
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkali metal decorated C60 fullerenes as promising materials for delivery of the 5-fluorouracil anticancer drug: a DFT approach†
Mehdi D. Esrafili *a and
Adnan Ali Khanbc
*a and
Adnan Ali Khanbc
aDepartment of Chemistry, Faculty of Basic Sciences, University of Maragheh, P. O. Box 55136-553, Maragheh, Iran. E-mail: esrafili@maragheh.ac.ir
bCentre for Computational Materials Science, University of Malakand, Chakdara, Pakistan
cDepartment of Chemistry, University of Malakand, Chakdara, Pakistan
First published on 1st February 2022
Abstract
The development of effective drug delivery vehicles is essential for the targeted administration and/or controlled release of drugs. Using first-principles calculations, the potential of alkali metal (AM = Li, Na, and K) decorated C60 fullerenes for delivery of 5-fluorouracil (5FU) is explored. The adsorption energies of the 5FU on a single AM atom decorated C60 are −19.33, −16.58, and −14.07 kcal mol−1 for AM = Li, Na, and K, respectively. The results, on the other hand, show that up to 12 Li and 6 Na or K atoms can be anchored on the exterior surface of the C60 fullerene simultaneously, each of which can interact with a 5FU molecule. Because of the moderate adsorption energies and charge-transfer values, the 5FU can be simply separated from the fullerene at ambient temperature. Furthermore, the results show that the 5FU may be easily protonated in the target cancerous tissues, which facilitates the release of the drug from the fullerene. The inclusion of solvent effects tends to decrease the 5FU adsorption energies in all 5FU-fullerene complexes. This is the first report on the high capability of AM decorated fullerenes for delivery of multiple 5FU molecules utilizing a C60 host molecule.
1. Introduction
It is generally accepted that the mode of administration and dose of a drug have a significant impact on its efficacy. By discovering a variety of drug delivery systems (DDSs), it is now feasible to better regulate the pharmacokinetics, pharmacodynamics, toxicity, immunogenicity, and efficacy of medications.1–3 DDSs are developed technologies that allow for the targeted administration and/or controlled release of therapeutic drugs.4,5 In recent decades, DDSs based on nanotechnology have attracted substantial attention due to their essential role in improving the pharmacological properties and bioavailability of different medicines.6–9 Drugs can be adsorbed or conjugated onto the surface of nanoparticles, encapsulated in the core, or dissolved inside the particle matrix in nanodelivery systems.10,11 By incorporating targeting moieties onto the particle surface, drug-loaded nanoparticles may be further tailored to a specific disease site. DDSs based on nanostructures offer several advantages, including increased drug stability and water solubility, extended cycle duration, improved absorption rate of target cells or tissues, and reduced enzyme degradation, all of which improve drug safety and efficacy.12–14Nanotechnology has been proven to bridge the gap between biological and physical sciences by employing nanostructures in a variety of scientific areas such as drug delivery and nanomedicine.15,16 Carbon nanostructures have inspired significant scientific interest in recent decades due to their unusual and extraordinary properties in comparison to typical bulk materials. They have been used in a variety of disciplines, including electronics, sensing, catalysis, gas storage and the environment.17–24 The high surface-to-volume ratio, superior mechanical and thermal stability, and, most importantly, the ease of functionalization have made these materials an attractive candidate for drug delivery by encapsulating or attaching drugs to their exterior surfaces.6,25–27 Furthermore, the small size of these systems allows them to move more easily throughout the human body than larger materials. Drug delivery using carbon nanostructures has been widely investigated as a possible strategy for reducing toxicity and side effects, as well as improving therapeutic efficacy in cancer treatment.28–30 For example, extensive experimental31–33 and theoretical studies34–36 have revealed that different carbon nanostructures, such as carbon nanotubes, fullerenes, and graphene, have a considerable advantage in the treatment of various diseases due to their high drug loading capacity, long circulation time, and short desorption times. Moreover, density functional theory (DFT) studies showed that the addition of impurities by chemical doping may offer additional active sites to establish intermolecular interactions between the drug and the nanocarrier, and noncovalent interactions were the primary drivers for drug loading and release.37–39 The functionalization of carbon nanostructures with various metal atoms has been also utilized to trigger and regulate drug loading/releasing in order to adsorption properties of drugs. For example, a recent quantum mechanics investigation based on DFT calculations40 have revealed that doping C60 fullerene with a B atom can transform this inert material into an active and promising material for delivering aspirin molecules in a water medium.
