
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and magnetic properties of sub-nanosized iron carbides on a carbon support†
Masanori Wakizaka a,
Wang-Jae Chunb,
Takane Imaoka*a and
Kimihisa Yamamoto*a
a,
Wang-Jae Chunb,
Takane Imaoka*a and
Kimihisa Yamamoto*a
aLaboratory for Chemistry and Life Science Institute of Innovative Research, Tokyo Institute of Technology, Yokohama 226-8503, Japan. E-mail: timaoka@res.titech.ac.jp; yamamoto@res.titech.ac.jp
bGraduate School of Arts and Sciences, International Christian University, Tokyo 181-8585, Japan
First published on 25th January 2022
Abstract
Iron carbide clusters with near-sub-nanometer size have been synthesized by employing a tetraphenylmethane-cored phenylazomethine dendrimer generation 4 (TPM-DPAG4) as a molecular template. Magnetic measurements reveal that these iron carbide clusters exhibit a magnetization–field hysteresis loop at 300 K. The data indicate that these iron carbide clusters are ferromagnets at room temperature.
Iron carbide is a well-established material that is typically generated during the steelmaking process. Research into the phase diagram of the Fe–C system was conducted as early as the 1890s.1 According to this phase diagram, iron and carbon atoms can be mixed in arbitrary proportions up to 0.095 atom% of C at temperatures below 1000 K; above this ratio iron carbide cementite (Fe3C) is formed.2 As with metallic iron, iron carbides are also known to exhibit ferromagnetism;3,4 therefore, there have been many studies reported on the ferromagnetism of bulk iron carbides and iron carbide nanoparticles.5–19 The size effect in nanomaterials is also of particular interest because the properties of the bulk materials can be significantly changed. For example, melting-point depression,20 catalyst activation,21 and the alloying of non-mixable metals22 have been reported to occur as the particle size decreases into the nanosize range. We have recently reported atomicity-dependent changes in the catalytic activity23,24 and size-dependent phase transformations of near-sub-nanometer particles.25 The properties of many substances are thus sensitively affected by particle size, particularly in the near-sub-nanosize range. In this context, the smallest iron carbide nanoparticles reported to date are as small as ca. 2 nm,7,16 aside from the gas phase experiments26,27 and the theoretical studies.28–32 In these cases, the iron carbide nanoparticles exhibit superparamagnetism, i.e., they do not act as magnets at ambient temperature. However, sub-nanosized iron carbide particles have remained elusive to date. In the present study, we have synthesized near-sub-nanometer iron carbide particles/clusters, and these iron carbide clusters are ferromagnets, even at room temperature, thereby countering superparamagnetism. Bulk iron carbide is an old material; however, the iron carbide clusters synthesized in this work are the smallest room temperature magnets reported to date.
