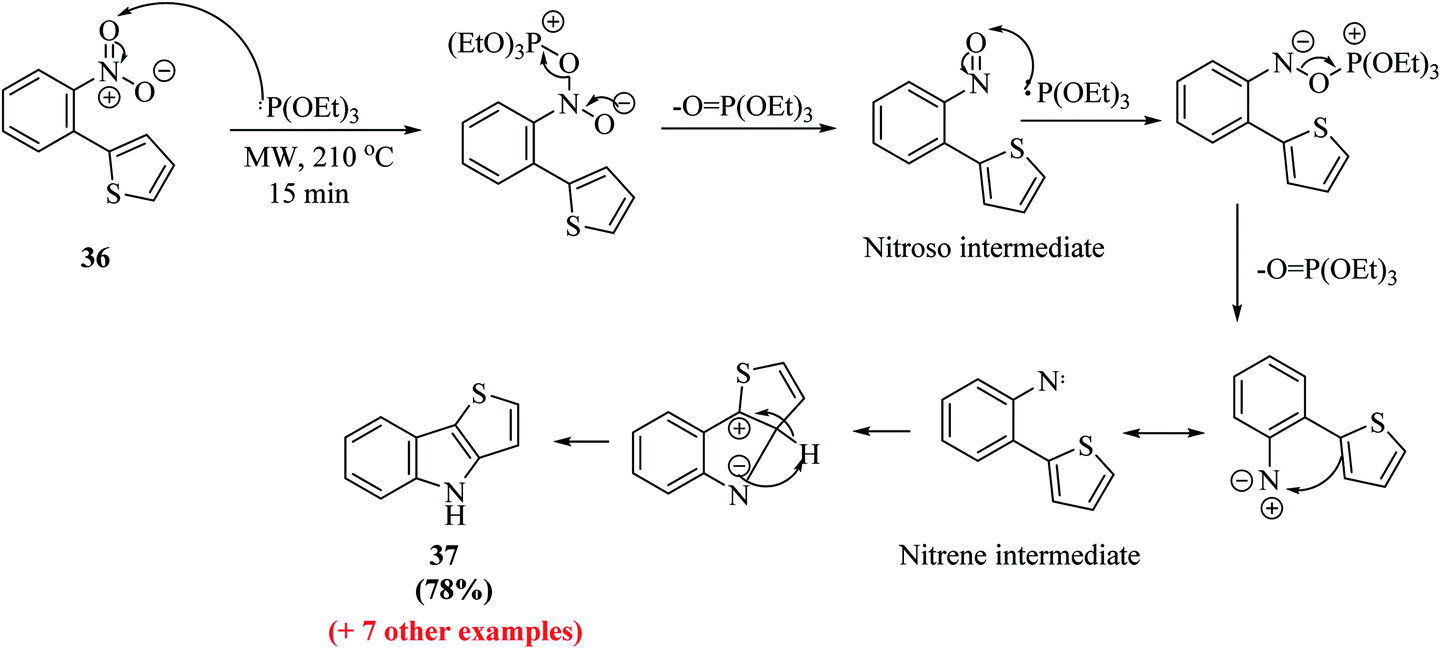 | ||
| Scheme 14 Cadogan reductive cyclization to thieno[3,2-b]indole 37. | ||
Dehaen et al. used 2-nitro-phenylboronic acid given that it gives access to the biaryl compounds needed for the Cadogan cyclization and also heterocyclic boronic acids are expensive and difficult to synthesize. Hence, it is a more versatile, efficient and economic method for synthesizing thieno[3,2-b]indoles.
2.5 Metal-free approach for synthesizing regioselective thieno[2,3-b]indole
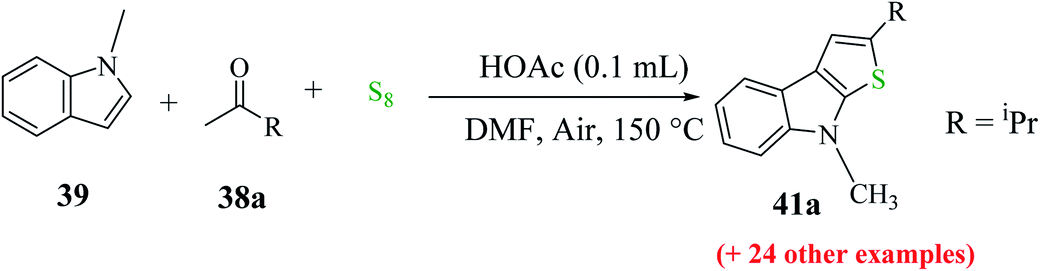
Penghui et al.24 described an effective metal-free approach for synthesizing substituted thieno[2,3-b]indole 40 and 41 with high regioselectivity and great functional group tolerance. In this approach, the cascade cyclization occurs via the acid-promoted annulation of ketone 38, indole 39 and sulfur powder, where the solvent DMF and the additive control the regioselectivity of the reaction (Scheme 15).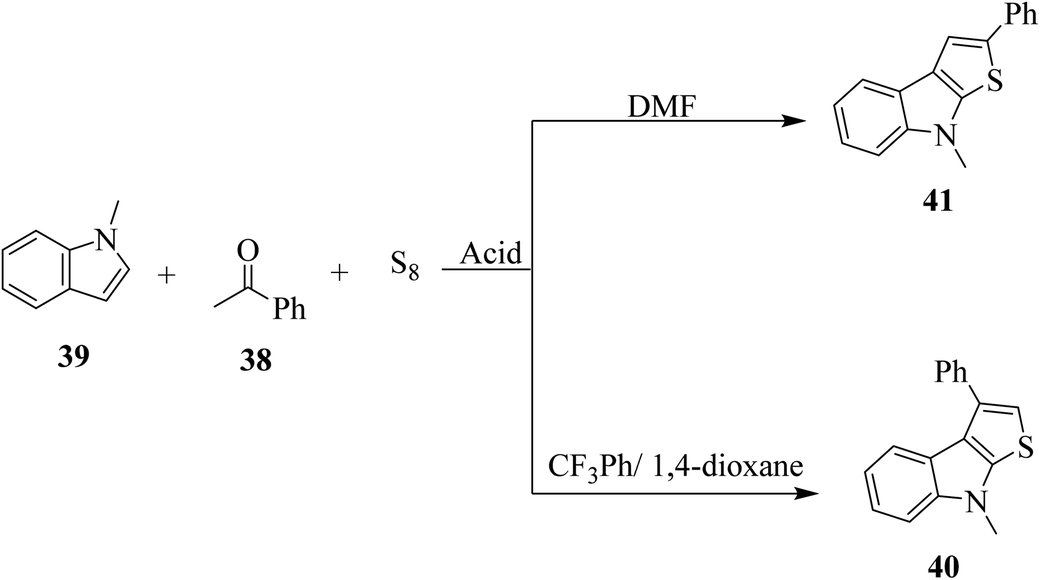 | ||
| Scheme 15 Methodology for synthesizing benzothieno[2,3-b]indole. | ||
Elemental sulfur was used as the source of sulfur given that it is abundant in nature, non-toxic and stable under normal conditions. Its low price and high purity make it a good choice. In recent years, elemental sulfur had found great applications in C–S bond-forming reactions. Most of the reported C–S bond formation reactions using elemental sulfur are catalyzed by transition metals. However, a few reactions are also available that do not need any transition metal.27 Our reaction is among these types of reactions (Scheme 16).
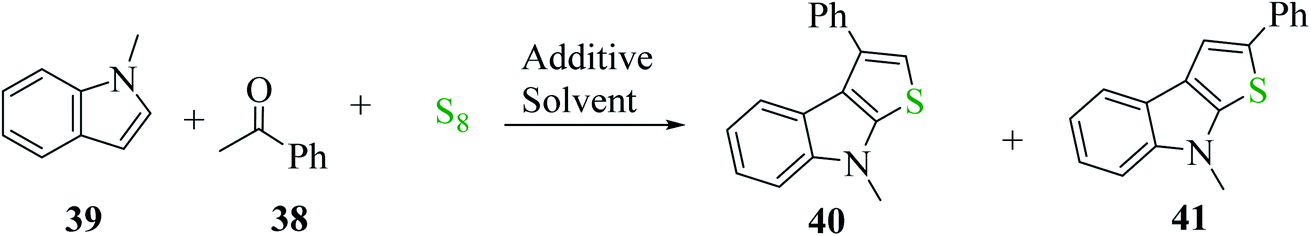 | ||
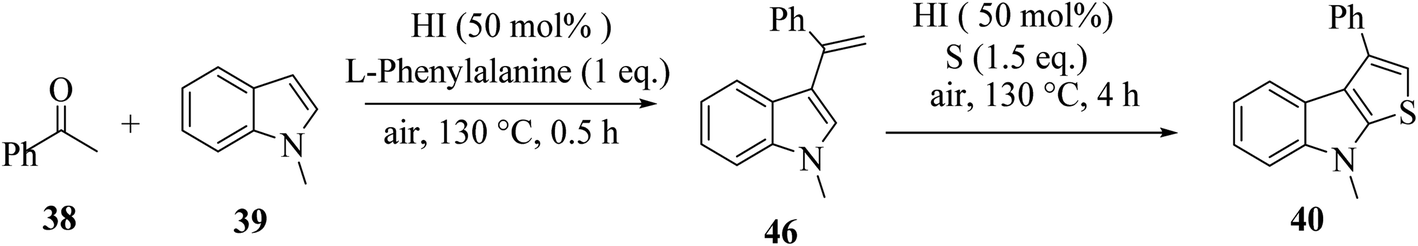
| Scheme 16 Schematic representation of the synthesis of 3-phenylthieno[2,3-b]indole 40 and 2-phenylthieno[2,3-b]indole 41. | ||
1-Methyl-1H-indole 39, acetophenone 38 and sulfur powder were reacted using different additives and solvents. The desired product, i.e., 3-phenylthieno[2,3-b]indole 40 was obtained when chlorobenzene was used as the solvent and 50 mol% HI as an additive at 130 °C. Further, the yield of product 40 was improved using PhCF3 and anisole as additives and 1,4-dioxane as the solvent. The yield of 40 was further improved using PhCF3/1,4-dioxane, which was further greatly enhanced when 1 equivalent of L-phenylalanine was used together with PhCF3 and 1,4-dioxane. 2-Phenylthieno[2,3-b]indole 41 was obtained when DMF was chosen as the solvent, whose yield was increased when acetic acid was used as the acid instead of HI. This is because when DMF was used as the solvent, the regioselectivity of the cyclization process switches as a result of the change in the polarity of solvent. This is a direct cyclization reaction. The reaction yield of 41 and the ratio of 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 further increased when the temperature was increased to 150 °C and the ratio of 39:38 changed to 1:2.3.
:40 further increased when the temperature was increased to 150 °C and the ratio of 39:38 changed to 1:2.3.
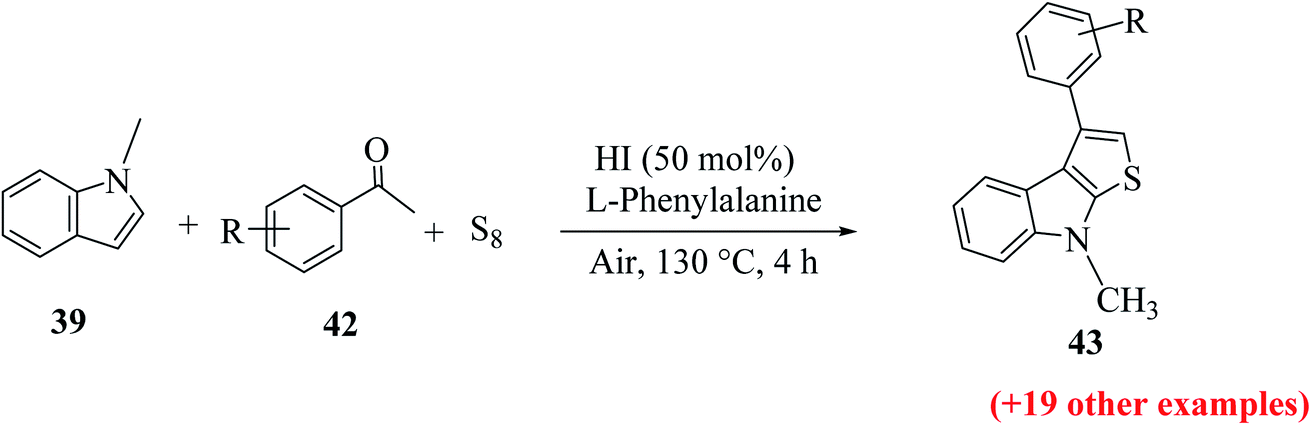 | ||
| Scheme 17 Synthesis of 3-substituted thieno[2,3-b]indole analogs 43 using different substituted indoles 42. | ||
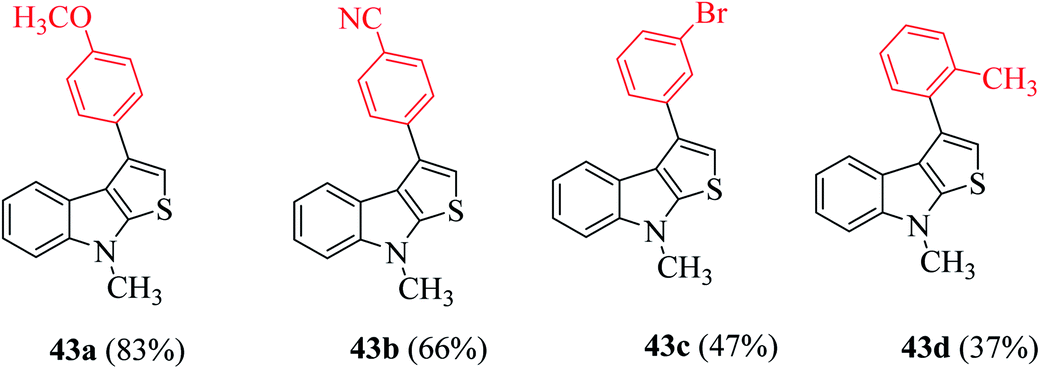 | ||
| Fig. 6 Analogs of 3-substituted thieno[2,3-b]indole. | ||
Further, the yield of product 44 was affected by the position of the functional group on the indole ring. When it was present on C-5, C-6 and C-7, then the yield of the product was good, whereas when it was present at the C-4 position, the yield decreased dramatically (Scheme 18).
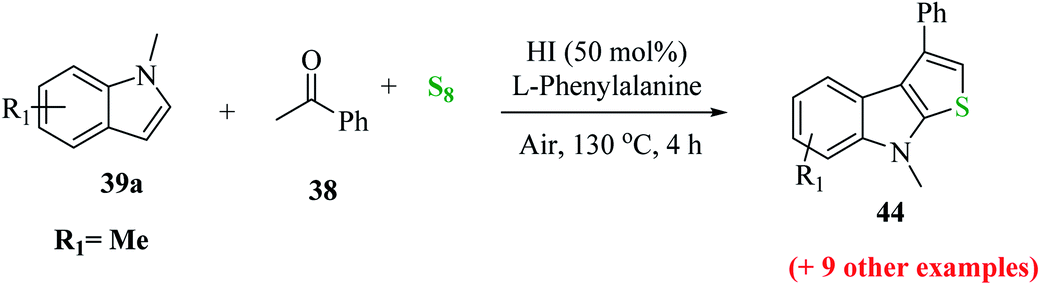 | ||
| Scheme 18 Synthesis of 3-substituted thieno[2,3-b]indole analogs by varying the position of the functional group on the indole ring. | ||
 | ||
| Scheme 19 Synthesis of 2-substituted thieno[2,3-b]indole analogs using different substituted ketones. | ||
Further, indoles bearing various substituents gave the product in a yield of up to 83%. When Me was at the C-6 or C-7 position of the indole moiety, the yield of product 45 decreased slightly to 67% and 73%, respectively, as shown in Scheme 20.