5-Fluorouracil (5FU) is an antimetabolite chemotherapeutic medication that is widely used to treat colorectal, breast, head and neck, pancreatic, and stomach cancers.41–43 Because 5FU is a water-soluble medication, it is administrated intravenously. However, the emergence of drug resistance is a significant restriction in the therapeutic use of 5FU. For example, because of its poor bioavailability, 5FU cure rates for advanced colorectal cancer are fewer than 15%.42 Furthermore, 5FU causes significant toxicological damage to the gastrointestinal system and blood factors, as well as neurological, dermatological, and cardiological responses.41,44 Thus, potential DDSs for 5FU must be developed in order to provide a greater therapeutic impact with less adverse effects. For example, previous DFT studies investigated the feasibility of different nanostructures for 5FU adsorption and delivery, such as carbon fullerenes and nanotubes or their BN analogues.45–47 However, one important weakness of these DFT works is that they only investigate the adsorption of a single 5FU molecule. Given the high surface-to-volume ratio of nanostructures, determining the maximum capacity of loading the 5FU over these systems would be very interesting.
The drug loading process is a very important component in DDSs that impacts therapeutic efficiency. It is commonly accepted that DDSs are based on reversible interactions between nanocarrier and drug, as such interactions facilitate drug release to the target cell.48,49 Noncovalent interactions like as van der Waals contacts, electrostatic interactions, hydrogen bonding, and π–π staking interactions are considered to be driving forces in drug delivery via nanostructures.50,51 Noncovalent interactions between drug and nanocarrier not only preserve the drug's purity but also improves therapeutic efficacy by boosting cellular uptake. As a result, investigating the nature of the drug–nanocarrier interaction and the mutual effect of the drug and nanocarrier is critical for the design and development of novel DDSs.
The main objective of this study is to investigate the potential of alkali metal decorated C60 fullerenes (AM/C60; AM = Li, Na, K) as a viable drug delivery vehicle for the 5FU drug. Dispersion-corrected DFT calculations are conducted to investigate the adsorption energies, charge transfers, and electronic structure of 5FU molecules adsorbed onto AM/C60 systems. The solvation effects on the adsorption energies of 5FU molecules are thoroughly studied. The results show that a C60 fullerene may be stably decorated with 12 Li and 6 Na or K atoms, and that each of these AM atoms can carry a 5FU molecule. The short desorption times and modest adsorption energies obtained indicate that AM/C60 fullerenes are ideal systems for the delivery of 5FU molecules.
2. Computational aspects
AM/C60 fullerenes were prepared by depositing AM atoms on the outer surface of pristine C60. The dispersion-corrected PBE-D2 (ref. 52 and 53) density functional was used in conjunction with a double numerical plus polarization function (DNP) basis set to minimize the structures. During the geometry optimizations, all atoms were free to move. A Fermi smearing of 0.005 Ha to the orbital occupancy was set to improve computational performance. All DFT calculations were performed within a cubic box with a = b = c = 30 Å. The solvent effects were included by the conductor-like screening model (COSMO)54 method. All the DFT calculations were conducted by the DMol.55,56To evaluate the stability of AM/C60 complexes, the adsorption energies of the AM atoms were calculated using the following equation:
| Eads (AM) = 1/n[Edecorated − EC60 – (n × EAM)] | (1) |
| Eads = 1/m[Ecomplex − Edecorated – (m × E5FU)] | (2) |
To obtain the degree of electron density redistribution (EDR) caused by the formation of 5FU–fullerene complexes, the following equation was used:
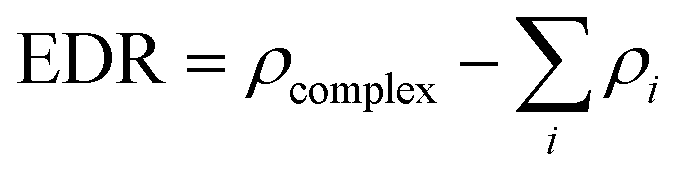 | (3) |
3. Results and discussion
3.1. Pristine and AM decorated C60 fullerenes
Previous studies21,57–61 have extensively investigated the geometry and electronic structure properties of pure C60. It consists of twelve pentagons and twenty hexagons, with two types of C–C bonds: one shared by two neighboring hexagons (R66) and the other by one pentagon and one hexagon (R65). The optimized R66 and R65 bond distances in C60 are 1.40 and 1.45 Å, respectively, which differ from the C–C bond length in graphene (≈1.42 Å).62 The charge density analysis reveals that the electronic charge is uniformly distributed throughout the C60, and there is no dipole moment in this material due to its high symmetry. Though there are plentiful of sites for adsorption of AM atoms, our DFT calculations show that the above of the center of pentagon and hexagon rings are the most active adsorption sites for the AM atoms. As a result, we initially put a single AM atom on the pentagon or hexagon rings of C60 to get the most stable geometry of AM/C60. In the