Fig. 1 shows the strategy employed for the synthesis of near sub-nanometer-sized iron carbide clusters. The macromolecular tetraphenylmethane-cored dendritic phenylazomethine dendrimer generation 4 (TPM-DPAG4) was used as a molecular template. This DPA-type dendrimer coordinates to metal ions in solution via its imine sites, and complexation proceeds stepwise from the center of the dendrimer to its periphery due to its basicity gradient.33–37 Stepwise complexation was confirmed in the present study by UV-Vis titrations. Upon the addition of FeCl3 to a solution of TPM-DPAG4, spectral changes and shifts in the isosbestic point were observed (Fig. S1†); these changes reached saturation after the combined addition of 60 eq. of FeCl3. Different isosbestic points were observed in the ranges of 0–4, 6–12, 16–28, and 32–60 eq., respectively, which is consistent with the number of imines at each type of site and reflects the stepwise complexation from the central to the peripheral sites. The in situ-prepared dendrimer complexes, i.e., TPM-DPAG4 with 4, 12, 28, or 60 eq. of FeCl3 incorporated were then adsorbed onto a graphitic carbon support (graphitized mesoporous carbon: GMC). Carbothermal hydrogen reduction (CHR), which is a synthetic method used to obtain metal carbides, was subsequently applied.25,38,39 After CHR at 773 K for 30 min, the samples (Fe12/C, Fe28/C, and Fe60/C) were examined using transmission electron microscopy (TEM), and the results are shown in Fig. 2 and S2.† There are several reports for TEM observations of iron carbide nanoparticles larger than 2 nm diameter without atomic-resolution.5–19 Very fine particles dispersed over the carbon support were observed as blurry black dots in the TEM images. The mean particle diameter and standard deviation of the size distribution were estimated to be 0.9 ± 0.2 nm (Fe12/C), 1.0 ± 0.3 nm (Fe28/C), and 1.3 ± 0.3 nm (Fe60/C), respectively. The average particle size consistently increased with the FeCl3 content in the TPM-DPAG4 template. These samples represent the first examples of near-sub-nanometer-sized iron carbide particles. However, we could not observe any individual particles in the Fe4/C sample, because the particle size was too small. In this case, the particle size was estimated to be ca. 0.6 nm using the tetra-nuclear cluster model of the [Fe4C(CO)12]2− carbidocarbonyl complex reported by Boehme et al. (Fig. S3a†)40 as well as the theoretical studies.28–32 It should be noted that atomic-resolution images that would project the clusters could not be obtained, because these samples exhibit ferromagnetism, even at room temperature (vide infra).
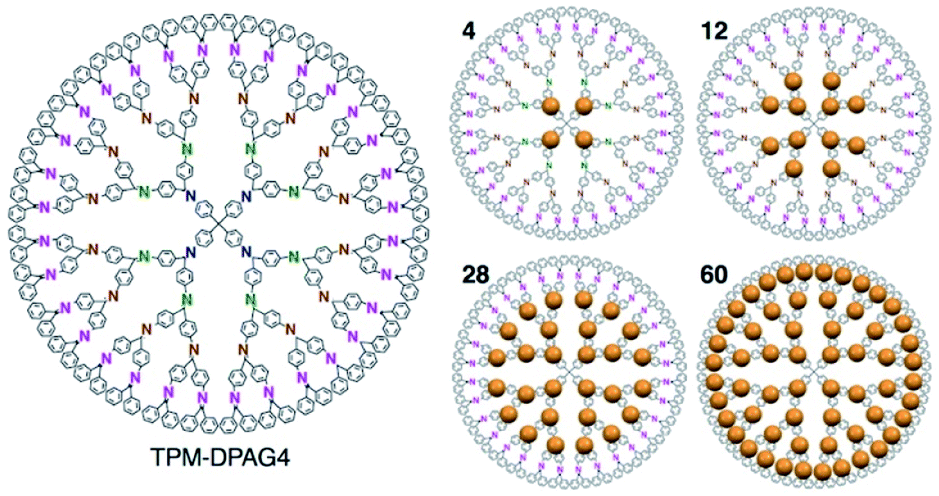 | ||
| Fig. 1 Chemical structure of the TPM-DPAG4 and illustration of metal ions assembly (4, 12, 28, 60 eq.). | ||
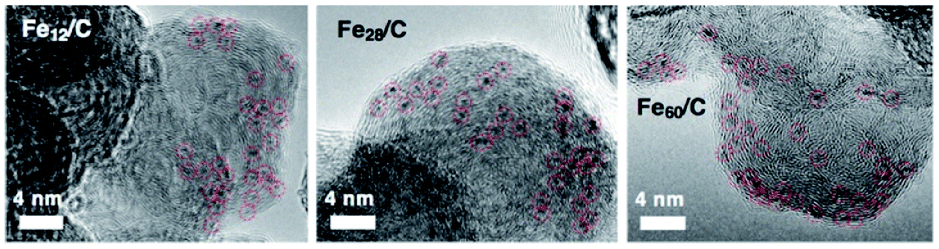 | ||
| Fig. 2 TEM images of Fe12/C, Fe28/C, and Fe60/C after 30 min of CHR at 773 K. | ||