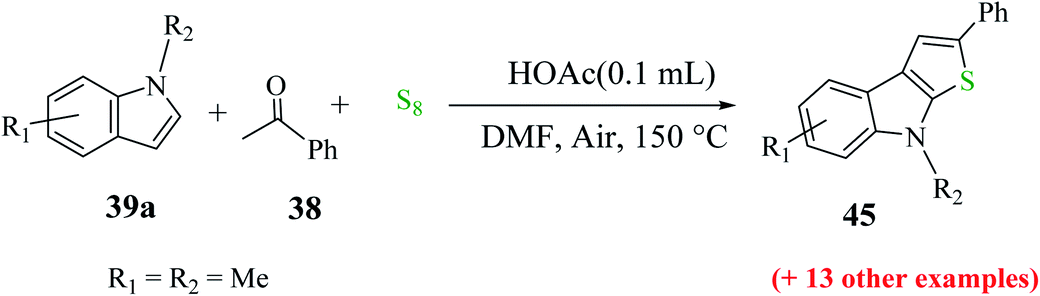 | ||
| Scheme 20 Synthesis of 2-substituted thieno[2,3-b]indole analogs using different indoles. | ||
 | ||
| Scheme 21 Schematic representation of 3-phenylthieno[2,3-b]indole synthesis. | ||
 | ||
| Scheme 22 Mechanism for the formation of 2-phenylthieno[2,3-b]indole. | ||
2.6 Three-component metal free synthesis of thieno[2,3-b]indole in sulfur powder
Li et al.31 synthesized thieno[2,3-b]indole from indoles and alkenes or alkynes in the presence of sulfur powder and in the absence of metal. This is a simple and efficient method in which a Brønsted acid promotes the formation of substituted thieno[2,3-b]indole, where DMF is essential for converting the reactants into the fused products. Substituted 1-methylindole 39a, substituted alkyne 50 and sulfur powder were treated in acid at 150 °C and in metal-free conditions using DMF as a solvent to obtain product 51. However, in the absence of DMF, no product was obtained (Scheme 23).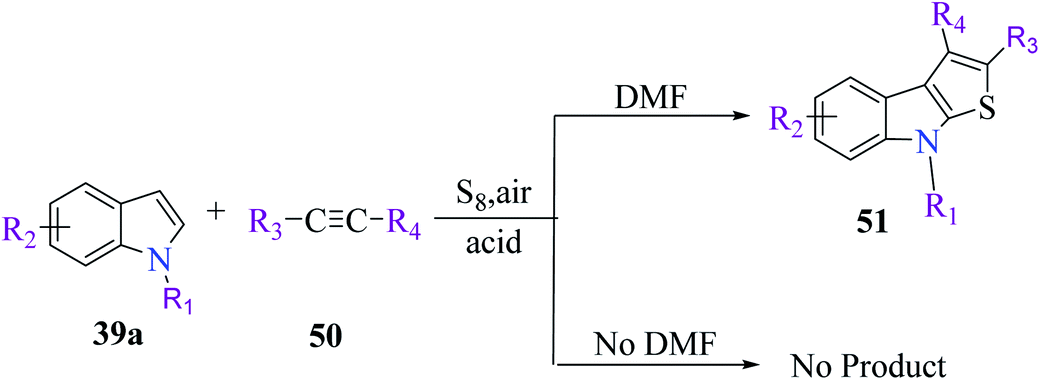 | ||
| Scheme 23 Strategy for the synthesis of indole-fused heterocycle. | ||
Further, product 41 was obtained in 70% yield when AcOH was used as an acid. Moreover, inorganic acids such as hydrochloric acid acted as the most efficient acid and gave the product in 86% yield. Reducing either the reaction temperature or HCl concentration reduced the yield of the product (Scheme 24).
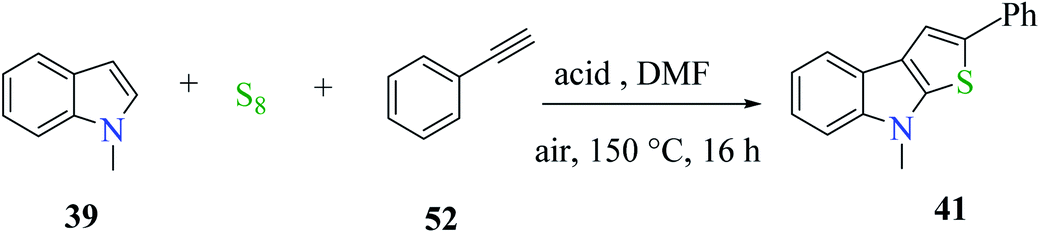 | ||
| Scheme 24 Optimization of the reaction conditions. | ||
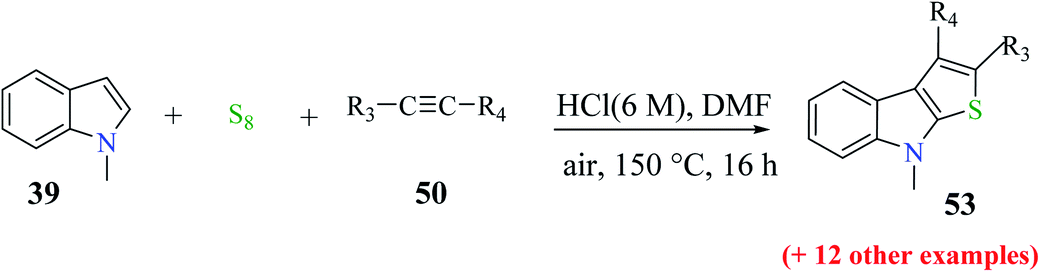 | ||
| Scheme 25 Synthesis of thieno[2,3-b]indole analogs by varying the substituents on the alkyne. | ||
53 was obtained in 31% to 82% when substituted alkynes were used. Para-substituted phenylacetylene such as p-ethoxyphenylacetylene reacted with 1-methylindole 39 in the presence of sulfur powder to give the product 2-(4-ethoxyphenyl)-8-methyl-8H-thieno[2,3-b]indole 53a in 82% yield. When 1-chloro-2-ethylbenzene was used as the substrate, 31% of 2-(2-chlorophenyl)-8-methyl-8H-thieno[2,3-b]indole 53b was formed due to the steric hindrance caused by the substrate and when aliphatic alkynes such as 1-octyne were used as the substrate, 31% of 2-hexyl-8-methyl-8H-thieno[2,3-b]indole 53c was formed because aliphatic alkynes do not favour annulation (Fig. 7).
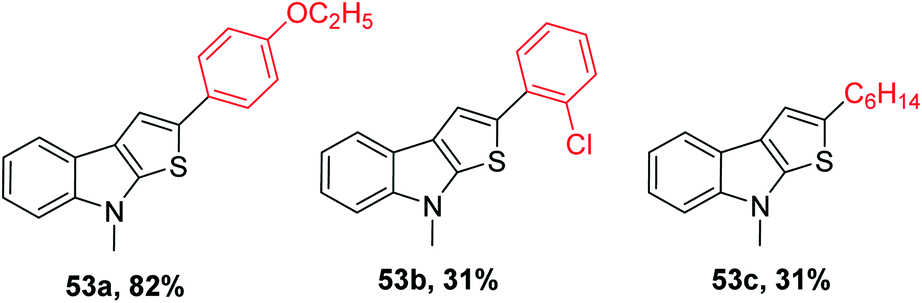 | ||
| Fig. 7 Analogs of 2-substituted thieno[2,3-b]indole. | ||
Further, different substituted indoles 39a were reacted with phenylacetylene 52 and sulfur powder and the product was obtained in yields of up to 81%. When methyl was substituted at the C-5 position of 1-methyl-1H-indole, 61% of 5,8-dimethyl-2-phenyl-8H-thieno[2,3-b]indole was obtained, and when it is substituted at the C-7 position, 56% of 7,8-dimethyl-2-phenyl-8H-thieno[2,3-b]indole was formed (Scheme 26).
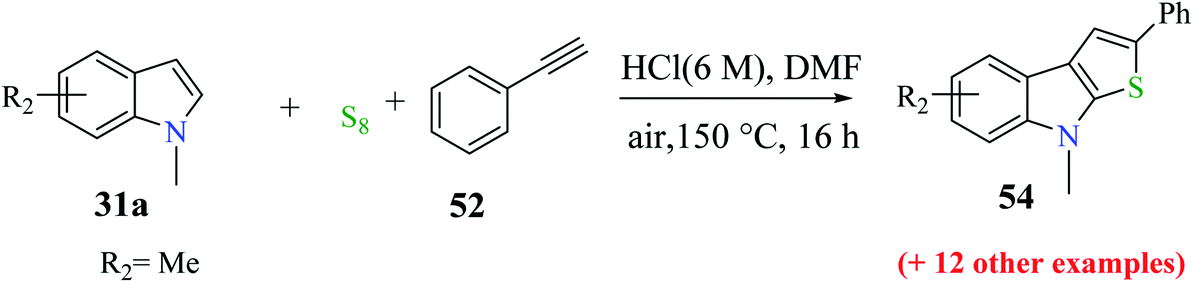 | ||
| Scheme 26 Extension of the substrate scope with respect to indoles. | ||
Moreover, 1-methylindole 39 was reacted with substituted styrene 55 and sulfur powder as the source of sulfur to get up to 76% of 2-substituted thieno[2,3-b]indole. Styrene derivatives with either an electron-donating group such as methoxy and methyl, or halogen (F, Cl or Br) gave the product in good yield upon reacting with 1-methylindole. Overall, the reaction yield was influenced by the placement of the substituent on the phenyl motif of styrene. The use of 1-chloro-2-vinyl benzene as the substrate gave product 56 in 30% yield (Scheme 27).
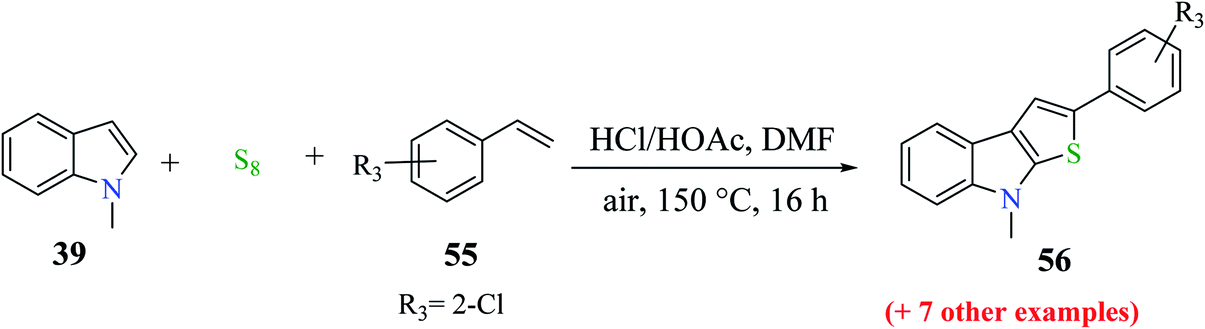 | ||
| Scheme 27 Synthesis of thieno[2,3-b]indoles using different substituted styrenes. | ||
The reactions of substituted indoles 39a such as methyl, methoxy, bromo, fluoro and chloro gave the product in good yield independent of the position of the substituent at the C-5, C-6 or C-7 position. Even good yield of the product was obtained when unprotected indole was used as the substrate (54–79%, Scheme 28).
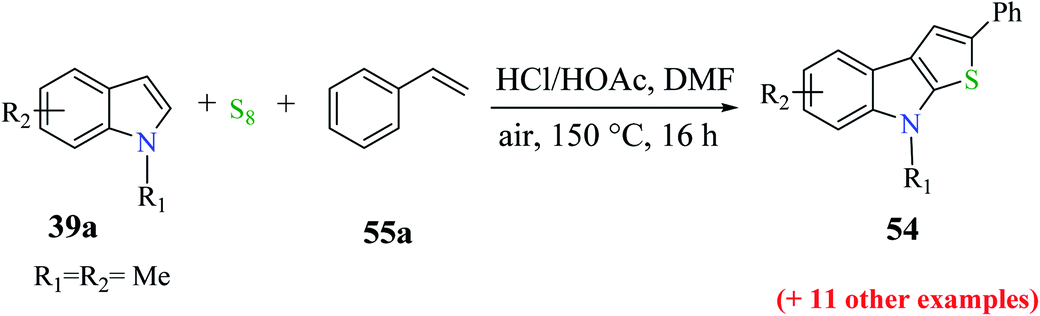 | ||
| Scheme 28 Reaction of different substituted indoles with styrene under the optimized reaction conditions. | ||
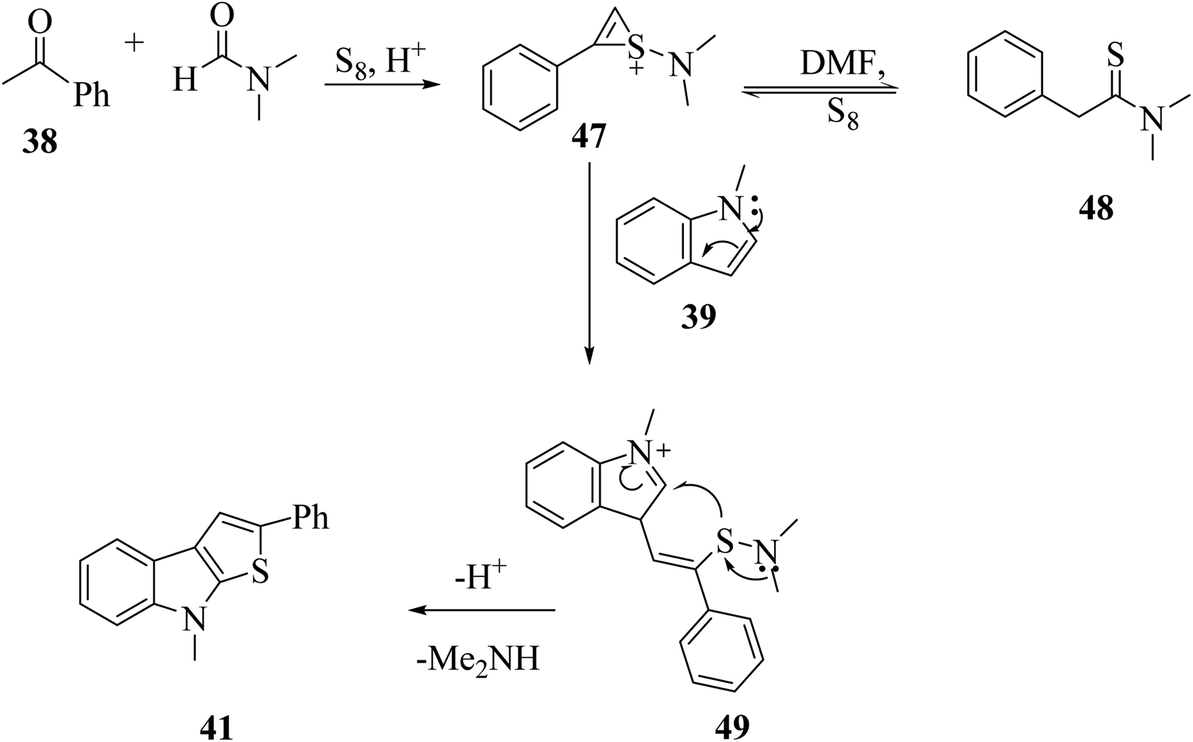
Plausible mechanism involves the reaction of phenylacetylene 52 with DMF and sulfur powder to form intermediate 47, which resonates to form 48. Further, 47 reacts with 1-methyl-indole 39 to form 49, which ultimately gets converted to 8-methyl-2-phenyl-8H-thieno[2,3-b]indole 41, the desired product32,33 in moderate yield (Scheme 29).