case of Li atom, it is revealed that the calculated adsorption energies on the center of a pentagon or a hexagon site differ by only 0.8 kcal mol−1. In accordance with earlier studies,63 the adsorption of the Li atom on the center of a hexagon is slightly more favorable. Likewise, the center of the hexagonal face is preferably the lowest energy binding site for the Na and K atoms (Fig. 1). From Table 1, one can see that the adsorption energies decrease as the size of the AM increases. The Eads values of the Li, Na and K atom adsorbed on the C60 fullerene are calculated to be −43.58, −36.86 and −36.22 kcal mol−1, respectively, which are in good agreement with those of other studies.63–65 This also implies that the interaction between the C60 fullerene and the AM atoms is rather strong. As a result, the C–C bond distances of the interacting hexagon ring are slightly elongated after the adsorption of the AM atom. Taking the Li/C60 as an example, the R65 and R66 distances of the fullerene increase by 0.01 and 0.02 Å, respectively, upon the Li adsorption. According to the Hirshfeld analysis, the charge on the AM atoms increases as their size grows. Though this observation is compatible with the reactivity of these atoms, it is not entirely consistent with the order of their adsorption energies. A similar discrepancy in charge transfer and adsorption energies of the AM atoms has been reported in several related investigations,64 as well as for AM/B80 complexes.66 It is most likely due to a more appropriate back-bonding interaction between the empty 2p states of the Li and the filled 2p states of the surrounding C atoms, which strengthens the Li–C interaction at the price of a smaller positive charge induced on the Li atom. A similar, albeit weaker, mechanism is also predicted for the Na/C60. Nonetheless, the presence of a significant positive charge on the AM atoms suggests a strong propensity for interaction with electron-rich species. The EDR analysis is used to gain a better understanding of the mutual polarization of the AM and C60 moieties during the formation of AM/C60 complexes. The approach of the AM atom to the C60 produces a significant electron density loss region on the AM atom, indicating its polarization in the presence of C60 (see Fig. 1). Likewise, the presence of the AM atom results in an area of increased electron density on the C60. It is worth noting that the emergence of such electron gain/loss areas can contribute to increased electrostatic attraction between the AM atom and C60, and hence the stability of the AM/C60 complex. Furthermore, the degree of the charge density difference in the AM/C60 systems is affected by the nature of the AM atom and becomes more significant as the size of the AM atom increases (Li < Na < K). | ||
| Fig. 1 The relaxed structure (above) and EDR isosurface (below, isovalue = 0.01 au) of (a) Li/C60, (b) Na/C60 and (c) K/C60. In the EDR isosurfaces, the gain and loss electron density areas are shown in green and yellow, respectively. | ||
| Species | Eads | QAM | HOMO(SOMO)–LUMO gap |
|---|---|---|---|
| Li/C60 | −43.58 | 0.47 | 1.48 |
| Na/C60 | −36.86 | 0.62 | 1.50 |
| K/C60 | −36.22 | 0.70 | 1.51 |
| Li12/C60 | −41.05 | 0.33 | 1.05 |
| Na6/C60 | −31.27 | 0.51 | 0.91 |
| K6/C60 | −30.90 | 0.59 | 0.89 |
The number of AM atoms that a C60 molecule may adsorb is a crucial topic here. This is significant because it enables maximum capacity for delivering 5FU molecules using only one C60. Therefore, additional AM atoms are added to the C60 in order to find the maximum coverage of these atoms. It should be noted that the cohesive energies of bulk Li, Na, and K are 37.8, 25.6, and 21.6 kcal mol−1,67 respectively, and thus the absolute value of average adsorption energy should be greater than these cohesive energies to avoid the “clustering” of AM atoms. Fig. 2 depicts the maximum number of AM atoms that C60 fullerene can hold before clustering occurs. Consistent with previous DFT calculations,68,69 it is found that when additional Li atoms are added to the fullerene, the pentagonal face definitely becomes the favored site. Indeed, as seen in Fig. S1 of the ESI,† the energy barrier for the migration of the Li atom from the hexagon ring into the neighboring pentagon is about 10 kcal mol−1. As a result, a maximum of 12 Li atoms can be adsorbed on pure C60 with an adsorption energy of −41.05 eV per Li atom (Table 1). However, in the case of Na and K, a maximum of 6 atoms may be attached to the C60 simultaneously, with average Eads values of −31.27 and −30.90 kcal mol−1, respectively. It is worth noting that the average adsorption energies of the AM atoms in Li12/C60, Na6/C60, or K6/C60 are less than those of a single AM atom adsorbed structures, which can be attributed to a decrease in the electron affinity of the C60 fullerene with stepwise addition of the AM atom. Meanwhile, the positive charge on the AM atoms decrease when more AM atoms are deposited over the C60 fullerene.