Powder X-ray diffraction (PXRD) analysis cannot be applied to the characterization of such sub-nanosized particles on solid supports, as they do not adopt any long-range-ordering crystal structure. On the other hand, X-ray absorption fine structure (XAFS) is a powerful tool to clarify the local structure around the metal atoms.41 We found that the X-ray absorption near edge structure (XANES) spectra of Fe60/C, Fe28/C, Fe12/C, and Fe4/C after CHR are very similar to those of metallic iron (Fe foil) and Fe3C, whereas they are substantially different from those of iron oxides such as Fe3O4 and α-Fe2O3 as well as from that of the FeCl3 starting material (Fig. S4†). Therefore, it can be concluded that these samples are not oxides. XANES spectrum of Fe3C and metallic iron can clearly be distinguished in their first derivatives form (Fig. 3). Metallic iron and Fe3C have a pre-edge peak in common at ca. 7111 eV (3s → 4d transitions). Metallic iron exhibits two maxima in the range of 7115–7130 eV (3s → 4p transitions), while Fe3C exhibits one maximum and several shoulders in this region. The spectra of Fe60/C, Fe28/C, Fe12/C, and Fe4/C after CHR had a pre-edge peak at ca. 7111 eV, together with a maximum peak in the 7115–7130 eV region, which indicates the iron carbide nature. Therefore, it can be concluded that Fe60/C, Fe28/C, Fe12/C, and Fe4/C are iron carbides rather than metals. The white-line peak was slightly shifted to the higher energy side with downsizing (Fig. 3b), i.e., 7129.4 eV (Fe3C), 7130.2 eV (Fe60/C), 7130.1 eV (Fe28/C), 7131.2 eV (Fe12/C), and 7131.4 eV (Fe4/C). The experimental error was estimated to be ±0.3 eV based on the applied energy resolution. This shift tendency supported that the cluster samples (Fe60/C, Fe28/C, Fe12/C, and Fe4/C) are very fine particles with high specific surface. In addition, the assignment as iron carbides is decisively supported by their Curie temperatures (TC), which were measured to be 483–488 K (Fig. 4). These TC values are comparable to that of Fe3C (483 K, Fig. S5 and S6†),9 which suggests that the ferromagnetic interactions originate from iron carbides. The Curie temperature of Fe3C is far from those of metallic iron (1043 K)3 and iron oxides e.g. Fe3O4 (850 K) and γ-Fe2O3 (820–986 K).42 It should also be noted that Fe3C forms a complicated crystal structure that involves nine types of Fe–Fe bonds (2.455–2.714 Å; Fig. S3b†).43,44 The presence of the corresponding Fe–Fe bonds in Fe60/C was suggested by extended X-ray absorption fine structure (EXAFS) measurements conducted in transmission mode (Fig. S7†). The Curie temperature for Fe3C mainly represents the average of the direct exchange interactions between the Fe–Fe bonds, similar to that in amorphous ferromagnets such as the Fe–C–P system,45,46 and thus, TC would be considered not to show a significant size dependence.