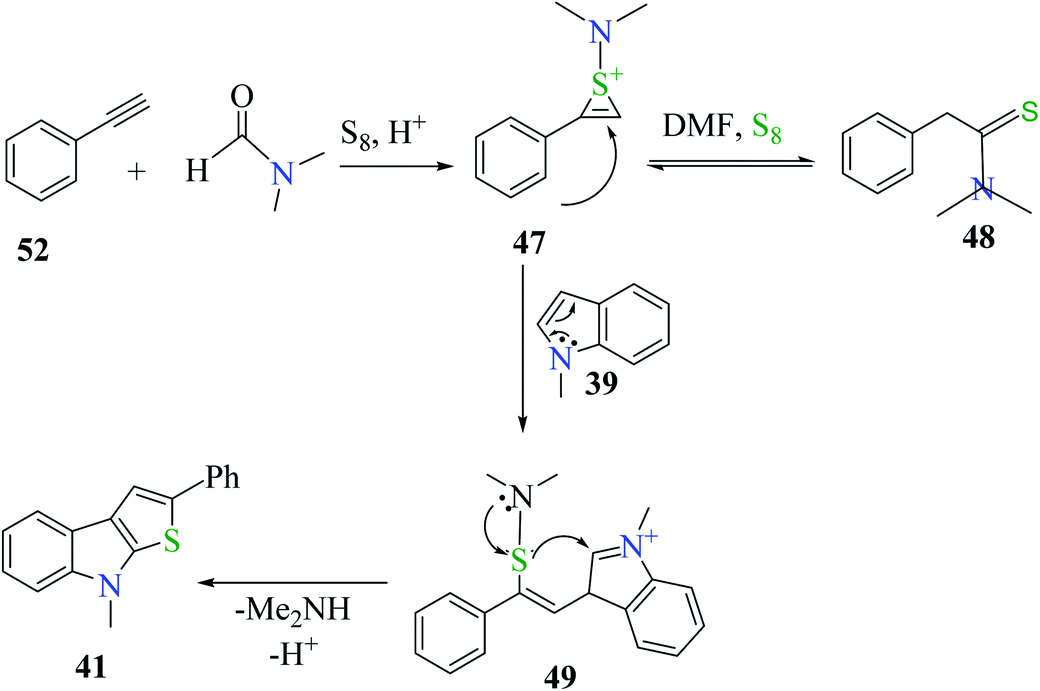 | ||
| Scheme 29 Proposed mechanism for the formation of thieno[2,3-b]indole derivative 41. | ||
2.7 Preparation of thieno[3,2-b]indole by halogen dance and sequential coupling reaction
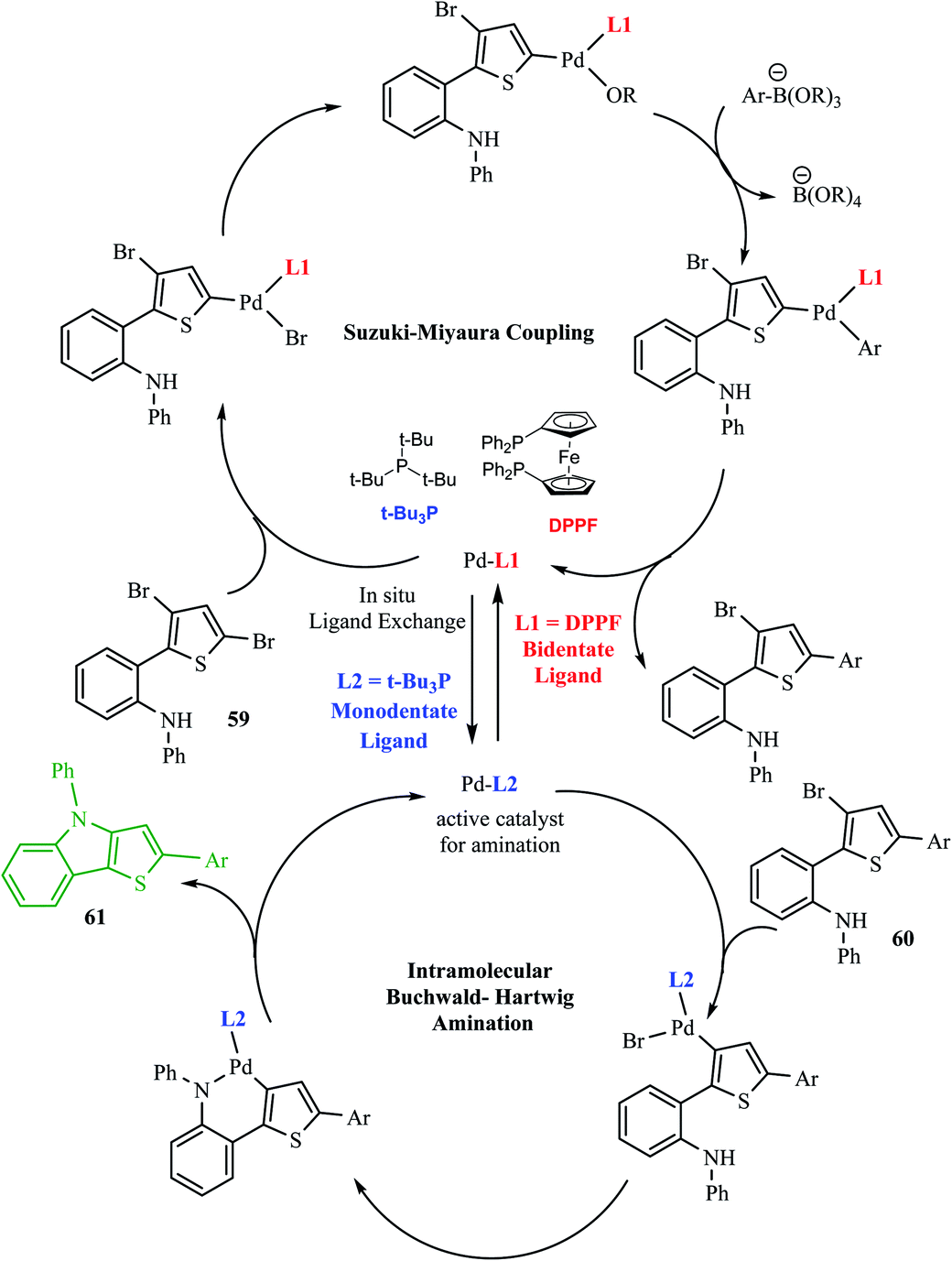
Hayashi et al.34 reported the synthesis of thieno[3,2-b]indole from 2,5 dibromothiophene 57 and lithium diisopropylamide (LDA), which resulted in the formation of transient thienyl anion species 58 via the mediated halogen dance reaction,35 which led to the development of two chemical bonds in one pot, followed by Negishi coupling36 and Suzuki–Miyaura coupling or Buchwald–Hartwig amination via tandem catalysis in the presence of 1,1′-bis(diphenylphosphino)ferrocene (dppf) and tri-tert-butylphosphine (tBu3P) ligand to give thieno[3,2-b]indole.The favourable environment for halogen dance with 2,5-dibromothiophene 57 occurred by its deprotonation at the 4-position with 1.3 eq. LDA at −78 °C for 5 min.37 Moreover, the lithiated species gets rearranged to 5-lithio-4-brominated thiophene 58 at −78 °C via halogen dance, which on treatment with ZnCl2 in tetramethylethylenediamine38 (TMEDA 1.4 equiv.) at 0 °C formed thienylzinc species 58a, which was further subjected to coupling conditions using the transition metal catalyst Pd(PPh3)4 and protected iodoaniline at 60 °C for 24 h, forming the coupled product 59 in 69% yield (Scheme 30).
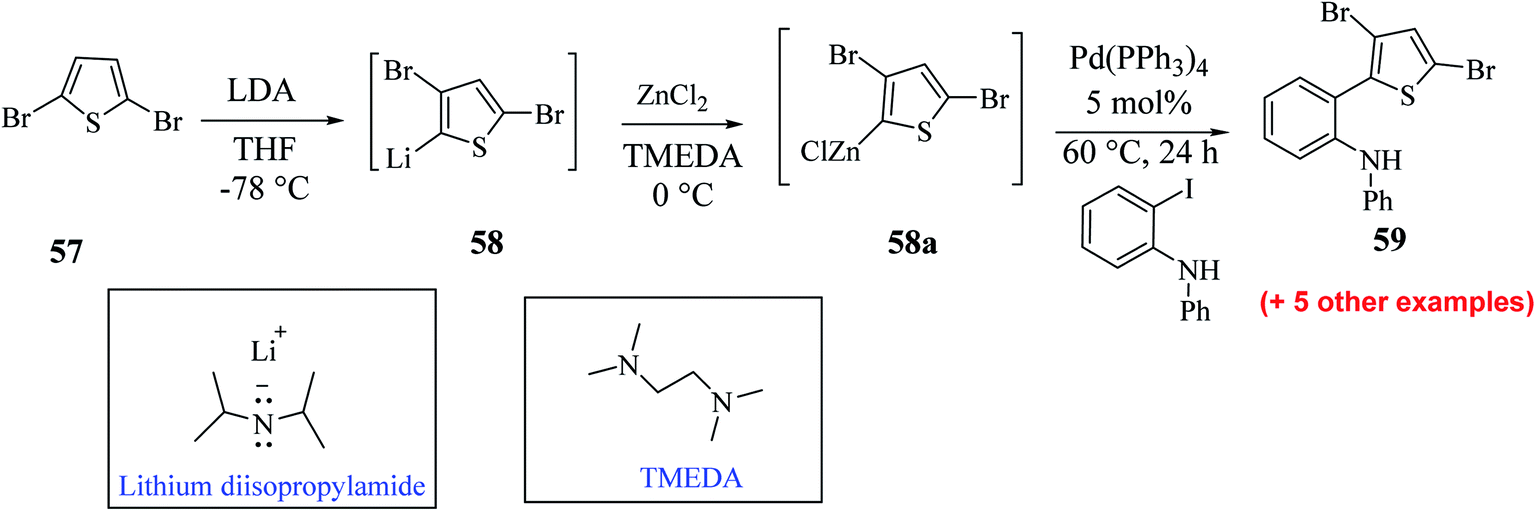 | ||
| Scheme 30 Synthetic route for 2-(3,5-dibromothiophen-2-yl)-N-phenylaniline. | ||
Hayashi et al.34 reported the synthetic approach of 59 towards ligand-controlled Suzuki–Miyaura coupling and Buchwald–Hartwig amination via assisted tandem catalytic transformation. Here, 2-(3,5-dibromothiophen-2-yl)-N-phenylaniline 59 gave π-conjugated thienoindole via Suzuki–Miyaura coupling39,40 and C–N bond formed by Pd-catalyzed reaction in the presence of aryl halide and amine groups in a stoichiometric amount of base (intramolecular Buchwald–Hartwig amination).41–43 Arylation at the α-position of thiophene resulted in the formation of 2-(3-bromo-5-(p-tolyl)thiophen-2-yl)-N-phenylaniline 60 and amination of the remaining β-bromo group with 2-aminophenyl group led to the formation of 4-phenyl-2-(p-tolyl)-4H-thieno[3,2-b]indole 61 (Scheme 31).
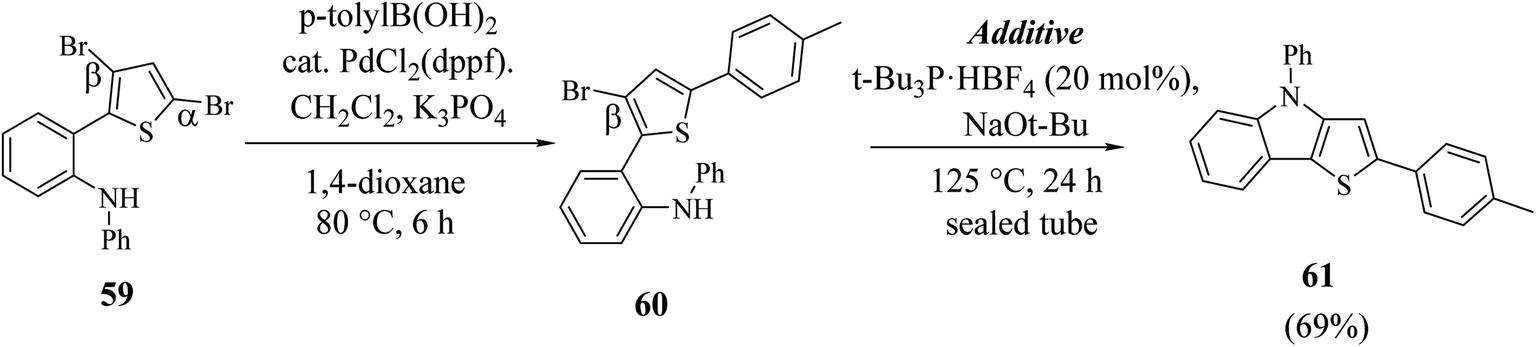 | ||
| Scheme 31 Synthetic route for the stepwise Suzuki–Miyaura coupling and intramolecular Buchwald–Hartwig amination reaction. | ||
The above-mentioned reaction failed in the absence of additive, as reported by Okano and coworkers,44 demonstrating that the additive plays an important role in significant amination for getting the desired product. Despite the longer response time of 24 h at 125 °C, additives such as NaOtBu were detected to be insufficient. The addition of tBu3P·HBF4 (20 mol%) notably promoted amination to form 4-phenyl-2-(p-tolyl)-4H-thieno[3,2-b]indole 61 in 69% yield. A reduced amount of the phosphorus ligand in the additive led to a lower yield of 61. This outcome suggests that tBu3P, a monodentate ligand, needs to coordinate Pd with dppf, a bidentate ligand to form the efficient catalyst in situ for intramolecular amination (Scheme 32).
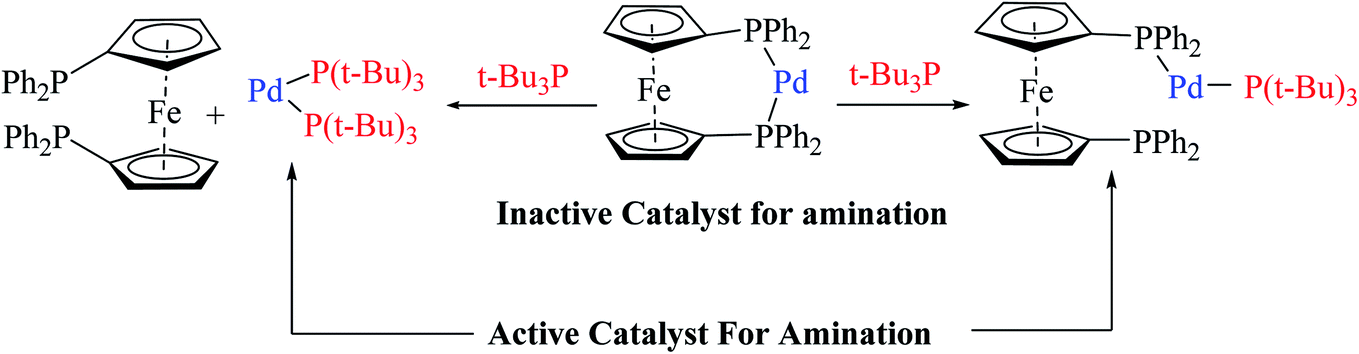 | ||
| Scheme 32 Action of additive t-Bu3P on activity of the Pd-dppf catalyst for amination. | ||
The one-pot reaction was also reported at 125 °C for 5 h by one-shot addition, which included all the required reagents for forming aryl substituted thieno[3,2-b]indole (Scheme 33).
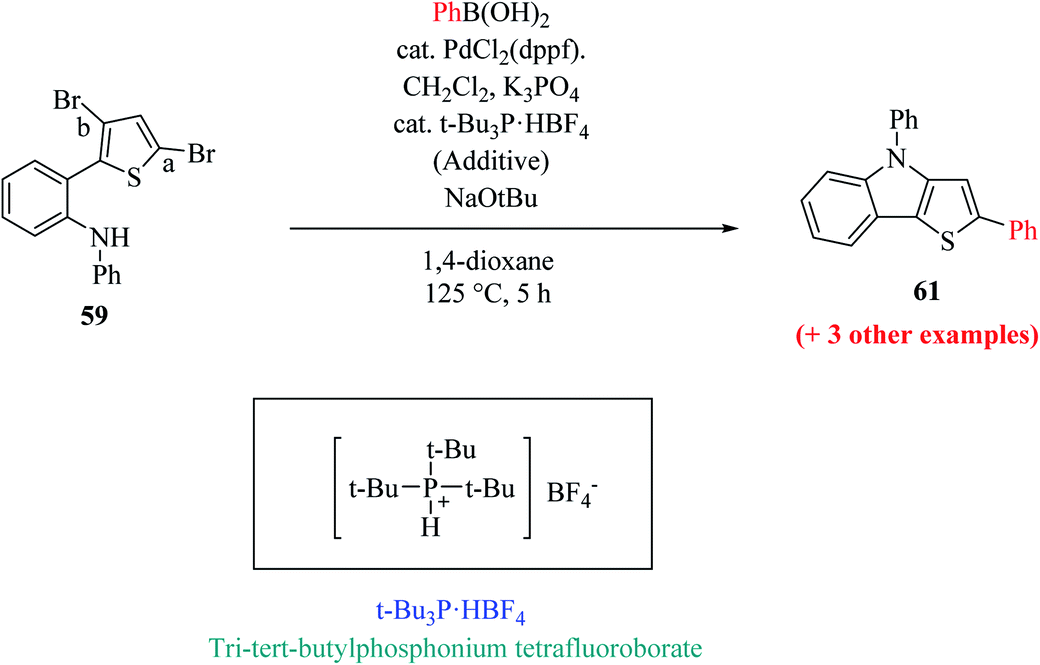 | ||
| Scheme 33 Synthetic route for one-pot single-shot addition. | ||
A few of the limitations of the above-mentioned one-pot reaction were justified based on the variation in the Ar group at the second position of thieno[3,2-b]indole 61. The above-mentioned reaction conditions (Scheme 20) were not applicable for Ar = 4-nitrophenylboronic acid given that it has low solubility in 1,4-dioxane, and thus water as the co-solvent in a ratio of 4:1 mainly helps to maximize the yield of 2-(2-nitrophenyl)-4-phenyl-4H-thieno[3,2-b]indole (Ar = 2-nitrophenyl) to 43%.
The Pd-dppf catalyst plays an important role in Suzuki–Miyaura coupling, which led to C–C bond formation at the α-position of thiophene by releasing the bromo group. Further, ligands were exchanged from Pd-dppf to Pd-(tBu3P)2 and the reaction moves towards C–N bond formation in which the β-bromo group undergoes oxidative addition of the Pd-(tBu3P)2 catalyst and reductive elimination to yield 4-phenyl-2-(aryl)-4H-thieno[3,2-b]indole 61. The maximum yield was obtained when tBu3P was used as an additive for amination. The following sequential coupling reactions (Scheme 34) were reported as tandem catalytic pathways.34
 | ||
| Scheme 34 Tandem catalytic pathway for sequential coupling reaction. | ||
2.8 Synthesis of 2-(hetero)aryl-substituted thieno[3,2-b]indole via Fisher indolization
Irgashev et al.45 reported the Fischer indolization46,47 methodology for the development of 2-(hetero)aryl-substituted thieno[3,2-b]indole 69 from 5-(hetero)arylthiophen-3(2H)-ones 68 and phenyl hydrazine 62. The intermediate 5-(hetero)arylthiophen-3(2H)-one was acquired in two steps, i.e., the reaction of α-bromocinnamates or their other hetero derivatives with methyl thioglycolate, followed by the base treatment of 5-(hetero)aryl-3-hydroxysubstituted 2-thenoates to yield 2-(hetero)aryl-substituted thieno[3,2-b]indole.Convenient and cheap synthetic approaches are urgent to meet the growing demand of hetero-arylated thieno[3,2-b]indoles, which are widely used in optoelectronic material engineering. In this context, the retro-synthetic approach towards the synthesis of thienoindoles is shown in the following scheme (Scheme 35).