 | ||
| Fig. 2 The optimized structures of (a) Li12/C60, (b) Na6/C60 and (c) K6/C60 fullerenes. | ||
We also performed ab initio molecular dynamics (MD) simulations at T = 300 K for 5000 fs to assess the stability of the obtained Li12/C60, Na6/C60, and K6/C60 systems. As seen in Fig. S2 of the ESI,† the AM decorated fullerenes remain stable following the MD simulations. The AM atoms, in particular, stay on the C60 and no clustering occurs, indicating that these structures are stable at ambient temperatures. It is worth mentioning that the calculated energy gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital (HOMO–LUMO gap) of pure C60 is 1.64 eV. The addition of AM atoms changes the electronic structure of the C60 and reduces its HOMO–LUMO gap. Because AM atoms have an unpaired electron, the formation of AM/C60 complexes results in the development of a singly occupied molecular orbital (SOMO) around the Fermi level. As shown in Table 1, the energy gap between the SOMO and LUMO of Li/C60, Na/C60 and K/C60 is 1.48, 1.50, and 1.51 eV, which is 0.16, 0.14, and 0.13 eV less than the HOMO–LUMO gap of C60. Given that a narrow HOMO(SOMO)–LUMO gap indicates low kinetic stability,70 this finding implies that AM/C60 complexes are more chemically reactive than pure C60.
3.2. Adsorption of 5FU on pristine and AM decorated C60
To evaluate the adsorption behavior of AM-decorated C60 fullerenes, a 5FU molecule is allowed to approach the exterior surfaces of these complexes. Consider the singly adsorbed AM complexes first. The optimized most stable structures of 5FU molecule absorbed on Li/C60, Na/C60, and K/C60 are shown in Fig. 3. Table 2 displays the adsorption energies and charge-transfer values of the 5FU molecule. According to our DFT calculations, the 5FU prefers to attach to the decorated AM atom from its O site. The binding distance between the drug molecule and AM/C60 is determined to be 1.82, 2.19, and 2.60 Å for AM = Li, Na, and K, respectively. The adsorption energy of the 5FU is −19.33, −16.58 and −14.07 kcal mol−1 on Li/C60, Na/C60 and K/C60, respectively, which falls within the range of chemisorption. The Eads values of the 5FU molecule decrease as the size of the AM atom increases, which is inconsistent with the degree of polarization and the Hirshfeld charge on these atoms. It should be noticed that the calculated Eads values of the 5FU are more negative than those calculated on other surfaces such as B24N24 (−11.90 kcal mol−1)45 and Ge-doped BN nanotubes (−12.20 kcal mol−1).47 The 5FU molecule serves as the electron donor in all optimized complexes, with a charge-transfer value of 0.20, 0.13, and 0.08 electrons for AM = Li, Na, and K, respectively. These findings clearly show that, in addition to electrostatic attraction between positively charged AM atom and negatively charged O atoms of 5FU, orbital interactions also play an essential role in the formation of these complexes. In fact, the adsorption energies are well correlated with the amount of the charge-transfer values between the 5FU and the AM/C60 fullerenes, suggesting that the charge-transfer is more essential for the stability of these complexes than the electrostatic interactions. This is confirmed further by the PDOS analysis, which shows that there is substantial overlap between the empty p orbital of the AM atom and the O-2p orbitals of the drug molecule around the Fermi level. Note that compared to Na/C60 and K/C60, the degree of such orbital overlap is more important in Li/C60. | ||
| Fig. 3 The relaxed structure (left), corresponding EDR isosurface (middle, isovalue = 0.01 au) and PDOS plots (right) of the 5FU molecule adsorbed on (a) Li/C60, (b) Na/C60 and (c) K/C60. All bond distances are in Å. In the EDR isosurfaces, the gain and loss electron density areas are shown in green and yellow, respectively. The Fermi energy is set to zero in the PDOS plots. | ||
| Species | Eads | QCT | τ |
|---|---|---|---|
| Li/C60 | −19.33 | 0.20 | 147.50 |
| Na/C60 | −16.58 | 0.13 | 1.42 |
| K/C60 | −14.07 | 0.08 | 0.02 |
| Li12/C60 | −16.06 | 0.16 | 0.59 |
| Na6/C60 | −14.69 | 0.09 | 0.06 |
| K6/C60 | −12.34 | 0.06 | <0.01 |
To get additional information about the charge density rearrangement upon the adsorption of 5FU on AM/C60 systems, the EDR analysis is performed. Fig. 3 shows the obtained isosurfaces, with the gain and loss electron density areas shown in green and yellow, respectively. As seen, the adsorption of 5FU is associated with a considerable charge density rearrangement around the interacting atoms, suggesting that the mutual polarization of 5FU and AM/C60 subunits upon the complex formation. In particular, the appearance of a large electron density loss region on the 5FU confirms that this molecule serves as the electron donor. Note also that the amount of the charge density rearrangement is not same for these complexes; the stronger interaction between the 5FU and Li/C60 induces a more substantial rearrangement of the electron density. That is to say, the EDR analysis can be used as a suitable tool for assessing the adsorption strength between the 5FU molecule and AM/C60 fullerenes.