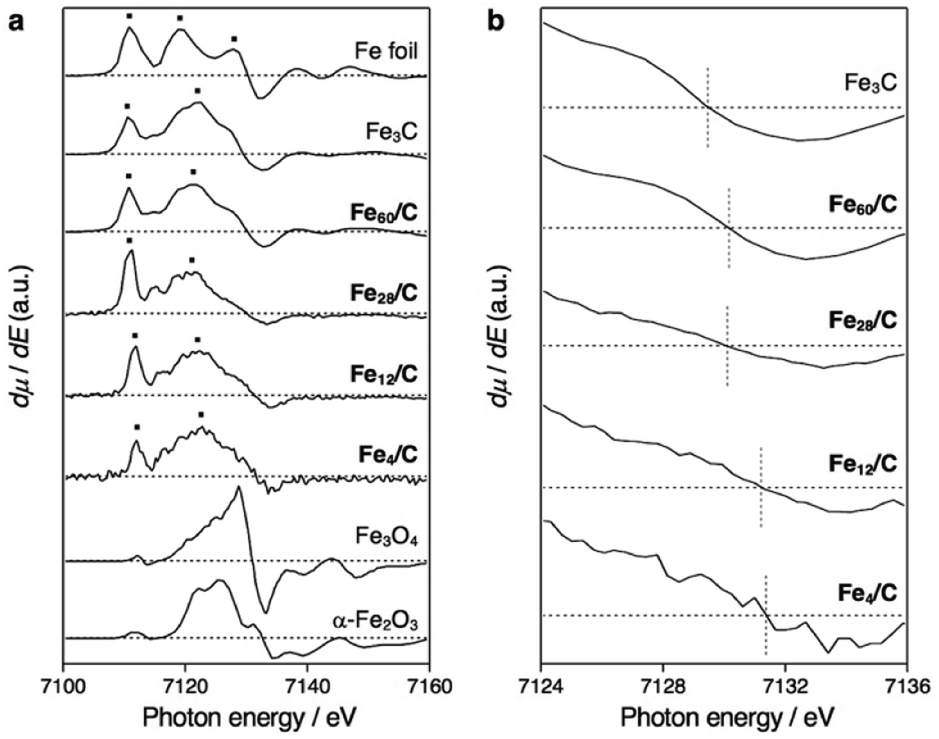 | ||
| Fig. 3 Fe K-edge XANES spectra. (a) First derivatives of normalized XANES spectra for Fe60/C, Fe28/C, Fe12/C, and Fe4/C after CHR at 773 K for 30 min, together with those for Fe foil (metallic iron), Fe3C, Fe3O4, and α-Fe2O3. The spectra for Fe28/C, Fe12/C, and Fe4/C were recorded in fluorescence mode, whereas the others were recorded in transmission mode. (b) Magnifications around the white-line peak. The experimental error was estimated to be ±0.3 eV based on the applied energy resolution. | ||
 | ||
| Fig. 4 Temperature-dependent magnetization curves for (a) Fe60/C, (b) Fe28/C, (c) Fe12/C, and (d) Fe4/C obtained by application of a magnetic field (5000 Oe) and measurement of the magnetization in increments of 10 K (300–420 K) or 5 K (420–600 K). The blue lines are smoothed trend lines. The Curie point (TC) was determined from the maximum of the second derivative (insets) and calibrated using TC = 483 K for Fe3C.9 The error in the maxima of the second derivatives was estimated to be 5 K. | ||
Fig. 5 shows magnetization–field (M–H) loops for the iron carbide clusters, and the magnetic data are summarized in Table S1.† The M per the sample weight data are shown in Fig. S8–S12.† The four cluster samples show hystereses in their M–H loops at 1.9 K (Fig. 5a), which indicates that they are ferromagnets with an associated coercivity (Hc). The Hc value increased with a decrease in the cluster size, i.e., 603 Oe (Fe60/C), 939 Oe (Fe28/C), 1856 Oe (Fe12/C), and 2697 Oe (Fe4/C). In contrast, bulk iron carbide cementite (Fe3C) with an average crystal size of 39 nm has a smaller hysteresis with Hc values of 166 and 21 Oe at 1.9 and 300 K, respectively (Fig. S8†). The magnetic behavior of bulk Fe3C indicates ferromagnetism with a multi-magnetic-domain structure.3 On the contrary, iron carbide nanoparticles have been reported to exhibit more pronounced hysteresis at room temperature than bulk Fe3C, with Hc values of 700 Oe (15 nm) and 544 Oe (14.1 ± 0.8 nm) by Grimes et al.5 and Hou et al.,6 respectively, which suggests a single-magnetic-domain structure. Therefore, the smaller iron carbide clusters in this study are considered to have a single-magnetic-domain structure. The increase in coercivity with the decrease in single-magnetic-domain particle size has been reported by Lartigue et al. for iron carbide nanoparticles with sizes of 15.1 nm (Hc = 331 Oe), 7.4 nm (Hc = 405 Oe), 5.5 nm (Hc = 625 Oe), and 2.8 nm (Hc = 1009 Oe) at 2.5 K.7 On the other hand, the iron carbide clusters exhibit hysteresis loops at 300 K (Fig. 5b), i.e., Hc = 140 Oe (Fe60/C), 163 Oe (Fe28/C), 367 Oe (Fe12/C), and 666 Oe (Fe4/C), which indicates that they are ferromagnets, even at room temperature. Clusters or near-sub-nanosize particles generally exhibit superparamagnetism with a complete loss of coercivity at room temperature.3