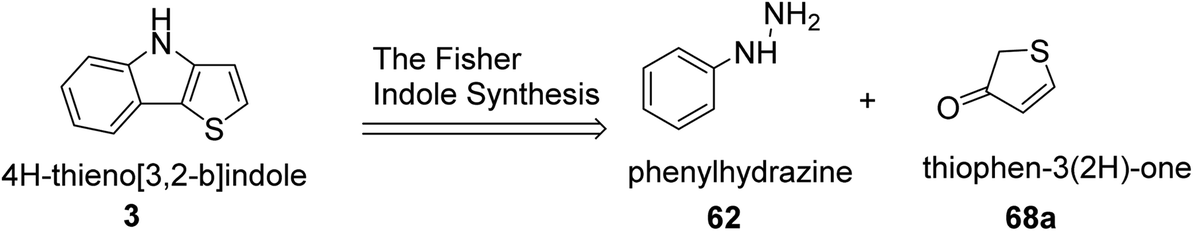 | ||
| Scheme 35 Retrosynthetic route of thieno[3,2-b]indole via Fisher indole synthesis. | ||
An appropriate route for the synthesis of alkyl 3-hydroxythiophene-2-carboxylates (2-thenoates) via Fiesselmann thiophene synthesis48 was reported. It is a condensation reaction of 1,3-C,C-dielectrophilic substrates and alkyl thioglycolates upon treatment with base. 5-(Hetero)aryl-3-hydroxy-substituted 2-thenoates were synthesized in two steps starting from substrate 2-(hetero)aroylacetates49 64 or 3-(hetero)arylpropiolates50 65 or 2-bromo-3-(hetero)arylacrylates 66. Moreover, 2-bromo-3-(hetero)arylacrylates 66 were easily available and also act as a 1,3-dielectrophilic three-carbon center for the Fiesselmann method. Further, 68 was formed by ester hydrolysis and in situ decarboxylation of alkyl 3-hydroxythiophene-2-carboxylates 67 (Scheme 36).
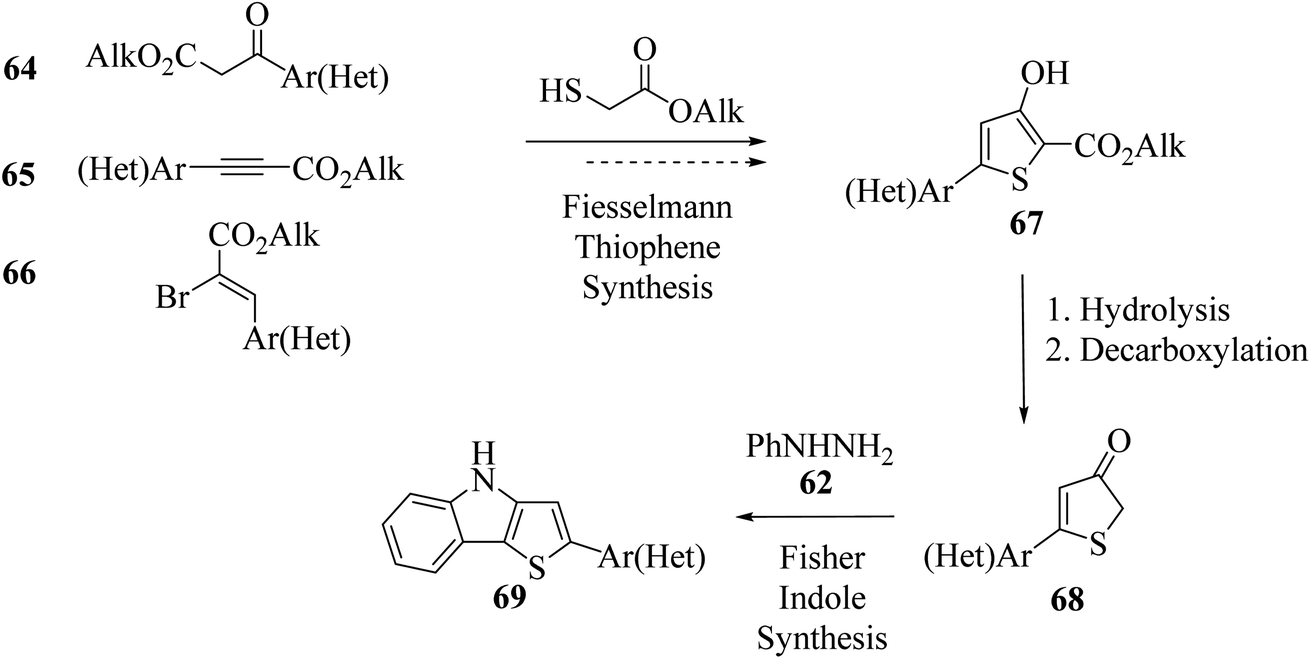 | ||
| Scheme 36 Synthetic route of 2-(hetero)aryl-substituted thieno[3,2-b]indoles via Fiesselmann and Fischer method. | ||
The maximum yield of the product was reported when 2-bromo-3-(hetero)arylacrylate 66 and methylthioglycolate were slowly added to NaOMe base (4 equiv.) in dry MeOH and refluxing the reaction mixture for 5 h resulted in the formation of methyl-5-aryl-3-hydroxythiophene-2-carboxylates 70 (Scheme 37).
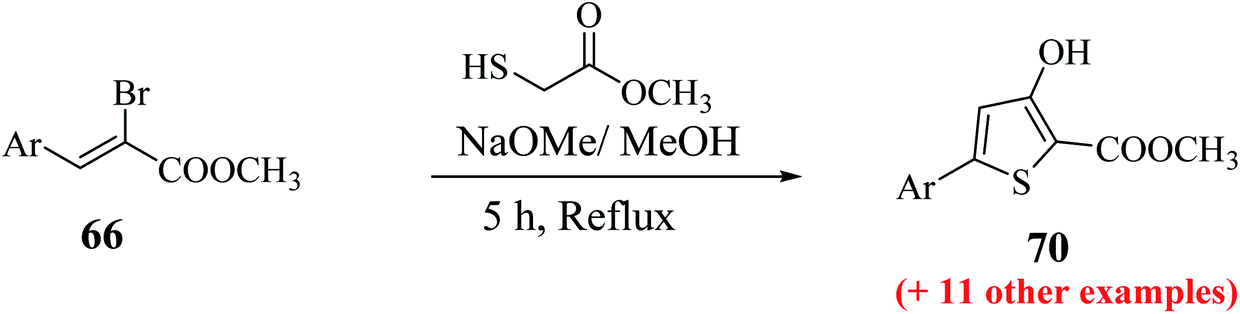 | ||
| Scheme 37 Synthetic route for methyl 5-aryl-3-hydroxythiophene-2-carboxylates. | ||
Methyl 5′-bromo-4-hydroxy-[2,2′-bithiophene]-5-carboxylate 70a was synthesized from methyl-2-bromo-3-(5-bromothiophen-2-yl)acrylate 66a, which was in turn prepared via the esterification of 2-bromo-3-(5-bromothiophen-2-yl)acrylic acid51 (Scheme 38). The ester having a Br group was changed to 5-phenylthien-2-yl-connected derivative 71 via Suzuki–Miyaura reaction.
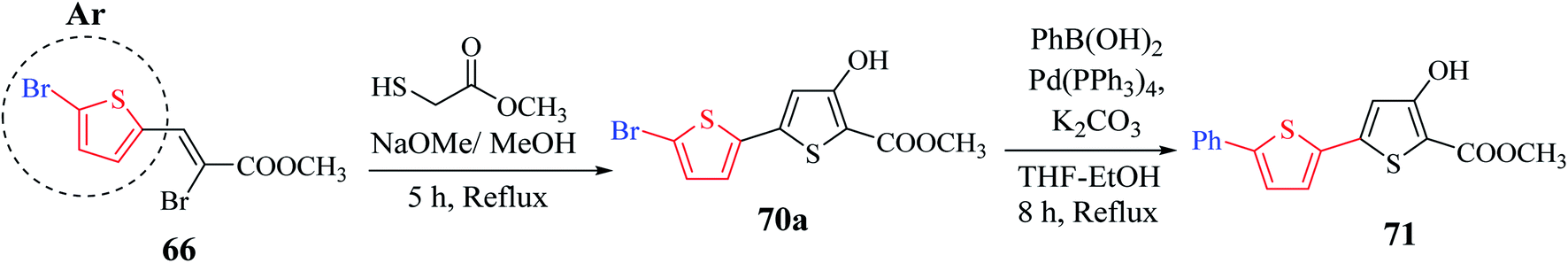 | ||
| Scheme 38 Synthetic route to methyl 4-hydroxy-5′-phenyl-[2,2′-bithiophene]-5-carboxylate 71. | ||
Further, saponification45 of methyl-4-hydroxy-5′-phenyl-[2,2′-bithiophene]-5-carboxylate 71 in the presence of NaOH and DMSO–H2O for 1.5 h at 140 °C under an argon atmosphere yielded 5′-phenyl-[2,2′-bithiophen]-4(5H)-one 72 in 94–95% yield, which on further treatment with phenylhydrazine 62 in glacial CH3COOH for 1 h at 120 °C, yielded 2-(5-phenylthiophen-2-yl)-4H-thieno[3,2-b]indole 73 in 58% yield (Scheme 39).
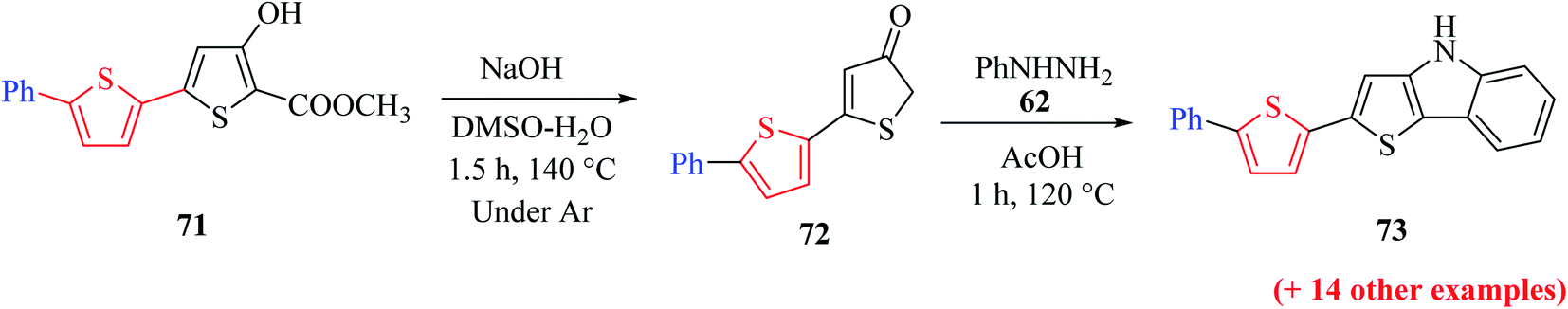 | ||
| Scheme 39 Synthetic approach towards the formation of 2-(5-phenylthiophen-2-yl)-4H-thieno[3,2-b]indole 73. | ||
2.9 One-pot approach for synthesizing thieno[2,3-b]indole starting from indoline-2-thiones and nitroalkene-based adducts
Mane et al.52 reported a novel approach for the synthesis of thieno[2,3-b]indoles via the base-assisted [3 + 2]annulation of indoline-2-thione involving nitroalkene adducts, i.e., Morita–Baylis–Hillman (MBH)53,54 76 and Rauhut–Currier (RC)55 78 adduct. The α-hydrazino-α,β-unsaturated nitroalkene intermediate was synthesized in excellent yields from imidazole or DMAP directed Morita–Baylis–Hillman reaction of nitroalkene and azodicarboxylates.56 Nitroalkene-based Rauhut–Currier adducts57 are effective synthons for synthesizing various functionalized heterocycles. Also, these synthons are great Michael acceptors, and also employed in Cascade Michael addition–cyclization reactions. The MBH adduct and RC adduct were synthesized, as shown in Scheme 40.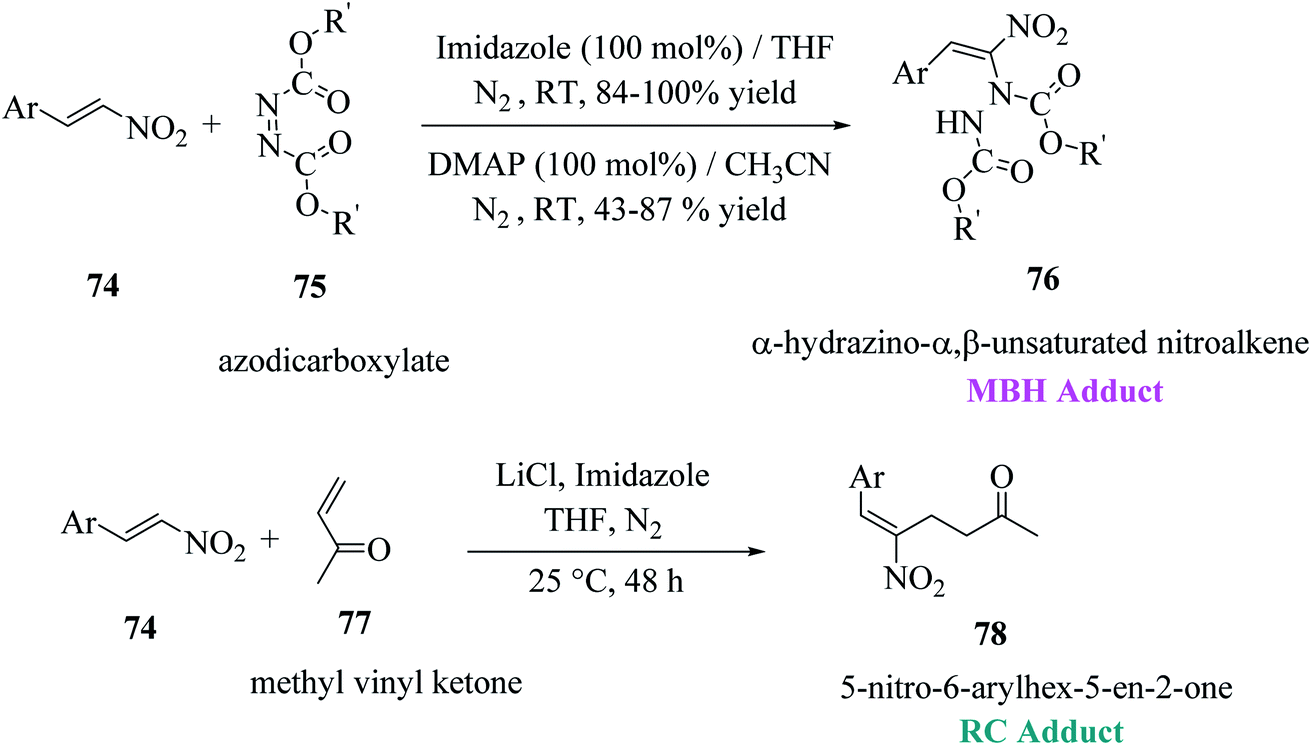 | ||
| Scheme 40 Synthesis of nitroalkene-based MBH and RC adducts. | ||
The reaction of 1-methylindoline-2-thione 79a and α-hydrazinonitroalkenes 76 was performed by using potassium acetate/acetic acid as an additive and CH3CN as the solvent to obtain diisopropyl-1-(8-methyl-3-(p-tolyl)-8H-thieno[2,3-b]indol-2-yl)hydrazine-1,2-dicarboxylate 80 in 78% yield.
Various organic and inorganic bases were tested as substitutes for KOAc to get the product in higher yield. Among them, K2CO3, NaOH, Cs2CO3 (inorganic bases) and Et3N (organic base) in the presence of CH3CN resulted in the formation of dihydrothienoindole 80a (Scheme 28), which was finally converted to thieno[2,3-b]indole derivative 80 (Scheme 41).
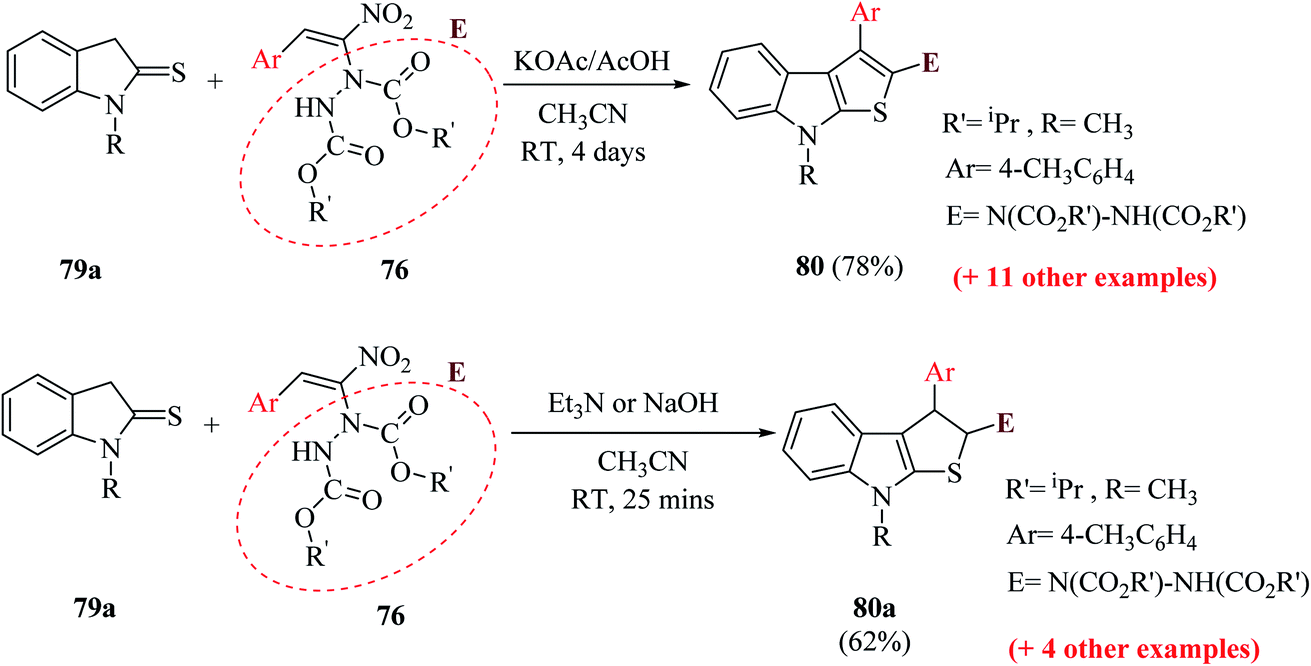 | ||
| Scheme 41 Synthesis of derivatives of thienoindole 80 and dihydrothienoindole 80a via MBH reaction. | ||
In the synthesis of thieno-[2,3-b]indole via MBH reaction, firstly proton abstraction at the C-3 position of indoline-2-thione 79 takes place to form an anion, which attacks hydrazinonitroalkene 76 via Michael addition reaction to form an intermediate that is further activated by the H-bonding property of acetic acid to form 81a. Subsequently, the removal of the nitro group was facilitated by the lone pair of the hydrazine moiety and an acyl iminium-type intermediate 81b was generated. Following this, intramolecular 5-exo-trig cyclization of 80 occurred to form dihydrothienoindole 81c, which underwent aerial oxidation to give aromatized thienoindole 80.