Using the above information, we placed a 5FU molecule on each AM atom of Li12/C60, Na6/C60, and K6/C60 and relaxed the resulting complexes. Fig. 4 illustrates the optimized structures of the derived complexes, and Table 2 lists the adsorption energies and charge-transfer values. The calculated adsorption energies per 5FU are −16.06, −14.69, and −12.34 kcal mol−1, indicating that the AM atoms in these systems can efficiently adsorb 5FU molecules. Due to repulsion effects between the adsorbed 5FU molecules, these average Eads values are slightly lower than the Eads value on the single AM decorated C60.
 | ||
| Fig. 4 (a) The optimized geometries of 5FU molecules adsorbed on (a) Li12/C60, (b) Na6/C60 and K6/C60 fullerenes. | ||
3.3. Drug release
Although the drug molecule must be stably adsorbed on the host material for effective drug delivery, this adsorption must not be so strong that it makes drug desorption difficult. As previously mentioned, the calculated adsorption energies of 5FU on the AM decorated C60 fullerenes range between −12.34 and −19.33 kcal mol−1, which is neither too strong nor too weak. That is, the decorated systems may be able to desorb the 5FU molecule due to the modest adsorption energies. However, as the ambient temperature rises, the 5FU bonding with the decorated AM atoms is expected to weaken owing to entropic effects. Hence, to more accurately assess this, the desorption time (τ) for the 5FU molecule is calculated using the following equation:72
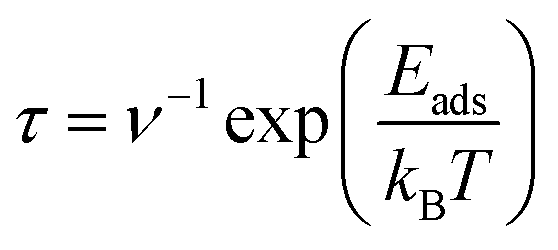 | (4) |
The 5FU, on the other hand, is intended to be delivered into cancerous cells. However, it is generally known that cancerous cells have a pH value less than that of normal cells (pH < 7).71 As a result, it is reasonable to anticipate that when the 5FU reaches the target cancer cell, it will react with the proton ion. To demonstrate this, we added a proton to different regions of the adsorbed 5FU molecule. Following careful geometry optimization, it is found that the proton is attached to the bridge O atom as a result of the larger negative charge concentrated on this atom (Fig. 5). As can be seen, the addition of the proton increases the binding distance of the 5FU relative to its neutral form. The calculated adsorption energies of protonated 5FU on Li/C60, Na/C60, and K/C60 fullerenes are −7.75, −5.64, and −3.90 kcal mol−1, respectively. Clearly, these Eads values are much smaller than those of neutral 5FU, indicating that the drug molecule can be readily separated from the fullerene when it reaches the targeted cancerous cell.