| τN ∝ exp(Ea/kBT) | (1) |
| Ea ≃ KeffV | (2) |
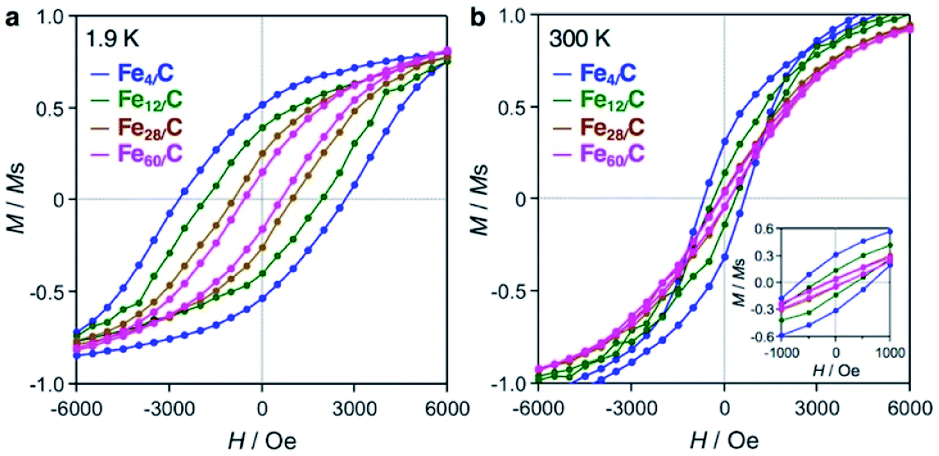 | ||
| Fig. 5 Magnetization–field (M–H) loops for Fe60/C (magenta), Fe28/C (brown), Fe12/C (green), and Fe4/C (blue) at (a) 1.9 K and (b) 300 K. Magnetization (M) was normalized with respect to the saturation magnetization (Ms). The inset shows the magnification in the region near zero field. | ||
Eqn (1) is the Néel–Arrhenius equation,47 where τN, Ea, kB, T, Keff, and V are the Néel relaxation time, magnetic anisotropy energy, Boltzmann constant, temperature, effective magnetic anisotropy constant, and volume of a single-magnetic-domain particle, respectively. The term for the angle between the magnetic moment and the easy magnetic axis was not introduced (eqn (2)), because these were powder samples. Therefore, superparamagnetism emerges with a decrease in size (V) because Ea becomes comparable to the thermal energy (kBT). Lartigue et al. have reported superparamagnetism for iron carbide nanoparticles with sizes less than 5.5 nm.7 Fig. S13† shows field-cooling (FC) and zero-field-cooling (ZFC) magnetization curves used to determine the blocking temperature (TB) at which the magnets completely lose their coercivity. Fe3C shows a TB of 467 K which is near the Curie point (483 K). On the other hand, the TB of Fe60/C is clearly lower (ca. 385 K) than the Curie point, which was attributed to the influence of superparamagnetism in light of the results of Lartigue et al. In contrast, Fe28/C has higher TB values close to the Curie point at 473 K. This behavior is contrary to superparamagnetism and cannot be explained without increased effective magnetic anisotropy (Keff). The interactions between the iron carbide clusters and the graphitic carbon surface may be a mechanism to afford large Keff, because the ratio of the interacting Fe atoms increases by decreasing size. The oxidation of Fe4/C in air at 553 K for 30 min significantly decreased the magnetization and coercivity (Fig. S14†). Carbides can be the magnets at sub-nano scale, while oxides would not. Due to the measurement sensitivity limit and noise, the TBs of Fe12/C and Fe4/C were roughly estimated to be 410–470 K and 350–470 K, respectively, which indicates that their TBs were at least above room temperature. It should also be noted here that the magnetic moment per Fe atom of these cluster samples (1.0–2.3 μB atomFe−1) was almost identical to that of Fe3C (1.5 μB atomFe−1), regardless of the Fe content (wt%) over an order of magnitude (Table S1†). The variation in the magnetic moment may involve not only experimental errors, but also atomicity. Becker et al. reported that Fe clusters exhibit an atomicity-dependent variation in their magnetic moment in the gas phase, especially below 100 atoms.48 Additionally, the density functional theory studies have reported that the iron carbide clusters show the magnetic