The reaction can also occur in a different manner, where thio-enolization of 81a takes place initially, and then intramolecular 5-exo-tet cyclization and aerial oxidation takes place to form thieno[2,3-b]indole 80 (Scheme 42).
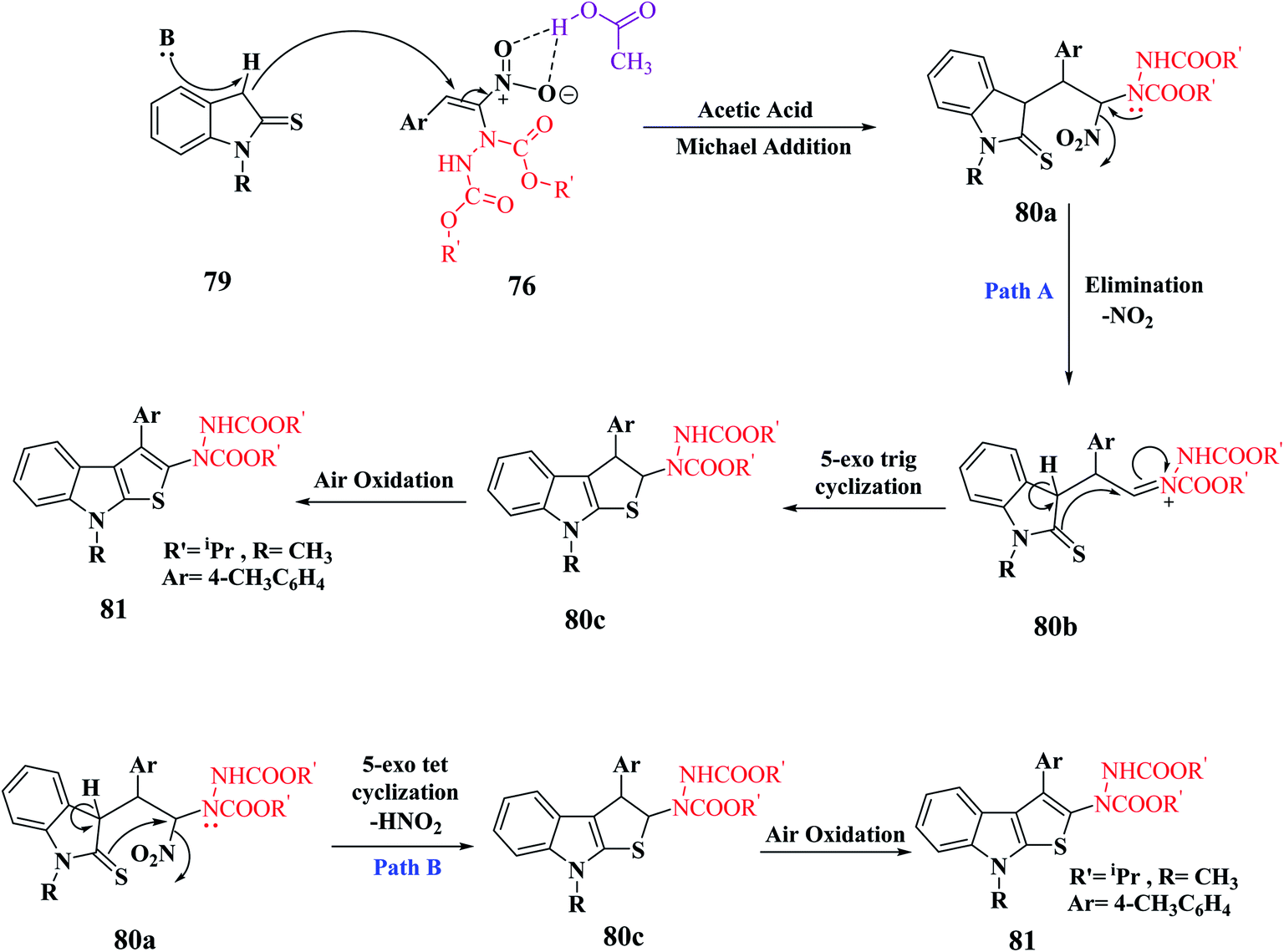 | ||
| Scheme 42 Mechanism for the synthesis of thieno[2,3-b]indole. | ||
Mane et al.52 also reported an alternative efficient approach for the synthesis of thieno[2,3-b]indole derivatives. They treated substituted RC adduct 78 with N-protected indoline-2-thione 79 in K2CO3 (1.0 eq.) and CH3CN:H2O solvent mixture to get 4-(8-methyl-3-phenyl-8H-thieno[2,3-b]indol-2-yl)butan-2-one 82 in 76% yield (Scheme 43). In addition, it was found that LiCl and H2O as additive enhanced the product yield.
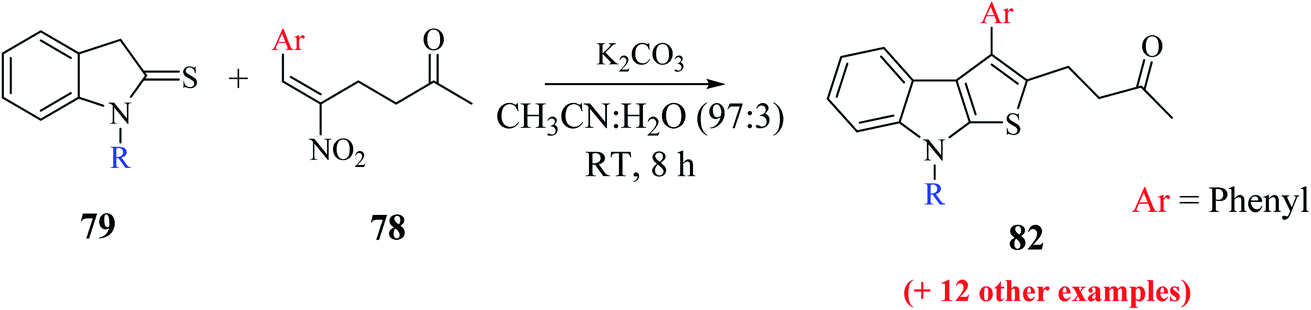 | ||
| Scheme 43 Synthesis of thieno[2,3-b]indole via RC adduct. | ||
The presence of electron-donating groups on the aryl ring of RC-adduct 78 significantly reduced the product (82b and 82c) yield with respect to 82a (Scheme 40, Fig. 8).
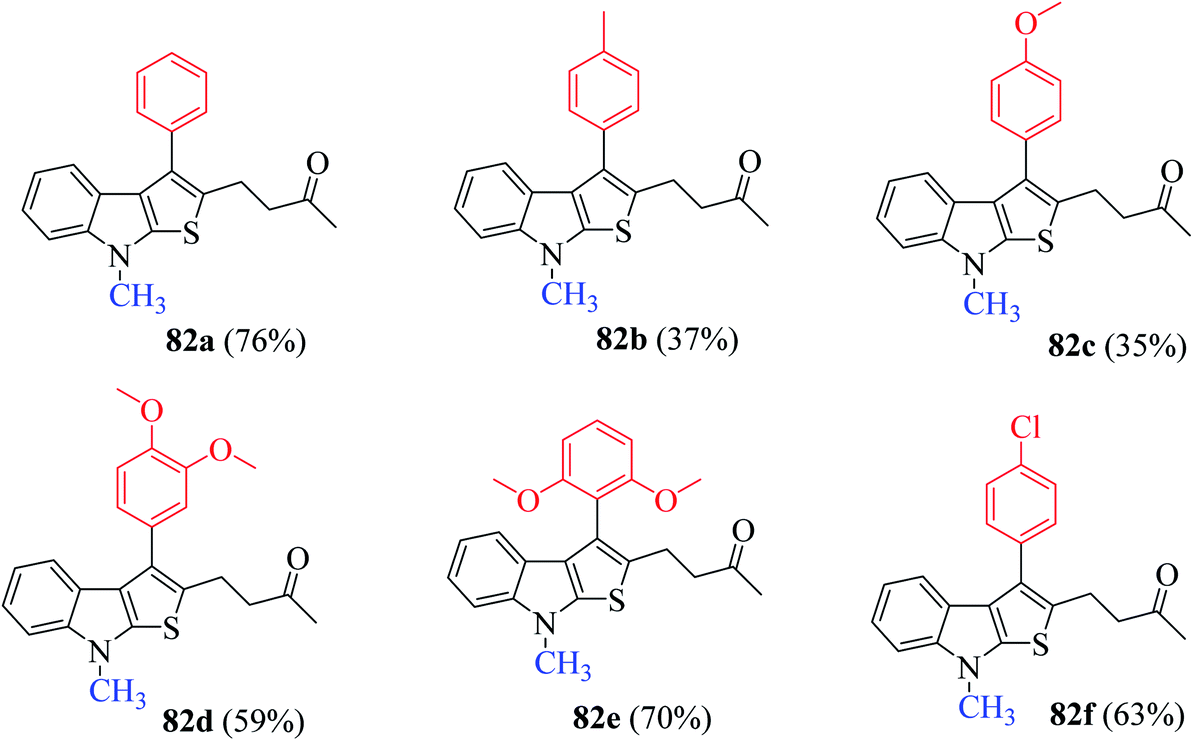 | ||
| Fig. 8 Substrate scope for synthesizing functionalized thieno[2,3-b] indoles using several substituted RC-adducts of nitroalkenes. | ||
(1) Presence of numerous electron-donating groups at several positions resulted in good yield of products 82d and 82e.
(2) Presence of weak electron-withdrawing group such as 4-chloro-substituted RC-adduct 82f led to moderate yield (63%) of the product (Fig. 9).
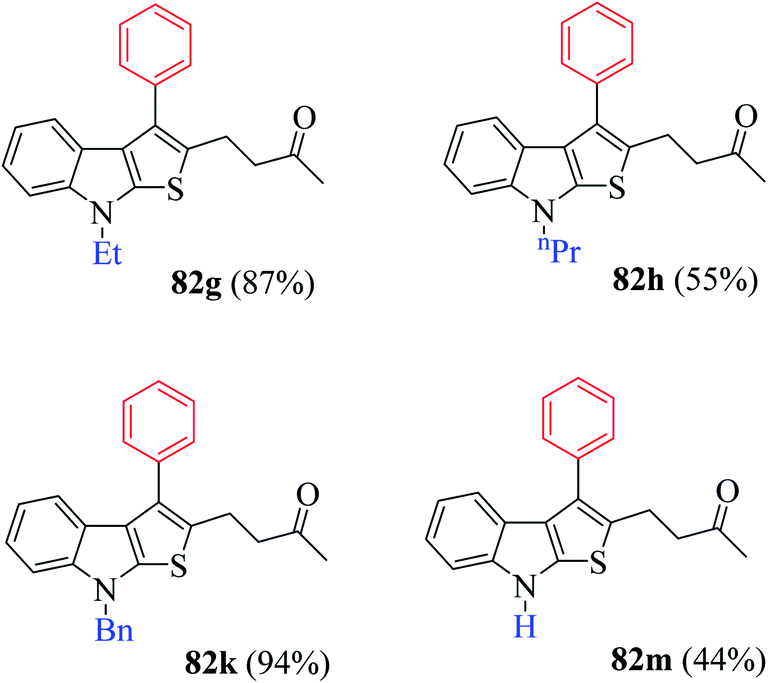 | ||
| Fig. 9 Reaction scope with distinctively substituted indoline-2-thiones. | ||
Several N-protecting groups such as ethyl and benzyl resulted in excellent yield of thieno[2,3-b]indole derivatives 82g and 82k, whereas groups such as H and n-propyl led to a much lower yield of derivatives 82h and 82m (Fig. 9).
The base-directed Michael addition of indoline-2-thione 79 to RC-adduct 78-formed intermediate 83a and intramolecular thio-Mannich-type reaction in 5-exo-trig fashion led to intermediate 83b, and further removal of HNO and H2O gave the aromatized product. The overall mechanism involved in the synthesis of the thienoindole derivative is depicted in Scheme 44.
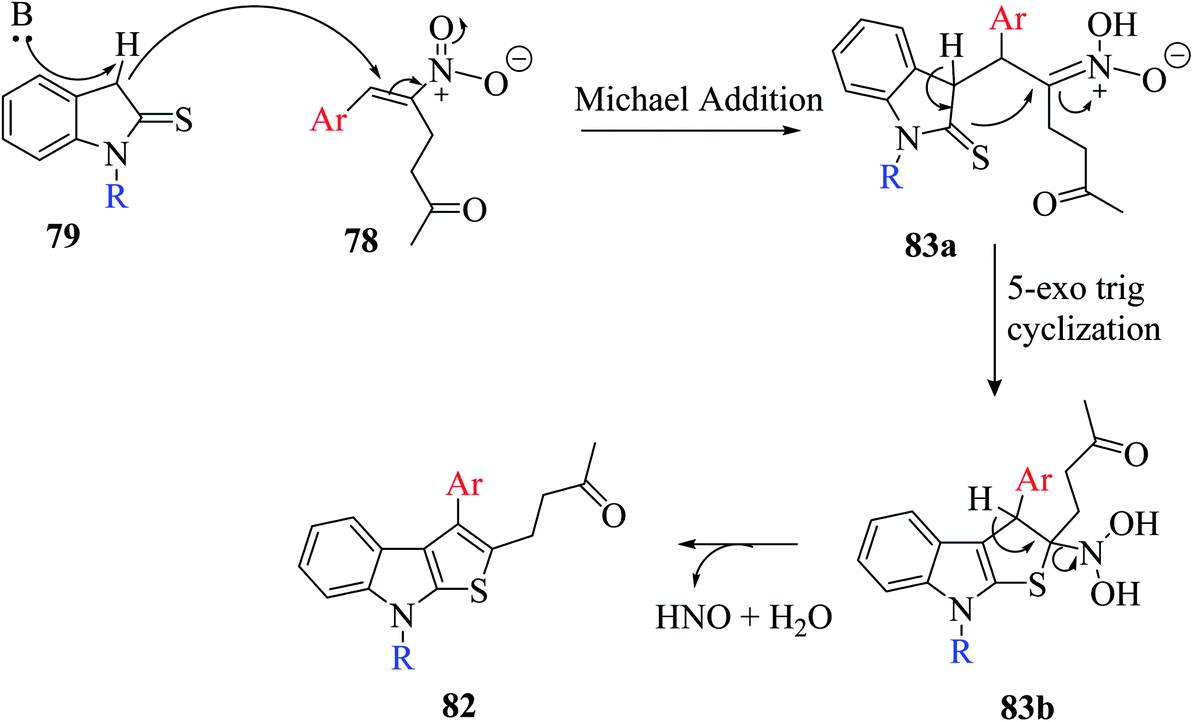 | ||
| Scheme 44 Mechanism for the formation of thieno[2,3-b]indole via the RC adduct. | ||
2.10 Synthesis of thieno[3,2-b]indole derivative via in situ generation of 3-aminothiophene
It is a convenient method to synthesize thieno[3,2-b]indole having a thien-2-yl, aromatic or styryl group at the C-2 position. This method uses 5-substituted-3-aminothiophene-2-carboxylate and proceeds via Fischer indolization. It involves two steps, where the first step is the saponification of 3-aminoester with NaOH, and in the second step, this sodium salt reacts with arylhydrazine in glacial CH3COOH. In the latter step, decarboxylation of 3-aminothiophene-2-carboxylic acid takes place to give 3-aminothiophene, which further reacts with arylhydrazines under acidic conditions to form arylhydrazone, ultimately undergoing Fischer indolization to give the desired product58 73.Thiophene derivatives with an amino group at the C-3 or C-3 and C-4 position have found numerous applications in the formation of thiophene-fused N-heterocycles such as thienoindoles.59
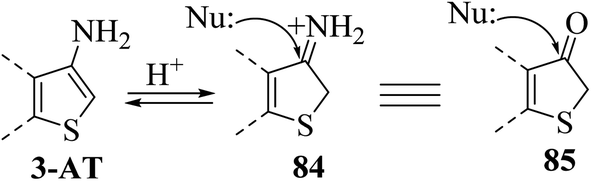 | ||
| Scheme 45 Nucleophilic attack on 3-aminothiophene. | ||
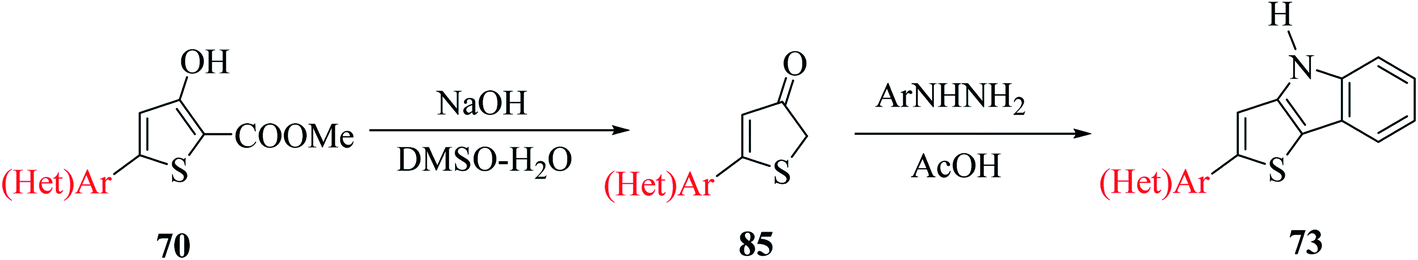 | ||
| Scheme 46 Synthesis of thieno[3,2-b]indole from 5(hetero)arylthiophene-3(2H)-ones. | ||
Later, they described another method for the synthesis of 2-substituted thieno[3,2-b]indoles starting from 5-substituted-methyl-3-aminothiophene-2-carboxylates 87 via the in situ generation of 3-aminothiophene, which further participates in Fischer indolization with arylhydrazines (Scheme 47).