 | ||
| Fig. 5 The adsorption of protonated 5FU on (a) Li/C60, (b) Na/C60 and (c) K/C60. All bond distances are in Å. | ||
3.4. Solvent effects
The solvation energies (Esolv) of the 5FU complexes with the AM anchored C60 fullerenes are calculated to determine drug solubility in the aqueous environment. The solvation energy is defined here as the difference between the total energy of the complexes in the gas and aqueous phases. Moreover, the adsorption energies and related charge-transfer values owing to the adsorption of 5FU on the decorated fullerenes are obtained in order to further analyze the solvation effects. Table 3 summarizes the obtained solvation energies, adsorption energies, charge-transfer and desorption time values per 5FU molecule. It is seen that the Esolv values are all negative, implying that the solvation of the derived complexes tends to stabilize these systems. As the size of the AM atom gets bigger, the solvation effects become more significant. When the adsorption energy values are considered, it is evident that the presence of water molecules tends to decrease the adsorption strength of the 5FU. This is because the solvation energies of bare AM/C60 fullerenes are larger than those of 5FU-AM/C60 adsorbed complexes due to the more polar AM-C bonds. The Hirshfeld study supports this conclusion, with calculated charge-transfer values in the aqueous medium are smaller than those in the gas phase. According to eqn (4), the incorporation of solvation effects can reduce the desorption time of 5FU molecules from AM decorated fullerenes. Hence, it may be inferred that the release of 5FU can be promoted inside the aqueous solution.| Species | Esolv | Eads | QCT | τ |
|---|---|---|---|---|
| Li/C60 | −30.70 | −13.73 | 0.14 | 1.17 × 10−2 |
| Na/C60 | −35.20 | −9.24 | 0.11 | 5.97 × 10−6 |
| K/C60 | −35.90 | −8.50 | 0.05 | 1.71 × 10−6 |
| Li12/C60 | −11.74 | −10.85 | 0.11 | 9.06 × 10−5 |
| Na6/C60 | −23.87 | −8.10 | 0.05 | 8.71 × 10−7 |
| K6/C60 | −24.58 | −6.88 | 0.03 | 1.11 × 10−7 |
4. Conclusions
In summary, a dispersion-corrected DFT investigation of the capability of AM decorated C60 fullerenes (AM/C60; AM = Li, Na, and K) was conducted. It was found that the AM atom may be decorated stably on the hexagon rings of C60 fullerene. The MD simulations revealed that the AM-decorated fullerenes are thermally stable. The decorated AM atoms in AM/C60 can serve as active sites for the adsorption of 5FU molecules. Our findings indicated that Li decorated C60 fullerene can adsorb up to 12 5FU molecules with an average adsorption energy of −16.06 kcal mol−1. In the case of the Na and K decorated systems, the results showed that a maximum of 6 Na or K may bind to the C60, each of which can adsorb one 5FU molecule. The Eads values for the adsorption of a protonated 5FU molecule are significantly smaller than those for a neutral one, suggesting that the drug molecule can be easily detached from the fullerene when it reaches the cancerous cell. The addition of solvent effects reduces the strength of 5FU-fullerene complexes, suggesting that the drug release process might be assisted by solvent molecules. This work demonstrated for the first time how the decorating of C60 fullerene with AM atoms may significantly improve the capability of this system for delivering multiple 5FU molecules.Conflicts of interest
There are no conflicts to declare.References
- V. V. Ranade and J. B. Cannon, Drug delivery systems, CRC Press, 2011 Search PubMed.
- C. Li, J. Wang, Y. Wang, H. Gao, G. Wei, Y. Huang, H. Yu, Y. Gan, Y. Wang and L. Mei, Acta Pharm. Sin. B, 2019, 9, 1145–1162 CrossRef PubMed.
- M. Naeem, U. A. Awan, F. Subhan, J. Cao, S. P. Hlaing, J. Lee, E. Im, Y. Jung and J.-W. Yoo, Arch. Pharmacal Res., 2020, 43, 153–169 CrossRef CAS PubMed.
- Y. K. Sung and S. W. Kim, Biomater. Res., 2020, 24, 1–12 CrossRef PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 1–19 CrossRef CAS PubMed.
- Q. Guo, X.-t. Shen, Y.-y. Li and S.-q. Xu, Curr. Med. Sci., 2017, 37, 635–641 CrossRef CAS PubMed.
- Z. Li, A. L. B. de Barros, D. C. F. Soares, S. N. Moss and L. Alisaraie, Int. J. Pharm., 2017, 524, 41–54 CrossRef CAS PubMed.
- G. Karthivashan, P. Ganesan, S.-Y. Park, J.-S. Kim and D.-K. Choi, Drug Delivery, 2018, 25, 307–320 CrossRef CAS PubMed.
- J. Kaur, G. S. Gill and K. Jeet, Characterization and Biology of Nanomaterials for Drug Delivery, Elsevier, 2019, pp. 113–135 Search PubMed.
- N. Zahin, R. Anwar, D. Tewari, M. Kabir, A. Sajid, B. Mathew, M. Uddin, L. Aleya and M. M. Abdel-Daim, Environ. Sci. Pollut. Res., 2020, 27, 19151–19168 CrossRef CAS PubMed.
- X. Chen, R. Tong, Z. Shi, B. Yang, H. Liu, S. Ding, X. Wang, Q. Lei, J. Wu and W. Fang, ACS Appl. Mater. Interfaces, 2018, 10, 2328–2337 CrossRef CAS PubMed.
- C. J. M. Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. G. Rodríguez, R. Á. Román, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef PubMed.