moment of ca. 1–3.5 μB atomFe−1,30–32 which is consistent with those in this study. Therefore, with consideration of the magnetic moment and the Curie temperature, it was concluded that the magnetic behavior of the iron carbide cluster samples is derived from the carbides themselves, and not from impurities. The reproducibility of the M–H hysteresis loop at 300 K for Fe4/C was certainly confirmed including another batch sample (sample B: Fig. S15†). It was also confirmed that a blank sample (GMC) showed diamagnetism measured at both 1.9 and 300 K (raw M data shown in Fig. S16–S23†).
Nanoparticle magnets have a single magnetic-domain structure and exhibit hysteresis at room temperature; however, they lose this hysteresis upon downsizing by superparamagnetism. There have been no reports of sub-nanoparticle magnets (diameter: ∼1 nm or less) that exhibit coercivity above room temperature;3–19,49 neither for e.g. Fe–Pt bimetallic nanoparticles50 nor iron oxide nanoparticles.51 The iron carbide clusters in this study are unique magnets that are different from both nanoparticle magnets. They do not have a long-range-ordering crystal structure such as nanoparticles and bulk substances on account of their sub-nanometer size. The iron carbide clusters in this study were carefully characterized by XAFS (Fig. 3) as well as by the Curie temperature (Fig. 4). The magnetic measurements (Fig. 5) revealed that the iron carbide clusters represent room-temperature magnets. Therefore, the iron carbide clusters discussed in this study can be regarded as a new class of magnets, i.e., sub-nano magnets.
This work has synthesized the first examples of sub-nanosized iron carbides on a graphitic carbon support. These iron carbide clusters act as magnets at room temperature. This study would open up the new research field of sub-nano magnets.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Ken Albrecht (Kyushu Univ.) for his support with the synthesis of the TPM-DPAG4, and Dr Y. Ida (Tokyo Tech.), Dr T. Otsuka (AIST), and Dr M. Fujiwara (IMS) for support with magnetic measurements, as well as Suzukakedai Materials Analysis Division (Tokyo Tech.) for support with PXRD and ICP-AES measurements. Magnetic measurements were supported by the Nanotechnology Platform Program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, (Grant No. JPMXP09F-19008715 and -20008747 for the AIST Nano-Processing Facility and No. JPMXP09-S19MS1098 for Molecule and Material Synthesis). This study was performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2019G655, BL9C). This study was supported by an ERATO Grant (No. JPMJER1503) from the Japan Science and Technology Agency (JST) and Kakenhi Grants-in-Aid (No. JP15H05757, JP18K14237, and JP21H01756) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- F. X. Kayser and J. W. Patterson, J. Phase Equilib., 1998, 19, 11 CrossRef CAS
.