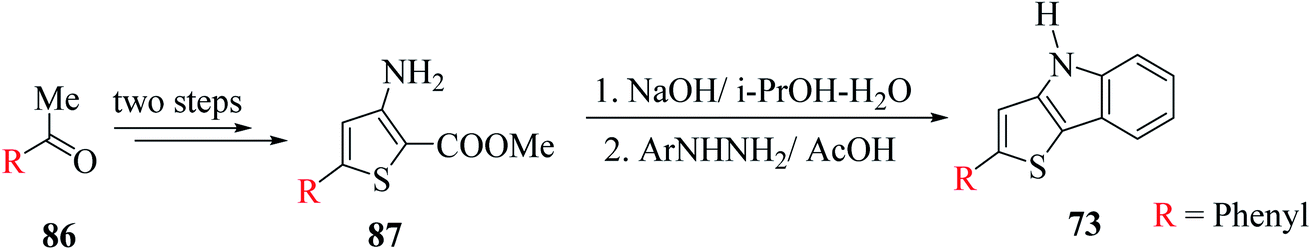 | ||
| Scheme 47 Synthesis of thieno[3,2-b]indole from 5-substituted-methyl-3-aminothiophene-2-carboxylates. | ||
3-Aminothiophene-2-carboxylate 87 was synthesized in two steps. Initially, methyl ketone 86 (R = aryl, styryl or thien-2-yl) was treated with Vilsmeier reagent (POCl3–DMF complex) and NH2OH·HCl at 30–60 °C to give 3-substituted-3-chloroacrylonitrile,61–64 which then reacted with methyl thioglycolate in NaOMe in CH3OH solution65 to form the product following Fiesselmann method for making the thiophene ring66 (Scheme 48).
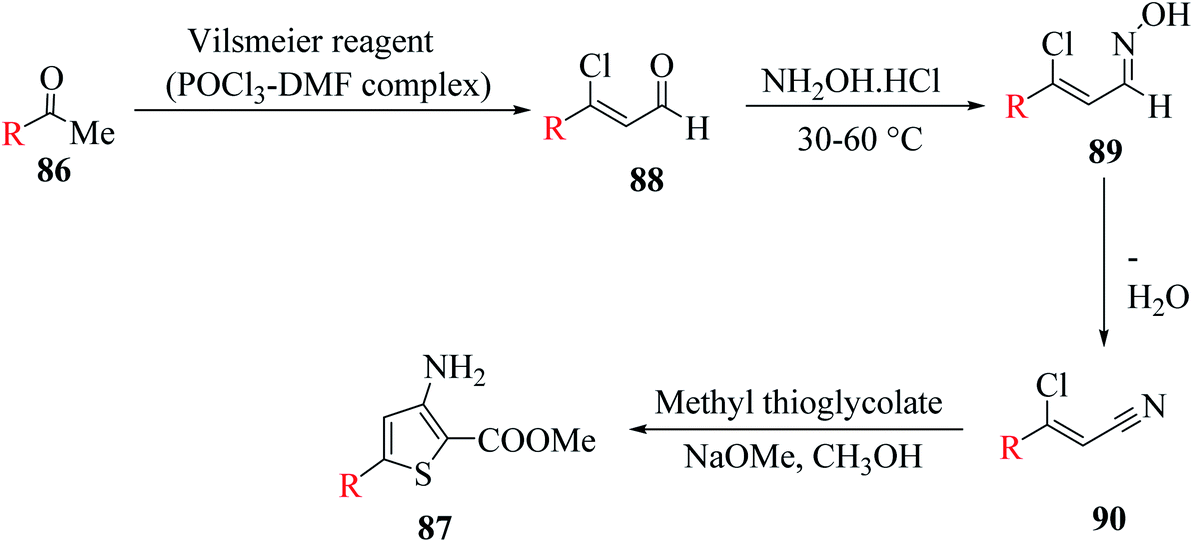 | ||
| Scheme 48 Formation of 5-substituted-3-aminothiophene-2-methylcarboxylate from methyl ketone. | ||
In the earlier method for the preparation of thieno[3,2-b]indole, DMSO was used for the saponification of 3-hydroxythiophene-2-carboxylate. However, the problem was the high temperature required for the reaction due to the initial occurrence of sodium-2-(methoxycarbonyl)thiophen-3-olate. Therefore, the direct conversion of 3-aminothiophene-2-carboxylate was carried out using NaOH in aqueous i-PrOH, without the isolation of the intermediate formed, 3-aminothiophene.
When 3-aminothiophene-2-carboxylates were substituted at the C-5 position by either aromatic or thien-2-yl groups, 55–77% of 2-substituted thieno[3,2-b]indole 73 was obtained (Scheme 49).
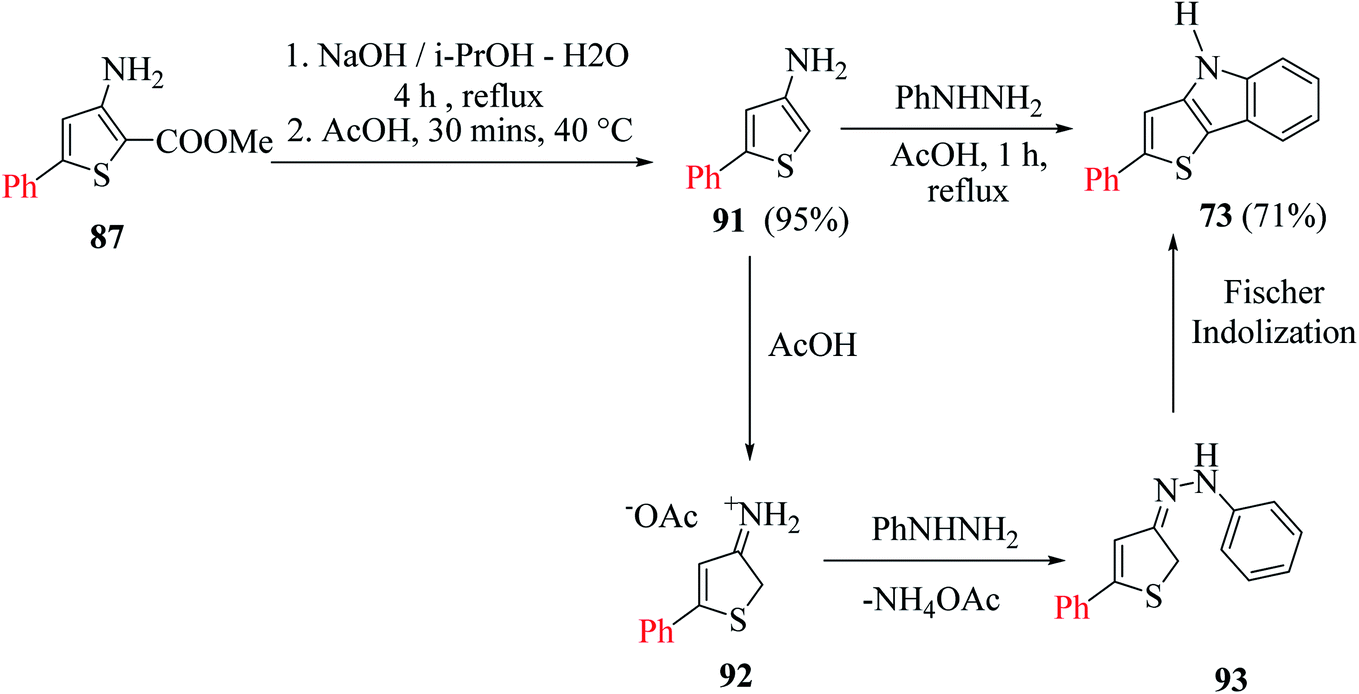 | ||
| Scheme 49 Synthesis of 2-phenylthieno[3,2-b]indole. | ||
2.11 Metal-free synthesis of thieno[2,3-b]indoles using elemental sulfur
Thieno[2,3-b]indoles have been synthesized via a base-assisted metal-free approach using cheap and readily available elemental sulfur.67 The development of attractive and valuable routes for forming carbonyl group-containing thieno[2,3-b]indoles represents several challenges. Here, β-indolyl ketone derivatives were used as effective and easily accessible substrates for the synthesis of carbonyl group-containing thieno[2,3-b]indoles through a CS bond formation reaction.
3-(1H-indol-3-yl)-3-phenyl-1-(o-tolyl)-propan-1-one 94 treated with elemental sulfur and base NaOtBu using anhydrous DMSO as the solvent at 120 °C under N2 for 24 h gave product 99 in 97% yield. Moreover, other organic bases, e.g., DBU, gave the product in a minute amount, whereas DABCO yielded the final product in 90%. The quantity of elemental sulfur did not affect the yield of the product68 (Scheme 50).
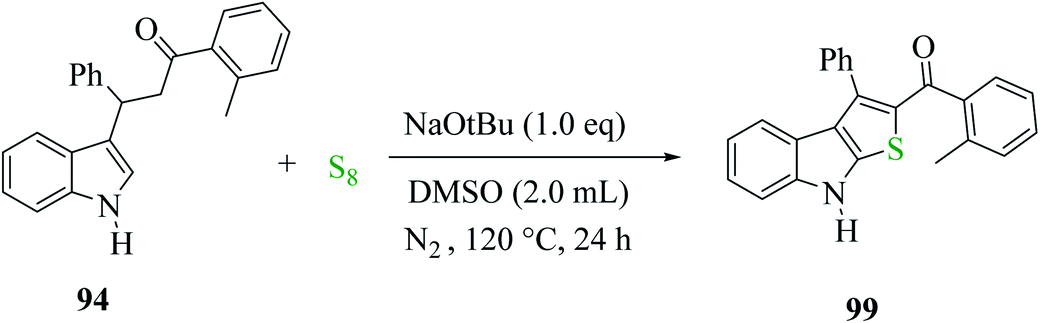 | ||
| Scheme 50 Strategy for preparing 2-substituted-3-phenyl-8H-thieno[2,3-b]indole 99. | ||
Substrate 3-(1H-indol-3-yl)-3-phenyl-1-(o-tolyl)-propan-1-one 94 was converted to intermediate 95 and remained in proper tautomeric equilibrium with 96. Further, the base abstracted the proton and carbon at the α-position of the CO group was electrophilically attacked by the elemental sulfur, giving intermediate 97. Later, intramolecular nucleophilic cyclization of 97 gave 98 by eliminating elemental sulfur (Sn−1) and oxidative aromatization of elemental sulfur formed 2-substituted-3-phenyl-8H-thieno[2,3-b]indole 99 (Scheme 51). Hence, a novel synthetic route to poly-substituted thienoindoles was achieved, where S8 promoted CS bond formation to form the desired product.
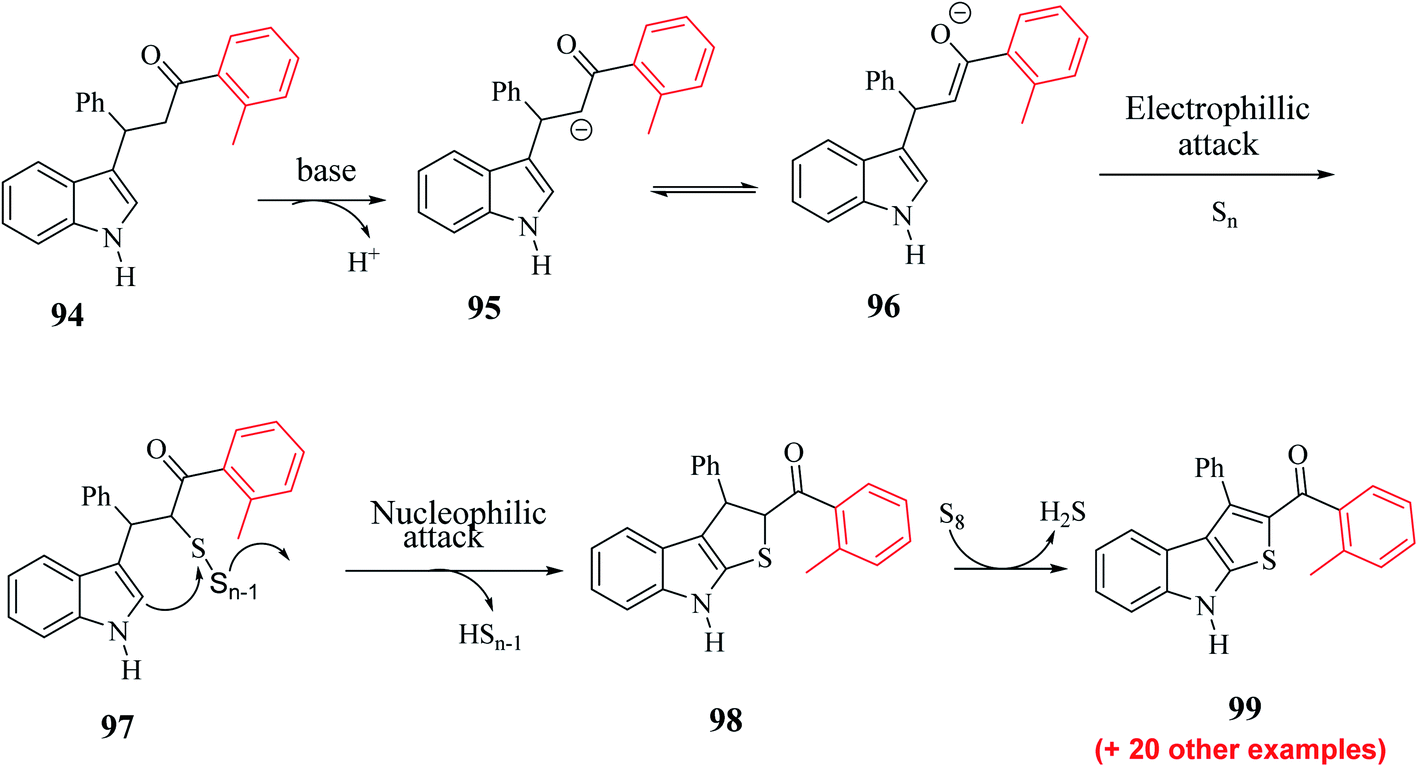 | ||
| Scheme 51 Plausible mechanism for the formation of the product. | ||
Moreover, the yield of the product decreased when an electron-withdrawing group was substituted at the p-position of an extra benzene ring (99a, Fig. 10) and increased in the case of an electron-donating group (99b, Fig. 10).
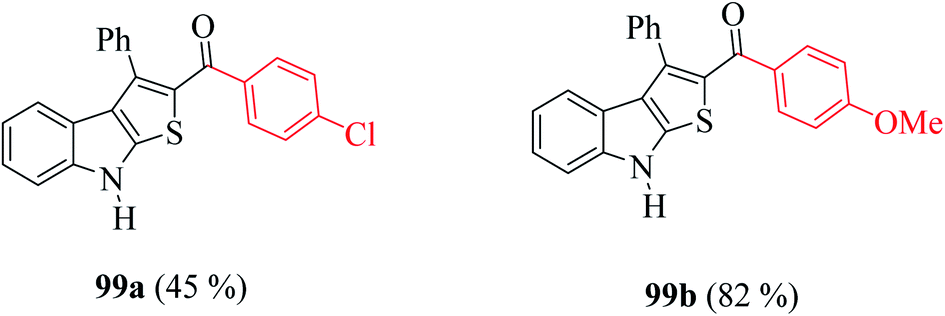 | ||
| Fig. 10 Analogs of thieno[2,3-b]indole. | ||
Furthermore, the presence of methyl, chloride, phenyl and nitrile groups at the p-position of the aromatic ring substituted at the C-3 position of the thienoindole derivative resulted in a high yield of the corresponding derivative products 99c–99f (Fig. 11).