- R. Kanwar, J. Rathee, D. B. Salunke and S. K. Mehta, ACS Omega, 2019, 4, 8804–8815 CrossRef CAS PubMed.
- M. Saeedi, M. Eslamifar, K. Khezri and S. M. Dizaj, Biomed. Pharmacother., 2019, 111, 666–675 CrossRef CAS PubMed.
- M. Mir, S. Ishtiaq, S. Rabia, M. Khatoon, A. Zeb, G. M. Khan, A. ur Rehman and F. ud Din, Nanoscale Res. Lett., 2017, 12, 1–16 CrossRef PubMed.
- J.-F. Hsu, S.-M. Chu, C.-C. Liao, C.-J. Wang, Y.-S. Wang, M.-Y. Lai, H.-C. Wang, H.-R. Huang and M.-H. Tsai, Cancers, 2021, 13, 195 CrossRef CAS PubMed.
- N. Martín, T. Da Ros and J.-F. Nierengarten, J. Mater. Chem. B, 2017, 5, 6425–6427 RSC.
- H. G. Shiraz and M. G. Shiraz, Int. J. Hydrogen Energy, 2017, 42, 11528–11533 CrossRef CAS.
- Y. Tang, J. Zhou, Z. Shen, W. Chen, C. Li and X. Dai, RSC Adv., 2016, 6, 93985–93996 RSC.
- E. Ashori, F. Nazari and F. Illas, Phys. Chem. Chem. Phys., 2017, 19, 3201–3213 RSC.
- M. D. Esrafili and H. Janebi, Mol. Phys., 2020, 118, e1631495 CrossRef.
- W. Huang, M. Shi, H. Song, Q. Wu, X. Huang, L. Bi, Z. Yang and Y. Wang, Chem. Phys. Lett., 2020, 758, 137940 CrossRef CAS.
- M. D. Esrafili, A. A. Khan and P. Mousavian, RSC Adv., 2021, 11, 22598–22610 RSC.
- E. Shakerzadeh and L. Azizinia, Chem. Phys. Lett., 2021, 764, 138241 CrossRef CAS.
- S. Kraszewski, E. Duverger, C. Ramseyer and F. Picaud, J. Chem. Phys., 2013, 139, 174704 CrossRef CAS PubMed.
- E. Alipour, F. Alimohammady, A. Yumashev and A. Maseleno, J. Mol. Model., 2020, 26, 1–8 CrossRef PubMed.
- M. Kumar and K. Raza, Pharm. Nanotechnol., 2017, 5, 169–179 CAS.
- R. Maleki, A. Khoshoei, E. Ghasemy and A. Rashidi, J. Mol. Graphics Modell., 2020, 100, 107660 CrossRef CAS PubMed.
- J. Saleem, L. Wang and C. Chen, Adv. Healthcare Mater., 2018, 7, 1800525 CrossRef PubMed.
- F. Niazvand, P. R. Wagh, E. Khazraei, M. B. Dastjerdi, C. Patil and I. A. Najar, J. Compos. Compd., 2021, 3, 140–151 Search PubMed.
- S. Augustine, J. Singh, M. Srivastava, M. Sharma, A. Das and B. D. Malhotra, Biomater. Sci., 2017, 5, 901–952 RSC.
- B. Hosnedlova, M. Kepinska, C. Fernandez, Q. Peng, B. Ruttkay-Nedecky, H. Milnerowicz and R. Kizek, Chem. Rec., 2019, 19, 502–522 CrossRef CAS PubMed.
- M. Serda, G. Szewczyk, O. Krzysztyńska-Kuleta, J. Korzuch, M. Dulski, R. Musioł and T. Sarna, ACS Biomater. Sci. Eng., 2020, 6, 5930–5940 CrossRef CAS PubMed.
- A. Hosseinian, E. Vessally, S. Yahyaei, L. Edjlali and A. Bekhradnia, J. Cluster Sci., 2017, 28, 2681–2692 CrossRef CAS.
- C. Parlak and Ö. Alver, J. Mol. Struct., 2019, 1184, 110–113 CrossRef CAS.
- H. Xu, X. Tu, G. Fan, Q. Wang, X. Wang and X. Chu, J. Mol. Liq., 2020, 318, 114315 CrossRef CAS.
- D. Shahabi and H. Tavakol, Appl. Surf. Sci., 2017, 420, 267–275 CrossRef CAS.
- R. Bagheri, M. Babazadeh, E. Vessally, M. Es' haghi and A. Bekhradnia, Inorg. Chem. Commun., 2018, 90, 8–14 CrossRef CAS.
- I. Ravaei, M. Haghighat and S. Azami, Appl. Surf. Sci., 2019, 469, 103–112 CrossRef CAS.