- J. Chipman, Metall. Mater. Trans. B, 1972, 3, 55 CrossRef CAS
- D. L. Huber, Small, 2005, 1, 482 CrossRef CAS PubMed
- Z. Ye, P. Zhang, X. Lei, X. Wang, N. Zhao and H. Yang, Chem.–Eur. J., 2018, 24, 8922 CrossRef CAS PubMed
- C. A. Grimes, J. L. Horn, G. G. Bush, J. L. Allen and P. C. Eklund, IEEE Trans. Magn., 1997, 33, 3736 CrossRef CAS
- Z. Yang, T. Zhao, X. Huang, X. Chu, T. Tang, Y. Ju, Q. Wang, Y. Hou and S. Gao, Chem. Sci., 2017, 8, 473 RSC
- L. Lartigue, J. Long, X. Dumail, S. I. Nikitenko, C. Cau, Y. Guari, L. Stievano, M. T. Sougrati, C. Guérin, C. Sangregorio and J. Larionova, J. Nanopart. Res., 2013, 15, 1490 CrossRef
- L. J. E. Hofer and E. M. Cohn, J. Am. Chem. Soc., 1959, 81, 1576 CrossRef CAS
- A. Tsuzuki, S. Sago, S.-I. Hirano and S. Naka, J. Mater. Sci., 1984, 19, 2513 CrossRef CAS
- S.-I. Hirano and S. Tajima, J. Mater. Sci., 1990, 25, 4457 CrossRef CAS
- S. I. Nikitenko, Y. Koltypin, O. Palchik, I. Felner, X. N. Xu and A. Gedanken, Angew. Chem., Int. Ed., 2001, 40, 4447 CrossRef CAS PubMed
- J. Geng, D. A. Jefferson and B. F. G. Johnson, Chem. Commun., 2004, 2442 RSC
- E. P. Sajitha, V. Prasad, S. V. Subramanyam, A. K. Mishra, S. Sarkar and C. Bansal, J. Phys.: Condens. Matter, 2007, 19, 046214 CrossRef
- N. A. Ivanova, A. A. Onischuk, S. V. Vosel, P. A. Purtov, N. T. Vasenin, V. F. Anufrienko and V. N. Ikorski, Appl. Magn. Reson., 2008, 33, 285 CrossRef CAS
- C. Giordano, A. Kraupner, S. C. Wimbush and M. Antonietti, Small, 2010, 6, 1859 CrossRef CAS PubMed
- B. Bittova, J. P. Vejpravova, M. Kalbac, S. Burianova, A. Mantlikova and S. Danis, J. Phys. Chem. C, 2011, 115, 17303 CrossRef CAS
- A. Meffre, B. Mehdaoui, V. Kelsen, P. F. Fazzini, J. Carrey, S. Lachaize, M. Respaud and B. Chaudret, Nano Lett., 2012, 12, 4722 CrossRef CAS PubMed
- Y. Gu, M. Qin, Z. Cao, B. Jia, X. Wang and X. Qu, J. Am. Ceram. Soc., 2016, 99, 1443 CrossRef CAS
- D. C. Fletcher, R. Hunter, W. Xia, G. J. Smales, B. R. Pauw, E. Blackburn, A. Kulak, H. Xin and Z. Schnepp, J. Mater. Chem. A, 2019, 7, 19506 RSC
- Ph. Buffat and J.-P. Borel, Phys. Rev. A, 1976, 13, 2287 CrossRef CAS
- A. Taketoshi and M. Haruta, Chem. Lett., 2014, 43, 380 CrossRef CAS
- K. Kusada, M. Yamauchi, H. Kobayashi, H. Kitagawa and Y. Kubota, J. Am. Chem. Soc., 2010, 132, 15896 CrossRef CAS PubMed
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089 CrossRef CAS PubMed
- T. Imaoka, Y. Akanuma, N. Haruta, S. Tsuchiya, K. Ishihara, T. Okayasu, W.-J. Chun, M. Takahashi and K. Yamamoto, Nat. Commun., 2017, 8, 688 CrossRef PubMed