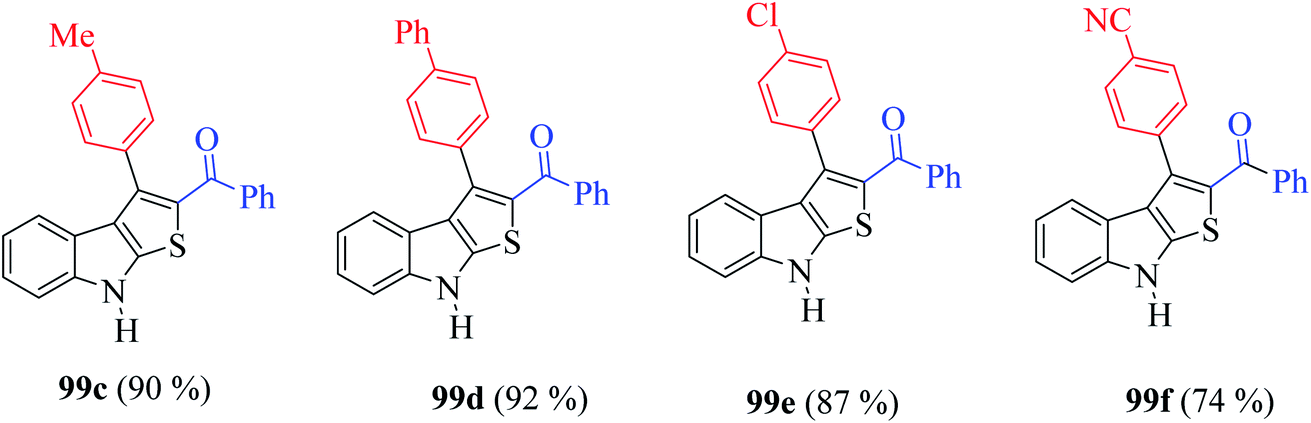 | ||
| Fig. 11 Derivatives of thieno[2,3-b]indole. | ||
2.12 Synthesis of thieno[2,3-b]indoles via green approach using magnetic nanoparticle-supported [urea]4[ZnCl2] deep eutectic solvent (DES@MNP)
Nguyen et al.69 reported a green approach for the one-pot three-component synthesis of functionalized thieno[2,3-b]indoles via the use of a magnetic nanoparticle-supported [urea]4[ZnCl2] deep eutectic solvent.The reaction was conducted in the presence of economical and widely accessible reagents, i.e., sulfur, acetophenone 38 and indole 39a, using the magnetic recyclable nanoparticle DES@MNP catalyst in N,N-dimethylformamide at 140 °C for 12 h. The deep eutectic solvent was prepared with 1.2 g urea in 0.68 g zinc chloride at 100 °C and further mixed with silica-coated Fe3O4 nanoparticles at 100 °C for 18 h and dried under low pressure at 60 °C for 6 h to form the DES@MNP catalyst. The deep eutectic solvent-covered nanoparticles were beneficial for easy handling, separation and recycling. Moreover, the magnetic nanoparticles have the advantages of easy preparation, high stability, low cost, availability, high surface area and easy separation by a magnet for reuse (Scheme 52).
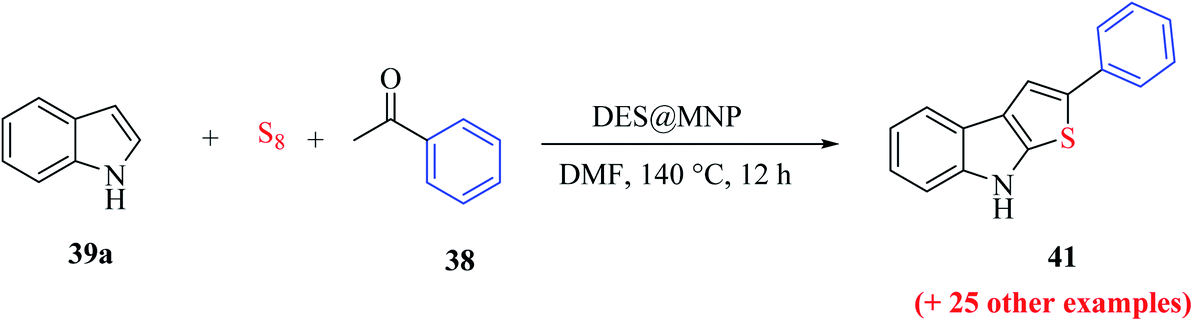 | ||
| Scheme 52 Synthesis of 2-phenylthieno[2,3-b]indole 41 from DES@MNP. | ||
The proposed mechanism for the synthesis of thieno[2,3-b]indole from acetophenone 38, N,N-dimethylformamide and sulfur aided by the DES@MNP catalyst suggests that the reaction takes place through the formation of 1-(dimethylamino)-2-phenyl-1H-thiiren-1-ium 47, which further forms 47a (65%). The resulting intermediate 47 further reacts with indole 39a via a ring-opening addition mechanism, ring closure and elimination of dimethyl-amine to form 47 in 87% yield. Furthermore, indole reacts with the reaction mixture, forming 87% product. Moreover, the proposed research led to an efficient protocol having merits, which include simple and efficient recyclable heterogeneous catalyst, vast substrate scope and regioselective product in high yield (Scheme 53).
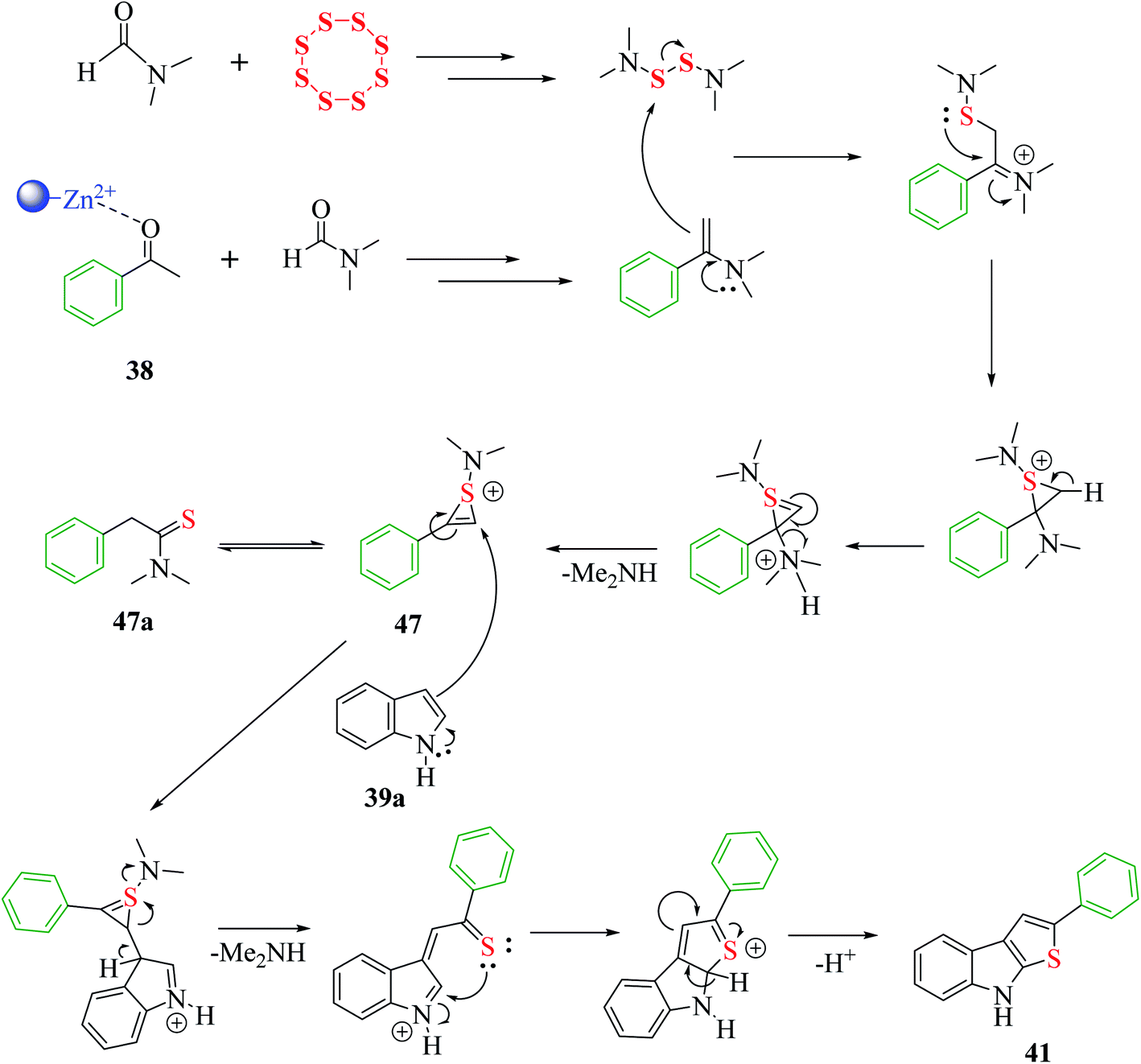 | ||
| Scheme 53 Mechanism for the synthesis of thieno[2,3-b]indole via nanoparticles. | ||
3 Applications of thienoindoles
3.1 Biological activities of thienoindoles
Thienoindole derivatives show a broad spectrum of biological activities such as anti-bacterial, anti-inflammatory, anti-allergic, anti-viral, anti-tuberculosis activities 100, 5-HT5A receptor binding inhibition 101, antitumor and anti-infective activities 102 (Fig. 12).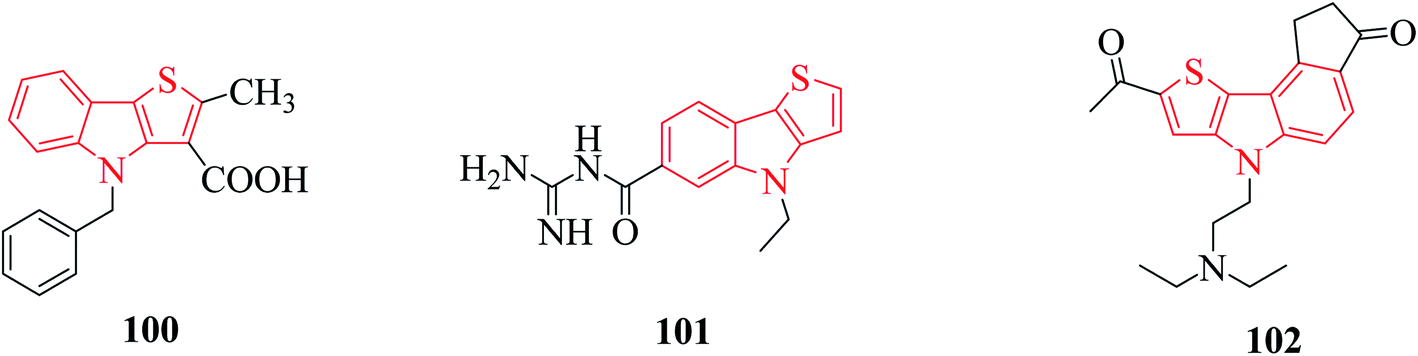 | ||
| Fig. 12 Examples of thienoindole analogs having biological activity. | ||
The alkaloid thienodolin70 103 is a natural derivative of thieno[2,3-b]indole obtained by the fermentation mixture of Streptomyces albogriseolus (Fig. 13). Kanbe et al. characterized its activity for plant growth regulation. Furthermore, some thienoindoles are used to treat diseases of the central nervous system and some are potential inhibitors of acetylcholine esterase and butyrylcholine esterase.
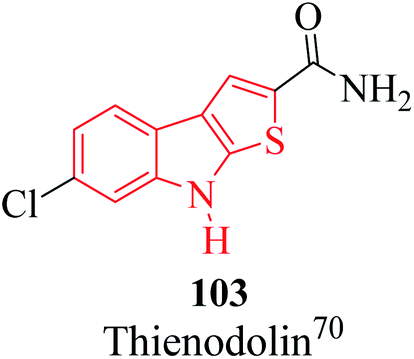 | ||
| Fig. 13 Thienodolin 103, a natural derivative of thieno[2,3-b]indole. | ||
3.2 Chemical activities of thienoindoles
Besides their therapeutic properties, thienoindoles are also used for designing molecules of photosensitive and photovoltaic devices because of their π-extended conjugation from electron-rich systems. They are reported to be effective photosensitizers for photothermal and photodynamic therapies and polymers such as PTTICN, PTTIF, and PTTI. Moreover, TI-DTBT3 104 is a donor–acceptor π-conjugated polymer with high charge carrier mobility.71 This moiety is interestingly available in several organic dyes such as MKZ-40 105 and DPP-r-TI. They are also used as precursors of polymers used in solar cell applications (Fig. 14).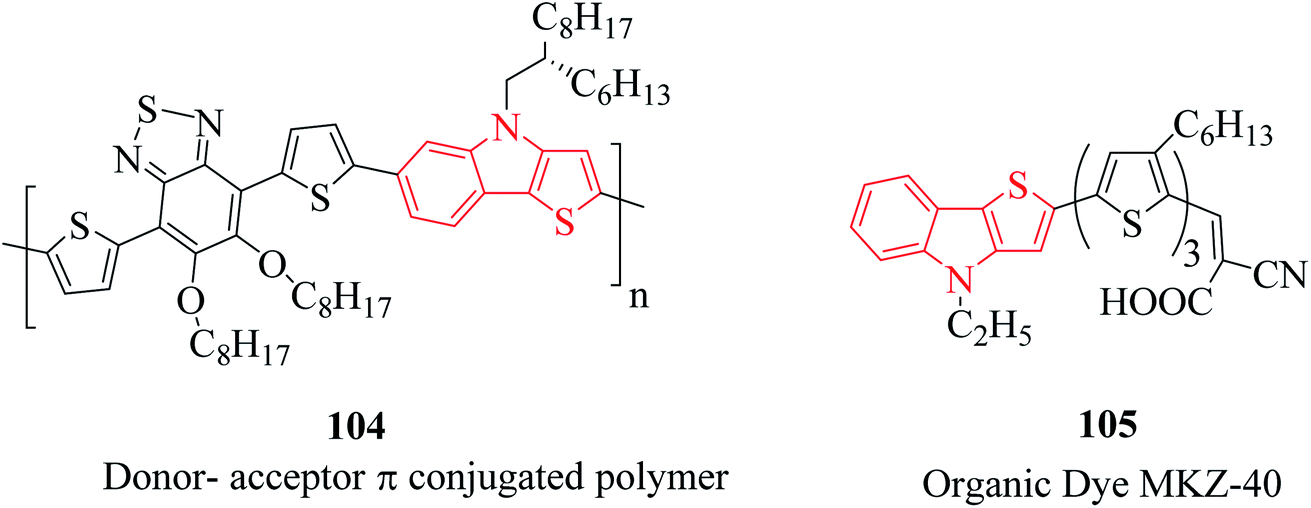 | ||
| Fig. 14 Thienoindole analogs having chemical activities. | ||
3.3 Electrical activities of thienoindoles
Derivatives of thieno[2,3-b]indole are used to design photo- and electroactive compounds, which have been recently assessed in dye-sensitized solar cells (DSSCs). Push–pull dyes IK-1,2 viz., 106 and 107 have been recently reported to be synthesized as a donor part of DSSCs72 (Fig. 15).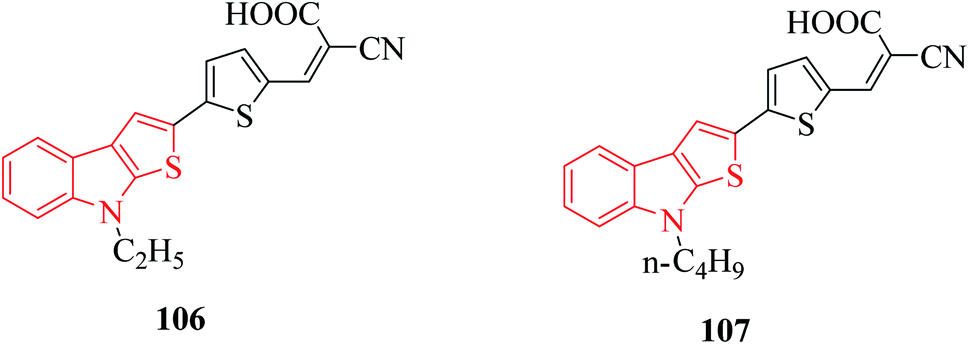 | ||
| Fig. 15 Thienoindole-based compounds having electrical activities in push–pull dyes. | ||
N-Methyl 109a- or N-phenyl 109b-substituted thieno[2,3-b]indoles are used as the electron donor part of push–pull D–π–A molecules, whereas 2,2-dicyanovinylmethyl (DCV) 115 and (1-(dicyanomethylene)-3-oxo-1-inden-2-ylidene)-methyl (DCI) 114 are used as the electron acceptor73 (Scheme 54 and Fig. 16).