- F. Nattagh, S. Hosseini and M. D. Esrafili, J. Mol. Liq., 2021, 117459 CrossRef CAS.
- J. D. Sara, J. Kaur, R. Khodadadi, M. Rehman, R. Lobo, S. Chakrabarti, J. Herrmann, A. Lerman and A. Grothey, Ther. Adv. Med. Oncol., 2018, 10, 1758835918780140 Search PubMed.
- K. R. Stefaniak, C. C. Epley, J. J. Novak, M. L. McAndrew, H. D. Cornell, J. Zhu, D. K. McDaniel, J. L. Davis, I. C. Allen and A. J. Morris, Chem. Commun., 2018, 54, 7617–7620 RSC.
- P. Alvarez, J. A. Marchal, H. Boulaiz, E. Carrillo, C. Vélez, F. Rodriguez-Serrano, C. Melguizo, J. Prados, R. Madeddu and A. Aranega, Expert Opin. Ther. Pat., 2012, 22, 107–123 CrossRef CAS PubMed.
- E. Carrillo, S. A. Navarro, A. Ramírez, M. Á. García, C. Griñán-Lisón, M. Perán and J. A. Marchal, Expert Opin. Ther. Pat., 2015, 25, 1131–1144 CrossRef CAS PubMed.
- M. K. Hazrati, Z. Javanshir and Z. Bagheri, J. Mol. Graphics Modell., 2017, 77, 17–24 CrossRef CAS PubMed.
- M. Vatanparast and Z. Shariatinia, J. Fluorine Chem., 2018, 211, 81–93 CrossRef CAS.
- A. Soltani, M. T. Baei, E. T. Lemeski, S. Kaveh and H. Balakheyli, J. Phys. Chem. Solids, 2015, 86, 57–64 CrossRef CAS.
- M. K. Hazrati, Z. Bagheri and A. Bodaghi, Phys. E, 2017, 89, 72–76 CrossRef CAS.
- U. Srimathi, V. Nagarajan and R. Chandiramouli, J. Mol. Liq., 2018, 265, 199–207 CrossRef CAS.
- J. Ding, L. Chen, C. Xiao, L. Chen, X. Zhuang and X. Chen, Chem. Commun., 2014, 50, 11274–11290 RSC.
- T. Doane and C. Burda, Adv. Drug Delivery Rev., 2013, 65, 607–621 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- A. E. Mahdy, Mol. Phys., 2015, 113, 3531–3544 CrossRef.
- M. K. Hazrati and N. L. Hadipour, Phys. Lett. A, 2016, 380, 937–941 CrossRef CAS.
- A. A. Khan, I. Ahmad and R. Ahmad, Chem. Phys. Lett., 2020, 742, 137155 CrossRef CAS.
- M. D. Esrafili and S. Heidari, ChemistrySelect, 2019, 4, 4308–4315 CrossRef CAS.
- M. D. Esrafili and S. Heydari, ChemistrySelect, 2019, 4, 2267–2274 CrossRef CAS.
- M. D. Esrafili and P. Mousavian, Appl. Surf. Sci., 2018, 455, 808–814 CrossRef CAS.
- A. Omidvar and A. Mohajeri, Int. J. Hydrogen Energy, 2017, 42, 12327–12338 CrossRef CAS.
- K. Chandrakumar and S. K. Ghosh, Nano Lett., 2008, 8, 13–19 CrossRef CAS PubMed.
- D. S. De, J. A. Flores-Livas, S. Saha, L. Genovese and S. Goedecker, Carbon, 2018, 129, 847–853 CrossRef CAS.
- Y. Li, G. Zhou, J. Li, B.-L. Gu and W. Duan, J. Phys. Chem. C, 2008, 112, 19268–19271 CrossRef CAS.
- A. M. Halpern, J. Chem. Educ., 2012, 89, 592–597 CrossRef CAS.
- Q. Wang and P. Jena, J. Phys. Chem. Lett., 2012, 3, 1084–1088 CrossRef CAS PubMed.
- Q. Sun, P. Jena, Q. Wang and M. Marquez, J. Am. Chem. Soc., 2006, 128, 9741–9745 CrossRef CAS PubMed.
- J.-i. Aihara, J. Phys. Chem. A, 1999, 103, 7487–7495 CrossRef CAS.
- G. Hao, Z. P. Xu and L. Li, RSC Adv., 2018, 8, 22182–22192 RSC.
- J. Beheshtian, A. Ahmadi and Z. Bagheri, Sens. Actuators, B, 2012, 171, 846–852 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra09153k |
| This journal is © The Royal Society of Chemistry 2022 |