- M. Wakizaka, A. Atqa, W.-J. Chun, T. Imaoka and K. Yamamoto, Nanoscale, 2020, 12, 15814 RSC
- J. S. Pilgrim and M. A. Duncan, J. Am. Chem. Soc., 1993, 115, 6958 CrossRef CAS
- G. von Helden, N. G. Gotts, P. Maitre and M. T. Bowers, Chem. Phys. Lett., 1994, 227, 601 CrossRef CAS
- H. H. Harris and I. G. Dance, Polyhedron, 2007, 26, 250 CrossRef CAS
- G. L. Gutsev, C. A. Weatherford, P. Jena, E. Johnson and B. R. Ramachandran, J. Phys. Chem. C, 2012, 116, 7050 CrossRef CAS
- L. Zheng, X. Liu, Y. Meng, Y. Zhou, W. Guo, Q. Peng, Y. Yang, H. Jiao, Y.-W. Liab and X.-D. Wen, Phys. Chem. Chem. Phys., 2016, 18, 32944 RSC
- P. Limon, A. Miralrio and M. Castro, Int. J. Quantum Chem., 2019, 119, e25932 CrossRef
- P. Limon, A. Miralrio and M. Castro, J. Phys. Chem. C, 2020, 124, 9484 CrossRef CAS
- K. Yamamoto, M. Higuchi, S. Shiki, M. Tsuruta and H. Chiba, Nature, 2002, 415, 509 CrossRef CAS PubMed
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397 CrossRef CAS PubMed
- R. Nakajima, M. Tsuruta, M. Higuchi and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 1630 CrossRef CAS PubMed
- M. Wakizaka, T. Imaoka and K. Yamamoto, Dalton Trans., 2019, 48, 14261 RSC
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397 CrossRef CAS PubMed
- C. Liang, P. Ying and C. Li, Chem. Mater., 2002, 14, 3148 CrossRef CAS
- Y. Shen, J. Mater. Chem. A, 2015, 3, 13114 RSC
- R. F. Boehme and P. Coppens, Acta Crystallogr., 1981, B37, 1914 CrossRef CAS
- H. Zhao, J.-X. Liu, C. Yang, S. Yao, H.-Y. Su, Z. Gao, M. Dong, J. Wang, A. I. Rykov, J. Wang, Y. Hou, W.-X. Li and D. Ma, CCS Chem., 2020, 2, 2712 Search PubMed
- A. S. Teja and P.-Y. Koh, Prog. Cryst. Growth Charact., 2009, 55, 22 CrossRef CAS
- D. Fruchart, P. Chaudouet, R. Fruchart, A. Rouault and J. P. Senateur, J. Solid State Chem., 1984, 51, 246 CrossRef CAS
- Y. Xu, M. Yamazaki and P. Villars, Jpn. J. Appl. Phys., 2011, 50, 11RH02 CrossRef
- A. I. Gubanov, Phys. Solid State, 1960, 2, 468 Search PubMed
- G. S. Cargill, AIP Conf. Proc., 1975, 24, 138 CrossRef
- G. F. Goya, F. C. Fonseca, R. F. Jardim, R. Muccillo, N. L. V. Carreño, E. Longo and E. R. Leite, J. Appl. Phys., 2003, 93, 6531 CrossRef CAS
- I. M. L. Billas, J. A. Becker, A. Chatelain and W. A. de Heer, Phys. Rev. Lett., 1993, 71, 4067 CrossRef CAS PubMed
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770 CrossRef CAS
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS PubMed
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Methods, UV-Vis spectra, TEM, SAFS, magnetic data, and PXRD. See DOI: 10.1039/d1ra09191c |
| This journal is © The Royal Society of Chemistry 2022 |