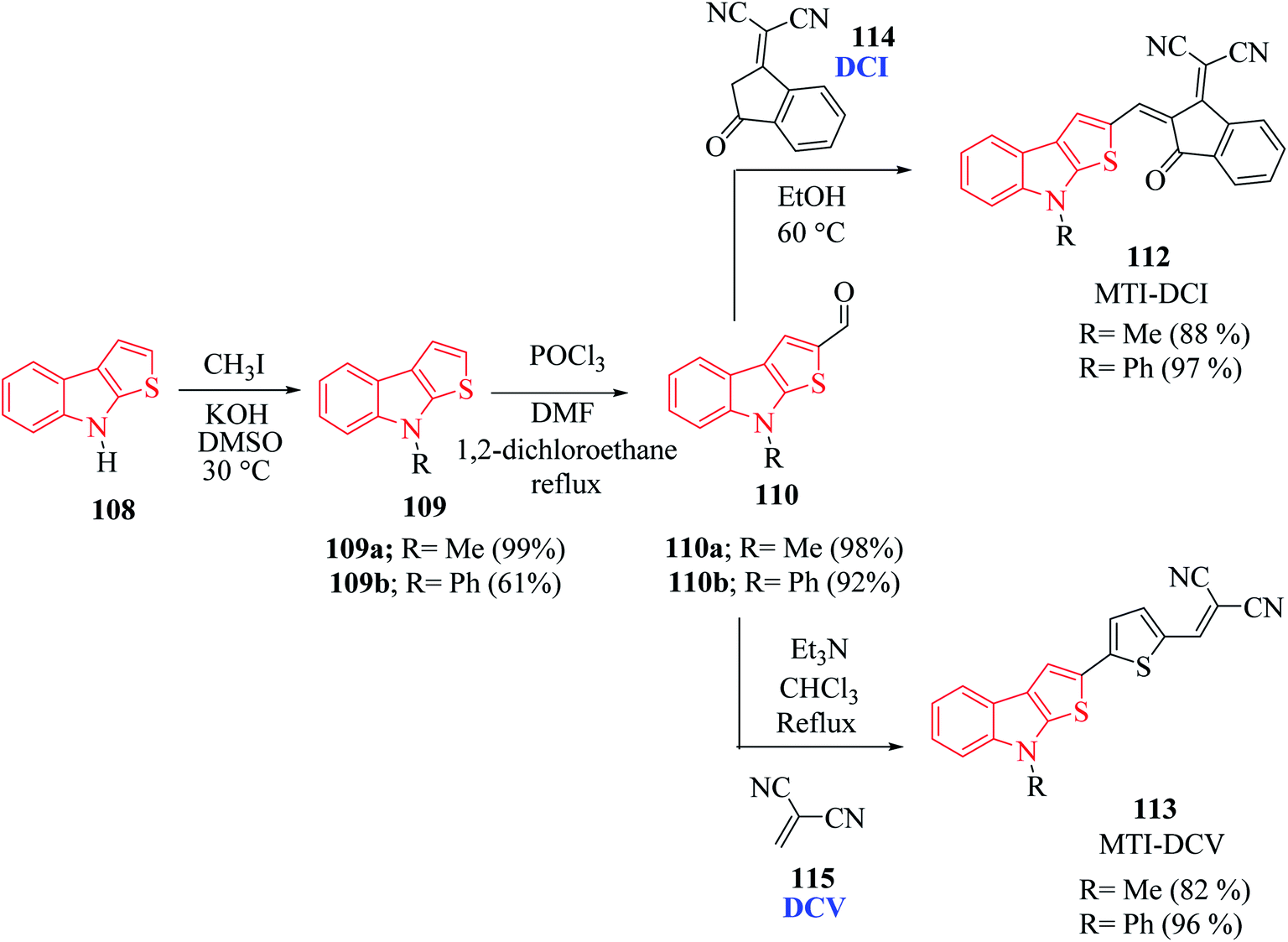 | ||
| Scheme 54 Synthetic approach of push–pull molecules having thieno[2,3-b]indole. | ||
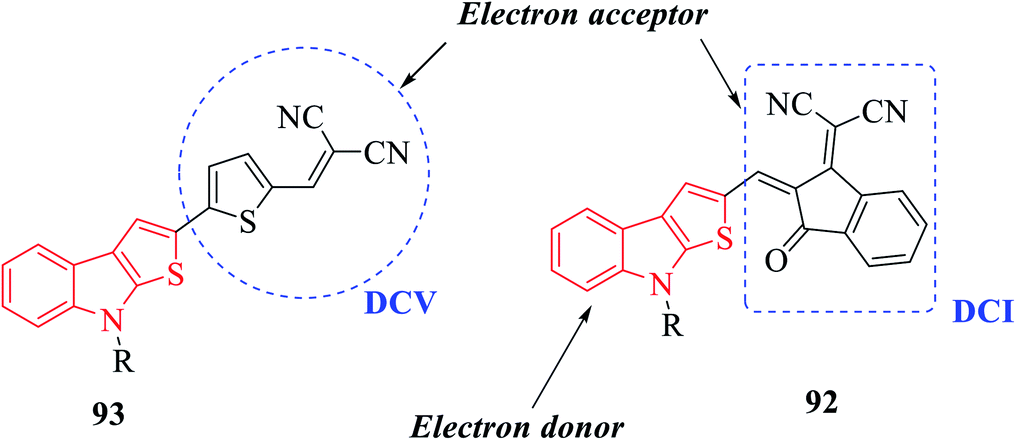 | ||
| Fig. 16 Diagram showing electron-donating and electron-withdrawing parts of push–pull D–π–A molecules. | ||
MTI-DCV 113 is the smallest push–pull molecule of the series. It has a high absorption coefficient, high thermal stability, good hole transport properties and good absorption in the visible region, and because of all these properties, a bilayer solar cell mostly composed of MTI-DCV showed a power conversion efficiency of more than 1%.
4 Conclusions
Thieno[2,3-b]indole and thieno[3,2-b]indole molecules have been extensively studied because of their wide range of acceptable biological and pharmaceutical applications and other characteristic uses such as in photothermal and organic photovoltaic cells (OPV), making them valuable heterocycles in synthetic organic chemistry, materials chemistry and chemical engineering. Over time, researchers have overcome multiple problems associated with their synthesis such as tedious product separation, problematic catalyst recovery and need for large stoichiometric amounts of solvent. Moreover, the use of highly volatile, toxic and explosive substrates, solvents, and additives for the preparation of thiophene-fused indoles limit their widespread application. Some synthetic protocols also use toxic and corrosive bases such as DABCO, DBU, and LDA and hazardous solvents such as 1,4-dioxane and DMF for the synthesis of thienoindoles. Moreover, a few of the reported methods for their synthesis require the use of functionalized furans, indoles, thiophenes, etc. as precursors, which are synthesized via multiple steps, making them inefficient. Thus, to overcome the aforementioned issues, newer and convenient synthetic strategies need to be developed, which involve environment-friendly solvents such as water, ethanol, and PEG and reduction in the use of workup solvents. Further, the electronic push–pull mechanism and extended conjugation indicate that thieno[2,3-b]indole-based polymers and dyes that display a large variation in properties are still to be discovered. Finally, the future prospects in the arena of thieno[2,3-b]indoles synthesis depends on the progress of competent synthetic procedures that can solve the above-mentioned concerns, while keeping environmental-friendliness a priority.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. A. acknowledges the financial support from I. O. E., University of Delhi, Delhi 110007, India. S. A. is thankful to CSIR for providing Junior-Research Fellowship.References
- H. Jiang, J. Sun and J. Zhang, A Review on Synthesis of Carbazole-based Chromophores as Organic Light-emitting Materials, 2012, vol. 16 Search PubMed.
- A. N. Grinev, Y. I. Trofimkin, E. V Lomanova, G. N. Pershin, L. M. Polukhina, I. S. Nikolaeva, T. V Pushkina, L. N. Filitis, E. A. Golovanova and O. V Okinshevich, Pharm. Chem. J., 1982, 16, 827–830 CrossRef.
- S. A. Al-Trawneh, J. A. Zahra, M. R. Kamal, M. M. El-Abadelah, F. Zani, M. Incerti, A. Cavazzoni, R. R. Alfieri, P. G. Petronini and P. Vicini, Bioorg. Med. Chem., 2010, 18, 5873–5884 CrossRef CAS PubMed.
- M. Shiozaki, K. Maeda, T. Miura, M. Kotoku, T. Yamasaki, I. Matsuda, K. Aoki, K. Yasue, H. Imai and M. Ubukata, J. Med. Chem., 2011, 54, 2839–2863 CrossRef CAS PubMed.
- S. A. Al-Trawneh, M. M. El-Abadelah, J. A. Zahra, S. A. Al-Taweel, F. Zani, M. Incerti, A. Cavazzoni and P. Vicini, Bioorg. Med. Chem., 2011, 19, 2541–2548 CrossRef CAS PubMed.
- M. S. C. Pedras and M. Suchy, Bioorg. Med. Chem., 2006, 14, 714–723 CrossRef CAS PubMed.
- X.-H. Zhang, Y. Cui, R. Katoh, N. Koumura and K. Hara, J. Phys. Chem. C, 2010, 114, 18283–18290 CrossRef CAS.
- Z.-S. Huang, T. Hua, J. Tian, L. Wang, H. Meier and D. Cao, Dyes Pigm., 2016, 125, 229–240 CrossRef CAS.
- Y. K. Eom, S. H. Kang, I. T. Choi, Y. Yoo, J. Kim and H. K. Kim, J. Mater. Chem. A., 2017, 5, 2297–2308 RSC.
- J. Kim, J. Lee, S. Chae, J. Y. Shim, D. Y. Lee, I. Kim, H. J. Kim, S. H. Park and H. Suh, Polymer, 2016, 83, 50–58 CrossRef CAS.
- I. Jeong, S. Chae, A. Yi, J. Kim, H. H. Chun, J. H. Cho, H. J. Kim and H. Suh, Polymer, 2017, 109, 115–125 CrossRef CAS.
- P. P. Singh, A. K. Yadav, H. Ila and H. Junjappa, Chem.–Eur. J., 2011, 2011, 4001–4007 CAS.
- E. M. Beccalli, A. Marchesini and T. Pilati, J. Chem. Soc., Perkin Trans. 1, 1994, 579–587 RSC.
- K. Albertshofer, B. Tan and C. F. Barbas III, Org. Lett., 2012, 14, 1834–1837 CrossRef CAS PubMed.
- S.-H. Cao, X.-C. Zhang, Y. Wei and M. Shi, Eur. J. Org. Chem., 2011, 2668–2672 CrossRef CAS.
- A. Shafiee and S. Sattari, J. Heterocycl. Chem., 1982, 19, 227–231 CrossRef CAS.
- P. A. S. Smith and J. H. Boyer, J. Am. Chem. Soc., 1951, 73, 2626–2629 CrossRef CAS.
- K. Takamatsu, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 2892–2895 CrossRef CAS PubMed.
- P. Appukkuttan, E. Van der Eycken and W. Dehaen, Synlett, 2005, 01, 127–133 Search PubMed.
- T. Q. Hung, T. T. Dang, A. Villinger, T. Van Sung and P. Langer, Org. Biomol. Chem., 2012, 10, 9041–9044 RSC.
- K. Nozaki, K. Takahashi, K. Nakano, T. Hiyama, H. Tang, M. Fujiki, S. Yamaguchi and K. Tamao, Angew. Chem., 2003, 115, 2097–2099 CrossRef.
- S.-M. T. Toguem, A. Villinger and P. Langer, Synlett, 2009, 20, 3311–3314 Search PubMed.
- J. I. G. Cadogan, M. Cameron-Wood, R. K. Mackie and R. J. G. Searle, J. Chem. Soc., 1965, 4831–4837 RSC.
- P. Ni, B. Li, H. Huang, F. Xiao and G.-J. Deng, Green Chem., 2017, 19, 5553–5558 RSC.
- V. Estevez, M. Villacampa and J. C. Menendez, Chem. Soc. Rev., 2010, 39, 4402–4421 RSC.
- H. Y. Cho and J. P. Morken, Chem. Soc. Rev., 2014, 43, 4368–4380 RSC.
- Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067–4072 RSC.
- Y. Kasano, A. Okada, D. Hiratsuka, Y. Oderaotoshi, S. Minakata and M. Komatsu, Tetrahedron, 2006, 62, 537–542 CrossRef CAS.
- C. Willgerodt, Ber. Dtsch. Chem. Ges., 1887, 20, 2467–2470 CrossRef.
- S. R. Pathipati, V. Singh, L. Eriksson and N. Selander, Org. Lett., 2015, 17, 4506–4509 CrossRef CAS PubMed.
- B. Li, P. Ni, H. Huang, F. Xiao and G. J. Deng, Adv. Synth. Catal., 2017, 359, 4300–4304 CrossRef CAS.
- S. Chen, Y. Li, P. Ni, B. Yang, H. Huang and G.-J. Deng, J. Org. Chem., 2017, 82, 2935–2942 CrossRef CAS PubMed.
- B. P. Mundy, M. G. Ellerd and F. G. Favaloro Jr, Name reactions and reagents in organic synthesis, John Wiley & Sons. 2005 Search PubMed.
- Y. Hayashi, K. Okano and A. Mori, Org. Lett., 2018, 20, 958–961 CrossRef CAS PubMed.
- K. Shono, Y. Sumino, S. Tanaka, S. Tamba and A. Mori, Org. Chem. Front., 2014, 1, 678–682 RSC.
- E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed.
- R. Levine and W. C. Fernelius, Chem. Rev., 1954, 54, 449–573 CrossRef CAS.
- K. Snégaroff, S. Komagawa, F. Chevallier, P. C. Gros, S. Golhen, T. Roisnel, M. Uchiyama and F. Mongin, Chem.–Eur J., 2010, 16, 8191–8201 CrossRef PubMed.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969–5970 CrossRef CAS.
- A. S. Guram and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 7901–7902 CrossRef CAS.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 12706 CrossRef CAS.
- K. Okano, K. Sunahara, Y. Yamane, Y. Hayashi and A. Mori, Chem.–Eur. J., 2016, 22, 16450–16454 CrossRef CAS PubMed.
- R. A. Irgashev, A. S. Steparuk and G. L. Rusinov, Org. Biomol. Chem., 2018, 16, 4821–4832 RSC.
- B. Robinson, Chem. Rev., 1963, 63, 373–401 CrossRef.
- G. W. Gribble, Indole ring synthesis: From natural products to drug discovery, John Wiley & Sons. 2016 Search PubMed.
- J. J. Li, Tebbe reagent, in Name Reactions, Springer. 2014, pp. 599–600 Search PubMed.
- H. Fiesselmann and F. Thoma, Chem. Ber., 1956, 89, 1907–1912 CrossRef CAS.
- Y. Sato, Y. Onozaki, T. Sugimoto, H. Kurihara, K. Kamijo, C. Kadowaki, T. Tsujino, A. Watanabe, S. Otsuki and M. Mitsuya, Bioorg. Med. Chem. Lett., 2009, 19, 4673–4678 CrossRef CAS PubMed.
- H. KESKIN, R. E. MILLER and F. F. Nord, J. Org. Chem., 1951, 16, 199–206 CrossRef CAS.
- V. Mane, T. V Baiju and I. N. N. Namboothiri, ACS Omega, 2018, 3, 17617–17628 CrossRef CAS PubMed.
- V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1–48 CrossRef CAS PubMed.
- G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614–4628 CrossRef CAS PubMed.
- P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 28, 6213–6226 CrossRef.
- M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2006, 4, 2525–2528 RSC.
- P. Shanbhag, P. R. Nareddy, M. Dadwal, S. M. Mobin and I. N. N. Namboothiri, Org. Biomol. Chem., 2010, 8, 4867–4873 RSC.
- R. A. Irgashev, A. S. Steparuk and G. L. Rusinov, Tetrahedron Lett., 2019, 60, 1–3 CrossRef.
- H. Zhang, M. S. Bednarz, N.-K. Lim, G. Hernandez and W. Wu, Org. Lett., 2014, 16, 2522–2525 CrossRef CAS PubMed.
- E. Campaigne and P. A. Monroe, J. Am. Chem. Soc., 1954, 76, 2447–2450 CrossRef CAS.
- J. Liebscher, B. Neumann and H. Hartmann, J. Prakt. Chem., 1983, 325, 915–918 CrossRef CAS.
- M. Pellet and F. Huet, Tetrahedron, 1988, 44, 4463–4468 CrossRef CAS.
- R. S. Al-awar, S. P. Joseph and D. L. Comins, Tetrahedron Lett., 1992, 33, 7635–7638 CrossRef CAS.
- Z. Mirjafary, M. Abdoli, H. Saeidian, S. Boroon and A. Kakanejadifard, RSC Adv., 2015, 5, 79361–79384 RSC.
- H. Hartmann and J. Liebscher, Synth, 1984, 275–276 CrossRef CAS.
- H. Fiesselmann and G. Pfeiffer, Chem. Ber., 1954, 87, 848–856 CrossRef CAS.
- J. Liu, Y. Zhang, Y. Yue, Z. Wang, H. Shao, K. Zhuo, Q. Lv and Z. Zhang, J. Org. Chem., 2019, 84, 12946–12959 CrossRef CAS PubMed.
- L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 10351–10355 CrossRef CAS PubMed.
- T. T. Nguyen and P. H. Tran, RSC Adv., 2020, 10, 9663–9671 RSC.
- R. Engqvist, A. Javaid and J. Bergman, Eur. J. Org. Chem., 2004, 12, 2589–2592 CrossRef.
- H. Huang, Q. Li, M. Qiu, Z. Wang, X. Zhang, S. Liu, N. Fu, B. Zhao, R. Yang and W. Huang, RSC Adv., 2016, 6, 45873–45883 RSC.
- R. A. Irgashev, A. A. Karmatsky, S. A. Kozyukhin, V. K. Ivanov, A. Sadovnikov, V. V Kozik, V. A. Grinberg, V. V Emets, G. L. Rusinov and V. N. Charushin, Synth. Met., 2015, 199, 152–158 CrossRef CAS.
- L. Ackermann and A. Althammer, Synfacts, 2007, 586 Search PubMed.
Footnote |
| † Equally contributed. |
| This journal is © The Royal Society of Chemistry 2022 |